
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switchable N–H vs. C3–H carboxylation of indoles using dual-function reagents
Katherine E.
Marris
 a,
Jazmine T.
Thorne
a,
Daniel J.
Ryder-Mahoney
a,
Ryan A.
Bragg
b,
Charles S.
Elmore
c and
Gregory J. P.
Perry
*a
a,
Jazmine T.
Thorne
a,
Daniel J.
Ryder-Mahoney
a,
Ryan A.
Bragg
b,
Charles S.
Elmore
c and
Gregory J. P.
Perry
*a
aSchool of Chemistry and Chemical Engineering, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: gregory.perry@soton.ac.uk
bEarly Chemical Development, Pharmaceutical Sciences, R&D, AstraZeneca, Cambridge, UK
cEarly Chemical Development, Pharmaceutical Sciences, R&D, AstraZeneca, Boston, USA
First published on 25th February 2026
Abstract
The carboxylation of indoles through a CO2 transfer reaction with carboxylate salts is described. By altering the reaction conditions either N–H or C3–H carboxylation can occur. The reaction is also applicable to the carboxylation of other indole derivatives and amines. The relevance of this procedure is further demonstrated through the preparation and carbon isotope labelling of several biologically relevant carboxylated indoles/amines.
Indoles are privileged structures in organic synthesis and widely used amongst various disciplines, from dyes and perfumes to medicines, agrochemicals and materials.1 Their ubiquity has spawned a strong interest in the development of methods for functionalization around the aromatic ring. In particular, the direct functionalization of C–H bonds presents an idealized route for indole modification.2
The carboxylation of indoles provides access to key motifs present in pharmaceuticals and natural products.3 For example, N–H carboxylation affords carbamates which are pro-drug elements in many pharmaceuticals.4 Alternatively, C–H carboxylation provides indole carboxylic acids, which form the core of several medicines.5 Considering current literature, the site selective carboxylation of indoles has thus far taken a stepwise approach in which distinct reaction conditions are required for carboxylating the various reactive sites around the indole ring.6,7 Authors have also been keen to discuss the difficulties associated with directing functionalisation between the N–H and C3–H positions.8,9 One of the most effective methods for the N–H carboxylation of indoles has recently been reported by Hopmann, Repo and co-workers (Scheme 1A).6d In this work, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was found to effect the N–H carboxylation of indoles 1 under relatively mild conditions (40 °C, 1 atm) and an excess of cesium carbonate (4.0 equiv.) to give the carbamate 2. However, directing the reactivity towards C3–H carboxylation was not investigated. Indeed, we were also unable to demonstrate C3–H carboxylation under similar conditions in our laboratory.10 Conversely, Kobayashi and co-workers developed a C3–H selective carboxylation of indoles 1 using an excess of lithium tert-butoxide (5.0 equiv.) to give indole carboxylic acid 3 (Scheme 1B).7b,7c The reaction again proceeded under an atmospheric pressure of CO2, though a higher temperature (100 °C) was required, presumably to overcome the higher energy barrier required for C3–H carboxylation. Notably, the Kobayashi group also demonstrated an N–H carboxylation to give carbamate 2, but the process was low yielding (15%).10 A unified approach for N–H vs C3–H carboxylation therefore presents a significant challenge.
 | ||
| Scheme 1 (A) and (B): previous N–H and C3–H carboxylations of indoles. (C) Previous carboxylations using dual-function reagents. (D) This work: N–H and C3–H carboxylation of indoles using dual-function reagents. | ||
We have recently demonstrated that carboxylates, for example the triphenylacetate salt 4, are able to promote the carboxylation of arenes bearing C–H bonds via CO2 transfer (Scheme 1C).11–13 We described carboxylate 4 as a dual-function reagent as it provided a combined source of base and CO2 for the reaction. We therefore questioned whether this reactivity could be applied to the carboxylation of indoles 1 (Scheme 1D). In this process, carboxylate 4 would initially undergo decarboxylation to provide the trityl anion 5 and CO2 (step i). The highly basic trityl anion 5 (pKa of Ph3CH = 30.6)14 would then be capable of deprotonating the indole 1 (pKa = 21.0)14 to deliver the intermediate 6 (step ii). At this point, we proposed to tune the conditions to deliver selective N–H or C3–H carboxylation. In this way, the deprotonated indole 6 would react with the in situ generated CO2 to give either N–H or C3–H carboxylated products 2 or 3. Here we report a unified approach to indole carboxylation and demonstrate the applicability of this carboxylation to other amine classes and carbon isotope labelling.
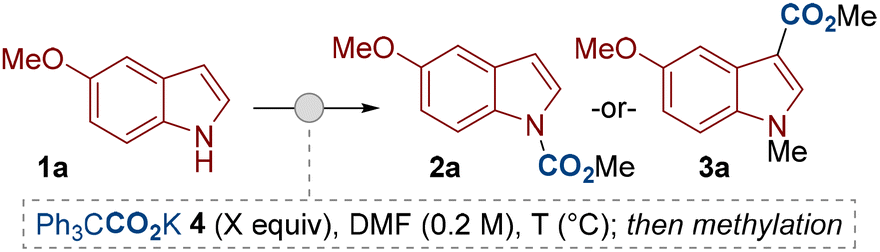
We began our study by subjecting indole 1a to our previously reported reaction conditions at 50 °C.11a The potassium salt of triphenylacetic acid 4 was used as the dual-function reagent to provide the source of base and CO2. (Table 1). After routine methylation with methyl iodide, the N-carboxylated product 2a was isolated in high yield (entry 1). Aware of the interest in directing functionalization around the indole ring, we were keen to explore the possibility of a related C3–H carboxylation. Indeed, by performing the reaction at higher temperature (100 °C) the C3-carboxylated product 3a was observed (entry 2). Optimal conditions for C3–H carboxylation were achieved by using 2.0 equivalents of the dual-function reagent 4 and by increasing the reaction temperature to 140 °C (entry 4). We chose to proceed with using 2.0 equivalents of the carboxylating agent 4 as our standard conditions for C3–H carboxylation as we were satisfied with the good yields this achieved with most substrates (vide infra), though higher yields can be achieved by further increasing the equivalents of 4 (entry 5). The C3-carboxylated product was isolated as the methylated derivative 3a to ease isolation and avoid costly deuterated solvents during analysis (e.g. acetone-d6), however, we have demonstrated that the free indole-3-carboxylic acid 3aH can be isolated if desired (entry 6). Other variables, such as other metal salts of 4 and solvents were also tested during optimisation, but no improvements in reactivity were observed.15
|
|
||||
|---|---|---|---|---|
| Entry | Ph3CCO2K 4 (equiv.) | T (°C) | 2a (%)a | 3a (%)a |
| DMF = N,N-dimethylformamide.a NMR yields using 1,1,2,2-tetrachloroethane as an internal standard.b Isolated yields.c The remaining mass balance was largely recovered starting material (observed as the free indole or N-methylindole). Indole recovery: entry 2 = 28%, entry 3 = 12%., entry 4 = 4%.d n.d. = not determined.e The product was isolated as the indole-3-carboxylic acid 3aH. After carboxylation, the methylation step was not performed. Instead, the reaction was quenched with 1 M HCl (aq.). See the SI for further details. | ||||
| 1 | 1.1 | 50 | 94 (87)b | 0 |
| 2c | 1.1 | 100 | 38 | 25 |
| 3c | 2.0 | 100 | 44 | 35 |
| 4c | 2.0 | 140 | 20 | 67 (66)b |
| 5 | 3.0 | 140 | 7 | 85 |
| 6 | 2.0 | 140 | n.d.d | 65be |
To gain some insight into the reaction mechanism, we subjected indole 1a to the standard conditions for N–H carboxylation in which the carbamate intermediate 2a′ presumably forms (Scheme 2, step i). However, instead of performing the alkylation step towards isolating the N–H carboxylated product 2a, we added additional Ph3CCO2K 4 and heated to 140 °C to mimic the C3–H carboxylation conditions (Scheme 2, step ii). This produced the same yields of 2a and 3a compared to our standard C3–H carboxylation conditions (c.f. Table 1, entry 4). This suggests that N–H carboxylation is a reversible process and that the C3–H carboxylated product 3a can be generated from the carbamate 2a′. Under N–H carboxylation conditions, the carbamate intermediate 2a′ can be trapped by the alkylating agent to give 2a. Presumably, more electron-rich substrates are better at N–H carboxylation as they push the carboxylation/decarboxylation equilibrium towards carbamate 2a′ (e.g. compare the yields of 2a and 2gin in Scheme 3). Under C3–H carboxylation conditions, we suggest that N–H carboxylation can occur, however this is a reversible process that is superseded by irreversible C3–H carboxylation at higher temperatures. The higher temperature for C3–H carboxylation is presumably required as the process involves an electrophilic aromatic substitution-type mechanism in which aromaticity is disturbed.
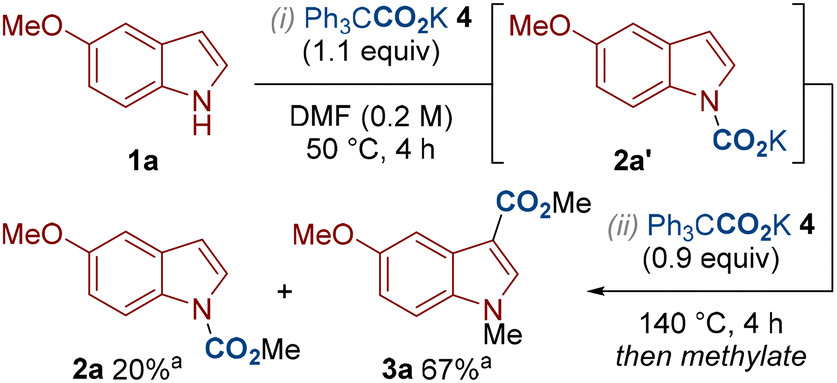 | ||
Scheme 2 Mechanistic investigation. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NMR yields using 1,1,2,2-tetrachloroethane as an internal standard. NMR yields using 1,1,2,2-tetrachloroethane as an internal standard. | ||
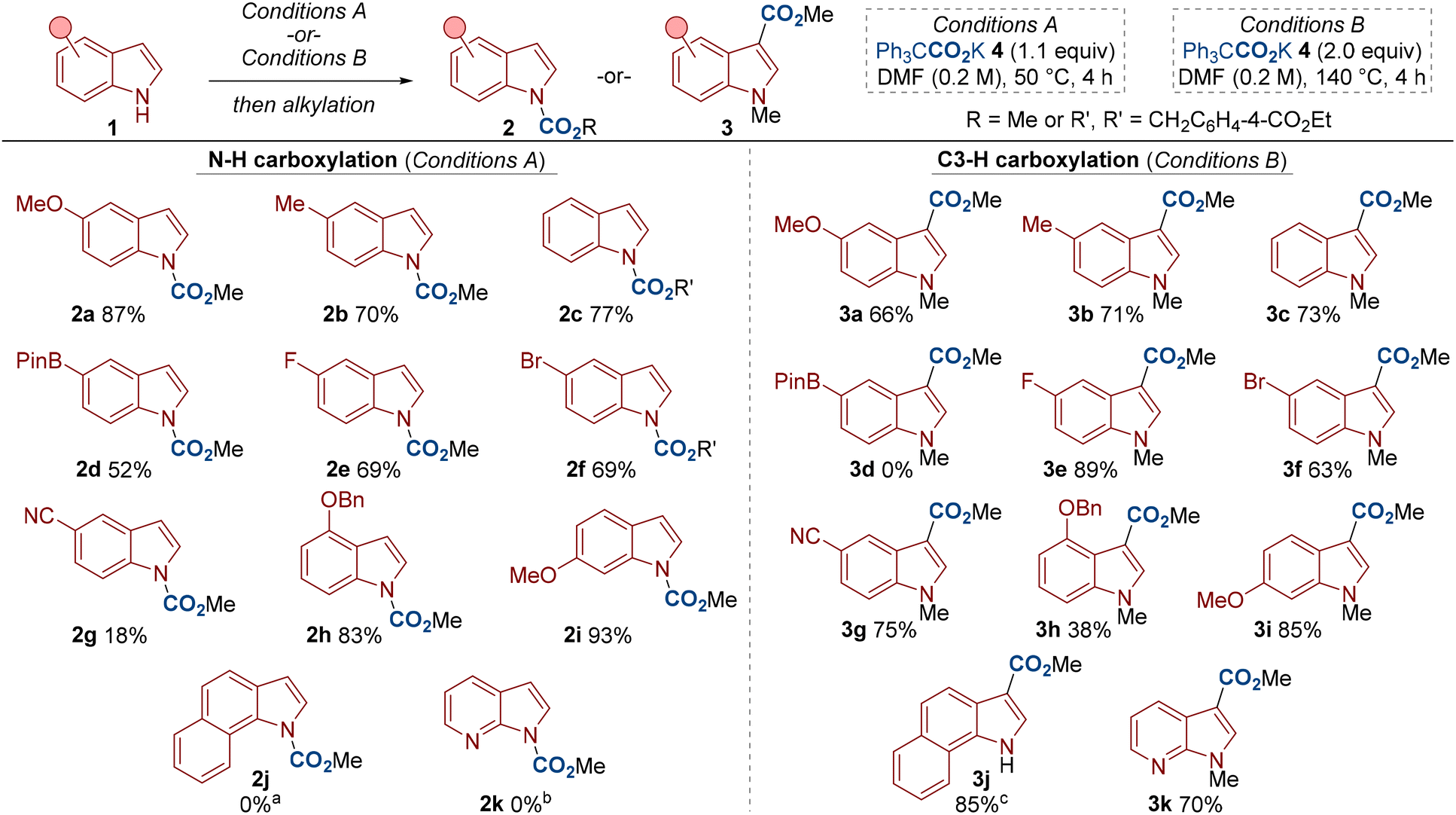 | ||
| Scheme 3 Scope of the N–H and C3–H carboxylation of indoles. a2j was not formed. Instead, C3–H carboxylation was observed, see compound 3j. bThe mass recovery was largely N-methylated indole (67%). cConditions A were used. | ||
With optimized conditions in hand, we went about investigating the scope of the reaction (Scheme 3). A range of electron-donating and electron-withdrawing substituents were compatible for both N–H and C3–H carboxylation, including halogen and cyano groups (see 1e–1g). For N–H carboxylation, an indole bearing a Bpin (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) substituent also gave respectable yield (see 2d). The N–H carboxylation with 5-cyanoindole 1g and 7-azaindole 2k were low yielding. We suggest that the lower nucleophilicity of these substrates disfavours carbamate formation, in agreement with studies by Repo and Hoppman.6d,16 Nonetheless, 5-cyanoindole 1g proved a good substrate for C3–H carboxylation to give 3g. Substituents at other positions around the indole ring were also compatible in both methods (see 1h and 1i). Finally, although indoles 1j and 1k did not undergo N–H carboxylation (to give 2j/2k), good yields of C3–H carboxylation were observed (see 3j/3k). Interestingly, C3–H carboxylated product 3j formed in excellent yield at 50 °C and with 1.1 equivalents of carboxylating agent 4. The high yield of 3k was also satisfying as 7-azaindole is a privileged motif in medicinal chemistry.17
We were keen to further test the limits of this methodology in related indole carboxylations (Scheme 4). We therefore demonstrated a C2–H carboxylation of indole 7.18 We have also applied our methodology to the carboxylation of 2-alkynylindole 9 to give the 6-endo-dig cyclized product 10.19 Notably, the formation of compound 10 previously required higher temperatures (100 °C vs. 50 °C).19a
 | ||
| Scheme 4 Other carboxylations of indoles. | ||
To further illustrate the generality of our procedure, we have also conducted a series of N–H carboxylations with various amines 11 (Scheme 5).20 Electron-rich and electron-deficient substrates, and anilines bearing ortho, meta and para substituents all displayed respectable reactivity (12a–12f). Secondary amine 11g and benzylamine 11h were also compatible.
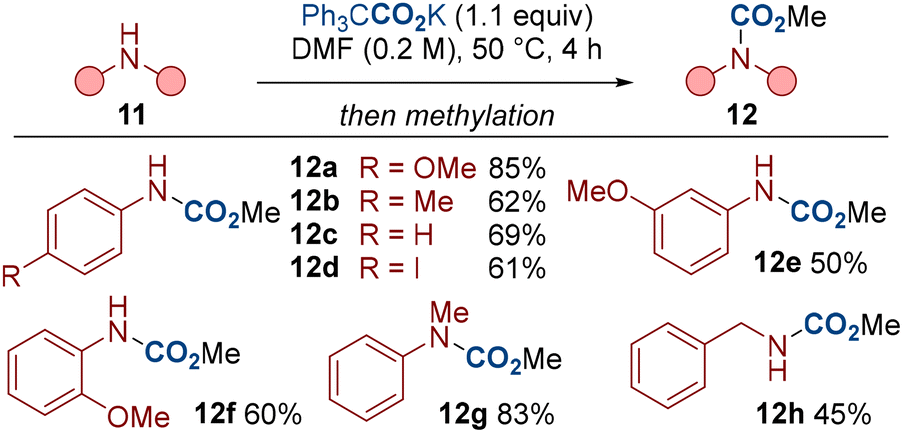 | ||
| Scheme 5 N–H carboxylation of amines. | ||
Our method has shown that a range of indoles and other amines can undergo carboxylation with near equimolar quantities of the carboxylating agent 4. This presents a useful strategy for carbon isotope labelling in which limiting the equivalents of the labelling reagent holds significant advantages.21 For example, labelled CO2 gases (e.g.14CO2, 13CO2) are more expensive and less available than non-labelled CO2 (for example, 14CO2 > £1000 mmol−1).22 The carboxylating reagent 4 is also a bench stable solid, thereby presenting practicality benefits over gaseous labelled reagents. We have therefore carboxylated a range of biologically relevant compounds, including the isotope labelled compounds 14d* and 3c-H* by using the labelled dual-function reagent 4* (Scheme 6). We also note that no erosion in the enantiomeric excess was observed in the preparation of the tryptophan derivative 14b, highlighting the mild conditions we have developed.
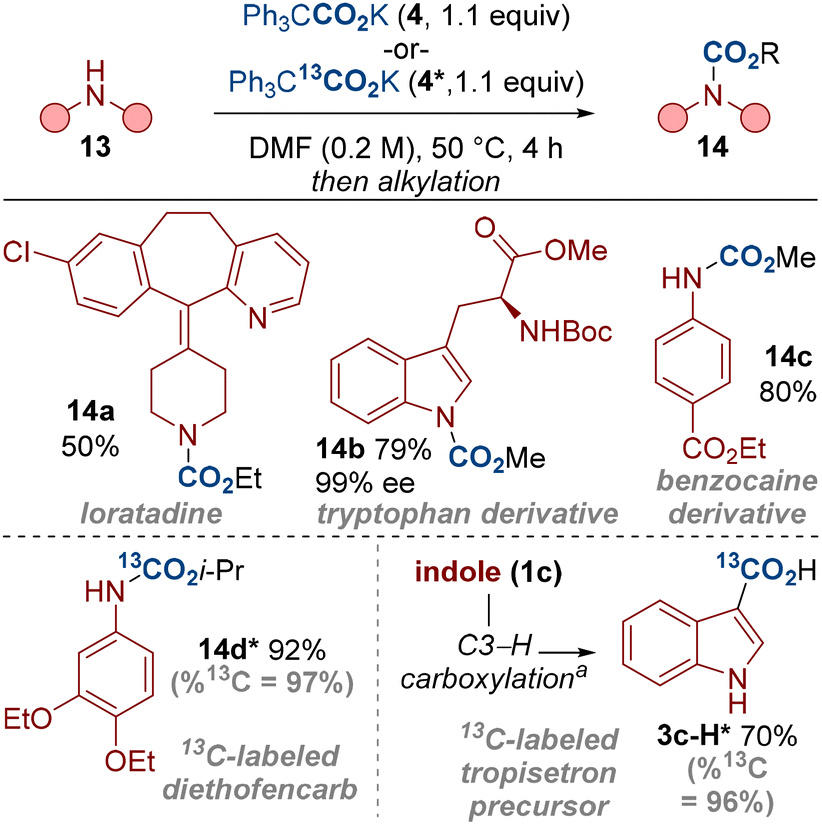 | ||
| Scheme 6 Carboxylation and isotope labelling of biologically relevant molecules. | ||
In summary, we have developed a CO2 transfer reaction of indoles and other amines using the dual-function reagent 4 as a combined source of base and CO2. Indoles can undergo N–H carboxylation or C3–H carboxylation depending on the reaction conditions. The carboxylation was also applicable to other amines and related carboxylation reactions with other indole architectures. Finally, we have applied this method to the incorporation of carbon isotopes into biologically relevant molecules using low equivalents of the labelled carboxylating reagent.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6cc00532b.Acknowledgements
This work was supported by a Royal Society Research Grant (RGS\R2\242219), an RSC Research Fund (R24-6800702932), an EPSRC New Investigator Award (UKRI1101/APP24094) and an EPSRC ICASE award in collaboration with AstraZeneca (WT8946489). We thank Dr Julie Herniman and Dr Neil J. Wells for leading the mass spectrometry and NMR spectroscopy facilities within the University of Southampton.References
- (a) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed; (b) Y. Wan, Y. Li, C. Yan, M. Yan and Z. Tang, Eur. J. Med. Chem., 2019, 183, 111691 Search PubMed; (c) D. Paul and J. John, Chem. Asian J., 2022, 17, e202200460 Search PubMed.
- (a) J. Kalepu, P. Gandeepan, L. Ackermann and L. T. Pilarski, Chem. Sci., 2018, 9, 4203–4216 Search PubMed; (b) K. Urbina, D. Tresp, K. Sipps and M. Szostak, Adv. Synth. Catal., 2021, 363, 2723–2739 Search PubMed; (c) J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723–1736 CrossRef CAS PubMed; (d) P. Kushwaha, Rashi and A. Bhardwaj, ChemistrySelect, 2025, 10, e02256 CrossRef CAS.
- (a) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (b) W. Schilling and S. Das, ChemSusChem, 2020, 13, 6246–6258 CrossRef CAS PubMed; (c) Y. Luo and W. Huang, Org. Biomol. Chem., 2023, 21, 8628–8641 Search PubMed.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 Search PubMed.
- (a) K. Simpson, C. M. Spencer and K. J. McClellan, Drugs, 2000, 59, 1297–1315 CrossRef CAS PubMed; (b) L. J. Scott and C. M. Perry, Drugs, 2000, 60, 1411–1444 CrossRef CAS PubMed; (c) J. Blaising, S. J. Polyak and E.-I. Pécheur, Antiviral Res., 2014, 107, 84–94 Search PubMed.
- For select examples of N–H carboxylation of indoles see: (a) D. L. Boger and M. Patel, J. Org. Chem., 1987, 52, 3934–3936 CrossRef CAS; (b) R. P. Davies, P. R. Raithby and R. Snaith, Organometallics, 1996, 15, 4355–4356 CrossRef CAS; (c) A. Ueno, Y. Kayaki and T. Ikariya, Organometallics, 2014, 33, 4479–4485 CrossRef CAS; (d) J. K. Mannisto, L. Pavlovic, J. Heikkinen, T. Tiainen, A. Sahari, N. M. Maier, K. Rissanen, M. Nieger, K. H. Hopmann and T. Repo, ACS Catal., 2023, 13, 11509–11521 CrossRef CAS.
- For select examples of C3–H carboxylation of indoles see: (a) K. Nemoto, S. Onozawa, N. Egusa, N. Morohashi and T. Hattori, Tetrahedron Lett., 2009, 50, 4512–4514 Search PubMed; (b) W.-J. Yoo, M. G. Capdevila, X. Du and S. Kobayashi, Org. Lett., 2012, 14, 5326–5329 Search PubMed; (c) S. Kobayashi, W.-J. Yoo, T. V. Q. Nguyen and M. Guiteras Capdevila, Heterocycles, 2015, 90, 1196 Search PubMed; (d) K. Nemoto, S. Tanaka, M. Konno, S. Onozawa, M. Chiba, Y. Tanaka, Y. Sasaki, R. Okubo and T. Hattori, Tetrahedron, 2016, 72, 734–745 CrossRef CAS; (e) L. C. Chetty, H. G. Kruger, P. I. Arvidsson, T. Naicker and T. Govender, Synthesis, 2022, 4827–4833 Search PubMed.
- For example, see ref. 6a, 6d and K. Xu, T. Gilles and B. Breit, Nat. Commun., 2015, 6, 7616 Search PubMed.
- For select reviews and examples of regioselective indole functionalization see: (a) N. Ahlsten, X. C. Cambeiro, G. J. P. Perry and I. Larrosa, in Au-Catalyzed Synthesis and Functionalization of Heterocycles, ed. M. Bandini, Springer International Publishing, Cham, 2016, vol. 46, pp. 175–226 Search PubMed; (b) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CAS; (c) M. Petrini, Chem. – Eur. J., 2017, 23, 16115–16151 CAS; (d) Y. Cheng, S. Yu, Y. He, G. An, G. Li and Z. Yang, Chem. Sci., 2021, 12, 3216–3225 CAS; (e) J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723–1736 CAS; (f) Y.-S. Yang, S. Lee, S. H. Son, H.-S. Yoo, Y. H. Jang, J.-W. Shin, H.-J. Won, J. Sim and N.-J. Kim, Org. Chem. Front., 2022, 9, 5906–5911 Search PubMed; (g) H. Xie, Y. Cheng and G. A. Chin, J. Org. Chem., 2025 DOI:10.6023/cjoc202510028.
- Attempting C3–H carboxylation using Hopmann, Repo and co-workers’ conditions (ref. 6d) at 140 °C gave only 8% of product 3a. Attempting N–H carboxylation using Kobayashi and co-workers’ conditions (ref 7b) at 50 °C gave only 50% product of 2a. See the supporting information for further details.
- (a) S. Wang, I. Larrosa, H. Yorimitsu and G. J. P. Perry, Angew. Chem., Int. Ed., 2023, 62, e202218371 CAS; (b) X. Liu, G. J. P. Perry and D. Kong, Angew. Chem., Int. Ed., 2026, 65, e22503 Search PubMed; (c) D. J. Ryder-Mahoney, K. Yamazaki and G. J. P. Perry, JACS Au, 2026, 6, 773–778 CAS; (d) The work presented in this manuscript was uploaded to ChemRxiv prior to submission: K. Marris, J. Thorne, D. Ryder-Mahoney, R. Bragg, C. Elmore and G. J. P. Perry, ChemRxiv, 2026 DOI:10.26434/chemrxiv-2026-6f3m5.
- For related CO2 transfer reactions see: (a) L.-L. Liao, G.-M. Cao, Y.-X. Jiang, X.-H. Jin, X.-L. Hu, J. J. Chruma, G.-Q. Sun, Y.-Y. Gui and D.-G. Yu, J. Am. Chem. Soc., 2021, 143, 2812–2821 CrossRef CAS PubMed; (b) X. Liu, S. Cao, C. Zhang, Y. Jiang and D. Kong, Org. Lett., 2024, 26, 8967–8972 CrossRef CAS PubMed; (c) B. M. Colella and J. Ohata, Synlett, 2025, 719–723 CAS; (d) K. Shimotai, O. Sasamoto and M. Shigeno, Org. Lett., 2025, 27, 352–356 CrossRef CAS PubMed; (e) X. Liu, H. Wang and D. Kong, Chem. Synth., 2025 DOI:10.20517/cs.2025.51; (f) During the submission of this manuscript, Kong and co-workers reported a related carboxylation: X. Liu, Z. Li, C. Song, G. Xu and D. Kong, ChemRxiv, 2026 DOI:10.26434/chemrxiv-2026-023mg.
- For reports on related base-mediated carboxylation see: (a) O. Vechorkin, N. Hirt and X. Hu, Org. Lett., 2010, 12, 3567–3569 CrossRef CAS PubMed; (b) S. Fenner and L. Ackermann, Green Chem., 2016, 18, 3804–3807 RSC; (c) S. Felten, C. Q. He, M. Weisel, M. Shevlin and M. H. Emmert, J. Am. Chem. Soc., 2022, 144, 23115–23126 Search PubMed; (d) M. Shigeno, K. Hanasaka, I. Tohara, K. Izumi, H. Yamakoshi, E. Kwon, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2022, 24, 809–814 CrossRef CAS PubMed; (e) M. Shigeno, I. Tohara, K. Sasaki, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2022, 24, 4825–4830 CrossRef CAS PubMed.
- The stated pKa values are in DMSO. See the tables made by F. G.Bordwell, H. J. Reich, D. H. Ripin and D. A. Evans available via the ACS organic division website https://organicchemistrydata.org/.
- See the supporting information for further details. NCCH2CO2K and EtO2CCH2CO2K were tested as alternative dual-function reagents. NCCH2CO2K showed poor reactivity in both N–H and C3–H carboxylation. EtO2CCH2CO2K was poorly reactive in the N–H carboxylation, but did show comparable reactivity to Ph3CCO2K in the C3–H carboxylation to give 60% of 3a.
- In the N–H carboxylation of 2g and 2k a significant amount of N-methylated indole was observed (59% and 67%) suggesting that deprotonation occurs (Scheme 1D, step ii), but not the carboxylation (Scheme 1D, step iii).
- (a) T. Irie and M. Sawa, Chem. Pharm. Bull., 2018, 66, 29–36 CrossRef CAS PubMed; (b) D. R. Motati, R. Amaradhi and T. Ganesh, Bioorg. Med. Chem., 2020, 28, 115830 CrossRef CAS PubMed.
- (a) T. Suga, H. Mizuno, J. Takaya and N. Iwasawa, Chem. Commun., 2014, 50, 14360–14363 RSC; (b) A. Ueno, M. Takimoto, W. N. O. Wylie, M. Nishiura, T. Ikariya and Z. Hou, Chem. – Asian J., 2015, 10, 1010–1016 CrossRef CAS PubMed; (c) C. Pei, J. Zong, S. Han, B. Li and B. Wang, Org. Lett., 2020, 22, 6897–6902 CrossRef CAS PubMed; (d) T. M. Porter and M. W. Kanan, Chem. Sci., 2020, 11, 11936–11944 RSC; (e) M. Shigeno, I. Tohara, K. Nozawa-Kumada and Y. Kondo, Eur. J. Org. Chem., 2020, 1987–1991 CrossRef CAS ; For select examples of carboxylations proceeding through classical C2-lithiation/magnesiation then carboxylation see: ; (f) R. J. Sundberg and H. F. Russell, J. Org. Chem., 1973, 38, 3324–3330 CrossRef CAS; (g) A. R. Katritzky and K. Akutagawa, Tetrahedron Lett., 1985, 26, 5935–5938 CrossRef CAS; (h) D. J. Hlasta and M. R. Bell, Heterocycles, 1989, 29, 849 CrossRef CAS.
- (a) Z. Xin, C. Lescot, S. D. Friis, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2015, 54, 6862–6866 Search PubMed; (b) S. Uema, K. Saito and T. Yamada, Org. Lett., 2022, 24, 4831–4834 CrossRef CAS PubMed.
- For select examples of N–H carboxylation beyond indoles see ref. 3b and: (a) I. I. F. Boogaerts, G. C. Fortman, M. R. L. Furst, C. S. J. Cazin and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674–8677 CrossRef CAS PubMed; (b) D. Riemer, P. Hirapara and S. Das, ChemSusChem, 2016, 9, 1916–1920 CrossRef CAS PubMed; (c) Y. Ren and S. A. L. Rousseaux, J. Org. Chem., 2018, 83, 913–920 CrossRef CAS PubMed; (d) Y. Guo, L. Wei, Z. Wen, C. Qi and H. Jiang, Acta Phys.-Chim. Sin., 2024, 40, 2307004 CrossRef; (e) J. K. Mannisto, L. Pavlovic, T. Tiainen, M. Nieger, A. Sahari, K. H. Hopmann and T. Repo, Catal. Sci. Technol., 2021, 11, 6877–6886 RSC.
- (a) R. Voges, J. R. Heys and T. Moenius, Preparation of Compounds Labeled with Tritium and Carbon-14, Wiley, 1st edn, 2009 CrossRef; (b) C. S. Elmore and R. A. Bragg, Bioorg. Med. Chem. Lett., 2015, 25, 167–171 CrossRef CAS PubMed; (c) R. A. Bragg, M. Sardana, M. Artelsmair and C. S. Elmore, J. Labelled Compd. Radiopharm., 2018, 61, 934–948 Search PubMed; (d) K. Hinsinger and G. Pieters, Angew. Chem., Int. Ed., 2019, 58, 9678–9680 Search PubMed; (e) V. Derdau, C. S. Elmore, T. Hartung, B. McKillican, T. Mejuch, C. Rosenbaum and C. Wiebe, Angew. Chem., Int. Ed., 2023, 62, e202306019 CrossRef CAS PubMed.
- A. Talbot, A. Sallustrau, A. Goudet, F. Taran and D. Audisio, Synlett, 2022, 171–176 CAS.
| This journal is © The Royal Society of Chemistry 2026 |