
Mn(II)-catalyzed switchable synthesis of P(O)–C and P(O)–O bonds from alcohols and diarylphosphine oxides via phospha-Brook rearrangement
Hirak Jyoti
Phukan
,
Arup
Samanta
,
Kailash
Mohar
,
Avijit
Mondal
,
Rinku
Dihingia
and
Dipankar
Srimani
 *
*
Department of Chemistry, Indian Institute of Technology-Guwahati, Kamrup, Assam 781039, India. E-mail: dsrimani@iitg.ac.in
First published on 14th April 2026
Abstract
We describe a Mn(II)-catalyzed, biocompatible, and economical strategy for selective and efficient synthesis of both P(O)–O and P(O)–C compounds via a dehydrogenative phospha-Brook rearrangement, with broad substrate scope and mechanistic validation from control and kinetic studies.
Organophosphorus compounds containing P(O)–O and P(O)–C bonds constitute an important class of phosphorus-based molecules. P(O)–O-containing compounds are widely used as herbicides and pesticides in agrochemistry,1 as well as flame retardants and plasticizers in industrial applications.2 They also play essential structural and chemical roles in DNA and RNA.3 In parallel, P(O)–C-bonded organophosphorus compounds are significant in medicinal chemistry as drug molecules and bioactive agents,4 and they serve as key synthetic intermediates and ligands in coordination and catalytic chemistry.5 Traditionally, organophosphorus compounds bearing P(O)–O bonds are synthesized via the Atherton–Todd reaction.6 However, this method suffers from the use of toxic CCl4 as both a halogenating agent and solvent, prompting the development of less toxic alternatives.7 In contrast, P(O)–C compounds are typically prepared through the Michaelis–Arbuzov reaction.8 Despite its importance, this transformation requires harsh conditions, shows limited functional-group tolerance, relies on toxic alkyl halides, and is generally ineffective for unactivated aryl halides. To address these issues, recent developments have been made to replace these toxic halide-based raw chemical materials with more efficient and environmentally friendly approaches for the construction of P(O)–O and P(O)–C bonds from sustainable feedstocks. In this context, the Yamaguchi group reported a palladium-catalyzed deoxygenative coupling of aromatic esters with H–P(O) compounds, providing an efficient route to construct P(O)–C bonds from readily available substrates9 (Scheme 1). Subsequently, the Han group introduced a nickel-catalyzed direct phosphorylation of sp3 C–CN bonds, enabling C–P(O) bond formation through an unconventional bond activation pathway.10 More recently, Han and co-workers further expanded this field by developing an iron-catalyzed strategy for P(O)–C bond formation directly from alcohols.11 H–P(O) compounds have also been efficiently coupled with carboxylic acid12 and aryl halide.13 On the other hand, for constructing P(O)–O bonds, the Chen group introduced a phosphoryl radical-triggered Atherton–Todd-type protocol using air as the radical source and CHCl3 as the halogenating reagent in the phosphorylation of alcohols.14 The Zhang group has developed another protocol to synthesize phosphinates from aldehydes with H–P(O) precursors.15 Other phosphorylating agents, such as P(O)–OH and P(OR)3 compounds, have also been coupled with alcohols to give similar products.16,17
 | ||
| Scheme 1 Schematic approaches of both P(O)–O and P(O)–C compound synthesis. | ||
While green strategies for constructing P(O)–C and P(O)–O bonds have progressed, developing a unified and selective method to access both bond types from the same starting materials would be both intriguing and practically valuable. Therefore, we envisioned a strategy to construct both P(O)–C and P(O)–O bonds from H–P(O) precursors and various alcohols, offering a versatile and efficient route to diverse organophosphorus compounds using a single set of starting materials. As part of our ongoing efforts18 in Mn-catalyzed green transformations,19 we have devised a Mn-catalyzed, switchable approach for the selective formation of P(O)–C and P(O)–O bonds from diverse alcohols and phosphine oxides.
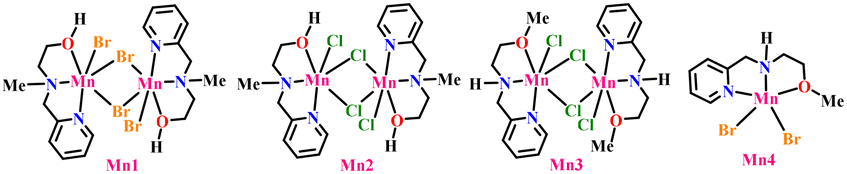
To accomplish this, we began screening using 4-methylbenzyl alcohol and diphenylphosphine oxide as model substrates with the Mn catalyst developed in our laboratory.20 After thorough investigations, we found that when 1a (0.5 mmol) and 2a (1 mmol) were heated at 140 °C in the presence of Cs2CO3 (50 mol%) and 5 mol% of Mn1 complex in 2 mL hexane in a 100 mL sealed tube under an argon atmosphere for 24 h, the reaction resulted in 90% of product 3a and 6% of product 4a (Table 1, entry 1). Again, we ran the reaction in the absence of Mn1 keeping all other reaction parameters the same, and only a trace amount of 3a and 26% of 4a were produced (Table 1, entry 2). Furthermore, amongst the bases of KOtBu, KOH, K2CO3, Na2CO3, CsF and Cs2CO3, Cs2CO3 was found to be the most suitable base under our reaction conditions. When we varied the amount of 1a (1.2 equiv.) and 2a (1 equiv.) from our optimized conditions, the reaction yielded 30% of 3a and 65% of 4a. The reaction was also attempted under similar conditions using the iron catalyst reported by the Han group; however, 4a was not formed, and further details are provided in the SI. Next, we investigated the substrate scope of our optimized conditions for P(O)–C bond formation. Initially, we evaluated various primary benzyl alcohols bearing different substituents at the C-4 position (Scheme 2, 3a–3j). Benzyl alcohols bearing methoxy, CF3, and halide groups underwent the reaction smoothly, yielding the desired products in good amounts, while those with nitro or cyano substituents remained completely silent under the established conditions. The nitro and cyano groups may coordinate to the metal center, poisoning the catalyst and deactivating the catalytic cycle. Similar catalyst poisoning was observed with indole-3-carbinol (3w), leading to decomposition and zero product yield. Benzyl alcohols bearing substituents at either the ortho or para positions were also well-tolerated, giving the desired P(O)–C products in good yields (Scheme 2, 3k–3n, 60–70%).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (in %) :4 (in %) |
| a Reaction conditions: 1a (0.5 mmol), 2a (1 mmol), base (50 mol%), Mn1 (5 mol%, i.e., 10 mol% with respect to the monomeric form of Mn1), under argon, 140 °C (oil bath), reactions were carried out in a 100 mL sealed tube. | ||
| 1 | None | 90:6 |
| 2 | Absence of catalyst | Trace:26 |
| 3 | KOtBu, KOH, K2CO3, and Na2CO3, CsF | — |
| 4 | Cyclohexane, xylene, decaline, THF, toluene | 0:6 |
| 5 | 1.2:1 equiv. of 1a:2a instead of 1:2 |
30:65 |
| 6 | Mn2, Mn3, and Mn4 | 60:5 |
|
||

 | ||
| Scheme 2 Substrate scope. aReaction conditions: 1 (0.5 mmol), 2 (1.0 mmol), Mn1 cat. (5 mol%, i.e., 10 mol% with respect to the monomeric form of Mn1), Cs2CO3 (0.5 equiv.), hexane (2 mL), under argon, 140 °C (oil bath), 24 h; b2 (4 mmol), Mn1 cat. (10 mol%, i.e., 20 mol% with respect to the monomeric form of Mn1, Cs2CO3 (2 equiv.), 36 h; isolated yield, reactions were carried out in a 100 mL sealed tube. | ||
To evaluate steric tolerance, highly substituted benzyl alcohols, such as 2,4,6-trimethoxybenzyl alcohol, were tested and afforded excellent yields, highlighting the reaction's ability to accommodate significant steric bulk. Furthermore, heteroatom-substituted primary alcohols afforded the corresponding products in moderate to good yields. (Scheme 2, 3t–3y, 55–78%). By slightly modifying our optimized conditions, naturally derived fatty alcohols such as stearyl, cetyl, myristyl, lauryl, and capric alcohols were successfully activated, delivering excellent yields (84–90%). It was observed that the product yield diminished progressively as the chain length of the alcohols decreased (3ah–3an). Next, a range of naturally occurring, synthetically useful alcohols were explored under this protocol. The medicinally important alcohol (S)-(−)-perillyl alcohol, widely studied in cancer research, furnished the desired product in 65% yield (3aq, 65%). Other naturally important cyclic and acyclic monoterpenoid-derived alcohols, including menthol, citronellol, nerol, and geraniol, also delivered good to excellent yields under the reaction conditions (3aq–3au, 60–70%). Subsequently, the substrate scope of phosphine oxides was explored, giving excellent results.
During our optimization study, we have observed the formation of phosphinate (4a) via P(O)–O formation in 65% yield together with 30% phosphine oxide (3a). To evaluate the scope, limitations, and selectivity of the reaction toward phosphinate formation, a variety of alcohols were investigated (Table 1, entry 5). Electronically rich para-substituted benzyl alcohols firmly coupled with the H–P(O) to give a good yield of the corresponding phosphinates (Scheme 3, 4a, 4c, and 4d, 60–65%). Pleasingly, halide substituted phosphinates were also generated with moderate to good yields (4e–4g) giving more scope for further derivatization. Consistent with P(O)–C bond formation, nitro- and cyano-substituted benzyl alcohols exhibited similar reactivity under the developed conditions, affording no desired product (4l, 4m). In the case of benzyl alcohol derivatives, moderate selectivity was observed, owing to the concurrent formation of the corresponding phosphine oxide (3) in 15–25% yield. Heteroatom-containing alcohols were moderately activated by the catalyst, affording the corresponding phosphinates (4n, 4o) in moderate yields. Challenging long-chain aliphatic fatty alcohols, as well as acyclic and cyclic branched aliphatic alcohols, smoothly delivered the desired products with excellent yields and selectivity under the modified reaction conditions. A diverse range of secondary phosphine oxides are well tolerated to deliver a good yield of the targeted product. The applicability of the synthesized phosphine oxides was demonstrated by gram-scale synthesis of 3a. Compound 3a was further converted to various organic compounds to showcase the applications (Scheme 4).
 | ||
| Scheme 3 Substrate scope. aReaction conditions: 1 (1.2 equiv.), 2 (1 equiv.), Mn1 cat. (5 mol%, i.e., 10 mol% with respect to the monomeric form of Mn1), Cs2CO3 (50 mol%), hexane (2 mL), 140 °C (oil bath), 24 h, isolated yield, b2 (3 equiv.), Mn1 cat. (10 mol%, i.e., 20 mol% with respect to the monomeric form of Mn1), Cs2CO3 (2 equiv.), 36 h, reactions were carried out in a 100 mL sealed tube. Yield of P(O)–C compounds in parentheses. | ||
 | ||
| Scheme 4 Post synthetic modification. | ||
To gain mechanistic insight, a series of control experiments were conducted (See Section 7. Mechanistic studies in the SI). To assess carbocation involvement,21tert-butanol (9a) was reacted with 2a under the optimized conditions. No desired product was formed, effectively ruling out a carbocation-mediated mechanism. Under Mn-catalysed conditions, dehydrogenation of alcohol to aldehyde was observed. Using 4-methylbenzaldehyde gave 3a (75%) and 4a (13%), supporting the role of the aldehyde as an intermediate. However, aliphatic aldehydes did not deliver any of the P(O)–C and P(O)–O bonds and instead we got the α-hydroxy phosphine oxides in 52% yield. Addition of cesium carbonate alone failed to convert the α-hydroxy phosphine oxides derived from aliphatic aldehydes into the corresponding Brook-type rearranged P(O)–O product. However, in the presence of Mn1 along with cesium carbonate, the P(O)–O product (4z) was obtained in 45% yield. These results suggest that the slow generation of the aldehyde, together with the involvement of the Mn catalyst in both the dehydrogenation and the phospha-Brook rearrangement, is key to achieving excellent yields with aliphatic alcohols.
Addition of radical scavengers like BHT or TEMPO did not hamper the reaction, indicating a non-radical pathway. Furthermore, the trityl cation quenching studies indicate the formation of Mn–H species (see SI). A detailed kinetic study of the reaction toward the selective formation of 3a revealed that both 4a and 3a are formed at the initial stage. The concentration of 4a increases and reaches a maximum at approximately 5 hours, after which it gradually decreases as 4a is converted into 3a (Scheme 5).
 | ||
| Scheme 5 Overall time profile of reaction progress. | ||
Summarising the details of the control experiment, mass analysis and literature support,22 we have established a plausible catalytic cycle (Scheme 6). The bimetallic Mn1 precatalyst is converted into a mononuclear Mn(II) species (I) under the reaction conditions. Then, the alcohol coordinates to the metal center as an alkoxide and ligand-based oxygen gets protonated. Subsequently, base-induced HBr elimination generates the active Mn species (III). Next, a second alcohol molecule binds to the vacant site on the metal center, forming the di-alkoxide complex Mn-IV. Through metal–ligand cooperation (MLC), the alcohol is dehydrogenated to produce the corresponding aldehyde and a reactive Mn–H species (V), and the subsequent release of dihydrogen regenerates the active catalyst (III).
 | ||
| Scheme 6 Plausible catalytic cycle. | ||
Furthermore, the control experiments underpin the involvement of the catalyst in the phospha-Brook arrangement, which is depicted in the catalytic cycle. The coordinated aldehyde species VI reacts with diphenyl phosphine oxide to form intermediate VII, which subsequently undergoes the phospha-Brook rearrangement. The resulting anion, stabilized at the α-position to the phosphinate group, is likely facilitated through the formation of intermediate IX.
In summary, we have developed a Mn(II)-catalyzed, biocompatible, and cost-effective protocol for the selective activation of alcohols to synthesize P(O)–O and P(O)–C compounds via a dehydrogenative phospha-Brook rearrangement. The protocol is effective for producing both of the compounds in good yield and selectivity. It is broadly applicable to challenging alcohols, including long-chain, branched, cyclic, acyclic, and naturally occurring alcohols such as citronellol, nerol, geraniol, and menthol. Various control experiments and kinetic studies suggest that the reaction proceeds through a dehydrogenative phospha-Brook rearrangement.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6cc01657j.CCDC 2525490 and 2525491 contain the supplementary crystallographic data for this paper.23a,b
Acknowledgements
This work is financially supported by SERB (CRG/2021/000402). The authors thank the Department of Chemistry, IIT Guwahati (COE-FAST:5-5/2014-TS VII and FIST:SR/FST/CS-II/2017/23C) and the Central Instrumentation Facility for the use of NMR and HRMS facilities. H. J. P. acknowledges UGC, A. S. thanks PMRF, and K. M., A. M. and R. D. thank IITG for their fellowship.References
- H. Saad, S. A. Elfeky, N. E. A. El-Gamel and A. S. Abo Dena, RSC Adv., 2025, 15, 40802–40822 RSC
.
-
(a) C.-S. Cho, L.-W. Chen and Y.-S. Chiu, Polym. Bull., 1998, 41, 45 CrossRef CAS
-
(a) J. J. Petkowski, W. Bains and S. Seager, Molecules, 2019, 24, 866 CrossRef PubMed
-
(a) S. Demkowicz, J. Rachon, M. Dasko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC
- W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed
- F. R. Atherton, H. T. Openshaw and A. R. Todd, J. Chem. Soc., 1945, 660–663 RSC
-
(a) J. Shen, Q.-W. Li, X.-Y. Zhang, X. Wang, G.-Z. Li, W.-Z. Li, S.-D. Yang and B. Yang, Org. Lett., 2021, 23, 1541–1547 CrossRef CAS PubMed
-
(a) A. Michaelis and R. Kaehne, Chem. Ber., 1898, 31, 1048 CrossRef
- M. B. Kurosawa, R. Isshiki, K. Muto and J. Yamaguchi, J. Am. Chem. Soc., 2020, 142, 7386–7392 CrossRef CAS PubMed
- J.-S. Zhang, T. Q. Chen, Y. B. Zhou, S.-F. Yin and L.-B. Han, Org. Lett., 2018, 20, 6746–6749 CrossRef CAS PubMed
- L. Gan, C. Ye, T. Pi, L. Wang, C. Li, L. Liu, T. Huang, T. Chen and L. B. Han, J. Org. Chem., 2024, 89, 7047–7057 CrossRef CAS PubMed
- C.-K. Li, Z.-K. Tao, A. Shoberu, W. Zhang and J.-P. Zou, Org. Lett., 2022, 24, 6083–6087 CrossRef CAS PubMed
- A. J. Bloomfield and S. B. Herzon, Org. Lett., 2012, 14, 4370–4373 CrossRef CAS PubMed
- Y. Ou, Y. Huang, Z. He, G. Yu, Y. Huo, X. Li, Y. Gao and Q. Chen, Chem. Commun., 2020, 56, 1357–1360 RSC
- Y. Qian, Q. Dai, Z. Li, Y. Liu and J. Zhang, Org. Lett., 2020, 22, 4742–4748 CrossRef CAS PubMed
- B. Xiong, X. Feng, L. Zhu, T. Chen, Y. Zhou, C.-T. Au and S.-F. Yin, ACS Catal., 2014, 5, 537–543 CrossRef
- X. Ma, Q. Xu, H. Li, C. Su, L. Yu, X. Zhang, H. Cao and L.-B. Han, Green
Chem., 2018, 20, 3408–3413 RSC
-
(a) A. Mondal, H. J. Phukan, K. Mohar, D. Pal, R. Sharma, S. Purkayastha, A. K. Guha and D. Srimani, Chem. Commun., 2025, 61, 4796–4799 RSC
-
(a) K. Das, S. Waiba, A. Jana and B. Maji, Chem. Soc. Rev., 2022, 51, 4386 RSC
- A. Mondal, H. J. Phukan, D. Pal, S. Kumar, M. Roy and D. Srimani, Chem. – Eur. J., 2024, 30, e202303315 CrossRef CAS PubMed
- H. F. Zhuang, P. Wan, C. X. Miao, Y. Yang, S. Y. Liang and F. Han, J. Org. Chem., 2024, 89, 2397 CrossRef CAS PubMed
- R. Sharma, K. Mohar, A. Mondal, E. Basumatary, K. Soni and D. Srimani, Organometallics, 2025, 44, 2172–2181 CrossRef CAS
-
(a)
CCDC 2525490: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qrzfx
| This journal is © The Royal Society of Chemistry 2026 |