
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBackbone nitrogen substitution restricts the conformation of glycine residues in β-turns
Fengyi
Gu
 ,
Diana
Thomas
and
Robert W.
Newberry
*
,
Diana
Thomas
and
Robert W.
Newberry
*
Department of Chemistry, The University of Texas at Austin, 105 E. 24th St., Austin, TX 78712, USA. E-mail: rnewberry@utexas.edu
First published on 1st December 2025
Abstract
Glycine adopts backbone conformations that are generally inaccessible to other amino acids, but specifying a particular conformation remains challenging. Inspired by studies of small-molecule models, we hypothesized that substituting the alpha carbon with nitrogen would bias glycine toward specific β-turn conformations, which we confirmed through biophysical analysis of backbone-modified peptides.
The diverse conformational repertoire of proteins is essential for their biochemical activity. Glycine residues, which lack a side-chain functionality, adopt conformations that are generally inaccessible to other amino acids and are therefore critical to a variety of protein structures. For example, whereas only 4% of residues in folded proteins adopt conformations with positive
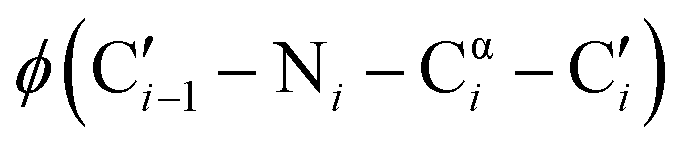 dihedral angles, 58% of those residues are glycine.1 The small size of glycine also enables close contacts between protein backbones, such as the intermolecular interactions in transmembrane helices2,3 and the collagen triple helix.4 The size and unique conformations of glycine together play a key role in protein β-turns, for example, where glycine is highly overrepresented in the tight i + 1 and i + 2 positions of type II and type I turns, respectively.5,6 Not only are the atypical dihedral angles of these positions (Table 1) readily accommodated by glycine residues, but the lack of a side chain avoids steric clashes that other amino acids would experience with neighboring residues.7
dihedral angles, 58% of those residues are glycine.1 The small size of glycine also enables close contacts between protein backbones, such as the intermolecular interactions in transmembrane helices2,3 and the collagen triple helix.4 The size and unique conformations of glycine together play a key role in protein β-turns, for example, where glycine is highly overrepresented in the tight i + 1 and i + 2 positions of type II and type I turns, respectively.5,6 Not only are the atypical dihedral angles of these positions (Table 1) readily accommodated by glycine residues, but the lack of a side chain avoids steric clashes that other amino acids would experience with neighboring residues.7
|
|
|
|
|---|---|---|
| Type I′ β-turn, i + 2 position | 90° | 0° |
| Type II′ β-turn, i + 1 position | 60° | −120° |
| Type II′ β-turn, i + 2 position | −80° | 0° |
| Aza-glycine conformational minima | ±90° | 0°/180° |
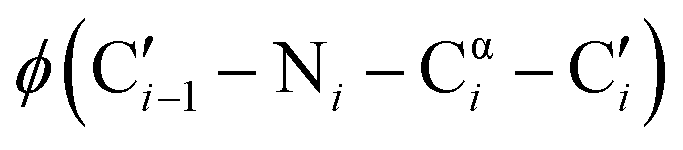
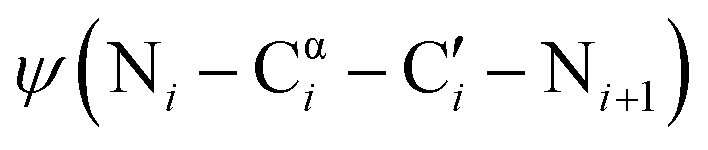
The uniquely permissive conformational landscape of glycine also presents challenges for the design of structured proteins and peptidomimetics. With so many relatively isoenergetic conformations, glycine readily changes structure, and as a result, glycine-rich protein regions are often intrinsically disordered.8 However, many potential applications of synthetic peptides require stable folds, such as for molecules designed to serve as ligands for protein binding or as antigens for generating antibodies. For structures that require glycine residues, this presents a conundrum: how to access the unique conformations of glycine while preventing adoption of confounding structures. Glycine's small size further complicates the problem, as other proteinogenic amino acids are typically poor substitutes and few synthetic analogs have been reported.
One promising strategy for specifying the conformation of glycine is to substitute the alpha carbon with nitrogen, thereby creating an azapeptide (Fig. 1A). This substitution affects the conformational landscape in two key ways. First, lone pair–lone pair repulsion between the adjacent nitrogen atoms restricts the 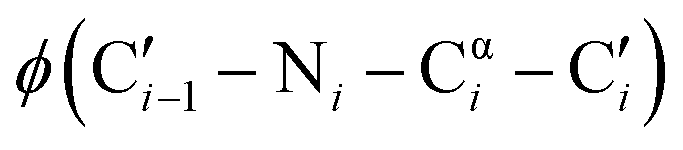 dihedral angle to approximately perpendicular arrangements (Fig. 1B).9–11 Second, resonance between the alpha nitrogen and the adjacent amide group creates a urea-like functionality (Fig. 1C) and thereby restricts the
dihedral angle to approximately perpendicular arrangements (Fig. 1B).9–11 Second, resonance between the alpha nitrogen and the adjacent amide group creates a urea-like functionality (Fig. 1C) and thereby restricts the 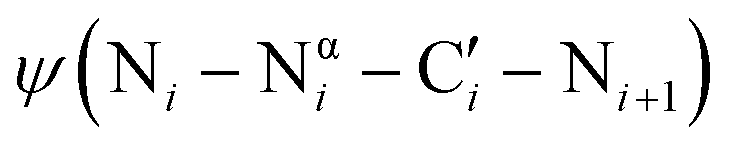 dihedral angle to planar conformations (ψ ∼ 0° or 180°).9–11 Consistent with these electronic features, computational studies have consistently identified the preferred backbone dihedral angles of azapeptides to include (ϕ = ±90 ± 30°, ψ = 0 ± 30°) or (ϕ = ±90 ± 30°, ψ = 180 ± 30°).10,12–16 These dihedral angles are found at the i + 1 and i + 2 positions of many β-turn structures,7 as well the polyproline type II helix. As a result, the conformational landscape of aza-glycine should be significantly restricted relative to glycine.
dihedral angle to planar conformations (ψ ∼ 0° or 180°).9–11 Consistent with these electronic features, computational studies have consistently identified the preferred backbone dihedral angles of azapeptides to include (ϕ = ±90 ± 30°, ψ = 0 ± 30°) or (ϕ = ±90 ± 30°, ψ = 180 ± 30°).10,12–16 These dihedral angles are found at the i + 1 and i + 2 positions of many β-turn structures,7 as well the polyproline type II helix. As a result, the conformational landscape of aza-glycine should be significantly restricted relative to glycine.
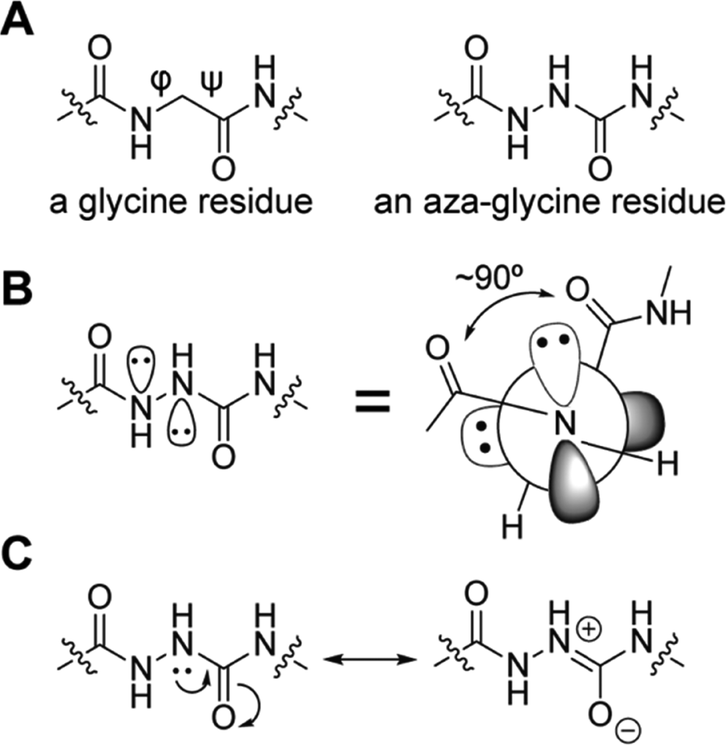 | ||
| Fig. 1 Aza-glycine restricts the backbone dihedral angles ϕ and ψ. (A) Chemical structures of glycine and aza-glycine. (B) Lone pair–lone pair repulsion forces the ϕ angle toward perpendicular geometries.12 (C) Urea-like resonance restricts the ψ angle toward planarity. | ||
Despite these observations, we are aware of few reports that have capitalized on the conformational restriction imparted by substituting glycine with aza-glycine to stabilize specific peptide structures selectively. Rather, most previous work has focused on the application of azapeptides to the optimization of peptide bioactivity, where they have shown great potential; for example, azapeptides have been used in drugs,17,18 hormone analogs,19,20 and supramolecular complexes.21–23
The clearest exception is application to the collagen triple helix,24–26 where substitution of the conserved, repeating glycine residues with aza-glycine increases thermal stability of collagen mimetic peptides.24–26 Not only is the conformation of aza-glycine compatible with the backbone conformation of the polyproline II secondary structure (ϕ ≈ −75°; ψ ≈ 150°) which comprises the collagen triple helix, the acidity of the Nα–H bond creates greater potential for backbone hydrogen bonding relative to the native backbone. Inspired by this result, we wondered if the restricted conformational landscape of aza-glycine alone, independent of any enhanced hydrogen-bonding interactions, would be sufficient to stabilize other peptide structures through backbone preorganization.
Spectroscopic and crystallographic studies of small molecules incorporating aza-amino acids frequently demonstrate β-turn formation.27–33 For example, Marraud and coworkers solved the crystal structures of model di- and tripeptide sequences containing azaAla, azaAsp, or azaAsn28 and analyzed the effects of aza-amino acids on peptide structure via proton nuclear magnetic resonance spectroscopy (1H NMR) and infrared (IR) spectroscopy;27 they found that aza-amino acid residues are strong β-turn inducers, with most of their derivatives adopting type I β-turn and type II β-turn. Furthermore, Lee and coworkers synthesized Boc–Phe–azaLeu–Ala–OMe and compared its structure to the peptide without the backbone nitrogen substitution;14 the results suggested that the azapeptide formed a type II β-turn while its parent peptide remained extended. Later, Tonali et al. incorporated two consecutive aza-amino acids into short peptides, resulting in β-turn formation.34 Inspired by these results, we hypothesized that incorporation of aza-glycine could stabilize peptides with specific β-turn geometries, thereby aiding in protein design.
To test this hypothesis, we compared the thermodynamic stabilities of four β-hairpins that feature glycine residues in key turn positions. Specifically, we focused on tryptophan zippers [TrpZips] with Gly–Asn (GN), Asn–Gly (NG), Aib–Gly (BG) and D-Pro–Gly (pG) turn sequences (Table 2), which lead to distinct conformations of the constituent glycine residues (Fig. 2). For example, TrpZip-GN and TrpZip-NG are isomeric, but the change in order of two amino-acid residues (Gly and Asn at positions 6 and 7) causes a change between type I′ and type II′ β-turns (Fig. 2 and Table 2).35 The i + 1 position of the type II′ turn in TrpZip-GN adopts a conformation that is disfavored by the stereoelectronics of aza-glycine, whereas the i + 2 position of the type I′ turn in TrpZip-NG adopts a conformation that is consistent with the known conformational minima of aza-glycine.
| Peptide | β-Turn type | G/aG position | Sequencea | T m (°C) |
|---|---|---|---|---|
| a Turn residues are underlined; aza-glycine (aG) residues are shown in bold; B represents 2-aminoisobutyric acid (Aib); p represents D-proline (D-Pro); all other amino acids are represented by standard one-letter codes. | ||||
| TrpZip-GN | Type II′ | i + 1 | Ac-S-W-T-W-E-G-N-K-W-T-W-K-NH2 | 41 |
| TrpZip-aGN | Type II′ | i + 1 | Ac-S-W-T-W-E-G-N-K-W-T-W-K-NH2 | 31 |
| TrpZip-NG | Type I′ | i + 2 | Ac-S-W-T-W-E-N-G-K-W-T-W-K-NH2 | 56 |
| TrpZip-NaG | Type I′ | i + 2 | Ac-S-W-T-W-E-N-G-K-W-T-W-K-NH2 | 59 |
| TrpZip-BG | Type I′ | i + 2 | Ac-A-W-A-W-E-B-G-K-W-A-W-K-NH2 | 50 |
| TrpZip-BaG | Type I′ | i + 2 | Ac-A-W-A-W-E-B-G-K-W-A-W-K-NH2 | 52 |
| TrpZip-pG | Type II′ | i + 2 | S-W-T-W-E-p-G-K-W-T-W-K-NH2 | 61 |
| TrpZip-paG | Type II′ | i + 2 | S-W-T-W-E-p-G-K-W-T-W-K-NH2 | 59 |
 | ||
| Fig. 2 Molecular structures of the β-turns studied herein.35 TrpZip-GN (black; 1le0) and TrpZip-pG adopt a type II′ turn, while TrpZip-NG (grey; 1le1) and TrpZip-BG adopt a type I′ turn. Note the distinct conformations of the relevant glycine residues. | ||
TrpZip-BG adopts a type I′ turn similar to TrpZip-NG, with glycine occupying the i + 2 position.36 In contrast, D-Pro–Gly turns, like those in TrpZip-pG, can adopt either type I′ or type II′ turn; however, most structurally characterized β-hairpins with D-Pro–Gly turns adopt the type II′ turn.37–40 Notably, the i + 2 position of both type I′ and type II′ turns adopts a conformation that should be compatible with the conformational landscape of aza-glycine (Table 1). We therefore predicted that aza-glycine substitution might stabilize the i + 2 positions of type I′ and type II′ turns (i.e., TrpZips-NG, -BG, and -pG) and destabilize the i + 1 position of type II′ turn (i.e., TrpZip-GN).
To test this prediction, we synthesized TrpZips-GN, NG, BG and pG, as well as backbone-modified variants in which glycine is substituted by aza-glycine (Fig. 3A and Table 2). TrpZips-pG and paG were synthesized without N-terminal acetylation to match prior reports.36 Far-UV circular dichroism (CD) spectra for all the peptides were qualitatively similar (see SI), confirming no overall changes in structure upon incorporation of aza-glycine. We then subjected these peptides to thermal denaturation to analyze the effect of aza-glycine on β-hairpin conformational stability.41 Contrary to our expectation, we observed only a minimal increase in melting temperature upon incorporation of aza-glycine at the i + 2 position of the type I′ β-turn (Fig. 3B and Table 2), despite the compatibility of this structure with the conformational landscape of aza-glycine. Specifically, aza-glycine substitution in the type I′ Asn–Gly turn caused only a ∼3 °C increase in melting temperature (Tm = 56 °C for TrpZip-NG vs. Tm = 59 °C for TrpZip-NaG). A similarly minimal ∼2 °C increase in melting temperature was observed for the type I′ Aib–Gly turn (Tm = 50 °C for TrpZip-BG vs. Tm = 52 °C for TrpZip-BaG). Incorporation of aza-glycine at the i + 2 position of the type II′ D-Pro–Gly β-turn led to a slight decrease in the melting temperature (Tm = 61 °C for TrpZip-pG vs. Tm = 59 °C for TrpZip-paG). Preorganization of glycine residues by substitution with aza-glycine therefore appears insufficient to stabilize these structures, despite their compatibility with the conformational minima of aza-glycine.
 | ||
| Fig. 3 (A) Chemical structures of the backbone-modified β-turns examined herein. (B) Thermal denaturation of TrpZip peptides based on measurements of mean-residue ellipticity (at 228 nm for TrpZip-GN/NG and 227 nm for TrpZip-BG/pG). | ||
In contrast, incorporation of aza-glycine at the i + 1 position of a type II′ β-turn, which lies outside the preferred conformational envelope of aza-glycine, was sufficient to dramatically destabilize the peptide (Fig. 3B and Table 2; Tm = 41 °C for TrpZip-GN vs. Tm = 31 °C for TrpZip-aGN). We questioned if the destabilization of this structure by aza-glycine was driven in part by side-chain–main-chain hydrogen bonding between the Asn side-chain carboxamide and the aza-glycine Nα–Hα group, which could promote conformations besides the β-turn. Several considerations led us to conclude that such a hydrogen bond is unlikely to contribute to the destabilization by aza-glycine. First, the NMR structure of TrpZip-GN with the Gly–Asn turn show that the asparagine side-chain carbonyl remains relatively far from the glycine methylene (>4.5 Å C′![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Cα), which is outside the range typically expected for meaningful hydrogen bonding (i.e., <3.5 Å O⋯Cα).42 Therefore, the hydrogen-bond in question is not significantly populated in the parent, glycine-containing hairpin. Second, asparagine in question is highly solvated (>120 Å2 side-chain solvent accessible surface area, corresponding to over 85% solvated; Table S1), so hydrogen bonds to water would likely compete with the putative side-chain–main-chain hydrogen. Third, the putative C′O⋯Hα–Nα hydrogen bonds in the isomeric Asn–azaGly and azaGly–Asn turns would both enclose eight-membered rings with similar composition and so should form with approximately equal probability. However, the effects of aza-glycine substitution on these two different turns are remarkably divergent (Table 2 and Fig. 3). If C′O⋯H–Nα hydrogen bonding contributed significantly to the destabilization of the Gly–Asn turn, then we would expect a similar effect on the Asn–Gly turn, which is not observed. Therefore, we conclude that the destabilization of TrpZip-GN upon aza-glycine incorporation likely results predominantly from the thermodynamic cost associated with adopting high-energy dihedral angles, rather than distracting hydrogen bonds.
O⋯Cα), which is outside the range typically expected for meaningful hydrogen bonding (i.e., <3.5 Å O⋯Cα).42 Therefore, the hydrogen-bond in question is not significantly populated in the parent, glycine-containing hairpin. Second, asparagine in question is highly solvated (>120 Å2 side-chain solvent accessible surface area, corresponding to over 85% solvated; Table S1), so hydrogen bonds to water would likely compete with the putative side-chain–main-chain hydrogen. Third, the putative C′O⋯Hα–Nα hydrogen bonds in the isomeric Asn–azaGly and azaGly–Asn turns would both enclose eight-membered rings with similar composition and so should form with approximately equal probability. However, the effects of aza-glycine substitution on these two different turns are remarkably divergent (Table 2 and Fig. 3). If C′O⋯H–Nα hydrogen bonding contributed significantly to the destabilization of the Gly–Asn turn, then we would expect a similar effect on the Asn–Gly turn, which is not observed. Therefore, we conclude that the destabilization of TrpZip-GN upon aza-glycine incorporation likely results predominantly from the thermodynamic cost associated with adopting high-energy dihedral angles, rather than distracting hydrogen bonds.
Although aza-glycine incorporation did not substantially increase the stability of turns with compatible conformations, aza-glycine substitution was sufficient to destabilize a conformation incompatible with the electronics of the aza-glycine residue. Therefore, our data demonstrates that aza-glycine substitution can be used as a tool to disfavor certain conformations of glycine over others, even in similar protein structural contexts (i.e., different types of protein β-turns). We speculate that this strategy can be extended to other contexts as well. For example, in misfolded proteins, which demonstrate significant polymorphism, glycine residues can adopt dramatically different conformations in misfolded species with distinct bioactivities.43 It is possible that incorporating aza-glycine into proteins that misfold could offer an opportunity to model and/or target specific misfolded structures. We also speculate that aza-glycine substitution might help overcome the aggregation potential of glycine-rich sequences,44 which tend to favor extended, β-sheet-like conformations that are destabilized by aza-amino acids.45 Other applications using aza-glycine to restrict glycine conformational space are also likely possible.
This work was supported by grants from the US National Institutes of Health (R00-NS116679 and R35-GM160477) and the Welch Foundation (F-2116) to R.W.N.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc06335c.Notes and references
- S. Hovmöller, T. Zhou and T. Ohlson, Acta. Crystallogr. D, 2002, 58, 768–776 CrossRef PubMed
.
- A. Senes, M. Gerstein and D. M. Engelman, J. Mol. Biol., 2000, 296, 921–936 CrossRef CAS PubMed
- A. Senes, I. Ubarretxena-Belandia and D. M. Engelman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9056–9061 CrossRef CAS
- B. Brodsky and A. V. Persikov, Adv. Protein Chem., 2005, 70, 301–339 CrossRef CAS
- E. G. Hutchinson and J. M. Thornton, Protein Sci., 1994, 3, 2207–2216 CrossRef CAS
- C. M. Wilmot and J. M. Thornton, J. Mol. Biol., 1988, 203, 221–232 CrossRef CAS PubMed
- J. S. Richardson, Adv. Protein Chem., 1981, 34, 167–339 CrossRef CAS PubMed
- R. Van Der Lee, M. Buljan, B. Lang, R. J. Weatheritt, G. W. Daughdrill, A. K. Dunker, M. Fuxreiter, J. Gough, J. Gsponer, D. T. Jones, P. M. Kim, R. W. Kriwacki, C. J. Oldfield, R. V. Pappu, P. Tompa, V. N. Uversky, P. E. Wright and M. M. Babu, Chem. Rev., 2014, 114, 6589–6631 CrossRef CAS PubMed
- C. Proulx, D. Sabatino, R. Hopewell, J. Spiegel, Y. García Ramos and W. D. Lubell, Future Med. Chem., 2011, 3, 1139–1164 CrossRef CAS PubMed
- M. Thormann and H. J. Hofmann, J. Mol. Struct. THEOCHEM, 1999, 469, 63–76 CrossRef CAS
- H. J. Lee, J. W. Song, Y. S. Choi, H. M. Park and K. B. Lee, J. Am. Chem. Soc., 2002, 124, 11881–11893 CrossRef CAS PubMed
- C. H. Reynolds and R. E. Hormann, J. Am. Chem. Soc., 1996, 118, 9395–9401 CrossRef CAS
- H. J. Lee, H. M. Park and K. B. Lee, Biophys. Chem., 2007, 125, 117–126 CrossRef CAS
- H. J. Lee, I. A. Ahn, S. Ro, K. H. Choi, Y. S. Choi and K. B. Lee, J. Pept. Res., 2000, 56, 35–46 CrossRef CAS
- H. J. Lee, K. H. Choi, I. A. Ahn, S. Ro, H. G. Jang, Y. S. Choi and K. B. Lee, J. Mol. Struct., 2001, 569, 43–54 CrossRef CAS
- H. J. Lee, H. J. Jung, J. H. Kim, H. M. Park and K. B. Lee, Chem. Phys., 2003, 294, 201–210 CrossRef CAS
- A. Zega, Curr. Med. Chem., 2005, 12, 589–597 CAS
- N. V. Hentig, Drugs Today, 2008, 44, 103–132 CrossRef
- D. Sabatino, C. Proulx, P. Pohankova, H. Ong and W. D. Lubell, J. Am. Chem. Soc., 2011, 133, 12493–12506 CrossRef CAS
- C. Proulx, É. Picard, D. Boeglin, P. Pohankova, S. Chemtob, H. Ong and W. D. Lubell, J. Med. Chem., 2012, 55, 6502–6511 CrossRef CAS PubMed
- X. S. Yan, K. Wu, Y. Yuan, Y. Zhan, J. H. Wang, Z. Li and Y. B. Jiang, Chem. Commun., 2013, 49, 8943–8945 RSC
- X. S. Yan, H. Luo, K. S. Zou, J. L. Cao, Z. Li and Y. B. Jiang, ACS Omega, 2018, 3, 4786–4790 CrossRef CAS PubMed
- Y. Yuan, X. S. Yan, X. R. Li, J. L. Cao, Z. Li and Y. B. Jiang, Chem. Commun., 2017, 53, 13137–13140 RSC
- Y. Zhang, R. M. Malamakal and D. M. Chenoweth, J. Am. Chem. Soc., 2015, 137, 12422–12425 CrossRef CAS PubMed
- T. Harris and D. M. Chenoweth, J. Am. Chem. Soc., 2019, 141, 18021–18029 CrossRef CAS
- A. J. Kasznel, Y. Zhang, Y. Hai and D. M. Chenoweth, J. Am. Chem. Soc., 2017, 139, 9427–9430 CrossRef CAS
- F. André, A. Vicherat, G. Boussard, A. Aubry and M. Marraud, J. Pept. Res., 1997, 50, 372–381 CrossRef
- F. André, G. Boussard, D. Bayeul, C. Didierjean, A. Aubry and M. Marraud, J. Pept. Res., 1997, 49, 556–562 CrossRef
- A. Lecoq, G. Boussard, M. Marraud and A. Aubry, Biopolymers, 1993, 33, 1051–1059 CrossRef CAS
- Z. Benatalah, A. Aubry, G. Boussard and M. Marraud, Int. J. Pept. Protein Res., 1991, 38, 603–605 CrossRef CAS
- M. Marraud and A. Aubry, Biopolymers, 1996, 40, 45–83 CrossRef CAS
- J. K. R. Deka, B. Sahariah and B. K. Sarma, J. Org. Chem., 2024, 89, 10419–10433 CrossRef
- J. K. R. Deka, B. Sahariah, K. Baruah, A. K. Bar and B. K. Sarma, Chem. Commun., 2020, 56, 4847–4877 RSC
- N. Tonali, I. Correia, J. Lesma, G. Bernadat, S. Ongeri and O. Lequin, Org. Biomol. Chem., 2020, 18, 3452–3458 RSC
- A. G. Cochran, N. J. Skelton and M. A. Starovasnik, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5578–5583 CrossRef CAS
- L. Wu, D. McElheny, V. Setnicka, J. Hilario and T. A. Keiderling, Proteins: Struct., Funct., Bioinf., 2012, 80, 44–60 CrossRef CAS
- R. Rai, S. Raghothama, R. Sridharan and P. Balaram, Biopolymers, 2007, 88, 350–361 CrossRef CAS PubMed
- I. L. Karle, S. K. Awasthi and P. Balaram, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8189–8193 CrossRef CAS PubMed
- I. L. Karle, H. N. Gopi and P. Balaram, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5160–5164 CrossRef CAS
- P. Chen, C. Lin, C. Lee, H. Jan and S. I. Chan, Protein Sci., 2001, 10, 1794–1800 CrossRef CAS PubMed
- A. D. Richaud, G. Zhao, S. Hobloss and S. P. Roche, J. Org. Chem., 2021, 86, 13535–13547 CrossRef CAS PubMed
- Z. S. Derewenda, L. Lee and U. Derewenda, J. Mol. Biol., 1995, 252, 248–262 CrossRef CAS
- R. Gallardo, N. A. Ranson and S. E. Radford, Curr. Opin. Struct. Biol., 2020, 60, 7–16 CrossRef CAS
- S. Ohnishi, H. Kamikubo, M. Onitsuka, M. Kataoka and D. Shortle, J. Am. Chem. Soc., 2006, 128, 16338–16344 CrossRef CAS
- M. A. Mcmechen, E. L. Willis, P. C. Gourville and C. Proulx, Molecules, 2019, 24, 1919–1931 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2026 |