
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganophotocatalysis using red to near-infrared light
Heng
Yang
a,
Si-Rui
Xiang
a,
Meng-Ru
Zhai
a,
Kun
Shen
 *a and
You-Quan
Zou
*ab
*a and
You-Quan
Zou
*ab
aDepartment of Radiation and Medical Oncology, Zhongnan Hospital of Wuhan University, School of Pharmaceutical Sciences, Wuhan University, Wuhan, Hubei 430071, China. E-mail: kun.shen@whu.edu.cn; youquanzou@whu.edu.cn
bTaiKang Center for Life and Medical Sciences, Wuhan University, Wuhan, Hubei 430071, China
First published on 24th February 2026
Abstract
Over recent years, photocatalysis has progressed substantially with the utilization of red to near-infrared (NIR) light to drive chemical transformations. Nevertheless, metal-based complexes continue to dominate this spectral region, despite their high cost and potential toxicity, which restrict their application in large-scale processes and biological settings. The development of efficient metal-free organic photocatalysts capable of operating under red and NIR light therefore represents a compelling research direction. This review surveys recent advances in red- and NIR-light-promoted reactions enabled by metal-free organic photocatalysts.
 Heng Yang | Heng Yang graduated from Shenyang Pharmaceutical University in 2023 with a bachelor degree. He received his master degree from Wuhan University in 2025, and in the same year, he continued to pursue his doctoral degree at Wuhan University under the supervision of Prof. You-Quan Zou. His research focuses on NIR bioorthogonal photocatalysis. |
 Si-Rui Xiang | Si-Rui Xiang obtained her Bachelor's degree from Wuhan Institute of Technology in 2023, and she received her Master's degree from Wuhan University in 2025 under the supervision of Prof. You-Quan Zou. Her research focuses on NIR bioorthogonal photocatalysis. |
 Meng-Ru Zhai | Meng-Ru Zhai obtained a Bachelor's degree from Jilin University in 2025. In the same year, she joined Wuhan University as a master's student under the supervision of Prof. You-Quan Zou. Her research focuses on NIR bioorthogonal photocatalysis. |
 Kun Shen | Kun Shen received his PhD in 2013 from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the supervision of Professor Xiyan Lu. He then conducted postdoctoral research at Duke University with Professor Qiu Wang. In 2018, he joined the School of Pharmaceutical Sciences at Wuhan University as an Associate Professor. His research focuses on organometallic chemistry and synthetic methodology development. |
 You-Quan Zou | You-Quan Zou completed his PhD studies in 2014 under the supervision of Professor Wen-Jing Xiao at the Central China Normal University. From November 2014 to October 2017 he worked as an Alexander von Humboldt postdoctoral fellow with Professor Thorsten Bach at the Technische Universität München. He then took up a SAERI postdoctoral position with Professor David Milstein at the Weizmann Institute of Science from November 2017 until October 2019. He has continued to work as a SAERI research associate with Professor Jonathan Nitschke at the University of Cambridge till April 2021. You-Quan Zou is currently working as a professor at the Wuhan University. His research interests focus on bioorthogonal photocatalysis and medicinal chemistry. |
1. Introduction
Photocatalysis has garnered significant attention over the past decade owing to its exceptionally mild operating conditions.1–10 The use of light as an abundant and renewable energy source aligns closely with the core principles of green chemistry.11–20 Both metal complexes (e.g., iridium, ruthenium) and organic dyes (e.g., Eosin Y) have been demonstrated to effectively harness visible light to drive catalytic transformations. However, visible-light-driven catalysis faces inherent limitations, including low natural abundance of suitable wavelengths,21 poor penetration in scattering media, and competitive absorption by substrates. These factors can promote side reactions and hinder scale-up, ultimately restricting wider adoption in synthesis and biological settings.To address these challenges, research focus has shifted toward longer-wavelength red and near-infrared (NIR) light. Owing to its deep tissue penetration and low phototoxicity,22 red and NIR irradiation enables scalable transformations and intracellular catalysis,23–27 yet demands photocatalysts with both appropriate excited-state properties and strong absorption in this spectral range.28–32 Two principal strategies are employed in red and NIR photocatalysis: (1) Triplet–triplet annihilation upconversion (TTA-UC):33,34 an upconversion system comprising a photosensitizer and an annihilator absorbs NIR light and converts it into higher-energy visible light. The process relies on multiple energy-transfer steps and requires precise energetic matching between the sensitizer and annihilator for efficient triplet–triplet energy transfer via collisions.35–38 (2) Direct red and NIR light absorption: photocatalysts directly absorb red or NIR photons to reach an excited state, subsequently driving reactions via energy transfer (EnT) or single-electron transfer (SET). This approach is generally simpler and more operationally straightforward than TTA-UC.
While metal complexes are widely employed as red and NIR photocatalysts, their high cost and inherent toxicity limit their utility in large-scale industrial processes and intracellular catalysis.39,40 In contrast, metal-free organic photocatalysts offer three distinct advantages: cost-effectiveness, biocompatibility, and synthetic accessibility (Fig. 1). This review highlights recent advances in red- and NIR-light-driven photocatalytic reactions catalyzed by metal-free organic photocatalysts. It is organized into two main sections, focusing on red and NIR light respectively, and each section is further subdivided according to the structural class of the organic photocatalyst employed.
 | ||
| Fig. 1 Organic photocatalysts used in Red/NIR photocatalysis. | ||
2. Red-light-driven organophotocatalysis
2.1. Porphyrins and porphyrinoids
Porphyrins and porphyrinoids represent a prominent class of metal-free organic photocatalysts for visible-light-driven transformations.41,42 Their extended conjugated macrocyclic architectures provide intense Soret-band absorption in the blue region and characteristic Q-band absorption in the 600–700 nm range, enabling efficient harvesting of red photons. These compounds typically feature long-lived triplet excited states and can participate in both SET and EnT pathways, supporting diverse photocatalytic reactions including photooxidations, photoreductions, and radical-based transformations.Structural modification of the porphyrin core provides a straightforward strategy to enhance red-light absorption and catalytic reactivity.42 For example, Derksen and co-workers demonstrated that thiophene-substituted thiaporphyrins (S1TPP, S2TPP) catalyze the photoreductive dehalogenation of α-haloketones under red light irradiation (λ > 645 nm). The reaction proceeds with catalyst loadings as low as 0.1 mol% in the absence of any transition metal, employing Hantzsch ester (HEH) as the reducing agent and DIPEA as an additive (Scheme 1).43 A range of aromatic substrates bearing electron-donating or electron-withdrawing substituents are well tolerated under the optimized conditions. Mechanistic studies indicate that the reaction proceeds via a sequential SET/hydrogen atom transfer (HAT) pathway rather than a radical-chain process, establishing metal-free porphyrinoids as competent photocatalysts for reductive transformations under red light. Under standard conditions, bromoacetophenone (1a) was converted to acetophenone (2a) in 25% yield after 5 min of red-light irradiation, with no further conversion was observed after an additional 17 h in the dark. Based on these experimental observations, a plausible catalytic cycle was proposed. Upon red-light excitation, the porphyrin photocatalyst S1TPP is promoted to its excited state S1TPP*, which subsequently undergoes SET with HEH to generate the radical anion S1TPP˙− and the radical cation HEH˙+. The α-haloketone 1a is then reduced by S1TPP˙−via SET to afford a carbon-centered radical intermediate and regenerate the photocatalyst S1TPP. Hydrogen atom transfer from HEH˙+ to this carbon-centered radical intermediate ultimately yields the corresponding dehalogenated ketone product 2a.
 | ||
| Scheme 1 Photoreductive dehalogenation of α-haloketones catalyzed by S1TPP. | ||
The Gryko group systematically expanded the scope of red-light photocatalysis using metal-free porphyrins, demonstrating their dual capacity to function as both photooxidants and photoreductants within photoredox cycles.44 These catalysts enable a diverse array of transformations, including the single-electron reduction of aryl diazonium salts for heteroarene arylation (Scheme 2), α-alkylation of aldehydes (Scheme 3), thiol–yne additions (Scheme 4a), and decarboxylative alkynylation of N-hydroxyphthalimide (NHPI) esters (Scheme 4b).
 | ||
| Scheme 2 Red-light-induced C–H arylation catalyzed by porphyrins. | ||
 | ||
| Scheme 3 Red-light-induced α-alkylation of aldehydes catalyzed by porphyrins. | ||
 | ||
| Scheme 4 Porphyrins-catalyzed red-light-induced photoredox reactions. | ||
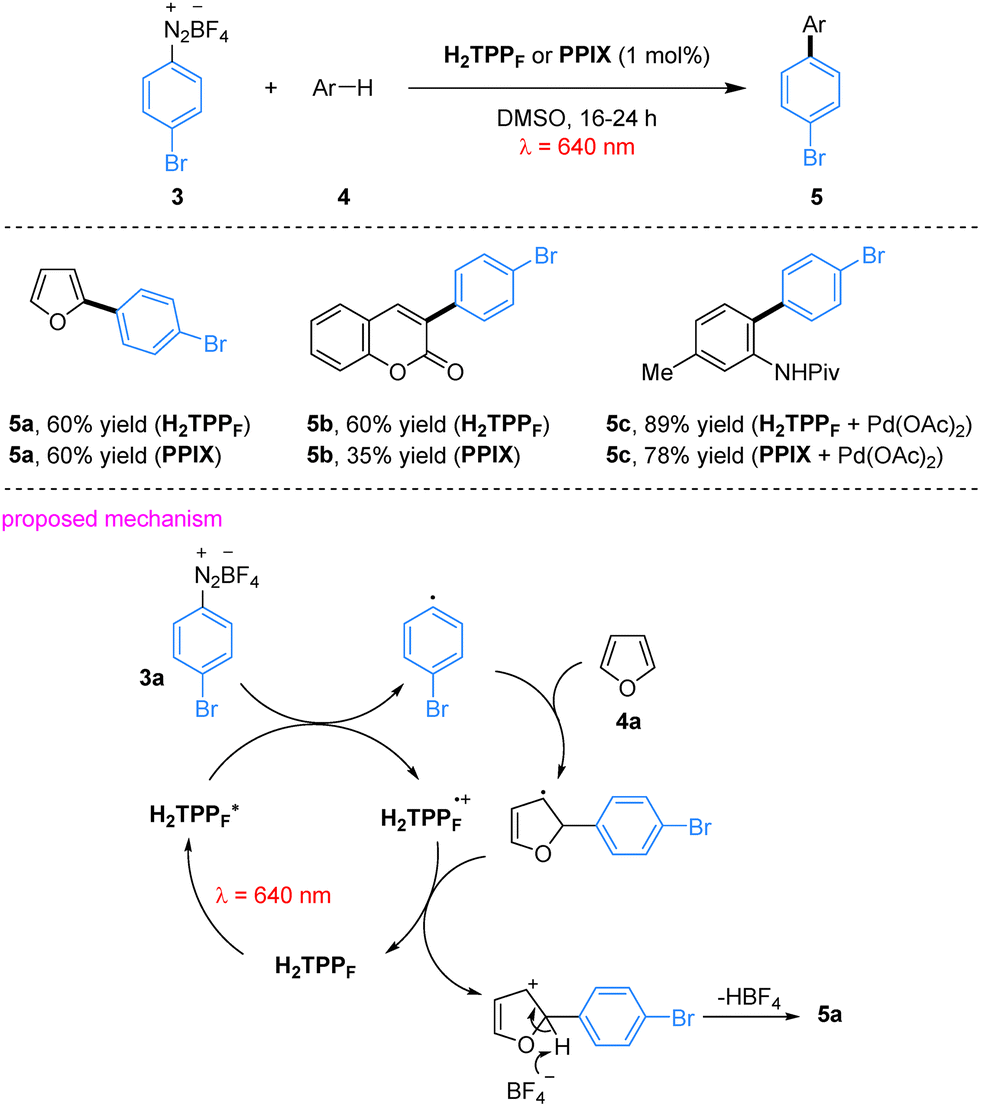
In their red-light-induced C–H arylation of heteroarenes, porphyrin derivatives such as H2TPPF serve as efficient photocatalysts, overcoming the traditional limitation that porphyrin-based systems are predominantly employed under blue or green light. This protocol provides a mild and operationally simple strategy for heteroarene C–H arylation. The system exhibits a broad substrate scope, accommodating heteroarenes including furan and coumarin. In this transformation, aryl diazonium salts serve as aryl radical precursors; aryl radicals are generated via SET from the excited-state porphyrin to the diazonium salts, followed by addition to the heteroarene to furnish the arylated product.45 Notably, naturally occurring protoporphyrin IX (PPIX) could replace synthetic porphyrins to afford comparable yields, substantially reducing catalytic cost. Importantly, porphyrin catalysts operate synergistically with palladium in dual catalytic C–C bond-forming reactions without undergoing metallation-induced deactivation, as exemplified by the formation of product 5c, highlighting their stability in multi-catalytic environments (Scheme 2).
Metal-free porphyrins also serve as efficient photooxidants under red light irradiation. Using 3-phenylpropanal (6) and ethyl diazoacetate (7) as model substrates in a DMSO/acidic buffer medium, the desired α-alkylated product 8 was obtained in 75% yield (Scheme 3), which is comparable to that achieved under traditional blue light irradiation.46 The reaction proceeds via a reductive quenching pathway. Upon red-light irradiation, ground-state H2TPP is promoted to its excited state (H2TPP*), which initiates two key parallel processes simultaneously. First, the enamine intermediate formed in situ via the condensation of aldehyde 6 with morpholine is oxidized through SET, generating the H2TPP radical anion (H2TPP˙−) and an enamine radical cation. Secondly, it interacts with ethyl diazoacetate (7) via EnT, triggering nitrogen extrusion and generating a triplet carbene intermediate. The enamine radical cation subsequently couples with this carbene to form a new carbon-centered radical intermediate. The H2TPP radical anion (H2TPP˙−) then transfers an electron to this intermediate, accompanied by protonation, delivering the desired α-alkylated aldehyde product 8 and regenerating the H2TPP photocatalyst, thus closing the catalytic cycle.
The Gryko group further extended this red-light-driven porphyrin photocatalytic system to thiol–yne addition reactions (Scheme 4a). The system exhibits excellent substrate compatibility and scalability; primary, secondary and tertiary thiols all participate smoothly, with secondary and tertiary thiols affording higher yields than primary thiols. The reaction proceeds via a reductive quenching pathway. Upon red-light irradiation, ground-state H2TPP is promoted to its excited state (H2TPP*) and undergoes SET with the thiol, followed by deprotonation to generate a thiyl radical. This radical adds to the alkyne triple bond to form a vinyl radical intermediate, which is subsequently converted to the corresponding addition product through hydrogen atom transfer.47
This catalytic system is also applicable to decarboxylative alkynylation reactions. The addition of Hantzsch ester (HE) and N,N-diisopropylethylamine (DIPEA) as reducing agents significantly enhances reaction efficiency (Scheme 4b). Notably, this system exhibits excellent biocompatibility: catalytic activity is retained when synthetic porphyrins are replaced with naturally occurring heme-derived protoporphyrin IX (PPIX) and when DMSO/PBS (v/v = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) is used as the reaction solvent. A variety of NHPI esters and p-toluenesulfonyl alkynes are well tolerated, demonstrating broad substrate scope. Moreover, this protocol is successfully applied to the late-stage decarboxylative alkynylation of bioactive molecules such as deoxycholic acid and indomethacin. The reaction follows a reductive quenching mechanism. Upon red-light excitation, the porphyrin photocatalyst H2TPP undergoes SET with HE to generate the HE radical cation, which subsequently reduces the NHPI ester, triggering decarboxylation to form a carbon-centered radical. This carbon-centered radical adds to the alkyne triple bond of the p-toluenesulfonyl alkyne, followed by desulfonylation to afford the alkynylated products.48
:1) is used as the reaction solvent. A variety of NHPI esters and p-toluenesulfonyl alkynes are well tolerated, demonstrating broad substrate scope. Moreover, this protocol is successfully applied to the late-stage decarboxylative alkynylation of bioactive molecules such as deoxycholic acid and indomethacin. The reaction follows a reductive quenching mechanism. Upon red-light excitation, the porphyrin photocatalyst H2TPP undergoes SET with HE to generate the HE radical cation, which subsequently reduces the NHPI ester, triggering decarboxylation to form a carbon-centered radical. This carbon-centered radical adds to the alkyne triple bond of the p-toluenesulfonyl alkyne, followed by desulfonylation to afford the alkynylated products.48
Gryko and co-workers further developed a porphyrin-mediated photosensitization strategy that activates aryl diazoesters via EnT under red light, generating reactive triplet carbenes.49 This approach enabled β-ketoester synthesis under aerobic conditions, as well as carboxylic acid O–H insertion and olefin cyclopropanation under anaerobic conditions (Scheme 5). Under aerobic conditions, the excited-state porphyrin converts ground-state molecular oxygen to singlet oxygen (1O2) via EnT. The resulting 1O2 undergoes cycloaddition with aryl diazoesters to form an unstable five-membered peroxyisoxazole intermediate, which rapidly decomposes with release of N2O and ultimately rearranges to afford β-ketoester products (Scheme 5a). Under anaerobic conditions, competitive oxidative pathways are suppressed. The excited porphyrin transfers energy directly to aryl diazoesters via EnT, generating triplet aryl diazoesters that extrude dinitrogen to form carbene species. These carbenes insert efficiently into carboxylic O–H bonds, exhibiting excellent substrate compatibility across diverse aryl diazoester scaffolds and delivering the corresponding O–H insertion products in high yields (Scheme 5b). This system is also applicable to olefin cyclopropanation. The mechanism parallels that of O–H insertion: triplet-state porphyrin engages in EnT with the aryl diazoester, triggering nitrogen extrusion to generate a carbene, which undergoes addition to alkene double bond to form cyclopropane rings. The reaction displays broad substrate scope, accommodating both electron-rich and electron-poor styrenes as well as internal olefins. Notably, cyclopropanation proceeds most efficiently with electron-poor aryl diazoesters (Scheme 5c).
 | ||
| Scheme 5 Red light-induced photosensitized transformations of diazo compounds. | ||
Leveraging the photoredox properties of porphyrins, the same team achieved radical activation of α-diazoesters via a proton-coupled electron transfer (PCET) mechanism (Scheme 6).49 This PCET strategy enabled the synthesis of γ-oximino esters from α-diazoesters, styrenes, and tert-butyl nitrite (TBN), as well as the construction of phenanthridines from α-diazoesters and isocyanobiphenyls. A wide range of α-diazoesters (20) and styrenes (21) delivered γ-oximino esters (22) in yields comparable to those obtained under blue light using Ru(bpy)3(PF6)2 as the photocatalyst.50 Similarly, diazoalkanes (23) and isocyanobiphenyls (24) furnished phenanthridines (25) with efficiency equal or superior to Xuan's conventional blue-light system employing Eosin Y as the photocatalyst, and with excellent substrate tolerance.51 Mechanistically, a reductive quenching cycle was proposed for the synthesis of γ-oximino esters. The excited state H2TPP* is reductively quenched by DIPEA to generate H2TPP˙−, which subsequently reduces α-diazoesters via PCET. This process triggers dinitrogen extrusion and proton abstraction from methanol, furnishing highly reactive carbon-centered radicals. These carbon radicals undergo addition to styrenes, and the resulting adducts are subsequently treated with tert-butyl nitrite to afford the γ-oximino esters via nitroso intermediates. In contrast, an oxidative quenching cycle was proposed for the construction of phenanthridines. The excited state H2TPP* mediates single-electron reduction of α-diazoesters through a PCET process, leading to dinitrogen release and generation of carbon-centered radicals. These radicals undergo a cascade of radical addition and intramolecular cyclization with 2-isocyanobiphenyl. Subsequent redox events ultimately deliver the phenanthridine products and regenerate the photocatalyst.
 | ||
| Scheme 6 Diazo compounds as radical precursors in red light-mediated photochemical transformations. | ||
Furthermore, diazo compounds participate in oxidative coupling reactions with tetrahydroisoquinolines and reductive coupling reactions with N-hydroxyphthalimide (NHPI) esters, enabling the efficient construction of β-amino-α-diazoesters and hydrazones (Scheme 7).49 Under porphyrin-catalyzed red-light photooxidation, tetrahydroisoquinolines 26 undergo single-electron oxidation followed by dehydrogenation to generate iminium ion intermediates. These intermediates undergo nucleophilic addition by ethyl diazoacetate (7), and subsequent protonation affords the corresponding β-amino-α-diazoester products 27. Tetrahydroisoquinolines bearing various substituents furnished the target products in good yields (53–78%). In parallel, alkyl radicals generated from NHPI esters were added to various donor/acceptor diazoalkanes to form hydrazones. This transformation exhibits excellent substrate compatibility, delivering the target products in moderate to good yields. Upon red-light irradiation, the porphyrin photocatalyst H2TPP is promoted to its excited state H2TPP* and undergoes reductive quenching by HEH to generate H2TPP˙−, which induces the formation of carbon-centered radicals via single-electron transfer with NHPI esters. These radicals undergo addition to the diazo double bond, and the target products are formed through subsequent protonation, with the diazo group remaining intact throughout the reaction.
 | ||
| Scheme 7 Diazo compounds as radical acceptors in red light-mediated photocatalyzed transformations. | ||
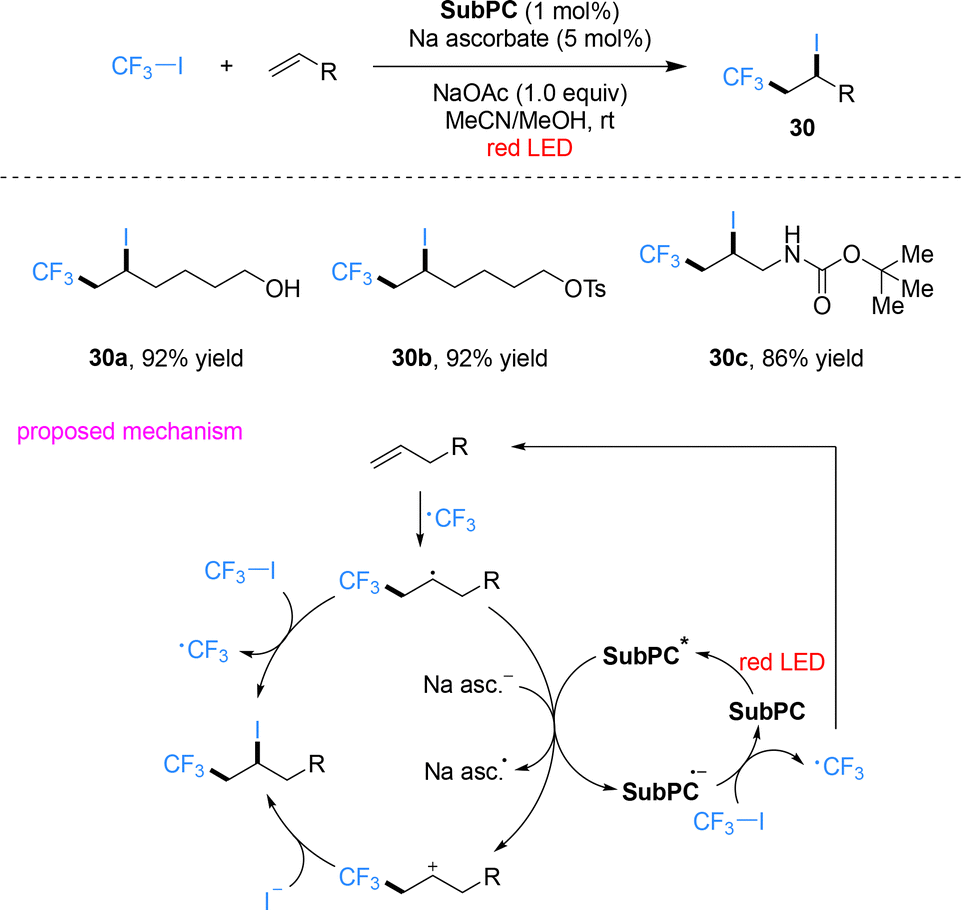
Beyond conventional porphyrins, their structural analogues such as phthalocyanines and subphthalocyanines also exhibit strong absorption in the red-light region. Shibata and co-workers designed a metal-free trifluoroethoxy-decorated boron subphthalocyanine (SubPC) that acts as an efficient red-light photocatalyst for trifluoromethylation and perfluoroalkylation of alkenes and alkynes (Scheme 8).52 Using CF3I as the fluoroalkyl source, a wide variety of terminal, internal and exocyclic alkenes bearing diverse functional groups, such as hydroxyl, tosylate and carbamate substituents, as well as alkynes, underwent smooth atom-transfer radical addition (ATRA) under red-light irradiation. The corresponding trifluoromethylethyl iodides and perfluoroalkylated products were obtained in good to high yields with excellent regioselectivity. This work constitutes the unprecedented example of a metal-free organic photocatalyst enabling red-light-driven fluoroalkylation, overcoming the long-standing reliance on metal complexes in this field. Mechanistic investigations confirm that the transformation requires both photocatalyst and light irradiation. Photoexcitated subphthalocyanine catalyst SubPC* accepts an electron from sodium ascorbate to form the radical anion SubPC˙−, which reduces CF3I to generate the CF3 radical. This radical adds to the carbon–carbon double bond of alkene to form a carbon-centered radical intermediate, which then reduces photoexcited SubPC* to regenerate SubPC˙− while forming a carbocation intermediate. The carbocation is trapped by iodide anion to afford the final product. An alternative mechanism involves a self-propagating radical chain: the carbon-centered radical abstracts the iodine atom from CF3I to generate the product and a CF3 radical, with light required for both initiation and maintenance of chain propagation.
 | ||
| Scheme 8 Trifluoromethylation of alkenes catalyzed by trifluoroethoxy-coated subphthalocyanine under red-light irradiation. | ||
Amara and co-workers demonstrated that silicon phthalocyanines could serve as highly efficient and sustainable metal-free photocatalysts for red-light-driven aerobic photooxidations, offering a robust alternative to conventional precious metal complexes.53 In the model oxidation of β-citronellol, a key step in the synthesis of rose oxide, representative silicon phthalocyanine derivatives (Si(Cl)2PCytBu4 and Si(OH)2PCytBu4) achieved 87% and 84% conversion, respectively, at sub-ppm catalyst loadings (Scheme 9). Despite possessing relatively low singlet oxygen quantum yields (ΦΔ ≈ 0.27), these silicon phthalocyanines exhibit exceptional photostability and pronounced resistance to photobleaching, enabling prolonged catalytic activity without degradation. This robustness translates into outstanding turnover numbers (TONs) of 52500–54375, significantly exceeding the performance of both ZnPCytBu4 (TON = 3750) and H2PCytBu4 (TON = 11380). Moreover, these catalysts operate effectively under solvent-free conditions and maintain high efficiency in multigram-scale continuous-flow processes, highlighting their potential for scalable, sustainable industrial photooxidations.
 | ||
| Scheme 9 Red-light-driven aerobic photooxidations of β-citronellol. | ||
Overall, owing to their tunable electronic structures, strong red-light absorption, and ability to operate via both energy- and electron-transfer pathways, porphyrins and their analogues have emerged as one of the most representative classes of photosensitizers in metal-free red-light photocatalysis. Future advances through precise structural modifications to regulate their redox potentials, triplet energy levels, and solubility are poised to further broaden their utility across organic synthesis, chemical biology, and sustainable chemistry.
2.2. Methylene blue
Methylene blue (MB), a classic metal-free organic photosensitizer, has attracted sustained interest in red-light-driven photocatalysis owing to its favorable excitation profile (λ ≈ 630–660 nm), excellent photostability, commercial availability, and low cost.54,55 Early work by Ferroud and co-workers demonstrated that MB could catalyze the photocyanation of tertiary amines using trimethylsilyl cyanide (TMSCN) as the cyanide source and molecular oxygen as the oxidant under red light irradiation (λ > 630 nm).56 Under these conditions, N-arylaminopiperidines and N-arylaminopyrrolidines (34) undergo photocyanation to afford the corresponding α-hydrazinonitriles (35) in moderate to good yields. The reaction proceeds with high regio- and stereoselectivity; notably, in unsymmetrical hydrazine substrates, cyanation occurs preferentially at the more substituted carbon atom (e.g., 35a). Mechanistically, the transformation is proposed to involve a single-electron transfer (SET) pathway initiated either by the excited photosensitizer (MB*) or by singlet oxygen (1O2), the latter generated through energy transfer (EnT) from excited triplet-state methylene blue to ground-state molecular oxygen (Scheme 10a). | ||
| Scheme 10 Red-light-driven photooxidation of N-arylaminopiperidines and N-arylaminopyrrolidines catalyzed by methylene blue. | ||
Subsequently, the same group extended this methodology to the MB-catalyzed ring-opening of N-arylaminopiperidines and N-arylaminopyrrolidines (34) under analogous conditions.57 Employing water or an alcohol (ROH) as the nucleophile in place of TMSCN, the SET-mediated photooxidation delivered the corresponding aminoaldehydes or aminodialkylacetals (36) in good yields with high regioselectivity. In unsymmetric substrates, ring-opening occurred preferentially via cleavage of the C–N bond adjacent to the less-substituted carbon. Mechanistically, these one-pot transformations are proposed to involve three distinct steps. Initially, oxidation at the carbon α to the ring nitrogen generates a hydrazinium alkylidene cation. Nucleophilic attack by ROH on this electrophilic intermediate then furnishes an oxidized hemiaminal. In the subsequent steps, the reaction pathway is proposed to involve cleavage of the hemiaminal C–N bond to afford an arylhydrazine, which is then photooxidized in situ to yield the final product. However, the precise sequence of these latter two steps remains to be conclusively established (Scheme 10b).
This reactivity was further extended to a two-step sequential photooxidation.58 Employing TMSCN followed by water as nucleophiles, the substrates (34) were converted to the corresponding lactams (37) in 45–95% yields. The process proceeds via initial MB-mediated SET to generate an α-hydrazinonitrile intermediate, which undergoes aerobic photooxidation to furnish the lactam. Specifically, oxidation of the hydrazino group forms a hydrazinium radical cation. Deprotonation of this species by O2˙− generates an α-hydrazino (or hydrazyl) radical, which is rapidly oxidized to the hydrazinium alkylidene cation. This cation exists in tautomeric equilibrium with the 1,1-dialkyl-2-phenyldiazenium cation. Subsequent nucleophilic attack by water yields a cyanohydrin intermediate, which affords the target lactam product with concomitant elimination of HCN (Scheme 10c).
Beyond amine functionalization, MB has also been applied in photouncaging chemistry. Hosoya and co-workers developed 3-acyl-2-methoxyindolizine-based caged compounds for the controlled release of carboxylic acids and alcohols (Scheme 11).59 Upon irradiation with red light at 660 nm, MB sensitizes the generation of singlet oxygen (1O2), which undergoes electrophilic addition to the C3 position of the indolizine core. The resulting adduct undergoes intramolecular rearrangement, resulting in oxidative ring cleavage and the rapid release of the caged substrates. This system enables the efficient release of functional molecules, including the anticancer agent bexarotene and the fluorescent dye 4-methylumbelliferone within five minutes. Notably, it retains high performance even in serum-containing media, highlighting its potential as a biocompatible tool for light-controlled release in biomedical applications.
 | ||
| Scheme 11 Uncaging of alcohols and carboxylic acids by red light-induced photooxidation. | ||
2.3. Helical carbenium ions
As an emerging class of metal-free red-light photocatalysts, helical carbenium ions have garnered significant attention owing to their distinctive conjugated helical architecture and versatile photoredox properties.60,61 Their rigid helical frameworks not only promote strong absorption of red light, but also modulate the excited-state electronic structure, endowing them with tunable redox activity that enables both oxidative and reductive quenching pathways under mild conditions.In 2020, the Gianetti group reported the helical carbenium ion N,N′-di-npropyl-1,13-dimethoxyquinacridinium (nPr-DMQA+) tetrafluoroborate as a versatile organic photocatalyst operating under red-light (λmax = 640 nm) (Scheme 12).62 Employing dual catalytic systems, nPr-DMQA+ enabled Pd-catalyzed C(sp2)–H arylation and Au-catalyzed terminal alkyne C(sp)–H arylation via oxidative quenching, as well as intermolecular atom-transfer radical addition (ATRA) reactions. Furthermore, the catalyst mediated aerobic oxidative hydroxylation of arylboronic acids and oxidative amidation of benzylic C(sp3)–H bonds through reductive quenching pathways, demonstrating broad applicability across both oxidative and reductive photocatalytic manifolds.
 | ||
| Scheme 12 [nPr-DMQA+][BF4−]-Catalyzed C–H arylation and aerobic oxidative hydroxylation under red light. | ||
Two distinct catalytic cycles involving oxidative and reductive quenching have been proposed for [nPr-DMQA+][BF4−]. In the oxidative pathway for Pd-catalyzed C(sp2)–H arylation, photoexcited nPr-DMQA+* reduces an aryl diazonium salt via oxidative quenching, generating an aryl radical and the radical cation nPr-DMQA˙++. The aryl radical adds to a Pd(II) intermediate (formed by C–H activation), yielding a Pd(III) species, which is oxidized by nPr-DMQA˙++ to regenerate the ground-state photocatalyst and produce a Pd(IV) complex. Reductive elimination from this Pd(IV) intermediate delivers the arylated product. In the reductive pathway for aerobic oxidative hydroxylation, photoexcited nPr-DMQA+* undergoes reductive quenching by iPr2NEt, affording the helicene radical nPr-DMQA˙ and an ammonium radical cation. The helicene radical then reduces O2 to superoxide (O2˙−) while regenerating nPr-DMQA+. Subsequent oxidative attack of the superoxide on the arylboronic acid, followed by hydrolysis, yields the phenol product.
They also utilized Umemoto's reagent as the trifluoromethyl source to achieve [nPr-DMQA+][BF4−]-catalyzed red-light-mediated trifluoromethylation/dearomatization cascade reactions of indole derivatives. This process efficiently affords CF3-containing 3,3-spiroindoline skeletons, a privileged scaffold in drug discovery (Scheme 13). The reaction tolerates indole derivatives bearing functional groups such as cyano, nitro, and halides, delivering the desired products in yields of 35–87% with excellent diastereoselectivity (exclusively trans isomers).63 Mechanistically, under red light irradiation, the organic photocatalyst [nPr-DMQA+][BF4−] is excited to nPr-DMQA+*, which reduces Umemoto's reagent via an oxidative quenching pathway to generate the key CF3 radical and the oxidized catalyst species nPr-DMQA˙++. The CF3 radical adds to the indole derivative, forming the dearomatized radical intermediate I. Subsequent single-electron transfer (SET) between I and nPr-DMQA˙++ regenerates the ground-state photocatalyst and affords the carbocation intermediate II. Intermediate II then undergoes a stereoselective intramolecular cyclization to yield the CF3-containing 3,3-spirocyclic indoline product as a single anti diastereomer (path a). An alternative oxidation pathway for the conversion of radical I to carbocation II involving direct oxidation by Umemoto's reagent cannot be entirely excluded (path b).
 | ||
| Scheme 13 Red-light-induced [nPr-DMQA+][BF4−]-catalyzed cascade trifluoromethyl-ation/dearomatization of indole derivatives. | ||
To construct cyclopentane/cyclopentene scaffolds, the group developed a [nPr-DMQA+][BF4−]-catalyzed, red-light-mediated [3+2] cycloaddition between N-aryl-cyclopropylamines and alkenes/alkynes (Scheme 14).64 The reaction tolerated a variety of electronic substituents on the cyclopropylamine aryl ring, including methyl, trifluoromethyl and chloro, as well as alkenes/alkynes bearing functional groups such as cyano, ester and pyridine, affording cycloadducts in 60–95% yields for alkenes and 33–81% yields for alkynes. Mechanistically, photoexcitation of nPr-DMQA+ generates the excited state nPr-DMQA+*, which undergoes single-electron transfer with the cyclopropylamine 46a to form a nitrogen radical cation. The inherent strain of the cyclopropane ring promotes β-scission of this intermediate, yielding a β-carbon radical iminium species. This species adds to styrene 47a to generate a distonic radical cation, which then undergoes intramolecular radical addition to the iminium moiety to furnish a second nitrogen radical cation. Reduction of this intermediate by the helicene radical nPr-DMQA˙ delivers the final product 48a and regenerates ground-state nPr-DMQA+, thereby completing the photocatalytic cycle.
 | ||
| Scheme 14 Red-light-induced nPr-DMQA+-catalyzed [3+2] cycloaddition of cyclopropylamines with alkenes or alkynes. | ||
In 2024, the Gianetti group reported a wavelength-regulated chromoselective C(sp2)–X bond activation strategy employing the helicenium photocatalyst [nPr-DMQA+][BF4−] (Scheme 15).65 Under red light irradiation (640 nm), low-bond-energy C(sp2)–I bonds are selectively cleaved via a halogen-atom-transfer (XAT) pathway initiated by an α-amino radical, generating key aryl-radical intermediates while leaving C(sp2)–Br bonds intact. Conversely, blue light irradiation triggers a consecutive photoinduced electron transfer (conPET) mechanism that reduces and activates the more challenging C(sp2)–Br bonds. This chromoselectivity enables divergent functionalization of multihalogenated arenes, as demonstrated in the wavelength-dependent α-arylation of cyclic ketones. The approach facilitates sequential, light-gated dual functionalization: red light-mediated XAT first functionalizes the iodo site, after which blue light-driven conPET or transition metal catalysis can modify the retained bromo substituent. The preserved bromo functionality thus serves as a handle for subsequent diversification, providing modular access to differentially substituted aromatic scaffolds from a common halogenated precursor and demonstrating light wavelength as a precise external control in photocatalytic bond activation.
 | ||
| Scheme 15 Chromoselective C(sp2)–X bond activation by nPr-DMQA+. | ||
In 2025, the Shaikh group reported a red light-mediated C3 formylation of indoles catalyzed by the helical carbenium ion nPr-DMQA+ using 2,2-dimethoxy-N,N-dimethylethanamine as the formyl source (Scheme 16).66 This protocol exhibits broad substrate scope under mild conditions and is readily scalable for the synthesis of C3-formylated indoles. It accommodates various N-protected (alkyl, benzyl, allyl) and N–H indoles bearing halogen, methyl, or methoxy substituents, and produces 3-formylindoles in 50–95% yields. Mechanistic investigations, including radical-trapping, Stern–Volmer quenching and light on/off experiments, supported a pathway initiated by reductive quenching of the photoexcited nPr-DMQA+*via SET with the amine 53. This generates an amine radical cation that undergoes β-scission to release a dimethoxymethyl carbon radical, which adds to the C3 position of the indole. The resulting adduct then aromatizes and hydrolyzes to deliver the 3-formylindole product 54. HRMS data also suggested an alternative pathway, whereby the amine radical cation loses a hydrogen to form a dimethoxymethyl iminium ion. This species attacks the indole electrophilically, and after a second photoredox cycle, yields a more labile iminium intermediate that hydrolyzes to produce the same aldehyde product 54.
 | ||
| Scheme 16 Red-light mediated formylation of indoles using a helical carbenium ion as a photoredox catalyst. | ||
3. NIR-light-driven photocatalysis
3.1. Cyanine dyes
Cyanine dyes are widely used in photodynamic therapy.67,68 In 2021, Goddard and co-workers pioneered the use of cyanine dyes as metal-free photoredox catalysts under NIR irradiation.69 A panel of stable commercial cyanines was evaluated, with the aza-Henry-type reaction selected as the model system. Among these catalysts, Cy746 exhibited exceptional efficiency in the reaction of tetrahydroisoquinolines 55 with diverse nucleophiles under mild 810 nm irradiation, enabling the formation of various C–C and C–P bonds (Scheme 17). Mechanistically, this reaction proceeds via SET process: under 810 nm NIR irradiation, Cy746 is excited to its excited state Cy746*, which undergoes reductive quenching by tetrahydroisoquinolines 55 to generate the Cy746˙− radical anion and amino radical cations. Cy746˙− is then reoxidized to ground state by O2, with the concomitant generation of superoxide anion O2˙−. The superoxide anion subsequently abstracts hydrogen from amino radical cations to form iminium salts, and nucleophilic addition to these iminium salts affords the final tetrahydroisoquinoline products 56. | ||
| Scheme 17 The aza-Henry-type reaction catalyzed by the photocatalyst Cy746. | ||
Furthermore, Cy746 proves effective in diverse other organic transformations, including photoredox oxidation, photosensitization, and alkene trifluoromethylation, highlighting its versatility as a NIR photocatalyst (Scheme 18).69 Specifically, it catalyzes the oxidative cycloaddition of N,N-dimethylaniline 57 with maleimide 58 to afford tetrahydroquinoline 59, oxidative dimerization of thioamide 60 to 1,2,4-thiadiazole 61, and oxidation of boronic acid 42a to phenol 43a. Under 810 nm NIR irradiation and O2 atmosphere, it mediates the oxidation of furfural 62 and tetraphenylcyclopentadienone 64via singlet oxygen generation, giving products 63 and 65 in 45% and 81% yields, respectively. It also enables the photosensitization of vinyl azide 66, producing disubstituted pyrrole 67 in 68% yield. Notably, Cy746 also mediates reductive processes: under anaerobic conditions, it reduces Umemoto's reagent to generate trifluoromethyl radicals for trifluoromethylation, with yields of 50–66% depending on the olefin substrate. This NIR-photoredox system is compatible with aqueous media, enabling the formation of hydroxytrifluoromethylation adduct 69 in 46% yield via a CF3 radical addition/oxidation/water addition sequence. Collectively, these results establish cyanines as one of the first classes of metal-free NIR photocatalysts to promote organic transformations.
 | ||
| Scheme 18 Selected examples of NIR-triggered reactions catalyzed by Cy746. | ||
To further optimize catalytic performance and expand structure–activity insights, a library of cyanine-based near-infrared photocatalysts was constructed and fully characterised, including the determination of photophysical and redox properties.70 The performance of the catalysts was evaluated in two model redox reactions under NIR irradiation. Cyanines with an amino group on the heptamethine chain (e.g., Cy637) were found to be the most effective catalyst for the oxidative aza-Henry reaction. In contrast, the extended-conjugation derivative Cy997 was identified as the optimal catalyst for alkene trifluoromethylation via Umemoto salt reduction due to its enhanced stability under reaction conditions. These findings collectively delineate clear structure–activity relationships: amino substitution on the heptamethine chain enhances oxidative photocatalytic activity, whereas extended conjugation is pivotal for maintaining stability and efficiency in reductive transformations.
3.2. Squaraine dyes
To further expand the toolbox of organic NIR photocatalysts, Goddard and co-workers investigated squaraine dyes (Sq) as effective candidates.71 Among the evaluated derivatives, Sq687 emerged as the most efficient photocatalyst, successfully mediating a range of transformations including the oxidative aza-Henry-type tetrahydroisoquinolines, [4+2] cycloadditions, and trifluoromethylation reactions under NIR irradiation (Scheme 19).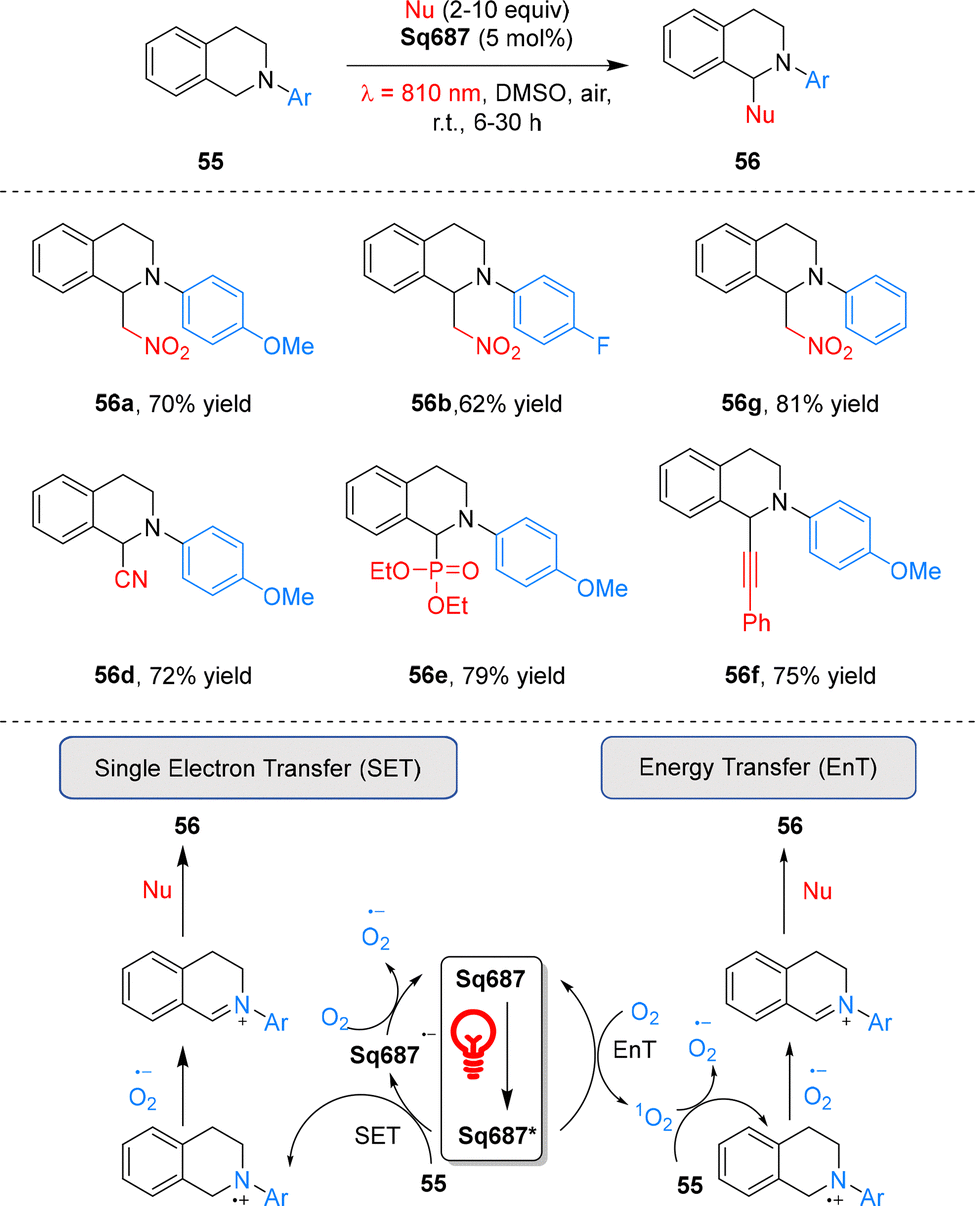 | ||
| Scheme 19 The aza-Henry-type reaction catalyzed by the photocatalyst Sq687. | ||
Mechanistic studies established SET as the dominant catalytic pathway, with EnT playing a negligible role in these processes. To investigate the potential involvement of singlet oxygen in the photocatalytic transformation, the authors examined the [4+2] cycloaddition between tetraphenylcyclopentadienone and 1O2. The starting material was nearly fully recovered, indicating that Sq687 does not generate significant concentrations of 1O2 under the reaction conditions. To further confirm that a SET mechanism predominates, molecular dioxygen was replaced with 1,3-dinitrobenzene as an alternative co-oxidant, which can regenerate the photocatalyst without producing 1O2. After 24 h of irradiation at 810 nm under these conditions, conversion of 55a to 56a reached 62%, supporting a SET-based pathway.
3.3. Planarized xanthene derivatives
Building upon cyanine dyes, structural modification of established organic scaffolds provides an alternative strategy for achieving NIR photoredox activity. Eosin Y (EY), a widely used visible-light photocatalyst lacks NIR absorption due to the orthogonal arrangement of its xanthene and benzene rings, which disrupts full π-conjugation.72 To overcome this limitation, Kamino and co-workers synthesized a planarized, fully bridged analogue BEY from EY in concentrated sulfuric acid at 180 °C in 95% yield (Scheme 20).73 This structural modification extends the conjugated system, resulting in a significant bathochromic shift in its absorption and emission spectra and imparting solvent-dependent photophysical properties, thereby activating BEY for NIR photocatalysis. As a NIR-active photocatalyst, BEY efficiently mediates the arylation of arenes with aryldiazonium salts, affording biaryl products in up to 82% yield under 521–730 nm light irradiation, with appreciable yields retained at longer wavelengths (57% at 830 nm and 28% at 940 nm). A key advantage of this NIR-driven system is its compatibility with colored substrates, such as fluorescein and rhodamine B, under 830 nm light irradiation. The transformations in conventional visible-light photocatalysts are hindered by competitive light absorption. | ||
| Scheme 20 Arylation of arenes with aryldiazonium salts catalyzed by BEY under NIR light. | ||
Employing a planarization strategy analogous to that used for BEY, the Kamino group synthesized coerulein B (CB) from fluorescein (FL) via heating in sulfuric acid; CB exhibits strong NIR absorption. As an effective NIR photocatalyst, CB mediated the arylation of furan with aryl diazonium salt 70a in organic media, affording product 71a in 63% yield at 730 nm and 42% yield at 830 nm (Scheme 21).74 Owing to its compact structure and inherent water solubility, CB also served as a competent aqueous-phase photocatalyst, enabling the NIR-light-driven arylation of pyridine hydrochloride with aryl diazonium salt 70a to furnish product 71h in 43% yield, albeit with an extended reaction time.
 | ||
| Scheme 21 Arylation of arenes with aryldiazonium salts catalyzed by CB under NIR light irradiation. | ||
3.4. BODIPY derivatives
Boron-dipyrromethene (BODIPY) derivatives constitute a prominent class of metal-free organic photocatalysts, distinguished by their high molar extinction coefficients, excellent photostability, and readily tunable photophysical properties.75,76 Their conjugated heterocyclic framework provides a flexible structural basis for the precise regulation of frontier molecular orbital energies. Strategic modifications such as terminal substitution, conjugation extension or ring fusion can be employed to tailor these catalysts to meet NIR light response requirements.In 2022, the Duan group reported carbazole-substituted iodinated BODIPY (BDP) as an efficient catalyst for NIR-driven photooxidation reactions under 720 nm irradiation (Scheme 22).77BDP mediated the benzylamine coupling reaction with high efficiency (Scheme 22a), and its superiority was highlighted by scale-up experiments: whereas the conversion rate of the benchmark Ru(bpy)3Cl2 catalyst dropped sharply to 38% when scaling the reaction from 1 mL to 20 mL, BDP retained a high conversion rate of 88%. This performance benefit stems from the excellent penetration ability of NIR light in reaction solutions. BDP also exhibited remarkable solvent tolerance for benzylamine coupling, achieving high conversion rates across diverse polar solvents-outperforming Ru(bpy)3Cl2, which showed 100% conversion in acetonitrile but only 5% in dichloromethane. Substrate scope exploration revealed that the reaction proceeded smoothly regardless of whether electron-donating or electron-withdrawing groups were attached to the benzene ring. Notably, BDP was also integrated with copper catalysis to couple NIR-generated Schiff bases with 4-tert-butylphenylacetylene in toluene, affording alkyne-substituted secondary amines in 80% isolated yield, demonstrating the compatibility of NIR photoredox catalysis with other catalytic systems.
 | ||
| Scheme 22 NIR-driven BDP-catalyzed photooxidation. | ||
Mechanistic studies indicated that BDP can generate singlet oxygen via energy transfer or superoxide anion via electron transfer upon NIR excitation. Upon absorption of NIR light, BDP is promoted to its singlet excited state (1[BDP]*), which undergoes intersystem crossing (ISC) to the triplet excited state (3[BDP]*). Triplet–triplet energy transfer from 3[BDP]* to molecular oxygen yields singlet oxygen. Benzylamine is then oxidized by 1O2, producing hydrogen peroxide and an imine intermediate that condenses with a second equivalent of benzylamine to form the final Schiff base.
Beyond benzylamine coupling, BDP also oxidizes sulfides to sulfoxides and arylboronic acids to phenols (Schemes 22b and c) and enables traceless deprotection of prodrugs under NIR irradiation. This photolytic activation represents a clean, green alternative to conventional acid/base- or redox-mediated deprotection methods.
3.5. Two-photon absorption (TPA) sensitizers
In addition to catalysts that operate via direct NIR absorption, two-photon absorption (TPA) offers an alternative strategy for NIR photoredox catalysis. The Sun group developed a series of benzothiazole-core organic TPA sensitizers (dBIP, dBIP-NMe2 and dBIP-OMe) featuring electron-donating termini (Fig. 1).78 Photophysical characterization showed that dBIP exhibits a two-photon absorption cross-section (σ2) > 1200 GM in the 825–855 nm range, whereas dBIP-NMe2 displays a higher σ2 of 2000 GM under the same conditions. The latter also possesses a longer fluorescence lifetime and a higher singlet oxygen yield, which are attributed to the stronger electron-donating character of its terminal groups. Both dBIP and dBIP-NMe2 efficiently catalyze benzylamine coupling under 850 nm irradiation, with the reaction tolerating electron-donating and electron-withdrawing substituents on the benzylamine aromatic ring (Scheme 23). On/off irradiation cycles confirmed the excellent photo-responsiveness and stability of dBIP: product formation occurred only during light-on periods. A ten test-tube penetration experiment further highlighted the advantage of NIR light: whereas 456 nm visible light failed to drive the reaction beyond the third tube, 850 nm NIR light still afforded 26% yield in the tenth tube. Moreover, dBIP-OMe was employed as a catalyst for the hydroxytrifluoromethylation of styrene under 720 nm irradiation.79 | ||
| Scheme 23 Photocatalytic homocoupling of benzylamines using dBIP as the photosensitizer. | ||
4. Summary and outlook
The photocatalysts discussed in this review exhibit marked differences in commercial availability and synthetic complexity. Within the porphyrin and porphyrinoid family, H2TPP, H2TPPF, and the naturally occurring protoporphyrin IX (PPIX) are readily obtainable from commercial suppliers. In contrast, S1TPP, S2TPP, the subphthalocyanine SubPC, and the silicon phthalocyanines (Si(Cl)2PCytBu4 and Si(OH)2PCytBu4) have limited commercial sources and typically require custom synthesis via cyclization of thiophene precursors or multi-step functionalization at the silicon center, presenting moderate to high synthetic difficulty. Methylene blue (MB), a thiazine dye, stands out as one of the most cost-effective and commercially accessible photosensitizers. Among cyanine dyes, Cy746 is commercially available. The squaraine dye Sq687 can be obtained commercially or synthesized through a two-step condensation reaction between squaric acid and aniline derivatives, with moderate operational simplicity. For BODIPY-based photocatalysts, while basic derivatives are commercially accessible and can be prepared via condensation of pyrroles with aldehydes followed by borylation, the long-chain-substituted BODIPY derivative BDP highlighted in this review possesses structural specificity that necessitates in-house synthesis. Other important classes, including the helical carbenium salt [nPr-DMQA+][BF4−], the planarized xanthlene derivatives (BEY and CB), and various two-photon absorption (TPA) sensitizers, currently lack commercial sources. Their preparation involves multi-step sequences.In summary, significant advances have been made in the field of metal-free red-to-NIR photocatalysis, driven by the development of various organic photocatalysts that facilitate efficient photon capture and redox cycling under long wavelength light. Representative structural scaffolds, including porphyrinoids, methylene blue, helical carbenium ions, cyanines, squaraines, planarized xanthenes, BODIPYs and two-photon absorption sensitizers, have been adapted or engineered to mediate a wide array of synthetically useful transformations. These systems exploit the intrinsic benefits of red and NIR irradiation, including deep penetration, low phototoxicity and minimal competitive absorption, thereby facilitating the development of scalable and potentially biocompatible photocatalytic protocols.
Several strategic directions will be pivotal for further progress. Currently, the field still relies heavily on the modification of existing dye frameworks. The de novo design of organic chromophores with tailored excited-state properties, strong absorption across the red-to-NIR window, and improved photostability remains a core challenge. Future work should prioritize establishing clear structure–property–activity relationships to guide rational catalyst development. It is essential to expand the reaction scope to include more demanding bond-forming and activation processes, especially those pertinent to complex-molecule synthesis and late-stage functionalization. Integrating red-to-NIR photocatalysis with other catalytic processes, such as transition-metal catalysis, organocatalysis or biocatalysis, could lead to new synergistic reactions. Furthermore, translating these methodologies into practical settings, such as continuous-flow reactors, industrial-scale synthesis and spatially resolved biological applications, is a significant frontier. Overcoming these challenges will establish red-to-NIR organophotocatalysis as an indispensable tool in sustainable chemical synthesis and beyond.
Author contributions
Heng Yang: writing – original draft. Si-Rui Xiang: writing – original draft. Meng-Ru Zhai: writing – original draft. Kun Shen: writing – original draft, visualization, funding acquisition. You-Quan Zou: supervision, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare no competing interests.Data availability
All data discussed in this review originate from the studies cited throughout the manuscript. Readers seeking detailed experimental procedures, analytical methods or supplementary data sets can refer to the corresponding publications listed in the References section.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22271225), the National Key R&D Program of China (No. 2021YFA0910500), the Fundamental Research Funds for the Central Universities (No. 2042025kf0067), the start-up funding from Wuhan University (No. 691000002), and the Wuhan Institute of Photochemistry and Technology Grant (No. GHY2024PY002).Notes and references
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- W. Liu, J. Li, C.-Y. Huang and C.-J. Li, Angew. Chem., Int. Ed., 2020, 59, 1786–1796 CrossRef PubMed.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- Y.-Z. Cheng, Z. Feng, X. Zhang and S.-L. You, Chem. Soc. Rev., 2022, 51, 2145–2170 RSC.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- Y.-W. Su, C. Pan, T.-Y. Sun and Y.-Q. Zou, Chem. Synth., 2025, 5, 10 CAS.
- Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- M. Rivas, V. Palchykov, X. Jia and V. Gevorgyan, Nat. Rev. Chem., 2022, 6, 544–561 CrossRef PubMed.
- Y.-W. Su, Y.-Q. Zou and W.-J. Xiao, Chin. J. Org. Chem., 2022, 42, 3201–3212 CrossRef CAS.
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS PubMed.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- B. Chen, L.-Z. Wu and C.-H. Tung, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS PubMed.
- F. Srieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- D. P. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958–2961 CrossRef CAS PubMed.
- K. Szaciłowski, W. Macyk, A. Drzewiecka-Matuszek, M. Brindell and G. Stochel, Chem. Rev., 2005, 105, 2647–2694 CrossRef PubMed.
- G. Chen, Y. Cao, Y. Tang, X. Yang, Y. Liu, D. Huang, Y. Zhang, C. Li and Q. Wang, Adv. Sci., 2020, 7, 1903783 CrossRef CAS PubMed.
- M. Russo, H. Janeková, D. Meier, M. Generali and P. Štacko, J. Am. Chem. Soc., 2024, 146, 8417–8424 CrossRef CAS PubMed.
- X. Liang, S. Qian, Z. Lou, R. Hu, Y. Hou, P. R. Chen and X. Fan, Angew. Chem., Int. Ed., 2023, 62, e202310920 CrossRef CAS PubMed.
- Z. Deng, H. Li, S. Chen, N. Wang, G. Liu, D. Liu, W. Ou, F. Xu, X. Wang, D. Lei, P.-C. Lo, Y. Y. Li, J. Lu, M. Yang, M.-L. He and G. Zhu, Nat. Chem., 2023, 15, 930–939 CrossRef CAS PubMed.
- J. Shen, B. Xu, Y. Zheng, X. Zhao, H. Qi, Y. Tang, W. Lin, S. Li and Z. Zhong, Angew. Chem., Int. Ed., 2025, 64, e202425309 CrossRef CAS PubMed.
- G. He, M. He, R. Wang, X. Li, H. Hu, D. Wang, Z. Wang, Y. Lu, N. Xu, J. Du, J. Fan, X. Peng and W. Sun, Angew. Chem., Int. Ed., 2023, 62, e202218768 CrossRef CAS PubMed.
- D. C. Cabanero and T. Rovis, Nat. Rev. Chem., 2025, 9, 28–45 CrossRef CAS PubMed.
- H. Li, Y. Kim, H. Jung, J. Y. Hyun and I. Shin, Chem. Soc. Rev., 2022, 51, 8957–9008 RSC.
- B. M. Vickerman, E. M. Zywot, T. K. Tarrant and D. S. Lawrence, Nat. Rev. Chem., 2021, 5, 816–834 CrossRef PubMed.
- T. Zhang, S. Pradhan and S. Das, Angew. Chem., Int. Ed., 2025, 64, e202501194 CrossRef CAS PubMed.
- N. Sellet, J. Frey, M. Cormier and J.-P. Goddard, Chem. Sci., 2024, 15, 8639–8650 RSC.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343 CrossRef CAS PubMed.
- L. Huang, W. Wu, Y. Li, K. Huang, L. Zeng, W. Lin and G. Han, J. Am. Chem. Soc., 2020, 142, 18460–18470 CrossRef CAS PubMed.
- L. Hou, A. Olesund, S. Thurakkal, X. Zhang and B. Albinsson, Adv. Funct. Mater., 2021, 31, 2106198 CrossRef CAS.
- H. Liang, X. Liu, L. Tang, Z. Mahmood, Z. Chen, G. Chen, S. Ji and Y. Huo, Chin. Chem. Lett., 2023, 34, 107515 CrossRef CAS.
- E. Madbak, D. J. Osborn, T. Small, T. Ishwara, T. W. Schmidt, K. Domen and G. F. Metha, Chem. Commun., 2025, 61, 157–160 RSC.
- T. Yu, Y. Liu, Y. Zeng, J. Chen, G. Yang and Y. Li, Chem. – Eur. J., 2019, 25, 16270 CrossRef CAS PubMed.
- D. C. Cabanero, J. A. Nguyen, C. S. J. Cazin, S. P. Nolan and T. Rovis, ACS Catal., 2023, 13, 4384–4390 CrossRef CAS.
- B. D. Ravetz, N. E. S. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. D. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053–2059 CrossRef CAS PubMed.
- L. R. Milgrom, The Colour of Life: An Introduction to the Chemistry of Porphyrins and Related Compounds, Oxford University Press, Oxford, UK, 1997 Search PubMed.
- R. Costae Silva, L. O. da Silva, A. de Andrade Bartolomeu, T. J. Brocksom and K. T. de Oliveira, Beilstein J. Org. Chem., 2020, 16, 917–955 CrossRef PubMed.
- J. G. Lee, J. W. Papatzimas, A. D. Bromby, E. Gorobets and D. J. Derksen, RSC Adv., 2016, 6, 59269–59272 RSC.
- K. Rybicka-Jasinska, T. Wdowik, K. Łuczak, A. J. Wierzba, O. Drapała and D. Gryko, ACS Org. Inorg. Au, 2022, 2, 422–426 CrossRef CAS PubMed.
- K. Rybicka-Jasińska, B. König and D. Gryko, Eur. J. Org. Chem., 2017, 2104–2107 CrossRef.
- K. Rybicka-Jasińska, W. Shan, K. Zawada, K. M. Kadish and D. Gryko, J. Am. Chem. Soc., 2016, 138, 15451–15458 CrossRef PubMed.
- E. L. Tyson, M. S. Ament and T. P. Yoon, J. Org. Chem., 2013, 78, 2046–2050 CrossRef CAS PubMed.
- J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275–5278 RSC.
- K. Orłowska, K. Łuczak, P. Krajewski, J. V. Santiago, K. Rybicka-Jasińska and D. Gryko, Chem. Commun., 2023, 59, 14649–14652 RSC.
- Y. Liu, K. Zhu, J. Zhao and P. Li, Org. Lett., 2022, 24, 6834–6838 CrossRef CAS PubMed.
- H.-B. Ye, X.-Y. Zhou, L. Li, X.-K. He and J. Xuan, Org. Lett., 2022, 24, 6018–6023 CrossRef CAS PubMed.
- K. Matsuzaki, T. Hiromura, E. Tokunaga and N. Shibata, ChemistryOpen, 2017, 6, 226–230 CrossRef CAS PubMed.
- M. Lancel, T. Golisano, C. Monnereau, C. Gomez, M. Port and Z. Amara, ACS Sustainable Chem. Eng., 2023, 11, 15674–15684 CrossRef CAS.
- M. Zhong and Y. Sun, Chem. Catal., 2024, 4, 100973 CAS.
- A. H. Schade and L. Mei, Org. Biomol. Chem., 2023, 11, 2472–2485 RSC.
- C. Ferroud, G. Cocquet and A. Guy, Tetrahedron Lett., 1999, 40, 5005–5008 CrossRef CAS.
- G. Cocquet, C. Ferroud and A. Guy, Tetrahedron, 2000, 56, 2975–2984 CrossRef CAS.
- G. Cocquet, C. Ferroud, P. Simon and P.-L. Taberna, J. Chem. Soc., Perkin Trans., 2000, 2, 1147–1153 RSC.
- K. Watanabe, N. Terao, I. Kii, R. Nakagawa, T. Niwa and T. Hosoya, Org. Lett., 2020, 22, 5434–5438 CrossRef CAS PubMed.
- B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS PubMed.
- I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guénée, C. Nançoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC.
- L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056–12061 CrossRef CAS PubMed.
- L. Mei, J. Moutet, S. M. Stull and T. L. Gianetti, J. Org. Chem., 2021, 86, 10640–10653 CrossRef CAS PubMed.
- S. M. Stulla, L. Meib and T. L. Gianetti, Synlett, 2022, 1194–1198 Search PubMed.
- M. M. Hossain, A. C. Shaikh, R. Kaur and T. L. Gianett, J. Am. Chem. Soc., 2024, 146, 7922–7930 CrossRef CAS PubMed.
- N. Lal, D. P. Singh and A. C. Shaikh, Chem. Commun., 2025, 61, 3005–3008 RSC.
- P. C. A. Swamy, G. Sivaraman, R. N. Priyanka, S. O. Raja, K. Ponnuvel, J. Shanmugpriya and A. Gulyani, Coord. Chem. Rev., 2020, 411, 213233 CrossRef.
- Q. An, S.-R. Xiang and Y.-Q. Zou, Pharm. Sci. Adv., 2024, 2, 100040 CrossRef CAS PubMed.
- A. R. O. Kosso, N. Sellet, A. Baralle, M. Cormier and J.-P. Goddard, Chem. Sci., 2021, 12, 6964–6968 RSC.
- N. Sellet, L. Clement-Comoy, M. Elhabiri, M. Cormier and J.-P. Goddard, Chem. – Eur. J., 2023, 29, e202302353 CrossRef CAS PubMed.
- N. Sellet, M. Sebbat, M. Elhabiri, M. Cormier and J.-P. Goddard, Chem. Commun., 2022, 58, 13759–13762 RSC.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- M. Tanioka, A. Kuromiya, R. Ueda, T. Obata, A. Muranaka, M. Uchiyama and S. Kamino, Chem. Commun., 2022, 58, 7825–7828 RSC.
- M. Tanioka, M. Oyama, K. Nakajima, M. Mori, M. Harada, Y. Matsuya and S. Kamino, Phys. Chem. Chem. Phys., 2024, 26, 4474–4479 RSC.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc.Rev., 2015, 44, 8904–8939 RSC.
- A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef CAS.
- L. Zeng, Z. Wang, T. Zhang and C. Duan, Molecules, 2022, 27, 4047 CrossRef CAS PubMed.
- B. K. Kundu, G. Han and Y. Sun, J. Am. Chem. Soc., 2023, 145, 3535–3542 CrossRef CAS PubMed.
- B. K. Kundu, C. Han, P. Srivastava, S. Nagar, K. E. White, J. A. Krause, C. G. Elles and Y. Sun, ACS Catal., 2023, 13, 8119–8127 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |