
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction of dearomatized heterocycles with sulfur hexafluoride (SF6) and the pentafluorosulfanyl (SF5) group
Emily K.
Burke
and
Alexander W. H.
Speed
 *
*
Department of Chemistry, Dalhousie University, Halifax, Nova Scotia B3H 4R2, Canada. E-mail: aspeed@dal.ca
First published on 3rd March 2026
Abstract
A Hantzsch ester and other dearomatized heterocycles undergo aromatization in the presence of sulfur hexafluoride (SF6) upon irradiation with 390 nm LEDs, reducing SF6 and releasing fluoride ion. Two aryl SF5 compounds are reduced under the same conditions. No added photocatalyst is necessary for these transformations.
Sulfur hexafluoride (SF6) and the pentafluorosulfanyl (SF5) group are highly oxidized sulfur species traditionally thought of as highly unreactive. The pentafluorosulfanyl (SF5) group is a bulky, electron-withdrawing fluorinated substituent, often described as a super-CF3 group, similarly chemically robust, but with enhanced electronegativity, lipophilicity, and larger size.1 While the challenging introduction of the SF5 group has limited exploration, new introduction protocols are resulting in increasing investigation of the SF5 group and its chemistry.2 As more research groups explore SF5 groups, unexpected reactivity patterns may emerge, and SF5 may prove to be an attractive replacement for CF3 in some molecules as regulatory restrictions around fluorocarbon use grows.
SF6 is an attractive starting material for SF5 chemistry, as it is inexpensive, and is non-toxic, compared with more commonly used SF5 precursors such as SF5Cl. Processes that consume SF6 are also of special interest for environmental reasons, as it is used as a dielectric in the electrical industry, despite having a global warming potential over 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times that of CO2 per unit mass.3 Accordingly, decomposition of surplus SF6 is of interest, even if SF5-containing products do not result from the process. SF6 chemistry has been challenging, despite these attractions, due to its low reactivity. A pioneering example from Beier added SF5 to styrenes from SF6 in the presence of TEMPO-lithium in low yield,4 while Hoge reported decomposition of SF6 with weakly-coordinated phenoxides.5 The most prominent example of SF6 functionalization to SF5 are works from Wagenknecht and Rombach, where phenyl phenothiazine (PTZ) photocatalysts were used to oxidatively add SF5 to diphenylethylene, and methylstyrene derivatives (Scheme 1 and eqn (1)).6
000 times that of CO2 per unit mass.3 Accordingly, decomposition of surplus SF6 is of interest, even if SF5-containing products do not result from the process. SF6 chemistry has been challenging, despite these attractions, due to its low reactivity. A pioneering example from Beier added SF5 to styrenes from SF6 in the presence of TEMPO-lithium in low yield,4 while Hoge reported decomposition of SF6 with weakly-coordinated phenoxides.5 The most prominent example of SF6 functionalization to SF5 are works from Wagenknecht and Rombach, where phenyl phenothiazine (PTZ) photocatalysts were used to oxidatively add SF5 to diphenylethylene, and methylstyrene derivatives (Scheme 1 and eqn (1)).6
 | ||
| Scheme 1 Selected chemistry of SF6. | ||
During the course of our work, a preprint from Wang's group reported conditions using the PTZ catalyst for pentafluorosulfanylation beyond styrene derivatives.7 Other processes to consume SF6 without generating organic SF5 groups have been reported recently, predominantly using nitrogen and phosphorus compounds. Reduction of SF6 by N-heterocyclic carbenes,8 diaminoethylenes and tetra aminoethylenes,9 in some cases stopping at the SF5 anion have been reported by Braun, Rueping, Kirsch, and Tlili (eqn (2)). Alternatively, Dielmann's group has reported the reduction of SF6 with electron-rich phosphines and irradiated triphenylphosphine (eqn (3)).10 Our group has also showed metal phosphanides are reactive with SF6.11 Recent examples of SF6 consumption also include a series of reactions with photocatalysts using SF6 as a deoxyfluorination reagent.12 Dielmann very recently showed even isopropanol can cause an autocatalytic degradation of SF6 under energetic 280 nm light.13
Photoexcited Hantzsch esters and their conjugate bases have been reported as potent reductants without external photocatalysts by various groups for dehalogenations,14 radical cyclization reactions,15 and reduction of CF3 groups.16 Being interested in SF6 reactivity, we sought to explore if photoexcited Hantzsch esters and related compounds were reactive with SF6. The redox potential  of the photoexcited anion of Hantzsch ester 1a has been reported by Xu and co-workers as −3.094V vs. Fc+/Fc (Fc = ferrocene)in acetonitrile (MeCN).16 A potential of −2.17 V vs. Fc+/Fc to reduce SF6 has been measured in MeCN by Goncalves, Magnier, and co-workers, when SF6 was decomposed electrochemically in a cell at a constant potential of −2.3 V vs. Fc+/Fc.17 These values suggest that the photoexcited Hantzsch ester anion is sufficiently reducing to transfer electrons to SF6.
of the photoexcited anion of Hantzsch ester 1a has been reported by Xu and co-workers as −3.094V vs. Fc+/Fc (Fc = ferrocene)in acetonitrile (MeCN).16 A potential of −2.17 V vs. Fc+/Fc to reduce SF6 has been measured in MeCN by Goncalves, Magnier, and co-workers, when SF6 was decomposed electrochemically in a cell at a constant potential of −2.3 V vs. Fc+/Fc.17 These values suggest that the photoexcited Hantzsch ester anion is sufficiently reducing to transfer electrons to SF6.
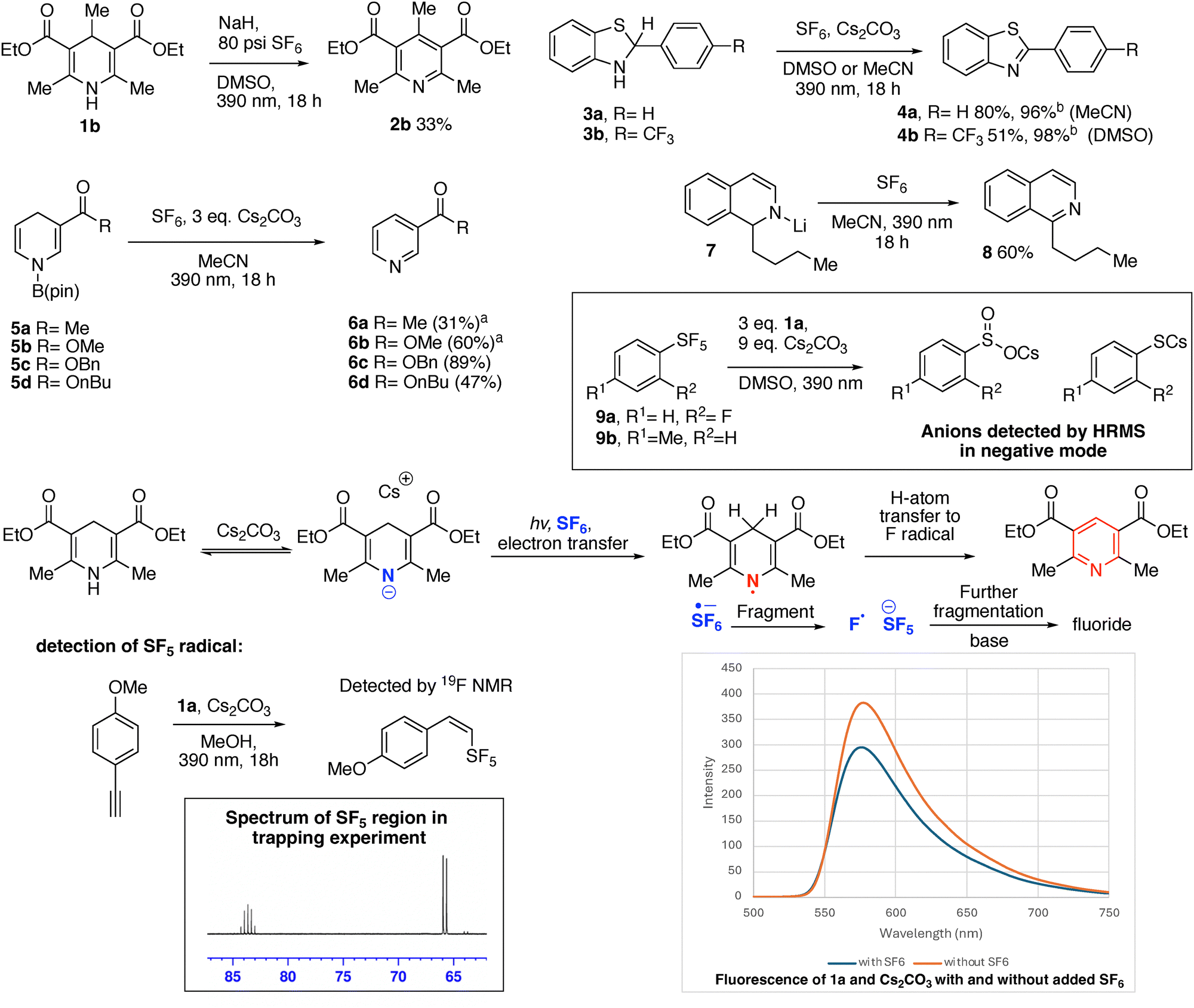
We observed that Hantzsch ester 1a was cleanly oxidized to the corresponding pyridine 2a by SF6, in MeCN in the presence of base, with attendant formation of a large fluoride signal observed in the 19F NMR spectrum (Scheme 2). Product 2a could readily be isolated by hexanes extraction and column chromatography. During the preparation of this manuscript, a pre-print from Rombach and Wagenknecht appeared, showing the pentafluorosulfanylation of alkynes from SF6 using an iridium phenylpyridine photocatalyst with Hantzsch esters as the terminal reductant, which also leads to 2a. However, the work reported here is distinct, as no added photocatalyst is required.18
 | ||
| Scheme 2 Reaction of SF6 with a Hantzsch esters (a) reaction conducted with 80 psi SF6. | ||
Both MeCN (entry 1) and toluene (entry 2), gave limited yield of 2a. Switching to DMSO, in which the Hantzsch ester has greater solubility, led to a more efficient reaction (entry 3). Cesium carbonate was the most efficient base (Entry 3), with potassium carbonate (entry 4) and sodium hydride (entry 5), giving reduced yields. Hypothesizing complete reduction of SF6 could give as many as six fluorides per equivalent of Hantzsch ester, an increase of the amount of base to three equivalents to provide six cesium counterions provided a slight increase in yield of 2a (entry 6).
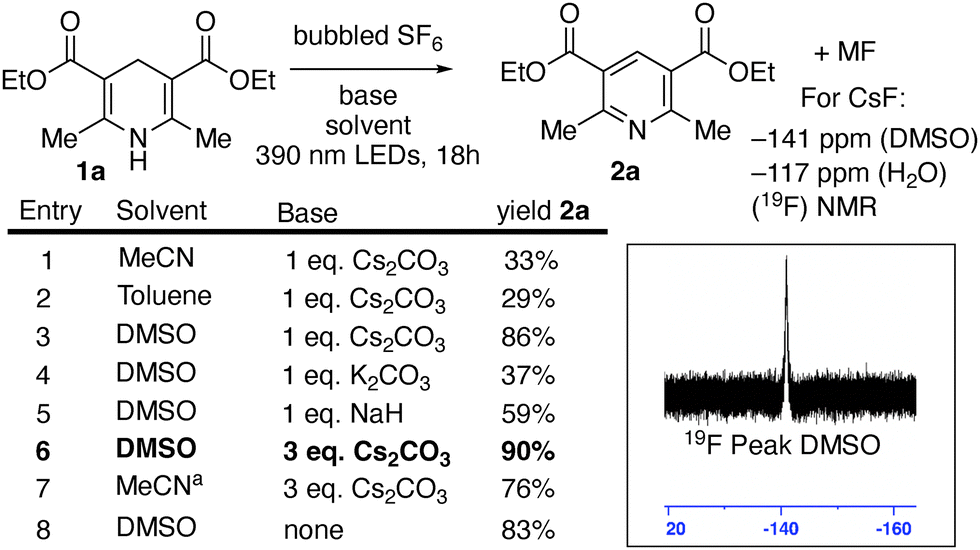
We speculated that the high solubility of the Hantzsch ester and moderate solubility of cesium carbonate in DMSO might explain the superiority of the optimal system. While MeCN had not been an ideal solvent in entry 1, conducting the reaction in MeCN under an increased pressure of SF6 (80 psi), resulted in an increase of the yield of 2a to 76% (entry 7). Interestingly, the Hantzsch ester was also oxidized by SF6 in DMSO under irradiation in the absence of base (entry 8). A strong HF peak was observed in 19F NMR spectroscopy of that crude reaction mixture.19 The photoexcited neutral Hantzsch ester 1a has been reported to have  vs. Fc+/Fc in MeCN, corroborating the feasibility of this electron transfer.16 While the isolated yield of 2a was slightly lower than with three equivalents of cesium carbonate, it was superior to the yields obtained in entries 5 and 6, where alternate bases were explored. We explored the reaction of other dearomatized compounds (Scheme 3). More hindered and less reactive Hantzsch ester 1b showed some aromatization to 2b, but forcing conditions of NaH base and 80 psi SF6 were required for good conversion. Pitre and co-workers observed that 4-substituted dihydropyridines were less reactive in reduction reactions, potentially due to steric hinderance slowing hydrogen atom transfer from the 4-position.14e
vs. Fc+/Fc in MeCN, corroborating the feasibility of this electron transfer.16 While the isolated yield of 2a was slightly lower than with three equivalents of cesium carbonate, it was superior to the yields obtained in entries 5 and 6, where alternate bases were explored. We explored the reaction of other dearomatized compounds (Scheme 3). More hindered and less reactive Hantzsch ester 1b showed some aromatization to 2b, but forcing conditions of NaH base and 80 psi SF6 were required for good conversion. Pitre and co-workers observed that 4-substituted dihydropyridines were less reactive in reduction reactions, potentially due to steric hinderance slowing hydrogen atom transfer from the 4-position.14e
 | ||
| Scheme 3 Substrate scope and proposed mechanism (a) Yield determined by 1H NMR and internal standard (b) Yield without base. | ||
Akiyama has shown that benzothiazolines can replace Hantzsch esters in reductive processes with carbonyls and imines.20 We found that benzothiazolines 3a and 3b were also oxidized to the corresponding benzothiazoles 4a and 4b, upon reaction with SF6.21 Reactivity both with and without cesium carbonate was observed, with cleaner and higher yielding reactions in the absence of base. Borylated dihydropyridines (5a–5d) underwent oxidation to the pyridines 6a–6d in the presence of SF6 in MeCN solvent with added cesium carbonate, however no productive reactivity was observed in the absence of cesium carbonate.22 Fluoride was observed bound to boron by 19F NMR in the crude reaction mixtures of these compounds. This shows the dual electron-withdrawing groups of the Hantzsch esters are unnecessary for this reactivity, and raises the possibility that someday nicotinamide (NAD-H)-dependent enzymes may be purposed to have reactivity with SF6.23
Meisenheimer intermediate 7, prepared by addition of butyllithium to isoquinoline, is reported by Gualandi, Negri, Ceroni, and Cozzi to be a potent photoreductant (with a reported 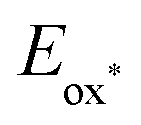 of < −3.8 V vs. SCE, therefore < −4.2 V vs. Fc+/Fc).24 Compound 7 was oxidized to the corresponding butyl-isoquinoline under SF6 atmosphere in MeCN with 390 nm irradiation.25
of < −3.8 V vs. SCE, therefore < −4.2 V vs. Fc+/Fc).24 Compound 7 was oxidized to the corresponding butyl-isoquinoline under SF6 atmosphere in MeCN with 390 nm irradiation.25
We extended this reactivity to the SF5 group. Commercially available o-fluorophenyl 9a and p-tolyl SF59b underwent reduction using 1a and cesium carbonate in DMSO or MeCN. Fluoride was again observed in the crude 19F NMR spectra, while the corresponding arylsulfinate and thiophenolate anions were detected by high-resolution mass-spectrometry. Only sluggish reaction between 9a and 1a under irradiation was observed in the absence of base, with some formation of fluoride, but the SF5 group remained substantially intact after a three-day irradiation, suggesting photoexcited 1a has insufficient reducing power to reduce aryl SF5 without deprotonation. DBU could also be used as a base rather than cesium carbonate to obtain complete decomposition of the SF5 group with 1a in an 18-hour irradiation. Several new SF5-introduction reagents have recently been disclosed, which will increase the accessibility of the SF5 group.26 Our results with Hantzsch esters should be considered in the design of reductive transformations in the presence of the SF5 group. While the SF5 group is considered chemically robust, its decomposition has been reported in a few limited examples, including by Lewis acids and bismuth-based reductive catalysts.27,28 Akiyama recently reported the reduction of aryl SF5 compounds bearing an additional electron withdrawing group to thiolates using potassium iodide via a charge transfer complex.29 Aryl SF5 reduction has also been reported in electrochemical devices.30
Based on the catalyst-free photoreductive behavior of Hantzsch esters, and precedent from solution phase reduction of SF6, we propose that the photoexcited anion of 1a (or photoexcited neutral 1a species in reactions without base) transfers an electron to SF6 or the pentafluorosulfanyl group under irradiation.31 This could be followed by fragmentation of the resulting sulfur-radical anion. Fragmentation to either fluorine radical and SF5 anion, or to fluoride and SF5 radical is feasible, with higher excess electron energy favouring the latter pathway.6a,13 Subsequent hydrogen atom transfer from the Hantzsch ester radical to the fluorine radical would aromatize the ring. The SF5 anion is fragile without a diffuse supporting counter-cation and readily releases fluoride and SF4.32 If the initial fragmentation to form SF5 radical occurs, the Hantzsch system is likely also reducing enough to reduce the SF5 radical to the SF5 anion, with subsequent fragmenation.5,9b SF4 would likely react with base present under the reaction conditions, and we did not detect partially fluorinated intermediates such as SOF2 (thionyl fluoride). We did detect hydrosulfite (HSO3−) anion in the crude reaction mixtures by low-resolution mass-spectrometry.33 We observed no significant change in the wavelength in the fluorescence spectrum corresponding to the maxima of fluorescence intensity 1a and cesium carbonate in the presence or absence of SF6, suggesting a charge transfer complex is not formed, however a decrease in fluorescence intensity of a mixture of 1a and cesium carbonate was observed when the mixture was sparged with SF6.34 Evidence for the formation of the SF5 radical intermediate in a different solvent was obtained by adding ethynylanisole as a SF5 radical trap to the reaction mixture in methanol with base, leading to the detection of a SF5-containing product. Ethynylanisole did not form any appreciable trapping product in MeCN or DMSO, nor was any SF5 trapping product observed in methanol in the absence of base. α-methylstyrene could also trap the SF5 radical in MeCN, but formed a complex mixture of products. Due to the observed instability of aryl-SF5 groups under the reaction conditions, it is unlikely this system will readily result in a preparative method for SF5 compounds.
In conclusion, we show that photoexcited Hantzsch esters and related dearomatized compounds undergo oxidation in the presence of SF6. Given the growing interest in SF6 chemistry, and introduction of the SF5 group, these findings may suggest new approaches to decomposing SF6, or alternatively, cautionary precedent for undesirable reactions that may occur in the design of radical reactions with Hantzsch esters or related reductants in the presence of the SF5 group.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: synthetic procedures, NMR Data, UV/Vis studies. See DOI: https://doi.org/10.1039/d5cc07159c.Acknowledgements
This work was supported by NSERC of Canada through a Discovery Grant (2024-04054), the Net Zero Atlantic Emerging Concepts & Technologies Nova Scotia Research Program “Electricity System Challenges Post 2030” Funding Stream, and MITACS Accelerate. The Canada Foundation for Innovation John R. Evans Leaders Fund (39824 and 43840) and Research Nova Scotia Research Opportunities Fund (2020-1208 and 2023-2797) are thanked for infrastructure funding. Dr Michael Lumsden (Dalhousie University NMR-3 Core Facility) and Mr Xiao Feng (Dalhousie University Chemistry Mass Spectrometry Core Facility) are thanked for assistance with NMR spectroscopy and mass spectrometry, respectively. Professor Alison Thompson and Dr Liandrah Gapare (Dalhousie University) are thanked for the use of UV equipment. Professor Rachel Chang (Dalhousie University) and Professor Nadine Borduas-Dedekind (University of British Columbia) are thanked for suggestions on the use and sourcing of gas-sampling bags to hold and dispense SF6.References
- P. R. Savoie and J. T. Welch, Chem. Rev., 2015, 115, 1130 CrossRef CAS PubMed.
- (a) Y. Yang, L. Han, L. Brettnacher, L. Canavero and A. Tlili, Chem. Rev., 2025, 125, 8426 CrossRef CAS PubMed; (b) T. M. Nguyen, F. René, V. Bizet and D. Cahard, Chem. Soc. Rev., 2026, 55, 1333 RSC.
- S. Shi, Y. Li, Z. Cui, Y. Yan, X. Zhang, J. Tang and S. Xiao, Chem. Eng. J., 2023, 470, 144166 CrossRef CAS.
- G. Iakobson, M. Pošta and P. Beier, J. Fluorine Chem., 2018, 213, 51 CrossRef CAS.
- R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Chem. – Eur. J., 2021, 27, 6460 CrossRef CAS PubMed.
- (a) D. Rombach and H.-A. Wagenknecht, ChemCatChem, 2018, 10, 2955 CrossRef CAS; (b) D. Rombach and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2020, 59, 300 CrossRef CAS PubMed; (c) S. Klehenz, H. Kucher and H.-A. Wagenknecht, Eur. J. Org. Chem., 2025, e202500478 CrossRef CAS; (d) D. Rombach and H.-A. Wagenknecht, Synthesis, 2022, 4883 CrossRef CAS.
- C.-H. Jiang, H. Xu, Y.-G. Yang, M.-M. Zheng, Y. Zhao, Y.-W. Zuo, W.-R. Ren, S. Zhu, R.-X. Jin, X.-S. Xue and X.-S. Wang, ChemRxiv, 2025, preprint, chemrxiv-2025-230kr DOI:10.26434/chemrxiv-2025-230kr.
- P. Tomar, T. Braun and E. Kemnitz, Chem. Commun., 2018, 54, 9753 RSC.
- (a) P. Kirsch, G.-V. Röschenthaler, D. Sevenrard and A. Kolometisev, DE10220901A1, 2023; (b) M. Rueping, P. Nikolaienko, Y. Lebedev and A. Adams, Green Chem., 2017, 19, 2571 RSC; (c) A. Taponard, T. Jarrosson, L. Khrouz, M. Médebielle, J. Broggi and A. Tlili, Angew. Chem., Int. Ed., 2022, 61, e202204623 CrossRef CAS PubMed.
- (a) F. Buß, C. Mück-Lichtenfeld, P. Mehlmann and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 2018 Search PubMed; (b) P. Rotering, C. Mück-Lichtenfeld and F. Dielmann, Green Chem., 2022, 24, 8054 RSC.
- B. S. N. Huchenski and A. W. H. Speed, Chem. Commun., 2021, 57, 7128 RSC.
- (a) T. A. McTeague and T. F. Jamison, Angew. Chem., Int. Ed., 2016, 55, 15072 CrossRef CAS PubMed; (b) Y.-L. Huang, Q.-Q. Zhang, C.-Y. Wang, Y. Zhao and X.-S. Wang, Org. Lett., 2024, 26, 5776 CrossRef CAS PubMed; (c) Y.-X. Bi, Y. Zhao, Q. Q. Zhang, X.-Y. Guo, J.-C. Dai, R.-X. Jin and X.-S. Wang, Eur. J. Org. Chem., 2024, e202400451 CrossRef CAS; (d) Y. Zhao, F. Ma, Y. Chen, S. Gu, F. Zhu, J. Cao, S. Zhu and L.-G. Xie, Org. Biomol. Chem., 2025, 23, 1094 RSC.
- A. Sietmann, P. Heinzel, J. Gamper, D. Leitner, L. C. Pasqualini, F. R. S. Purtscher, H. Kopacka, T. S. Hofer, A. Zemann and F. Dielmann, Nat. Commun., 2026, 17, 465 CrossRef CAS PubMed.
- (a) W. Chen, H. Tao, W. Huang, G. Wang, S. Li, X. Cheng and G. Li, Chem. – Eur. J., 2016, 22, 9546 CrossRef CAS PubMed; (b) D.-L. Zhu, Q. Wu, H.-Y. Li, H.-X. Li and J.-P. Lang, Chem. – Eur. J., 2020, 26, 3484 CrossRef CAS PubMed; (c) M. O. Konev, L. Cardinale and A. Jacobi von Wangelin, Org. Lett., 2020, 22, 1316 CrossRef CAS PubMed; (d) G. S. Yedase, S. Venugopal and V. R. Yatham, Asian J. Org. Chem., 2022, 11, e202200478 CrossRef; (e) P. C. Gallage, M. G. McKee and S. P. Pitre, Org. Lett., 2024, 26, 1975 CrossRef CAS PubMed.
- J. Xu, Y. Lan and B. Liu, J. Org. Chem., 2024, 89, 599 CrossRef CAS PubMed.
- N. Li, J.-L. Si and M.-Y. Xu, Chem. – Eur. J., 2025, 31, e202404116 CrossRef CAS PubMed.
- S. Bouvet, B. Pegot, S. Sengmany, E. Le Gall, E. Leonel, A.-M. Goncalves and E. Magnier, Beilstein J. Org. Chem., 2020, 16, 2948 CrossRef CAS PubMed.
- M. Flügge, S. Klehenz, S. Leidenheimer, D. Rombach and H.-A. Wagenknecht, JACS Au, 2026, 6, 965 Search PubMed.
- M. Hudlicky, J. Fluorine Chem., 1985, 28, 461 CrossRef CAS.
- (a) C. Zhu and T. Akiyama, Org. Lett., 2009, 11, 4180 CrossRef CAS PubMed; (b) T. Uchikura, N. Kamiyama, T. Mouri and T. Akiyama, ACS Catal., 2022, 12, 5209 CrossRef CAS; (c) C. Zhu, K. Saito, M. Yamanaka and T. Akiyama, Acc. Chem. Res., 2015, 48, 388 CrossRef CAS PubMed.
- J. Chen, W. Huang, Y. Li and X. Cheng, Adv. Synth. Catal., 2018, 360, 1466 CrossRef CAS.
- T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Organometallics, 2018, 37, 841 CrossRef CAS.
- (a) T. Hyster, Synlett, 2020, 248 CAS; (b) A. Bhunia and A. Studer, Chemistry, 2021, 7, 2060 CrossRef CAS.
- F. Calogero, L. Wilczek, E. Pinosa, A. Gualandi, R. Dorta, A. Herrera, Y. Dai, A. Rossignol, F. Negri, Z. Ziani, A. Fermi, P. Ceroni and P. G. Cozzi, Angew. Chem., Int. Ed., 2024, 63, e202411074 CrossRef CAS PubMed.
- Lithium fluoride has limited solubility, and was not detected. See: D. A. Wynn, M. M. Roth and B. D. Pollard, Talanta, 1984, 31, 1036 CrossRef CAS PubMed.
- (a) Y. Yang, L. Han, A. Taponard, L. Khrouz, C. Bucher, C. Monnereau, M. Médebielle and A. Tlili, Angew. Chem., Int. Ed., 2025, 65, e202505146 Search PubMed; (b) R. Li, C. Hu., C. Liu, T. Lyness, W. Li, C.-Y. Cai, E. Crossley, Y. Kanda, R. R. Merchant, B. S. Matsuura, N. S. Williams and T. Qin, J. Am. Chem. Soc., 2025, 147, 34218 CrossRef CAS PubMed; (c) J.-Y. Shou and F.-L. Qing, Chin. J. Chem., 2025, 43, 3033 CrossRef CAS.
- L. Köring, A. Stepen, B. Birenheide, S. Barth, M. Leskov, R. Schoch, F. Krämer, F. Breher and J. Paradies, Angew. Chem., Int. Ed., 2023, 62, e202216959 CrossRef PubMed.
- V. A. Béland, N. Nöthling, M. Leutzsch and J. Cornella, J. Am. Chem. Soc., 2024, 146, 25409 Search PubMed.
- T. Uchikura, F. Akutsu and T. Akiyama, Chem. Commun., 2025, 61, 6328 Search PubMed.
- H. Gao, A. R. Sevilla, G. M. Hobold, A. M. Melemed, R. Guo, S. C. Jones and B. M. Gallant, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2121440119 CrossRef CAS.
- M.-Z. Jin, L. Yang, L.-M. Wu, Y.-C. Liu and Z.-L. Liu, Chem. Commun., 1998, 2451 RSC.
- K. Matsumoto, Y. Haruki, S. Sawada, S. Yamada, T. Konno and R. Hagiwara, Inorg. Chem., 2018, 57, 14882 CrossRef CAS PubMed.
- The m/z ratio of sulfite (SO32−) fell below the range accessible on our instrumentation. Attempt to quantify sulfite/bisulfite were complicated due to matrix effects (carbonate and fluoride appear to interfere with precipitation or derivatization as hydroxymethylsulfonate with formaldehyde). See: C. R. Warner, D. H. Daniels, D. E. Pratt, F. L. Joe, T. Fazio and G. W. Diachenko, Food Addit. Contam., 1987, 4, 437 CrossRef CAS PubMed.
- A similar effect was observed when ArSF59a was added. Attempts to construct a Stern–Volmer plot with varying SF6 or aryl-SF5 were unsuccessful, as the anion of 1a had limited stability at low concentrations, even in absence of quencher.
| This journal is © The Royal Society of Chemistry 2026 |