
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative cleavage of α-substituted styrenes using excited dibenzothiophene S-oxide and DMSO
Yuto
Tamba
,
Saki
Maejima
* and
Hideki
Yorimitsu
 *
*
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: maejima.saki.4j@kyoto-u.ac.jp; yori@kuchem.kyoto-u.ac.jp
First published on 6th April 2026
Abstract
Visible-light-induced oxidative cleavage of α-substituted styrenes has been achieved using a photocatalyst, dibenzothiophene S-oxide, and DMSO. The reaction proceeds via an addition of a methyl radical to the styrenic double bond, representing a mechanistically distinct pathway compared with conventional methods for oxidative cleavage that rely on toxic and/or explosive oxidants. This unique radical mechanism enables interesting chemoselectivities such as preferential oxidation of electron-deficient alkenes over electron-rich ones as well as provides an environmentally benign alternative for oxidative cleavage.
The oxidative cleavage of alkenes is a fundamental transformation in organic synthesis, providing straightforward access to carbonyl compounds from readily available alkenes. Conventional methods for the cleavage utilize toxic and/or explosive oxidants such as ozone,1 heavy-metal oxides,2 and peroxides,2h,i,3 which often raise concerns regarding safety and environmental impact (Fig. 1A). Recently, photocatalytic oxidative cleavage of alkenes using safe and easy-to-handle molecular oxygen has been reported.4 However, overoxidation and poor site-selectivity remain major limitations. Consequently, the development of new oxidation methods that are safe, mild, and highly selective remains an important topic, particularly for molecules bearing multiple olefinic moieties. In this context, Parasram4g,5 and Leonori6 independently reported oxidative cleavage of alkenes employing nitroarenes as stoichiometric oxidants (Fig. 1B). Upon photoexcitation, nitroarenes transfer oxygen atoms in an electrophilic manner via a radical pathway, which is a mechanistically distinct process that enables selective oxidation of more electron-rich alkenes. Accordingly, the development of oxidative cleavage that operates through mechanisms fundamentally different from conventional approaches is crucial for achieving site-selective alkene oxidation.
 | ||
| Fig. 1 (A) Conventional oxidative cleavage. (B) Oxidative cleavage of alkenes using photoexcited nitroarenes. (C) Our previous work. Oxomethylation of styrenes using photoexcited DBTSO. (D) This work. Oxidative cleavage of α-substituted styrenes using photoexcited DBTSO. | ||
We have recently reported that dibenzothiophene S-oxide (DBTSO) can be excited via triplet energy transfer using visible light and an Ir photocatalyst, enabling oxomethylation of styrenes via methyl radical generation (Fig. 1C).7 Mechanistic investigations revealed that the excited DBTSO undergoes rapid fragmentation in DMSO to produce a methyl radical, which adds to the styrene to generate a benzylic radical intermediate. Subsequent oxidation of this radical by the excited Ir photocatalyst affords the corresponding benzylic cation, which is trapped by DMSO to form a sulfonium intermediate. This intermediate then undergoes Swern-type oxidation to afford propiophenone.
Notably, benzaldehyde was detected as a minor side product (<10%). We presumed that this aldehyde arises from β-scission of the alkoxy radical generated by single-electron reduction of the sulfonium intermediate by the Ir photocatalyst. On this basis, we envisioned that applying the excited DBTSO/DMSO system to α-substituted styrenes would enable exclusive formation of alkoxy radicals, ultimately leading to oxidative cleavage (Fig. 1D). Herein, we report a new oxidative cleavage process initiated by methyl radical addition, utilizing the DBTSO/DMSO system.
 | ||
| Scheme 1 Oxidative cleavage of α-methylstyrene using excited DBTSO. Conditions: DBTSO (0.10 mmol, limiting agent), 1a (0.30 mmol), fac-Ir(ppy)3 (0.0050 mmol), DMSO (1.0 mL), blue LED (427 nm, 20 W), r.t., 3 h. | ||
Under similar conditions to the oxomethylation reaction,7 a solution of DBTSO (ET = 54.4 kcal mol−1), α-methylstyrene (1a, 3 equiv.), and fac-Ir(ppy)3 photocatalyst (5 mol%, ET = 58.1 kcal mol−1) in DMSO (0.1 M) was irradiated with blue LED (427 nm, 20 W) for 3 h (Scheme 1). The expected oxidative cleavage proceeded smoothly to give acetophenone (2a) in 80% yield. We confirmed that none or a small amount of 2a was observed in the absence of DBTSO, fac-Ir(ppy)3, or light irradiation.
We next investigated the scope of the oxidative cleavage of various α-substituted styrenes 1 (Scheme 2). α-Methylstyrenes 1b-1d bearing a halogen substituent at the para-position afforded the corresponding acetophenones 2 in moderate to high yields, except for p-bromo-α-methylstyrene (1d). The reaction also proceeded smoothly on a 1-mmol-scale to afford 2c in 66% yield. Strongly electron-withdrawing p-CF3 group markedly lowered the yield of 2e. Substrates 1f and 1g having an electron-donating group at the para-position gave the corresponding products 2f and 2g in moderate yields. Unfortunately, the reaction of p-methoxy-α-methylstyrene (1h) provided acetophenone 2h in only 7% yield. These results suggest that pronounced electronic perturbations relative to unsubstituted α-methylstyrene (1a) adversely affect the efficiency of the oxidative cleavage.8 For reactions that afforded moderate to low yields, one-carbon-homologation products, including 2-aryl-2-butenes and α-ethylstyrenes, were observed as side products in approximately 10–30% yields. These side products likely arose from deprotonation of the corresponding benzylic cations.
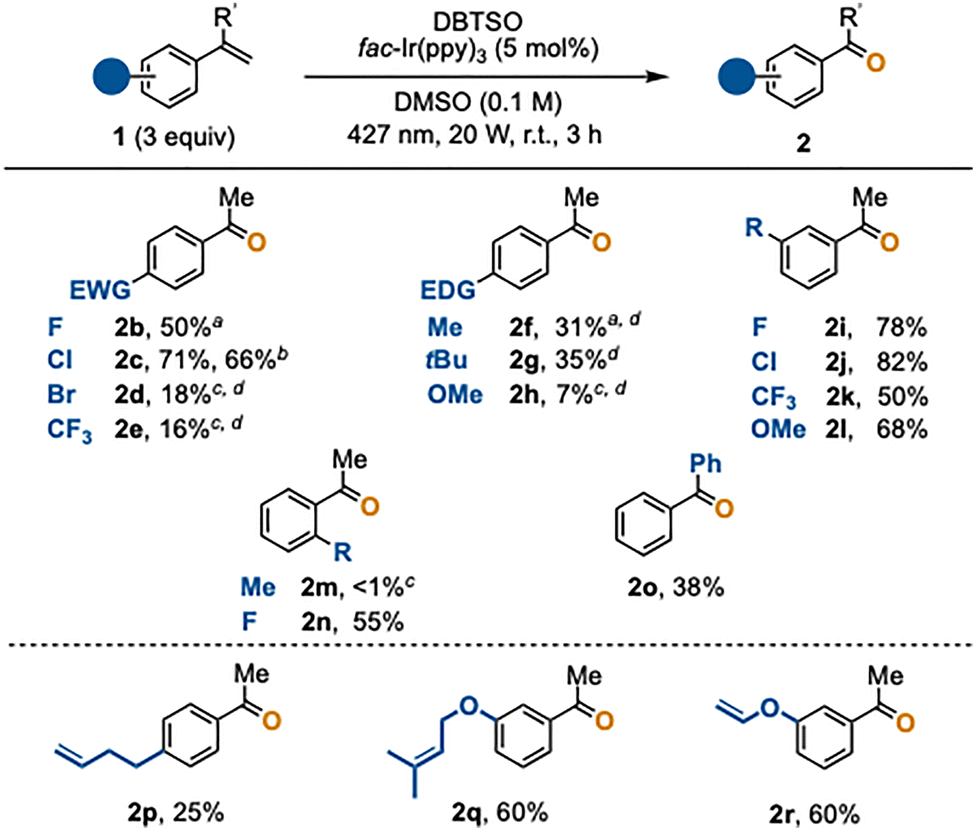 | ||
| Scheme 2 Oxidative cleavage of various α-substituted styrene 1. Standard conditions: DBTSO (0.20 mmol, limiting agent), 1 (0.60 mmol), fac-Ir(ppy)3 (0.010 mmol), DMSO (2.0 mL), blue LED (427 nm, 20 W), r.t., 3 h. aPerformed on half the scale of the standard conditions with the same reaction time of 3 h. bPerformed on a 1 mmol scale (5 × standard scale) with two LED lamps and the reaction time of 5 h. cNMR yield. d2-Aryl-2-butenes and α-ethylstyrenes were observed as side products (ca. 10–30% yields). | ||
In the case of meta-substituted substrates, where electronic perturbation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is minor, the oxidative cleavage proceeded smoothly to afford the corresponding acetophenones 2i–2l in good yields (50–82%). On the other hand, o-methyl-α-methylstyrene (1m) did not react, probably because the conjugation between the CC bond and the phenyl group is hindered. In contrast, o-fluoro-α-methylstyrene (1n), which imposes less steric demand, underwent the reaction smoothly to give 2n in 55% yield. Benzophenone (2o) was obtained in 38% yield when 1,1-diphenylethylene (1o) was used.
C bond is minor, the oxidative cleavage proceeded smoothly to afford the corresponding acetophenones 2i–2l in good yields (50–82%). On the other hand, o-methyl-α-methylstyrene (1m) did not react, probably because the conjugation between the CC bond and the phenyl group is hindered. In contrast, o-fluoro-α-methylstyrene (1n), which imposes less steric demand, underwent the reaction smoothly to give 2n in 55% yield. Benzophenone (2o) was obtained in 38% yield when 1,1-diphenylethylene (1o) was used.
Remarkably, substrates 1p, 1q, and 1r, each bearing two distinct olefinic moieties, selectively gave acetophenones 2p, 2q, and 2r, leaving the non-arylated CC bonds intact. This high site-selectivity is due to the higher reactivity of the phenylated CC bonds toward the radical addition of a methyl radical (Fig. 1C). It is worth mentioning that, based on a survey of the literature, research on conventional oxidative cleavage lacks general information on site-selectivity and that one could suffer from poor selectivity in performing site-selective oxidative cleavage. The present methyl-radical-based approach therefore offers unique selectivity,4g–6,9 being complementary to or advantageous over established oxidative cleavage.2–4
As shown in Scheme 2, the yields were strongly influenced by the electronic properties of the α-methylstyrenes. To further clarify the site-selectivity of this transformation, we conducted a competition experiment using two α-methylstyrenes with contrasting electronic properties (Scheme 3A). When a mixture of electron-deficient p-chloro-α-methylstyrene (1c) and electron-rich p-methoxy-α-methylstyrene (1h) was subjected to our photoinduced DBTSO/DMSO system, oxidative cleavage occurred preferentially at 1c, affording an 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 selectivity in favor of the electron-deficient alkene. This result is consistent with the substrate scope shown in Scheme 2 and demonstrates that the present method selectively oxidizes electron-deficient CC bonds. For comparison, the same substrate mixture was oxidized using ruthenium tetroxide2f (Scheme 3B). In line with the well-established behavior of conventional oxidative cleavage reactions,2 the more electron-rich alkene of 1h was preferentially oxidized (2c:2h = 21:79). Thus, the DBTSO-based photoinduced system exhibits a clear reversal of selectivity relative to traditional oxidative cleavage methods. These results suggest that the site-selectivity of the present oxidative cleavage is governed by the preferential addition of a nucleophilic methyl radical to the more electron-deficient CC bond, highlighting a mechanistic origin distinct from that of conventional electrophilic oxidation processes.
:11 selectivity in favor of the electron-deficient alkene. This result is consistent with the substrate scope shown in Scheme 2 and demonstrates that the present method selectively oxidizes electron-deficient CC bonds. For comparison, the same substrate mixture was oxidized using ruthenium tetroxide2f (Scheme 3B). In line with the well-established behavior of conventional oxidative cleavage reactions,2 the more electron-rich alkene of 1h was preferentially oxidized (2c:2h = 21:79). Thus, the DBTSO-based photoinduced system exhibits a clear reversal of selectivity relative to traditional oxidative cleavage methods. These results suggest that the site-selectivity of the present oxidative cleavage is governed by the preferential addition of a nucleophilic methyl radical to the more electron-deficient CC bond, highlighting a mechanistic origin distinct from that of conventional electrophilic oxidation processes.
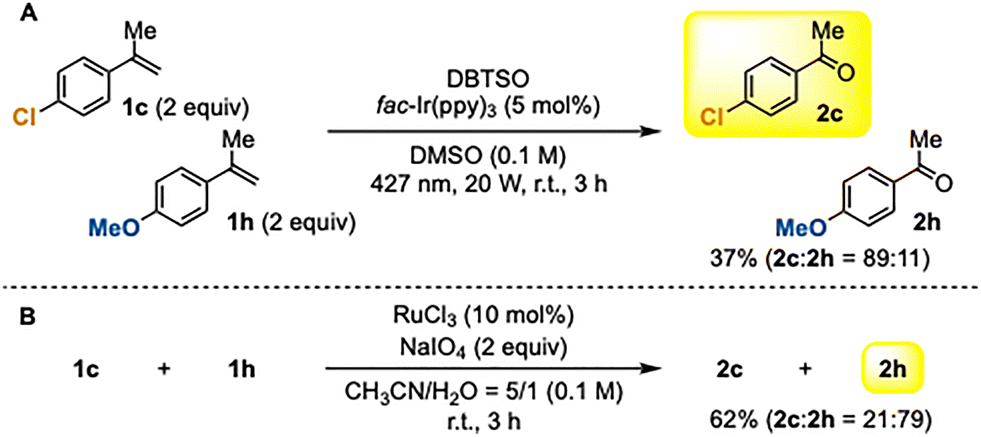 | ||
| Scheme 3 Competition experiments. (A) Oxidative cleavage by our photoinduced DBTSO/DMSO system. (B) Oxidative cleavage using ruthenium tetroxide. | ||
To further validate our mechanistic working hypothesis, several control experiments were performed. Addition of 1 equivalent of TEMPO completely suppressed the oxidative cleavage, affording methylated TEMPO 3 in 18% yield (Fig. 2A). This result is consistent with our previous study,7 supporting the essential role of the generation of methyl radicals. When 18O-labeled DBTSO was employed in the oxidative cleavage of 1a (Fig. 2B), only negligible incorporation of 18O into 2a was observed (18O:16O = 6:94) as previously noted in the previous oxomethylation.7 Given that the reaction was conducted entirely under an inert atmosphere, the oxygen atom in 2a most reasonably originated from DMSO. To determine whether the reaction proceeds via a radical chain or a non-chain process, a light on/off experiment was performed (Fig. 2C), wherein irradiation was alternately turned on and off every 10 min. No changes in the amounts of 2a, DBTSO, and DBT were observed during the light-off periods, which indicates that the oxidative cleavage reaction does not proceed via a radical chain mechanism.
 | ||
| Fig. 2 (A) Trapping of methyl radical. (B) 18O-labeling experiment using DBTSO-18O. (C) Light on/off experiment. (D) Investigation of the selectivity of β-scission. (E) Plausible mechanism. | ||
When 2-phenyl-1-hexene (1s) was subjected to the standard conditions (Fig. 2D), valerophenone (2s) and propiophenone (2s′) were formed in 30% and 12% yields, respectively. This product distribution supports the generation of a tertiary alkoxy radical via single-electron reduction of the corresponding sulfonium intermediate (also depicted in Fig. 1C) and subsequent β-scission of the alkoxy radical intermediate. Importantly, elimination of an ethyl radical was slightly favored over that of a butyl radical, in agreement with the previous report.10
Based on these mechanistic experiments, the site-selectivity observed in Schemes 2 and 3, and our previous report,7 we propose the reaction mechanism illustrated in Fig. 2E. Following a pathway analogous to that described in Fig. 1C, the reaction should proceed via the formation of the corresponding sulfonium intermediate 4. This intermediate is then reduced by the Ir(II) species via single-electron transfer to form alkoxy radical 5,11 which subsequently undergoes β-scission to afford acetophenone 2 with predominant release of the much more stable ethyl radical over a methyl radical.
In summary, we have developed photoinduced oxidative cleavage of α-substituted styrenes mediated by excited DBTSO. The reaction proceeds through generation of a methyl radical from the reaction between excited DBTSO and DMSO, followed by selective addition of the radical to the more electron-deficient styrenic CC bond, thereby enabling site-selective oxidative cleavage. This selectivity contrasts with conventional oxidative cleavage methods, which typically oxidize electron-rich alkenes preferentially. The distinct methyl-radical-based mechanism underlying this transformation provides a new strategy for controlling site-selectivity in alkene oxidation and is expected to be applicable to the selective functionalization of complex molecules bearing multiple olefinic moieties.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: general information, experimental procedure, characterization data, NMR spectra, together with References. See DOI: https://doi.org/10.1039/d6cc00617e.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP24K17679 and the Mitsui Chemicals Award in Synthetic Organic Chemistry, Japan (to S. M.) and by JSPS KAKENHI Grant Number JP24H02208 in Transformative Research Areas (A) JP24A202 Integrated Science of Synthesis by Chemical Structure Reprogramming (SReP) and JST CREST Grant Number JPMJCR19R4 (Innovative Reactions) (to H. Y.). We thank to Flugelz Co., Ltd, JAPAN, to provide the amine-modified monolithic silica column.References
- (a) P. S. Bailey, Chem. Rev., 1958, 58, 925 CrossRef CAS; (b) R. Criegee, Angew. Chem., Int. Ed. Engl., 1975, 14, 745 CrossRef; (c) T. Veysoglu, L. A. Mitscher and J. K. Swayze, Synthesis, 1980, 807 CrossRef CAS; (d) S. G. V. Ornum, R. M. Champeau and R. Pariza, Chem. Rev., 2006, 106, 2990 CrossRef PubMed.
- (a) R. Pappo, D. S. Allen, Jr., R. U. Lemieux and W. S. Johnson, J. Org. Chem., 1956, 21, 478 CrossRef CAS; (b) K. B. Wiberg and K. A. Saegebarth, J. Am. Chem. Soc., 1957, 79, 2822 CrossRef CAS; (c) L. M. Berkowitz and P. N. Rylander, J. Am. Chem. Soc., 1958, 80, 6682 CrossRef CAS; (d) P. Viski, Z. Szeverényi and L. I. Simándi, J. Org. Chem., 1986, 51, 3213 CrossRef CAS; (e) V. Piccialli, D. M. A. Smaldone and D. Sica, Tetrahedron, 1993, 49, 4211 CrossRef CAS; (f) D. Yang and C. Zhang, J. Org. Chem., 2001, 66, 4814 CrossRef CAS PubMed; (g) K. Miyamoto, N. Tada and M. Ochiai, J. Am. Chem. Soc., 2007, 129, 2772 CrossRef CAS PubMed; (h) A. Rajagopalan, M. Lara and W. Kroutil, Adv. Synth. Catal., 2013, 355, 3321 CrossRef CAS; (i) P. Spannring, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. K. Gebbink, Catal. Sci. Technol., 2014, 4, 2182 RSC.
- (a) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC; (b) D. J. Lippincott, P. J. Trejo-Soto, F. Gallou and B. H. Lipshutz, Org. Lett., 2018, 20, 5094 CrossRef CAS PubMed; (c) Z. Huang, X. Qi, J.-F. Lee and A. Lei, Organometallics, 2018, 37, 1635 CrossRef CAS; (d) T. Cousin, G. Chatel, N. Kardos, B. Andrioletti and M. Draye, Catal. Sci. Technol., 2019, 9, 5256 RSC.
- (a) A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994 CrossRef CAS PubMed; (b) G. Urgoitia, R. SanMartin, M. T. Herrero and E. Domínguez, ACS Catal., 2017, 7, 3050 CrossRef CAS; (c) Y. Zhang, N. Hatami, N. S. Lange, E. Ronge, W. Schilling, C. Jooss and S. Das, Green Chem., 2020, 22, 4516 RSC; (d) P. Xie, C. Xue, J. Luo, S. Shi and D. Du, Green Chem., 2021, 23, 5936 RSC; (e) Z. Huang, R. Guan, M. Shanmugam, E. L. Bennett, C. M. Robertson, A. Brookfield, E. J. L. McInnes and J. Xiao, J. Am. Chem. Soc., 2021, 143, 10005 CrossRef CAS PubMed; (f) Y.-L. Shih, Y.-K. Wu, M. Hyodo and I. Ryu, J. Org. Chem., 2023, 88, 6548 CrossRef CAS PubMed; (g) W. A. Hussain and M. Parasram, Synthesis, 2024, 1775 CAS.
- (a) D. E. Wise, E. S. Gogarnoiu, A. D. Duke, J. M. Paolillo, T. L. Vacala, W. A. Hussain and M. Parasram, J. Am. Chem. Soc., 2022, 144, 15437 CrossRef CAS PubMed; (b) T. Patra and T. Wirth, Angew. Chem., Int. Ed., 2022, 61, e202213772 CrossRef CAS PubMed.
- A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022, 610, 81 CrossRef CAS PubMed.
- S. Maejima, B. Kinoshita, R. Wakabayashi, M. Abe and H. Yorimitsu, Org. Lett., 2025, 27, 11678 CrossRef CAS PubMed.
- J. Ballesteros-Soberanas, C. Bilanin and A. Leyva-Pérez, ACS Org. Inorg. Au, 2023, 3, 13 CrossRef CAS.
- Oxidative cleavage of alkenes based on a different radical mechanism: K. Liao, Y. Fang, L. Sheng, J. Chen and Y. Huang, Nat. Commun., 2024, 15, 6227 CrossRef CAS PubMed.
- J. D. Bacha and J. K. Kochi, J. Org. Chem., 1965, 30, 3272 CrossRef CAS.
- (a) H. Zhao, D. Filippini, Y. Chen, A. Gallego-Gamo, L. S. Natrajan, L. R. E. Pantaine, C. Romano and D. J. Procter, Nat. Chem., 2025 DOI:10.1038/s41557-025-02003-7; (b) Y. Mao, X. Zhang, W.-Y. Shi, H. Guo and R. Zhou, Chem. Sci., 2026, 17, 1771 RSC.
| This journal is © The Royal Society of Chemistry 2026 |