
Triplet nitrene-mediated photocatalytic azidation of tertiary C(sp3)–H bonds
Noriaki
Shimamoto
,
Norihito
Arichi
 * and
Hiroaki
Ohno
*
* and
Hiroaki
Ohno
*
Graduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
First published on 25th November 2025
Abstract
A photocatalytic C(sp3)–H azidation involving a triplet nitrene as a hydrogen atom abstractor is described. The key nitrene intermediate is generated through energy transfer to a sulfonyl azide, which also functions as an azide source. This method exhibits excellent selectivity for tertiary C–H bonds and is applicable to leucine-containing dipeptides.
Organic azides serve as privileged building blocks in synthetic chemistry, providing access to a diverse array of nitrogen-containing functional groups and heterocycles.1 They also play a central role in azide–alkyne cycloaddition and Staudinger ligation, both of which enable the assembly of complex conjugates for applications in drug discovery, chemical biology, and materials science.2 The direct azidation of ubiquitous C(sp3)–H bonds is emerging as a powerful strategy to access these valuable molecules in a step-economical fashion, as it obviates the need for substrate pre-functionalization.3,4
Recent advancements in photocatalysis have led to the development of several methods for visible-light-mediated C(sp3)–H azidation under mild conditions (Fig. 1). Generally, photochemically generated heteroatom-centered radicals undergo hydrogen atom transfer (HAT) from alkane substrates to generate alkyl radicals. These intermediates are then trapped by electrophilic azides, such as azidoiodinanes and sulfonyl azides, to afford the corresponding alkyl azide products.
 | ||
| Fig. 1 Photocatalytic C(sp3)–H azidation. | ||
For instance, Chen and coworkers reported an azidation of tertiary aliphatic C–H bonds (Fig. 1a).5 The key HAT step is mediated by an iodanyl radical, which is likely generated by single electron transfer (SET) from the excited ruthenium-based photocatalyst to an azidoiodinane. A collaborative study by the Nicewicz and Alexanian groups demonstrated a C(sp3)–H azidation involving an oxygen-centered radical as a hydrogen atom abstractor, which is generated from a phosphate salt via single electron oxidation by an acridinium photocatalyst (Fig. 1b).6 More recently, West and coworkers developed a C(sp3)–H azidation using earth-abundant tetra-n-butylammonium decatungstate (TBADT) as a photo-HAT catalyst, in which the electrophilic photoexcited decatungstate directly abstracts a hydrogen atom from the substrate (Fig. 1c).7 Despite these significant achievements, the photochemical C(sp3)–H azidation of complex molecular scaffolds remains a considerable challenge. Therefore, the development of new methods that offer versatility and robustness is highly desirable.
On this basis, we envisioned a novel strategy for C(sp3)–H azidation involving energy transfer (EnT) photocatalysis (Fig. 1d).8 In this approach, a sulfonyl azide serves the dual role of terminal azide source and precursor to a triplet nitrene, a reactive species possessing two unpaired electrons with parallel spin. Owing to its N-centered diradical character, the triplet nitrene could act as a hydrogen atom abstracting species. The reactivity of triplet nitrenes has been demonstrated in transformations9 such as alkene aziridination,10 [2+2+1] cycloaddition with 1,6-dienes,11 and sulfide imination recently reported by our group.12 However, their application in C(sp3)–H functionalization remains underdeveloped and is limited to intramolecular C–H insertion13 or HAT from a solvent.14 Herein, we report an EnT-mediated C(sp3)–H azidation through intermolecular HAT by N-sulfonyl triplet nitrenes. This reaction proceeds with excellent site selectivity for tertiary C(sp3)–H bonds and is applicable to the direct azidation of leucine side chains on dipeptides.
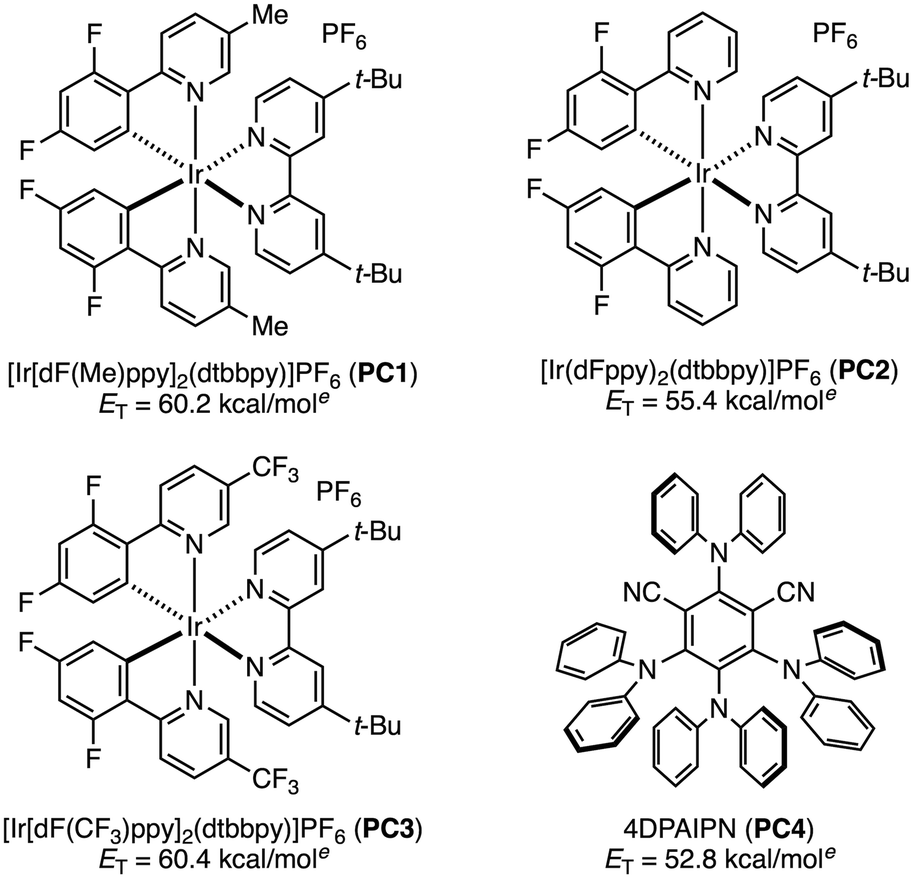
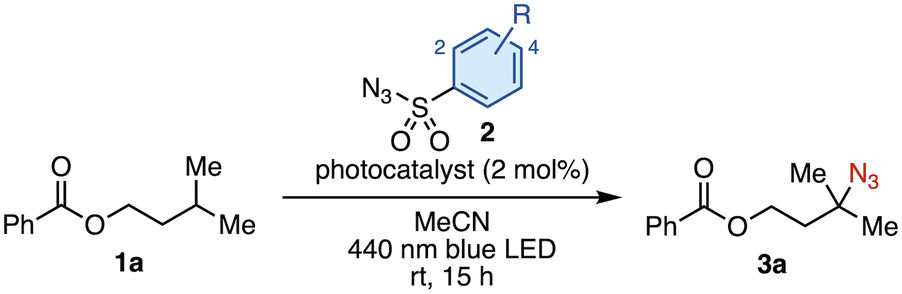
Our investigation commenced with isoamyl benzoate (1a) as a model substrate for the C(sp3)–H azidation (Table 1). We selected [Ir(dF(Me)ppy)2(dtbbpy)]PF6 (PC1) as a photocatalyst because it had proven optimal in our previous work, owing to its high triplet energy (ET = 60.2 kcal mol−1).12 The model reaction was first conducted using two equivalents of tosyl azide (2a) in acetonitrile under 440 nm blue LED irradiation, affording tertiary alkyl azide 3a in 27% yield (entry 1). Intrigued by this initial hit, we screened a series of sulfonyl azides. No products were obtained using benzenesulfonyl azides bearing electron-donating groups such as 4-acetamido (2b) or 4-methoxy (2c) (entries 2 and 3). This result is presumably due to a polarity-mismatched HAT between the less electrophilic nitrenes and the hydridic C–H bond. This outcome prompted us to evaluate sulfonyl azides bearing electron-withdrawing groups (entries 4–9), which revealed that 4-(trifluoromethyl)benzenesulfonyl azide (2g) provided the highest product yield (38%, entry 7). The electron-withdrawing group renders both the resulting triplet nitrene and the sulfonyl azide itself more electrophilic, accelerating both the HAT step and the azidation of the nucleophilic alkyl radical. Increasing the amount of 2g from 2 to 6 equivalents improved the yield of 3a to 53% (entry 10). Although 30% of 1a was recovered, no regioisomeric products were observed. The use of other common EnT photocatalysts (PC2–PC4) resulted in lower yields (entries 11–13). No improvement in the product yield was observed by testing different solvents, longer reaction time, or additives (see the SI). Based on these results, we selected the conditions in entry 10 as optimal. Control experiments confirmed that no reaction occurred in the absence of PC1 or light, which is consistent with the proposed EnT-based mechanism (entries 14 and 15).
|
|
|||
|---|---|---|---|
| Entry | Sulfonyl azide 2 | Photocatalyst | Yield of 3ab (%) |
| a Reaction conditions: 1a (0.2 mmol), 2 (0.4 mmol or 1.2 mmol) and photocatalyst (2 mol%) in MeCN (2 mL) under irradiation with a 45W Kessil 440 nm LED lamp at room temperature under N2 for 15 h. b Yields as determined by 1H NMR of the crude mixtures using dimethyl sulfone as an internal standard. c Isolated yield. d Reaction was performed in the dark. e Reported triplet energies.10b,15 | |||
| 1 | 2a, R = 4-Me (2 equiv.) | PC1 | 27 |
| 2 | 2b, R = 4-NHAc (2 equiv.) | PC1 | <5 |
| 3 | 2c, R = 4-OMe (2 equiv.) | PC1 | <5 |
| 4 | 2d, R = 4-CO2Me (2 equiv.) | PC1 | 27 |
| 5 | 2e, R = 4-CN (2 equiv.) | PC1 | 28 |
| 6 | 2f, R = 4-F (2 equiv.) | PC1 | 34 |
| 7 | 2g, R = 4-CF3 (2 equiv.) | PC1 | 38 |
| 8 | 2h, R = 2-CN (2 equiv.) | PC1 | 24 |
| 9 | 2i, R = 2-CF3 (2 equiv.) | PC1 | 34 |
| 10 | 2g (6 equiv.) | PC1 | 53 (46)c |
| 11 | 2g (6 equiv.) | PC2 | 46 |
| 12 | 2g (6 equiv.) | PC3 | 35 |
| 13 | 2g (6 equiv.) | PC4 | <5 |
| 14 | 2g (6 equiv.) | — | <5 |
| 15d | 2g (6 equiv.) | PC1 | <5 |
|
|||
With the optimized conditions in hand, we next investigated the substrate scope (Fig. 2). Introduction of various substituents on the para-position of benzoate 1a, including bromo (3b), alkyl (3c, 3d), methoxy (3e), trifluoromethyl (3f), and cyano (3g) groups, was well tolerated. The azidation was successfully applied to substrates bearing other functionalities, such as pyridine (3h), ketone (3i), phthalimide (3j), tosylate (3k), and carboxy group (3l). Replacing one of the geminal dimethyl groups of 1a with an ethyl group caused a slight decrease in the product yield (from 46% for 3a to 37% for 3m). A substrate with a longer n-propyl chain resulted in a further decrease in conversion, which was deemed insufficient for isolation of the product (see Table S4). The use of 1-chloroadamantane afforded 3n in 33% yield. This azidation was successfully applied to N-protected memantine, an oral treatment for Alzheimer's disease, providing 3o in 62% yield. For substrates bearing multiple tertiary C–H bonds, azidation occurred selectively at the terminal site (3p, 3q). The azidation of a protected leucine (N-Phth-L-leucine-OMe) occurred exclusively on the methine position of the side chain, affording 3r in 53% yield. To evaluate its applicability to peptide modification, leucine-containing dipeptides were subjected to the reaction conditions (3s–3w). In all cases, the methine C–H bond of the leucine side chain was selectively azidated in the presence of potentially competitive C–H bonds, such as the methine C–H of valine (3u) or the α-oxy C–H of serine (3w).
 | ||
Fig. 2 Substrate scope. Reaction conditions: 1 (0.2 mmol), 2g (1.2 mmol) and PC1 (2 mol%) in MeCN (2 mL) under irradiation with a 45W Kessil 440 nm LED lamp at room temperature under N2 for 15 h. Isolated yields are shown. b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction was run for 24 h. Reaction was run for 24 h. | ||
We then explored product derivatization (Fig. 3). The copper-catalyzed azide–alkyne cycloaddition (CuAAC) of azide 3r with alkyne 4 afforded triazole 5 in 80% yield. This result suggests that a tertiary alkyl azide retains sufficient reactivity as a Click handle despite its steric congestion. Furthermore, tertiary alkyl azides exhibit selective reactivity for bicyclononynes (BCN) over azadibenzocyclooctynes (ADIBO) in strain-promoted azide–alkyne cycloaddition (SPAAC).16 This unique property was applied to dual labelling through the sequential reaction of a primary azide with ADIBO, followed by the tertiary azide with BCN.
 | ||
| Fig. 3 Product derivatization. | ||
To gain insight into the reaction mechanism, we conducted a series of mechanistic experiments. The triplet energy of sulfonyl azide 2g was computed to be 50.8 kcal mol−1 by density functional theory (DFT) calculations at the SMD(MeCN)-(U)B3LYP-D3/6-311++G(2d,p) level of theory. This calculated value is lower than that of PC1 (ET = 60.2 kcal mol−1), indicating the thermodynamic feasibility of the EnT process. Another possible scenario involves single electron reduction of 2g by photoexcited PC1, generating a nitrene radical anion capable of abstracting a C–H bond.14b However, this pathway is considered less likely based on the redox potentials of 2g (Ered1/2 = −1.10 V vs. SCE in MeCN)10c and PC1 (E1/2[IrIV/*IrIII] = −0.92 V vs. SCE).15c Furthermore, a radical trap experiment using 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) prevented the formation of 3a (Fig. 4a). LC/MS analysis of this reaction mixture revealed the formation of bis-TEMPO adduct 6, supporting the intermediacy of the triplet nitrene.
 | ||
| Fig. 4 (a) Radical trap experiment. (b) Proposed mechanism. The BDE values were calculated at the SMD(MeCN)-(U)B3LYP-D3/6-311++G(2d,p)//(U)B3LYP-D3/6-31+G(d) level of theory. | ||
On the basis of the above results, we propose a plausible mechanism shown in Fig. 4b. First, PC1 is excited to triplet state PC1* upon blue light irradiation. EnT from PC1* to sulfonyl azide 2g generates the corresponding triplet sulfonyl azide, which rapidly extrudes nitrogen gas to form triplet nitrene A. This electrophilic diradical then undergoes intermolecular HAT from a hydridic C–H bond of substrate 1, generating alkyl radical B and sulfonamidyl radical C. We postulate that electrophilic N-centered radical C also abstracts a hydrogen from 1 to produce another equivalent of alkyl radical B and 4-(trifluoromethyl)benzenesulfonamide (7). The bond dissociation enthalpy (BDE) of the tertiary C–H bond of isobutane (93.0 kcal mol−1) is lower than the N–H BDEs of C (96.8 kcal mol−1) and 7 (99.9 kcal mol−1), confirming the thermodynamic feasibility of both HAT events. Alkyl radical B is then trapped by sulfonyl azide 2g to afford alkyl azide 3 and sulfonyl radical D. The quantum yield for the model reaction of 1a with 2g was measured to be 3.6 (see the SI). This value suggests that a radical chain mechanism is operative.17 Thus, sulfonyl radical D abstracts a hydrogen atom from another molecule of 1, thereby regenerating alkyl radical B. The concomitant formation of sulfinic acid 8 was observed by LC/MS analysis of the crude reaction mixture (see the SI). Although the O–H BDE of 8 (80.0 kcal mol−1) suggests that the HAT process is thermodynamically unfavored and reversible, alkyl radical B is irreversibly trapped by excess sulfonyl azide 2g. This productive step, which regenerates sulfonyl radical D, kinetically and thermodynamically outcompetes the reverse HAT, accounting for the chain mechanism propagated by D (see Fig. S5 in the SI).
In conclusion, we have developed an EnT-mediated C(sp3)–H azidation by successfully harnessing a triplet nitrene as a HAT reagent. This method offers operational simplicity because the sulfonyl azide plays the dual role of nitrene precursor and terminal azide source. The utility of this reaction was demonstrated in the functionalization of leucine-containing dipeptides. Further investigations to expand the scope to other complex molecules are currently underway.
This work was supported by JSPS KAKENHI (grant numbers JP22K15248 and JP25K09894), Daicel Award in Synthetic Organic Chemistry, Japan, JST SPRING (grant number JPMJSP2110), and Research Support Project for Life Science and Drug Discovery [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from AMED (grant numbers JP22ama121034 and JP22ama121042).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc05587c.Notes and references
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 Search PubMed.
- For recent reviews, see: (a) C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, Chem. Rev., 2020, 120, 4301–4354 CrossRef CAS PubMed; (b) R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed; (c) C. Yang, R. Tripathi and B. Wang, RSC Chem. Biol., 2024, 5, 189–197 RSC.
- For reviews, see: (a) L. Ge, M.-F. Chiou, Y. Li and H. Bao, Green Synth. Catal., 2020, 1, 86–120 Search PubMed; (b) P. Sivaguru, Y. Ning and X. Bi, Chem. Rev., 2021, 121, 4253–4307 CrossRef CAS PubMed; (c) A. A. Antonov and K. P. Bryliakov, Tetrahedron Chem., 2024, 12, 100114 CrossRef CAS.
- For selected examples of C(sp3)–H azidation, see: (a) X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300–5303 CrossRef CAS; (b) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef CAS; (c) C. S. Day, A. Fawcett, R. Chatterjee and J. F. Hartwig, J. Am. Chem. Soc., 2021, 143, 16184–16196 CrossRef CAS PubMed; (d) R. R. Karimov, A. Sharma and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 715–724 CrossRef CAS PubMed; (e) S.-E. Suh, S.-J. Chen, M. Mandal, I. A. Guzei, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 11388–11393 CrossRef CAS PubMed; (f) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS; (g) J. F. Hooson, H. N. Tran, K.-J. Bian and J. G. West, Chem. Commun., 2024, 60, 3705–3708 RSC.
- Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679–2683 RSC.
- K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213–4217 CrossRef CAS PubMed.
- Y.-C. Lu, S.-C. Kao and J. G. West, Chem. Commun., 2022, 58, 4869–4872 RSC.
- (a) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (b) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS; (c) F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CrossRef CAS; (d) S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- M. Yao, G. Chen, X. Zhang, A. Yusuf and X. Xu, Eur. J. Org. Chem., 2025, e202401404 CrossRef CAS.
- (a) S. O. Scholz, E. P. Farney, S. Kim, D. M. Bates and T. P. Yoon, Angew. Chem., Int. Ed., 2016, 55, 2239–2242 CrossRef CAS; (b) A. R. Meyer, M. V. Popescu, A. Sau, N. H. Damrauer, R. S. Paton and T. P. Yoon, ACS Catal., 2024, 14, 12310–12317 CrossRef CAS; (c) D. Dam, N. R. Lagerweij, K. M. Janmaat, K. Kok, E. Bouwman and J. D. C. Codée, J. Org. Chem., 2024, 89, 3251–3258 CrossRef CAS; (d) N. Baris, M. Dračínský, J. Tarábek, J. Filgas, P. Slavíček, L. Ludvíková, S. Boháčová, T. Slanina, B. Klepetářová and P. Beier, Angew. Chem., Int. Ed., 2024, 63, e202315162 CrossRef CAS PubMed; (e) C. Wu and Y. Zhu, Chem. Commun., 2024, 60, 12449–12452 RSC.
- F. Li, W. F. Zhu, C. Empel, O. Datsenko, A. Kumar, Y. Xu, J. H. M. Ehrler, I. Atodiresei, S. Knapp, P. K. Mykhailiuk, E. Proschak and R. M. Koenigs, Science, 2024, 383, 498–503 CrossRef CAS.
- N. Arichi, T. Amano, S. Wu, S. Inuki and H. Ohno, Chem. – Eur. J., 2024, 30, e202401842 CrossRef CAS.
- (a) H. Jung, H. Keum, J. Kweon and S. Chang, J. Am. Chem. Soc., 2020, 142, 5811–5818 CrossRef CAS; (b) S. Bouayad-Gervais, C. D.-T. Nielsen, A. Turksoy, T. Sperger, K. Deckers and F. Schoenebeck, J. Am. Chem. Soc., 2022, 144, 6100–6106 CrossRef CAS.
- (a) S. Zhu, A. Pathigoolla, G. Lowe, D. A. Walsh, M. Cooper, W. Lewis and H. W. Lam, Chem. – Eur. J., 2017, 23, 17598–17604 CrossRef CAS; (b) N. Shimamoto, N. Arichi, S. Inuki and H. Ohno, Chem. Pharm. Bull., 2025, 73, 831–834 CrossRef CAS PubMed.
- (a) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC; (b) A. Singh, K. Teegardin, M. Kelly, K. S. Prasad, S. Krishnan and J. D. Weaver, J. Organomet. Chem., 2015, 776, 51–59 CrossRef CAS; (c) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS.
- D. Svatunek, N. Houszka, T. A. Hamlin, F. M. Bickelhaupt and H. Mikula, Chem. – Eur. J., 2019, 25, 754–758 CrossRef CAS PubMed.
- J. C. Scaiano, Chem. Soc. Rev., 2023, 52, 6330–6343 CAS.
| This journal is © The Royal Society of Chemistry 2026 |