
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClick chemistry-driven heteromolecular integration into layered zeolite frameworks for photochemical upconversion applications
Fuminao
Kishimoto
 *a,
Kyohei
Hisano
b,
Toru
Wakihara
ac and
Tatsuya
Okubo
a
*a,
Kyohei
Hisano
b,
Toru
Wakihara
ac and
Tatsuya
Okubo
a
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kfuminao@chemsys.t.u-tokyo.ac.jp
bLaboratory for Chemistry and Life Science, Institute of Integrated Research, Institute of Science Tokyo, Yokohama 226-8501, Japan
cInstitute of Engineering Innovaion, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 18th February 2026
Abstract
Organic–inorganic hybrid layered porous materials enabling triplet–triplet annihilation upconversion (TTA-UC) were developed by integrating anthracene-based emitters and metal–complex sensitizers within zeolitic nanospace. Diphenylanthracene (DPA) units were covalently anchored between MWW-type layers via thiol–ene click chemistry, forming organic pillars that generated ordered interlayer micropores, confirmed by XRD and N2 adsorption. Incorporation of cationic chromophores and Pt(octaethylporphyrin) [Pt(OEP)] enabled efficient intermolecular energy transfer. Upon 535 nm excitation, Pt(OEP)-loaded samples exhibited phosphorescence at 650 nm and upconverted emission at 400–500 nm. This work highlights a rational strategy for tuning nanoscale photochemical behavior via host–guest engineering in layered hybrid systems.
The incorporation of photoresponsive molecules into porous materials via host–guest chemistry has been widely explored.1–5 Depending on the host's characteristics, this approach allows control over photoluminescence efficiency, emission color, and environmental stability.6–11 Molecules can be introduced into nanoscale spaces through ion exchange, diffusion, or covalent immobilization with linker molecules.12–14 Moreover, arranging multiple molecular species precisely within zeolitic micropores enables regulated photochemical energy transfer under nanoscale confinement.15–19
Among various energy-transfer-based photochemical processes, triplet–triplet annihilation upconversion (TTA-UC) of arene-type molecules20–25 has drawn particular attention for enhancing solar energy utilization26,27 and enabling advanced photochemical reactions,28 optical diagnostics,29,30 and biotechnological applications.31 Considerable efforts have focused on molecular design of arene-based emitters and metal-complex sensitizers to expand anti-Stokes shifts,32,33 improve quantum yields,34 and extend spectral response into the near-infrared region.35,36
However, TTA-UC performance remains strongly limited by oxygen quenching of triplet states, which suppresses upconversion under ambient conditions.37–39 To address this, diverse porous materials have been developed as hosts for TTA-UC systems.40–42 Efficient upconversion nonetheless requires effective triplet energy transfer between sensitizers and emitters, demanding precise molecular arrangement.43–47 Layered compounds provide attractive two-dimensional nanospace offering controlled mobility and spatial organization.48–50 Yet, achieving homogeneous dispersion of anthracene-based emitters and metal–complex sensitizers within interlayer regions remains difficult due to polarity differences that hinder uniform mixing under confined conditions. Thus, a methodology for uniformly and effectively arranging donor and acceptor molecules for TTA–UC within an inorganic host material should be highly required.
In this study, we designed a material system to achieve a molecularly homogeneous assembly of TTA-UC emitter and sensitizer molecules within the two-dimensional nanospace of a layered compound. Using click chemistry, we synthesized an organic–inorganic hybrid layered porous material in which diphenylanthracene (DPA) units—serving as TTA-UC emitters—act as molecular linkers between adjacent layers of an MWW-type zeolitic precursor (Fig. 1).53 Platinum octaethylporphyrin (PtOEP), a triplet sensitizer, was then introduced into the interlayer nanopores, forming a well-mixed DPA–PtOEP ensemble within the confined space. To clarify how framework composition affects upconversion efficiency, we prepared pure silica, borosilicate, and aluminosilicate analogues and examined the relationship between their compositions and TTA-UC intensities.
 | ||
| Fig. 1 Schematic illustration of the synthesis of an organic–inorganic hybrid porous material derived from an MWW-type zeolitic layered precursor. | ||
Fig. 2(a) shows XRD patterns obtained during the synthesis of the organic–inorganic hybrid layered porous material from the uncalcined borosilicate MWW-type precursor (ERB-1P).51 In ERB-1P, the (001) reflection appeared at 2θ = 3.30° (d = 2.68 nm), characteristic of its layered structure. After treatment with CTAB and TPAOH, this peak shifted to 2θ = 1.98°, indicating CTAB intercalation and interlayer expansion to form structure 1 (Fig. 1).52 The other reflections ((100), (220), (130)) remained unchanged, confirming that the layer framework was preserved.
 | ||
| Fig. 2 (a) XRD patterns of the organic–inorganic hybrid porous material synthesized from the ERB-1P precursor. (b) Raman spectra before and after the thiol–ene click reaction (λex = 532 nm). (c) N2 adsorption–desorption isotherms before and after the removal of interlayer CTA+ template molecules. (d) Absorption or diffuse-reflectance spectra (solid lines) and photoluminescence spectra under 380 nm excitation (dotted lines), together with the external quantum yield determined by the absolute method. (e) Photoluminescence decay profiles (excitation wavelength: 365 nm, emission wavelength: 450 nm). IRF denotes the instrument response function, which represents the time constant of the excitation light source itself. | ||
Subsequently, mercaptopropyltrimethoxysilane (MPTMS) was reacted with interlayer silanol groups to introduce thiol functionalities (structure 2, Fig. 1). The (001) reflection shifted to 2θ = 2.29°, indicating slight interlayer contraction due to partial CTAB desorption. The Raman spectrum (Fig. 2b) showed a sharp S–H peak around 2500 cm−1. CHN analysis (Table S1) quantitatively tracked changes in organic content during synthesis. The uncalcined ERB-1P contained about 6.0 wt% piperidine (0.7 mmol g−1) as the structure-directing agent. After CTAB treatment, carbon increased, corresponding to ∼2.6 wt% PI and 12 wt% CTAB, indicating partial exchange of interlayer PI with CTA+ while some PI remained in the layers. Following MPTMS reaction, CTA+ decreased to 4.0 wt% (0.14 mmol g−1) and 7.5 wt% (0.88 mmol g−1) of MPTMS was introduced, explaining the reduced basal spacing.
Subsequent thiol–ene coupling with DPA derivative (3) caused the (001) reflection to shift to lower angles, confirming interlayer expansion. CHN data indicated 3.6 wt% (0.10 mmol g−1) DPA incorporation, and Raman spectra showed disappearance of the S–H band (∼2500 cm−1) with new aromatic peaks (1400–1600 cm−1), confirming successful immobilization.
After mild acid treatment to remove remaining CTA+ (4), the (001) reflection shifted to higher angles, indicating contraction. Nitrogen content dropped to 0.22 mmol g−1, consistent with residual PI trapped in zeolitic layers, while carbon content changed little, showing DPA retention. N2 adsorption (Fig. 2c) revealed a transition from type III (3) to type I isotherm (4), with a micropore volume of 0.096 cm3 g−1, demonstrating formation of an organic–inorganic hybrid microporous framework.
The photophysical properties of the obtained materials were subsequently evaluated. Fig. 2(d) and (e) presents the absorption and emission spectra together with the photoluminescence lifetime data of the as-synthesized samples. DPA-del. in toluene solution exhibits multiple characteristic absorption peaks around 400 nm, similar to other anthracene derivatives, and shows an emission spectrum in the range of 400–600 nm (Fig. 2(d)). The external photoluminescence quantum yield (EQY) determined by the absolute method was 48%. The photoluminescence decay profile was well fitted by a single exponential component, giving an emission lifetime of approximately 3.0 ns (Fig. 2(e)).
Structures 3 and 4 containing DPA-del. showed diffuse reflectance and emission spectra similar to DPA-del. in toluene, though their EQY values followed the order: DPA in toluene > 3 > 4. The photoluminescence decay profiles were also comparable. EQY (φ) and emission lifetime (τ) are given by:
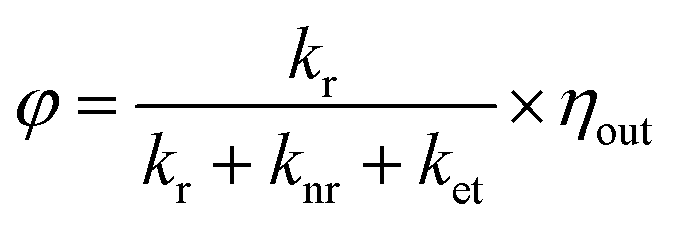 | (1) |
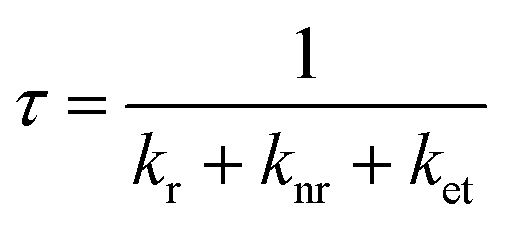 | (2) |
The cationic luminescent species Ru(dmb)32+ (dmb = dimethylbipyridine), auramine O (AuO+), and rhodamine B (RhB+) were incorporated into the nanospace of structure 4 through an ion-exchange process conducted in solution (Fig. S1(a)). Because the absorption spectra of these molecules overlap with the emission spectrum of DPA-del., photoexcitation of the DPA units is expected to induce Förster-type energy transfer to the introduced luminescent molecules. CHN elemental analysis revealed incorporation amounts of 0.032, 0.012, and 0.026 mmol g−1 for Ru(dmb)32+, AuO+, and RhB+, respectively.
Fig. S1(b) shows the XRD patterns of the samples after the introduction of the luminescent molecules. The (001) diffraction peak remained unchanged, indicating that the porous structure of structure 4 was preserved. In contrast, the absorption spectra shown in Fig. 3(a) exhibit, in addition to the absorption bands originating from the DPA units (highlighted in yellow), new absorption features attributable to the incorporated luminescent molecules (indicated in red).
 | ||
| Fig. 3 (a) Absorption or diffuse-reflectance spectra (solid lines) and photoluminescence spectra under 380 nm excitation (dotted lines). (b) Photoluminescence decay profiles (excitation wavelength: 365 nm, emission wavelength: 450 nm). | ||
The emission spectra of each sample were measured under 380 nm excitation, corresponding to the excitation of the DPA units (Fig. 3(a)). In the sample containing Ru(dmb)32+ (denoted as Ru(dmb)32+@4), multiple emission peaks were observed in the range of 400–750 nm. These peaks are attributed to the overlap of emissions from three sources: direct emission from the DPA units, Ru(dmb)32+ emission arising from energy transfer from the excited DPA units, and direct excitation of Ru(dmb)32+ itself. In contrast, in the AuO+@4 and RhB+@4 samples, the emission peaks originating from the DPA units were almost completely suppressed, and only sharp emission bands corresponding to AuO+ and RhB were observed, respectively. These results suggest that efficient energy transfer occurs from the DPA units within the host framework to the guest molecules (AuO+ and RhB+). The absence of the DPA emission peak indicates that the energy-transfer efficiency is close to unity.
To evaluate the kinetics of the energy-transfer process, photoluminescence decay profiles corresponding to DPA excitation and emission were measured (Fig. 3(b)). In all samples, the observed lifetimes were shorter than that of structure 4, indicating accelerated decay due to energy transfer (larger ket in eqn (2)). Because the emission decay of Ru(dmb)32+@4 involves multiple parallel emission pathways, the discussion focuses on AuO+@4 and RhB+@4. In these samples, the initial portion of the emission decay—down to 1% of the initial intensity—completely overlapped with the IRF. This observation indicates that the energy transfer occurs with a rate constant exceeding the detection limit of the instrument (ket ≈ 109 s−1). These results indicate that highly efficient energy transfer occurs from the DPA units to the guest molecules within the nanospace of structure 4.
Finally, the Pt(OEP) complex (OEP = octaethylporphyrin) was introduced into the nanospace by a diffusion method conducted in solution. Specifically, upon photoexcitation, triplet–triplet energy transfer (TTET) takes place from the excited Pt(OEP) to DPA, followed by triplet–triplet annihilation (TTA) between DPA-del units, resulting in the generation of a higher-energy photon from two low-energy photons (upconversion). For comparison, in addition to the borosilicate ERB-1P layered precursor used as the host porous material, two aluminosilicate materials with the same topology, MCM-22P54 and ITQ-1P,55 were also employed.
Fig. 4(a) shows the diffuse reflectance spectrum of the sample incorporating Pt(OEP). In addition to the absorption bands originating from the DPA units, a distinct peak attributed to Pt(OEP) was observed at 535 nm. This observation confirms the successful incorporation of Pt(OEP) into the nanospace. ERB-1P exhibits the strongest absorption peak in this region, whereas MCM-22P and ITQ-1P show approximately 70% and 60% of the intensity, respectively. In contrast, ERB-1P and ITQ-1P exhibit comparable peak intensities in Pt(OEP) region, whereas MCM-22P shows a relatively weaker absorption. Thus, the amount of introduced organic molecules estimated from the UV-Vis spectra follows the order shown below.
| DPA: ERB-1P > MCM-22P > ITQ1P |
| Pt(OEP): ERB-1P ∼ ITQ1P > MCM-22P |
 | ||
| Fig. 4 (a) Diffuse reflectance spectrum of Structure 4 after incorporation of Pt(OEP). (b) Emission spectra of Pt(OEP)-incorporated Structure 4 samples (derived from ERB-1P, MCM-22P, and ITQ-1P) under 535 nm excitation. | ||
Fig. 4(b) shows the emission spectra of the organic–inorganic hybrid porous material containing Pt(OEP) under 535 nm excitation. All the samples exhibited not only the normal Stokes-shifted emission of Pt(OEP), peaking at 650 nm, but also an anti-Stokes emission in the 400–500 nm region arising from the TTA-UC process. The quantum efficiency of ITQ-1P, MCM-22, and ERB-1P was determined to be 0.22%, 0.19%, and 0.17%, respectively. Compared with the samples derived from zeolite precursors possessing acid sites, such as ERB-1P and MCM-22P, the ITQ-1P-derived sample exhibited much stronger emission intensities for both the self-emission and the upconverted emission. This is probably because the pure-silica zeolite provides a relatively hydrophobic environment, allowing a larger amount of Pt(OEP) to be incorporated into the nanospace, which induced the efficient TTA-UC process. In contrast, ERB-1P exhibited the weakest emission intensity despite having the highest loading amounts of DPA and Pt(OEP). Thus the presence of heteroatoms in the host material tends to inhibit photochemical processes.
In conclusion, DPA-linked organic–inorganic hybrid layered materials were synthesized via thiol–ene click chemistry using MWW-type zeolitic precursors. The confined interlayer nanospace accommodated both emitter and sensitizer molecules, enabling efficient intermolecular energy transfer. Upon intercalation of cationic luminescent molecules, nearly 100% Förster-type energy transfer from photoexcited DPA units to the guest molecules was achieved, resulting in large Stokes-shifted emission. Incorporation of Pt(OEP) further generated visible-light TTA-UC emission. Investigation of the framework composition revealed that the pure-silica ITQ-1P host, with its hydrophobic environment, enabled higher sensitizer loading and enhanced upconversion efficiency. These results establish a clear structure–property relationship between framework composition and upconversion performance, providing design principles for photofunctional hybrid materials capable of efficient nanoscale energy conversion.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details and CHN organic analysis. See DOI: https://doi.org/10.1039/d5cc06270e.Acknowledgements
This work was financially supported by Grant-in-Aid for JSPS Fellows (SPD) 18J01320.References
- F. L. Cozens, H. Garcia and J. C. Scaiano, J. Am. Chem. Soc., 1993, 115, 11134 Search PubMed
.
- J. C. Scaiano and H. Garcia, Acc. Chem. Res., 1999, 32, 783 CrossRef CAS
- G. Calzaferri, S. Huber, H. Maas and C. Minkowski, Angew. Chem., Int. Ed., 2003, 42, 3732 CrossRef CAS PubMed
- S. Hashimoto, J. Photochem. Photobiol., C, 2003, 4, 19 CrossRef CAS
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2020, 32, 1905227 CrossRef CAS PubMed
- K. Weh, M. Noack, K. Hoffmann, K. P. Schröder and J. Caro, Microporous Mesoporous Mater., 2002, 54, 15 CrossRef CAS
- F. Marlow, M. D. McGehee, D. Zhao, B. F. Chmelka and G. D. Stucky, Adv. Mater., 1999, 11, 632 CrossRef CAS
- A. Corma, V. Fornes, H. Garcia, M. A. Miranda, J. Primo and M. J. Sabater, J. Am. Chem. Soc., 1994, 116, 2276 CrossRef CAS
- Y. Wada, T. Okubo, M. Ryo, T. Nakazawa, Y. Hasegawa and S. Yanagida, J. Am. Chem. Soc., 2000, 122, 8583 CrossRef CAS
- C. Xu, D. Mochizuki, M. M. Maitani and Y. Wada, Eur. J. Inorg. Chem., 2014, 1470 CrossRef CAS
- F. Kishimoto, K. Hisano, T. Yoshioka, K. Iyoki, T. Wakihara and T. Okubo, ACS Appl. Mater. Interfaces, 2023, 15, 49500 CrossRef CAS PubMed
- A. Moissette, S. Marquis, I. Gener and C. Brémard, Phys. Chem. Chem. Phys., 2002, 4, 5690 RSC
- A. Corma and H. Garcia, Eur. J. Inorg. Chem., 2004, 1143 CrossRef CAS
- Y. Li and J. Yu, Nat. Rev. Mater., 2021, 6, 1156 CrossRef CAS
- E. Pérez-Botella, S. Valencia and F. Rey, Chem. Rev., 2022, 122, 17647 CrossRef PubMed
- L. Brancaleon, D. Brousmiche, V. J. Rao, L. J. Johnston and V. Ramamurthy, J. Am. Chem. Soc., 1998, 120, 4926 Search PubMed
- M. Borja and P. K. Dutta, Nature, 1993, 362, 43 CrossRef CAS
- M. Sykora and J. R. Kincaid, Nature, 1997, 387, 162 CrossRef CAS
- K. Lutkouskaya and G. Calzaferri, J. Phys. Chem. B, 2006, 110, 5633 CrossRef CAS PubMed
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560 CrossRef CAS
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195112 CrossRef
- T. W. Schmidt and F. N. Castellano, J. Phys. Chem. Lett., 2014, 5, 4062 CrossRef CAS PubMed
- Y. Murakami and K. Kamada, Phys. Chem. Chem. Phys., 2021, 23, 18268 RSC
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345 RSC
- H. J. Feng, M. Y. Zhang, L. H. Jiang, L. Huang and D. W. Pang, Acc. Chem. Res., 2025, 58, 21791 CrossRef PubMed
- B. S. Richards, D. Hudry, D. Busko, A. Turshatov and I. A. Howard, Chem. Rev., 2021, 121, 9165 CrossRef CAS PubMed
- L. Wondraczek, E. Tyystjärvi, J. Méndez-Ramos, F. A. Müller and Q. Zhang, Adv. Sci., 2015, 2, 1500218 CrossRef PubMed
- L. Huang and G. Han, Nat. Rev. Chem., 2024, 8, 238 CrossRef CAS PubMed
- Q. Dou, L. Jiang, D. Kai, C. Owh and X. J. Loh, Drug Discovery Today, 2017, 22, 1400 CrossRef CAS PubMed
- A. Hlaváček, Z. Farka, M. J. Mickert, U. Kostiv, J. C. Brandmeier, D. Horák, P. Skládal, F. Foret and H. H. Gorris, Nat. Protoc., 2022, 17, 1028 CrossRef PubMed
- Y. Sasaki, M. Oshikawa, P. Bharmoria, H. Kouno, A. Hayashi-Takagi, M. Sato, I. Ajioka, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2019, 58, 17827 CrossRef CAS PubMed
- L. Huang, Y. Zhao, H. Zhang, K. Huang, J. Yang and G. Han, Angew. Chem., 2017, 129, 14592 CrossRef
- C. Fan, X. Wei, W. Wu and C. Yang, Org. Lett., 2024, 26, 950 Search PubMed
- Y. Zhou, F. N. Castellano, T. W. Schmidt and K. Hanson, ACS Energy Lett., 2020, 5, 2322 CrossRef CAS
- P. Bharmoria, H. Bildirir and K. Moth-Poulsen, Chem. Soc. Rev., 2020, 49, 6529 Search PubMed
- S. Amemori, Y. Sasaki, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2016, 138, 8702 Search PubMed
- D. Dzebo, K. Moth-Poulsen and B. Albinsson, Photochem. Photobiol. Sci., 2017, 16, 1327 CrossRef CAS PubMed
- S. H. Askes and S. Bonnet, Nat. Rev. Chem., 2018, 2, 437 Search PubMed
- X. Wang, F. Ding, T. Jia, F. Li, X. Ding, R. Deng and G. Chen, Nat. Commun., 2024, 15, 2157 CrossRef CAS PubMed
- S. H. Askes, V. C. Leeuwenburgh, W. Pomp, H. Arjmandi-Tash, S. Tanase, T. Schmidt and S. Bonnet, ACS Biomater. Sci. Eng., 2017, 3, 322 Search PubMed
- J. Park, M. Xu, F. Li and H.-C. Zhou, J. Am. Chem. Soc., 2018, 140, 5493 Search PubMed
- B. Zhang, K. D. Richards, B. E. Jones, A. R. Collins, R. Sanders, S. R. Needham, P. Qian, A. Mahadevegowda, C. Ducati, S. W. Botchway and R. C. Evans, Angew. Chem., Int. Ed., 2023, 62, e202308602 CrossRef CAS PubMed
- P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056 CrossRef CAS PubMed
- D.-G. Ha, R. Wan, C. A. Kim, T.-A. Lin, L. Yang, T. Van Voorhis, M. A. Baldo and M. Dincă, Nat. Mater., 2022, 21, 1275 CrossRef CAS PubMed
- S. Mattiello, S. Mecca, A. Ronchi, A. Calascibetta, G. Mattioli, F. Pallini, F. Meinardi, L. Beverina and A. Monguzzi, ACS Energy Lett., 2022, 7, 2435 CrossRef CAS
- L. Wei, C. Yang and W. Wu, Chem. Commun., 2025, 61, 7221 RSC
- K. Miwa, T. Okamoto, Y. Kobori and A. Ishizaki, J. Phys. Chem. C, 2025, 129, 12904–12915 CrossRef
- F. Kishimoto, T. Wakihara and T. Okubo, ACS Appl. Mater. Interfaces, 2020, 12, 7021 CrossRef CAS PubMed
- Y. Kageshima, S. Tateyama, F. Kishimoto, K. Teshima, K. Domen and H. Nishikiori, Phys. Chem. Chem. Phys., 2021, 23, 5673 RSC
- A. Yamagishi, K. Tamura, S. Yamamoto, F. Sato, J. Yoshida and H. Sato, Appl. Clay Sci., 2024, 255, 107397 CrossRef CAS
- X. Ouyang, S.-J. Hwang, R. C. Runnebaum, D. Xie, Y.-J. Wanglee, T. Rea, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2014, 136, 1449 CrossRef CAS PubMed
- P. Chlubná, W. J. Roth, A. Zukal, M. Kubů and J. Pavlatová, Catal. Today, 2012, 179, 35 CrossRef
- M. Álvaro, M. Benítez, J. F. Cabeza, H. García and A. Leyva, J. Phys. Chem. C, 2007, 111, 7532 CrossRef
- M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910 CrossRef CAS PubMed
- M. A. Camblor, C. Corell, A. Corma, M. J. Díaz-Cabañas, S. Nicolopoulos, J. M. González-Calbet and M. Vallet-Regí, Chem. Mater., 1996, 8, 2415 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2026 |