
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unconventional magnetism and residual entropy in La7Os3O18
Ashley T.
Brennan
 ab,
Maximilian T.
Pelly
ab,
Alexandra S.
Gibbs
*ac and
Andreas W.
Rost
b
ab,
Maximilian T.
Pelly
ab,
Alexandra S.
Gibbs
*ac and
Andreas W.
Rost
b
aEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: a.gibbs@st-andrews.ac.uk
bSchool of Physics and Astronomy, University of St Andrews, St Andrews, KY16 9SS, UK
cISIS Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
First published on 9th January 2026
Abstract
In this communication we describe the crystal structure and unusual magnetic properties of La7Os3O18, a new layered osmate with structurally discrete (non-corner-sharing) octahedra containing Os5+ with 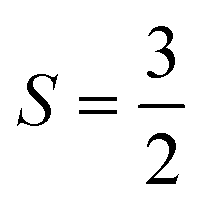 and strong spin–orbit coupling. Investigations through powder X-ray diffraction, magnetometry and heat capacity measurements demonstrate that the material undergoes an antiferromagnetic ordering transition at TN = 43 ± 1 K, with a further broad overturn in susceptibility at T ≈ 14 K indicating low dimensional short range magnetic ordering. Curie–Weiss fits give an effective moment of µeff = 3.26µB per Os5+ and Weiss constant of θW ≈ −92 K. Heat capacity data reveal that it is the low temperature feature with which the majority of the magnetic entropy is associated. However, between our lowest temperature of 2 K and well above TN we find only approximately half of the expected magnetic entropy to be released. Magnetisation versus field measurements at low temperature reveal a hysteretic metamagnetic transition above 5 T, further indicating that the magnetism below TN is non-trivial in nature.
and strong spin–orbit coupling. Investigations through powder X-ray diffraction, magnetometry and heat capacity measurements demonstrate that the material undergoes an antiferromagnetic ordering transition at TN = 43 ± 1 K, with a further broad overturn in susceptibility at T ≈ 14 K indicating low dimensional short range magnetic ordering. Curie–Weiss fits give an effective moment of µeff = 3.26µB per Os5+ and Weiss constant of θW ≈ −92 K. Heat capacity data reveal that it is the low temperature feature with which the majority of the magnetic entropy is associated. However, between our lowest temperature of 2 K and well above TN we find only approximately half of the expected magnetic entropy to be released. Magnetisation versus field measurements at low temperature reveal a hysteretic metamagnetic transition above 5 T, further indicating that the magnetism below TN is non-trivial in nature.
In comparison to 3d transition metal oxides, 5d compounds offer access to more spatially extended d orbitals and greater spin orbit coupling, which scales with atomic number Z between Z2 and Z4.1,2 These considerations frequently lead to physical properties that differ substantially from related compounds of the lighter transition metals, driving substantial research interest in the field.
Of the 5d metal oxides, the osmates have remained relatively understudied due to the practical implications of the toxicity of OsO4. However, discoveries of polar metallicity in LiOsO3,3 charge ordering in Sr5Os3O134 and superconductivity in the β-pyrochlores AOs2O6 (A = K, Rb, Cs)5–7 have demonstrated the diversity of thermodynamic phases accessible through osmium chemistry. Such states are supported by the variety of structures accessible when multiple oxidation states (+4, +5, +6, +7, +8) can be stabilised, and even mixed.
Amongst the osmates described in the literature, there are few with isolated OsO6 octahedra. Those which have been studied, such as Li5OsO6 and Ca3OsO6, tend to occupy the higher +6 and +7 Os oxidation states and thus have a smaller effective moment on each Os site. In this study we present the basic structural, magnetic and thermodynamic properties of the compound La7Os3O18, which has isolated octahedrally coordinated Os5+ sites within a complex layered crystal structure.
Initially La7Os3O18 was identified as an impurity during the synthesis of polycrystalline samples of other lanthanum osmates, but was subsequently isolated, and could be made using several different sets of conditions. Note that caution is required when handling osmium and its compounds because highly toxic and volatile OsO4is produced at elevated temperatures and under oxidising conditions. All handling of osmium and its compounds was carried out within an argon glove box or in a manner suited to air sensitive samples. The starting reagents (OsO2, min 83% Os, and La2O3, ≥99.9%) were ground together and pressed into a pellet which was then sealed under vacuum in a quartz ampoule, along with a small quantity of AgO as an oxidising agent. AgO decomposes in two steps at approximately 200 and 400 °C to yield O2, and was kept separate from the pelleted reagents by encasing it in Pt foil. Significant excesses of OsO2 were used (up to 5 times the stoichiometric quantity in the most extreme case) in order to counteract the loss of Os to OsO4 which occurs readily at temperatures in excess of 500 °C. The sealed ampoules were heated in a tube furnace at temperatures between 800 and 950 °C for times ranging from 4 to 48 hours. Full synthesis details for each sample can be found in the SI.
The structure of La7Os3O18 was identified by comparison with powder diffraction data from La7Ru3O18,9 which adopts space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. The related structure type of Sr5Re2O1210 was found to yield a worse fit to the data, with La occupancies on the additional site refining to zero. Detailed structural analysis was carried out using Mo Kα powder X-ray diffraction measured in Debye–Scherrer mode at room temperature with the sample sealed in a 0.3 mm diameter quartz capillary. No absorption correction was applied. A good fit to the data could be achieved via Rietveld refinement (see SI), giving a = 9.8639(5) Å, c = 56.3934(19) Å, Rwp = 0.05412, χ2 = 2.07 (see SI Fig. S1), with little change in structural parameters from those of the ruthenate analogue. The bond valence sum gives 4.91, 4.54 and 4.42 for the three symmetry-inequivalent osmium sites, consistent with a 5+ oxidation state.11 An impurity was identified as LaClO, with a refined phase fraction of 3.9% for a representative sample. The Cl in this phase was found to come from an impurity in the OsO2 starting reagent. LaClO is not magnetic and its contribution is negligibly small to the results we report here.
c. The related structure type of Sr5Re2O1210 was found to yield a worse fit to the data, with La occupancies on the additional site refining to zero. Detailed structural analysis was carried out using Mo Kα powder X-ray diffraction measured in Debye–Scherrer mode at room temperature with the sample sealed in a 0.3 mm diameter quartz capillary. No absorption correction was applied. A good fit to the data could be achieved via Rietveld refinement (see SI), giving a = 9.8639(5) Å, c = 56.3934(19) Å, Rwp = 0.05412, χ2 = 2.07 (see SI Fig. S1), with little change in structural parameters from those of the ruthenate analogue. The bond valence sum gives 4.91, 4.54 and 4.42 for the three symmetry-inequivalent osmium sites, consistent with a 5+ oxidation state.11 An impurity was identified as LaClO, with a refined phase fraction of 3.9% for a representative sample. The Cl in this phase was found to come from an impurity in the OsO2 starting reagent. LaClO is not magnetic and its contribution is negligibly small to the results we report here.
The structure adopted by La7Os3O18 is quite intricate with an unusually large unit cell (see Fig. 1). Considering only the Os positions, a distorted double hexagonal close packed structure emerges. The double hexagonal close packing sequence is ABAC, however in La7Os3O18, a distortion is applied to the ‘A’ layer, displacing the Os ions slightly from the plane. The resulting undulations produce a far more complex stacking sequence that expands the unit cell by  and 3c. This subcell-supercell relationship was previously observed in precession images from single crystal X-ray diffraction data by Mons and colleagues, working on the related structures of Sr5Re2O12 and Ca5Re2O12.10
and 3c. This subcell-supercell relationship was previously observed in precession images from single crystal X-ray diffraction data by Mons and colleagues, working on the related structures of Sr5Re2O12 and Ca5Re2O12.10
 | ||
| Fig. 1 A view of the structure of La7Os3O18 along the a-axis, with unit cell in black, OsO6 octahedra in blue and La in yellow. There are two principal layer types, each with two arrangements, pictured individually along the c-axis on the right. The ABab stacking sequence emerges when considering the structure in its entirety, and relates to the double hexagonal close packed ABAC sequence from osmium positions alone. Visualised using VESTA.8 | ||
By considering the OsO6 octahedra, it becomes clear that there are two principal types of layer in the material; one makes up the undulating layer ‘A’ of the distorted double hexagonal close packed structure, the other making up both layers ‘B’ and ‘C’. Layer ‘A’ has OsO6 octahedra arranged with faces aligned with the ab-plane, this layer having two of the three crystallographically inequivalent Os sites (panels A and a in Fig. 1). The remaining Os site is located in the other layer type and has OsO6 arranged without coplanar faces (panels B and b in Fig. 1).
Lanthanum atoms are present in both layer types, located above and below the Os sites. Incorporating the lanthanum positions into the description of the structure leads to a more complete, though more complex, picture. The two layer types stack alternately, however every second occurrence of each layer has a transformation applied, leading to an ABab stacking sequence where layer a is equivalent to layer A with a twofold rotation about the a-axis and layer b is equivalent to layer B after inversion through the centre of the unit cell. Layers B and b make up the B and C positions of the double hexagonal ABAC stacking of Os sites described above. The unit cell contains three of these stacks of four layers with the ABab sequence, each stack of four translated by 1/3 in a and −1/3 in b.
Layer A/a (see Fig. 1) has a La![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Os ratio of 2:1 and is thinner than layer B/b which has a La:Os ratio of
:Os ratio of 2:1 and is thinner than layer B/b which has a La:Os ratio of  . There are three crystallographically distinct La positions, two occurring in the relatively La rich layer B/b and one in the relatively La poor layer A/a. Aside from the ruthenate analogue other examples of this structure type exist, though it is relatively rare.12–14 Recently tungstates in this class have garnered interest within the luminescent materials community for their separation of WO6 octahedra and stability to dopants.15–17
. There are three crystallographically distinct La positions, two occurring in the relatively La rich layer B/b and one in the relatively La poor layer A/a. Aside from the ruthenate analogue other examples of this structure type exist, though it is relatively rare.12–14 Recently tungstates in this class have garnered interest within the luminescent materials community for their separation of WO6 octahedra and stability to dopants.15–17
Magnetic properties were investigated under an applied field µ0H = 1 T (Fig. 2) using the VSM options for the Quantum Design MPMS 3 and PPMS. The behaviour was consistent across samples synthesised under varying conditions. The susceptibility, M/H, of La7Os3O18 has paramagnetic behaviour at high temperatures, down to a transition temperature of TN = 43 ± 1 K (with the uncertainty indicating the degree of variation between different samples), where there is a maximum consistent with antiferromagnetic ordering. A second maximum is also observed centred on T ≈ 14 K. The field cooled and zero field cooled data diverge below the higher temperature transition. These three principal features of the susceptibility were quantitatively and qualitatively reproducible across samples of varying quality, indicating that they are intrinsic, which suggests further complexity in the magnetic structure beyond simple collinear antiferromagnetism. Further measurements at low applied fields (µ0H = 0.01 T, see SI Fig. S2) show an additional feature at T = 37 K and a greater divergence between the zero field cooled and field cooled data. These features at low fields were also reproducible (see SI).
 | ||
| Fig. 2 Upper panel: polycrystalline magnetic susceptibility data, for an applied field µ0H = 1 T. Dots indicate zero field cooled and crosses indicate field cooled data. Lower panel: Heat capacity divided by temperature for the same temperature interval, with the integrated entropy plotted on the right axis. | ||
A Curie–Weiss fit of further variable temperature magnetic studies carried out at µ0H = 1 T, with inclusion of a temperature-independent Van Vleck term and a core diamagnetism correction of −144 µemu mol-Os−118 (SI Fig. S3), yields θW ≈ −92 K, indicating relatively strong antiferromagnetic interactions, and moderate but not remarkable frustration. The effective moment is 3.26µB per Os5+, lower than the expected spin only value of 3.87µB for 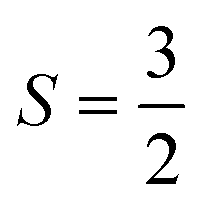 , likely the result of spin–orbit coupling, and the Van Vleck contribution 357 ± 3 µemu mol-Os−1. Measurements of magnetisation versus field (Fig. 3) reveal a metamagnetic transition in high field with hysteresis, resulting in a double hysteresis loop. In addition, a smaller hysteresis loop is also present at lower fields at 7.5 K and 10 K (see inset, Fig. 3).
, likely the result of spin–orbit coupling, and the Van Vleck contribution 357 ± 3 µemu mol-Os−1. Measurements of magnetisation versus field (Fig. 3) reveal a metamagnetic transition in high field with hysteresis, resulting in a double hysteresis loop. In addition, a smaller hysteresis loop is also present at lower fields at 7.5 K and 10 K (see inset, Fig. 3).
 | ||
| Fig. 3 Measurements of magnetisation versus field for a sample of La7Os3O18 at 2 K, 7.5 K and 10 K. A distinctive hysteretic field-induced magnetic phase transition is observed at 7.5 K and 10 K but is beyond the range of fields measured at 2 K. The inset shows a smaller hysteresis loop in the low-field data at the higher temperature points. A plot of the derivatives is available in the SI. | ||
In the case of La7Os3O18, there is an isostructural ruthenate analogue, La7Ru3O18,9,19 which is also a spin- system with µeff = 3.49µB/Ru5+, and θW = −58 K. The ruthenate was found to undergo an antiferromagnetic ordering transition at TN = 14 K, significantly lower than in La7Os3O18 and the authors observed field dependence, with the transition temperature suppressed as applied field was increased.
system with µeff = 3.49µB/Ru5+, and θW = −58 K. The ruthenate was found to undergo an antiferromagnetic ordering transition at TN = 14 K, significantly lower than in La7Os3O18 and the authors observed field dependence, with the transition temperature suppressed as applied field was increased.
To investigate the intriguing findings of the susceptibility measurements further, and to confirm that these features arise intrinsically from La7Os3O18, zero field heat capacity measurements were conducted on two polycrystalline samples of La7Os3O18 from 303 K to 1.8 K using the heat capacity option of a Quantum Design PPMS. These data were fitted between 49.4 and 303 K with a Debye–Einstein model in order to estimate and subtract the phonon contribution to the specific heat. It should be noted that the subtraction of phonon specific heat in the absence of a non-magnetic isostructural phononic reference sample is not trivial. Our data are well described by a roughly equal combination of one Debye and one Einstein mode with characteristic temperatures θD = (262 ± 3) K and TE = (586 ± 7) K respectively, with the remaining optical modes at too high an energy to contribute. We note that this widely used model does violate the total phonon mode count and we discuss alternatives for modelling the phonon specific heat in the SI. The resulting magnetic contribution to the heat capacity is shown in Fig. 2. The data show a sharp peak at 42 K and a broad crossover centred around 8.3 K, with a plateau between the two features. The peak at 42 K corresponds to TN from the susceptibility data, while the crossover centred on 8.3 K is associated with the temperature where there is the maximum change in the susceptibility as the temperature drops below the broad peak at 14 K. This crossover, with its characteristic behaviour in susceptibility and heat capacity, is potentially indicative of low-dimensionality driven short-range ordering.20 The small peak in low-field χ at T = 37 K is located in the plateau region of the magnetic heat capacity data.
From the C/T data, it becomes clear that the majority of the magnetic entropy is associated with the lower temperature, broad crossover. Integration yielded a magnetic contribution to the entropy of the system which approaches S = 4.6 J mol−1 K−1 for both peaks. The expected value for a 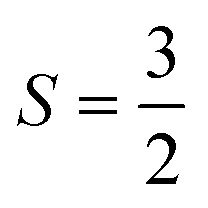 system calculated by Rln(2S + 1) is 11.5 J mol−1 K−1, more than double what we observe, indicating the potential for incomplete magnetic ordering within this system (see SI). This magnetic entropy depends relatively weakly on the low temperature cut-off of the fit (± 20% at the extreme ends of the range). When considering the various possible models for describing the phonon background this magnetic entropy at 60 K is at most 70% of Rln(2S + 1) per mole osmium. The same phonon fit was subtracted from data for another sample, yielding a value for magnetic entropy which approaches S = 5.0 J mol−1 K−1.
system calculated by Rln(2S + 1) is 11.5 J mol−1 K−1, more than double what we observe, indicating the potential for incomplete magnetic ordering within this system (see SI). This magnetic entropy depends relatively weakly on the low temperature cut-off of the fit (± 20% at the extreme ends of the range). When considering the various possible models for describing the phonon background this magnetic entropy at 60 K is at most 70% of Rln(2S + 1) per mole osmium. The same phonon fit was subtracted from data for another sample, yielding a value for magnetic entropy which approaches S = 5.0 J mol−1 K−1.
Taken together, the multiple magnetic transitions as a function of temperature, sharp metamagnetic transition with double hysteresis loop, and residual low-T entropy indicate a complex magnetic phase diagram with potential interplay between competing interactions. These features suggest a richer phase space than in the ruthenate analogue. The higher Néel temperature and greater Weiss constant compared to the ruthenate points to stronger antiferromagnetic interactions within La7Os3O18, likely arising due to the more spatially extended Os d-orbitals. The broad peak we observe in the susceptibility data of La7Os3O18 at 14 K and associated crossover in the heat capacity data constitute a marked departure from the ruthenate analogue, and taken together suggest the possibility of two interwoven magnetic sublattices, with one having low dimensionality. These surprising findings highlight the unusual phases which can emerge when Os, a 5d metal with strong spin–orbit coupling, is incorporated into complex oxide structures. Our findings motivate further studies on both La7Os3O18 and La7Ru3O18, in either case magnetic studies using single crystals would offer insights on the unusual magnetic properties observed. In the case of La7Os3O18, neutron powder diffraction will be used to shed light on its magnetic phase diagram.
Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc06230f.Acknowledgements
We acknowledge funding from the Engineering and Physical Sciences Research Council via EP/P024564/1 (AWR), EP/T011130/1 (ASG) as well as the University of St Andrews for a St Leonard's College World-Leading Doctoral Scholarship (ATB), the International Max Planck Research School for Chemistry and Physics of Quantum Materials (MTP), and the EPSRC for characterisation infrastructure via EP/V034138/1, EP/L017008/1, EP/R023751/1, EP/T019298/1 and EP/T031441/1.References
- A. J. Browne, A. Krajewska and A. S. Gibbs, J. Mater. Chem. C, 2021, 9, 11640–11654 RSC.
- D. I. Khomskii, Transition Metal Compounds, Cambridge University Press, Cambridge, 2014 Search PubMed.
- Y. Shi, Y. Guo, X. Wang, A. J. Princep, D. Khalyavin, P. Manuel, Y. Michiue, A. Sato, K. Tsuda, S. Yu, M. Arai, Y. Shirako, M. Akaogi, N. Wang, K. Yamaura and A. T. Boothroyd, Nat. Mater., 2013, 12, 1024–1027 CrossRef CAS PubMed.
- S. A. Mohitkar, J. Nuss, H. A. Höppe, C. Felser and M. Jansen, Dalton Trans., 2018, 47, 5968–5976 RSC.
- S. Yonezawa, Y. Muraoka and Z. Hiroi, J. Phys. Soc. Jpn., 2004, 73, 1655–1656 CrossRef CAS.
- S. Yonezawa, Y. Muraoka, Y. Matsushita and Z. Hiroi, J. Phys. Soc. Jpn., 2004, 73, 819–821 CrossRef CAS.
- S. Yonezawa, Y. Muraoka, Y. Matsushita and Z. Hiroi, J. Phys.: Condens. Matter, 2004, 16, L9 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- P. Khalifah, Q. Huang, D. M. Ho, H. W. Zandbergen and R. J. Cava, J. Solid State Chem., 2000, 155, 189–197 CrossRef CAS.
- H. A. Mons, M. S. Schriewer and W. Jeitschko, J. Solid State Chem., 1992, 99, 149–157 CrossRef CAS.
- O. C. Gagné and F. C. Hawthorne, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 562–578 CrossRef PubMed.
- F. C. Goerigk and T. Schleid, in 27th Annual Conference of the German Crystallographic Society, March 25–28, 2019, Leipzig, Germany, De Gruyter, Berlin, Boston, 2019, p. 93.
- T. Hummel, F. Salk, M. Ströbele, D. Enseling, T. Jüstel and H.-J. Meyer, Eur. J. Inorg. Chem., 2015, 977–981 CrossRef CAS.
- Q. Ye, M. Barré, K. Adil, A. Rousseau, E. Suard and F. Goutenoire, J. Solid State Chem., 2022, 313, 123310 CrossRef CAS.
- J. K. Lee, A. Nande, A. A. Bhat, S. Watanabe, T. K. Gundu Rao and V. Singh, Ceram. Int., 2024, 50, 17063–17074 CrossRef CAS.
- K. Li and R. Van Deun, Dyes Pigm., 2019, 162, 990–997 CrossRef CAS.
- K. Li and R. Van Deun, Mater. Chem. Front., 2019, 3, 403–413 RSC.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 Search PubMed.
- P. Khalifah, D. A. Huse and R. J. Cava, J. Phys.: Condens. Matter, 2003, 15, 5747 CrossRef CAS.
- A. Vasiliev, O. Volkova, E. Zvereva and M. Markina, npj Quantum Mater., 2018, 3, 18 CrossRef.
| This journal is © The Royal Society of Chemistry 2026 |