
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the limits of inductive electron withdrawal in fused bicyclic azaheterocycles
Sachin Suresh
Bhojgude
 a,
David R.
Turnbull
b,
Sudhakar
Pagidi
cd,
Amruth
C.
b,
Eli
Zysman-Colman
d,
Gregory C.
Welch
b and
Jeffrey F.
Van Humbeck
*b
a,
David R.
Turnbull
b,
Sudhakar
Pagidi
cd,
Amruth
C.
b,
Eli
Zysman-Colman
d,
Gregory C.
Welch
b and
Jeffrey F.
Van Humbeck
*b
aDepartment of Discovery Synthesis, Biocon Bristol Myers Squibb R&D Centre, Syngene International Ltd., Biocon Park, Plot No. 2 & 3, Jigani Link Road, Bommasandra IV, Bangalore, 560 099, India
bDepartment of Chemistry, University of Calgary, Calgary, Alberta T2N 1N4, Canada. E-mail: jeffrey.vanhumbec1@ucalgary.ca
cDepartment of Chemistry, Indian Institute of Technology Tirupati, Venkatagiri Road, Yerpedu Post, Tirupati District, Andhra Pradesh 517 619, India
dOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews KY16 9ST, Fife, UK
First published on 2nd December 2025
Abstract
The ability to easily modify the electronic properties of aromatic azaheterocycles has led to their ubiquitous application across all disciplines of chemistry. The electronic properties of the aromatic π-system are most often modified by π-electron donating and withdrawing groups, naturally. Nearly fifty years ago, an important exception to this trend was reported, when the single-step synthesis of 5,7-bis(trifluoromethyl)-1,8-naphthyridine was described—a highly electron deficient system based exclusively on inductive electron withdrawing groups (CF3) and the inherent effect of nitrogen for carbon substitution. Herein, we report on efforts to extend these substitutions to create maximally electron withdrawn fused bicyclic azaheterocycles using only inductive effects. DFT calculations support the assignment of these structures as being strongly electron withdrawing, with ground-state LUMO energies as low as −2.91 eV obtained. Host-free OLEDs featuring these acceptor structures as part of polymer emissive layers show colors consistent with their electron-deficient character.
In the 1970s, researchers at Merck reported the one-step synthesis of electron-deficient 1,8-naphthyridine 1 (Fig. 1),1 and described the use of this heterocycle for anti-hypertensive and bronchodilator development.2–4 Given its facile synthesis this heterocycle has found application in several fields, for example as a pH- and metal-sensitive fluorescent probe (2),5 as a large Stokes-shift fluorophore (3),6 and as an amine-reactive cell imaging agent (4).7 Specifically, we were most drawn to the fact that the aromatic system is rendered very electron poor without the use of π-electron withdrawing groups. One cannot discount the successful use of canonical π-electron withdrawing groups (e.g. –CN) in countless applications—but having alternatives that lack the functional group chemistry inherent to such groups would be useful for the synthetic chemist. Modifying the parent scaffold (1) to provide new electron deficient building blocks for a range of applications became our goal. Beginning with naphthalene itself, there are seven C–H positions available for modification (with one position reserved for connection to the rest of the molecule). While there are many permutations of N/CF3 substitution possible, we targeted the hypothetical molecule 5 (Fig. 1) with an alternating substitution pattern. Flanking –CF3 groups would reduce the availability of the nitrogen lone pairs, while the nitrogen atoms would alleviate potential steric clashes between –CF3 groups at adjacent positions on the same ring, or 1,3-related across the ring junction. Herein, we describe the incorporation of additional N/CF3 to the 1,8-naphthyridine scaffold and the production of model small molecules and polymers that are representative of potential applications for such systems.
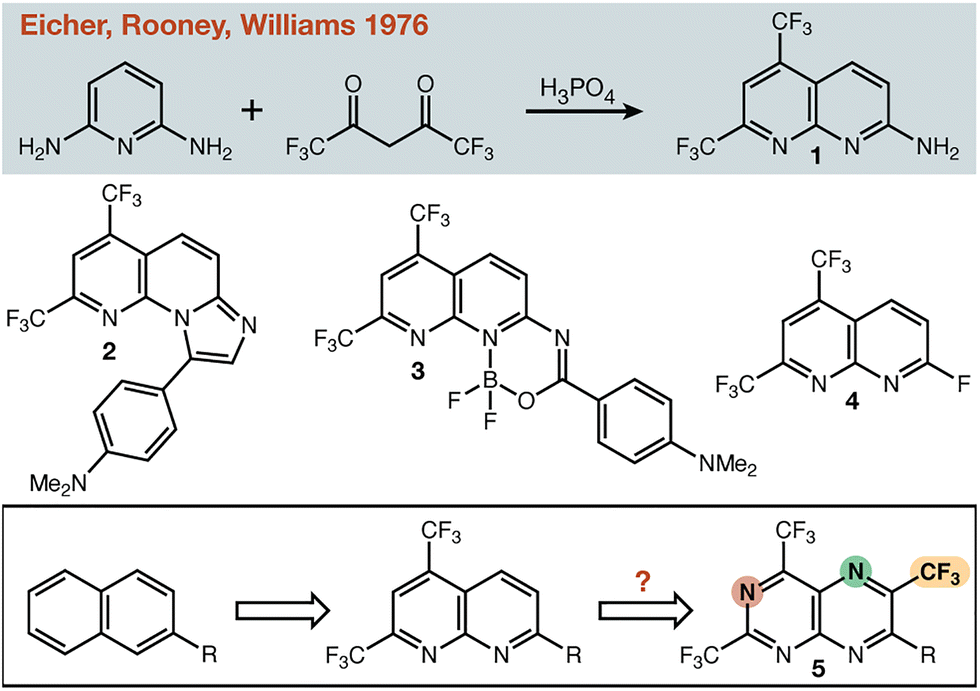 | ||
| Fig. 1 Representative applications of the 5,7-di-trifluoromethyl-1,8-naphthyridine scaffold and a hypothetical maximally withdrawn target. | ||
Synthetic approaches to building blocks: amino-naphthyridine 1 can be converted into the corresponding chloride through a two-step sequence (1 → 6 → 7) as previously reported (Fig. 2).8 While 7 has been known for more than a decade, its C–Cl bond has been used almost exclusively to make new heteroatom bonds, with only a single example of C–C bond formation (i.e. the addition of benzylcyanide anion) reported;9 developing C–C bond forming reactions for these heterocycles became our focus (vide infra). Other potential cross-coupling partners at this position (i.e. –Br, –I, –OSO2R) have never been reported. To achieve a nitrogen-for-carbon swap at the 6-position, we used commercially available triazene 10. This reagent has been reported to undergo cyclization reactions to yield bis(trifluoromethyl) pyrimidine derivatives.10–25 Our specific target (11) had not been described in the academic literature, though one patent report suggested the potential for success.26 We found 10 is reactive towards both diamino-(8) and amino-hydroxy pyridine (9), affording 11 and 12, respectively. Next, we installed an additional trifluoromethyl group in the 3-position using the radical strategy reported by Baran.27 We found these conditions delivered a mixture containing a small amount of unreacted starting material (1) that was difficult to separate from the desired product 14. The simplest solution proved to be carrying this mixture through diazotization (to 15 and 6), and finally chlorination (to 16 and 7), at which stage separation is easy. Addition at positions adjacent to—or across the ring junction from—existing –CF3 groups was less favourable as expected.
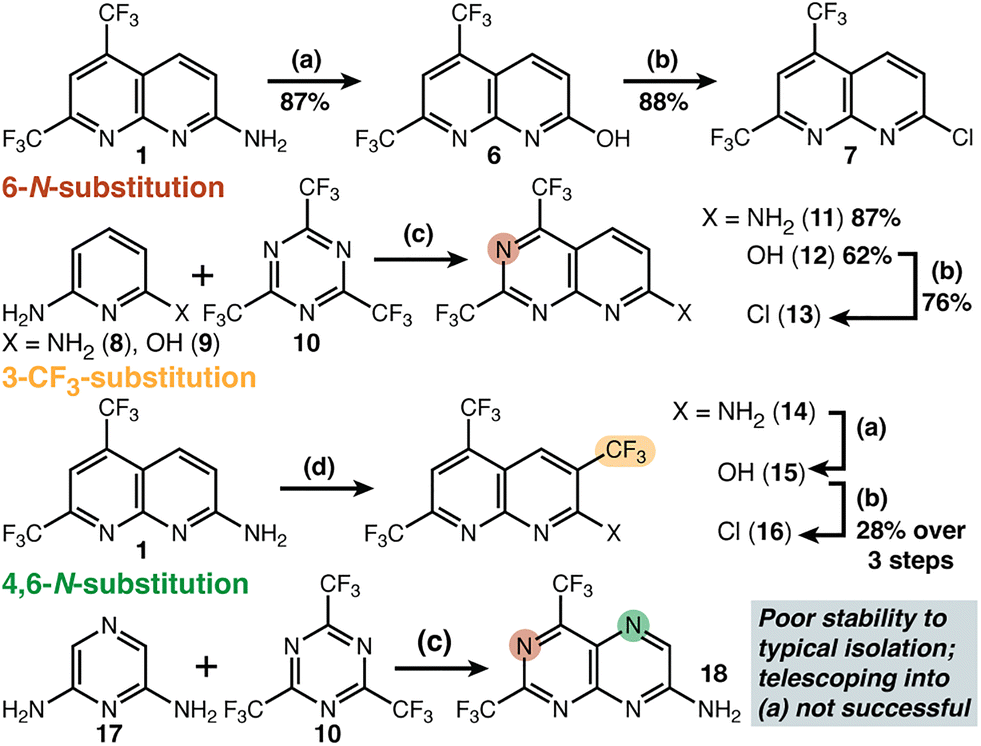 | ||
| Fig. 2 Synthetic strategy for C/N substitution and CF3 addition at remaining unsubstituted positions. (a) H2SO4, NaNO2, H2O 0 to 80 °C (b) POCl3, DMF 0 to 100 °C (c) AcOH, 0 to 80 °C (d) NaSO2CF3, tBuOOH (5.5 M in decane), H2O/CH2Cl2, rt. | ||
OH/NH2-containing compounds (i.e.1, 6, 11, 12, 14, and 15) displayed consistent colours (i.e. yellow to red fluorescence) and TLC behaviour. The exposure of 2,6-diaminopyrazine (17) to our standard cyclization conditions initially looked promising by similar TLC analysis. Isolation of the desired pteridine proved far more difficult. Compounds that were initially yellow fluorescent (on TLC) were isolated as black solids with complex spectra after either aqueous work-up or silica gel chromatography. Quick filtrations through alumina and telescoping into diazotization—including anhydrous conditions with iso-amyl nitrite—were also unsuccessful. We suspect that the fully-substituted target 5 would share similar sensitivity to (presumed) 18, which features only one unsubstituted position—and so did not further pursue that target here.
C–C bond formation and model molecules: we next sought to apply 7, 13, and 16 as building blocks in contexts where their electron deficient nature would be relevant. We began with cross-coupling onto 2,2′-bithiophene as a proxy for the canonical acceptor-donor–acceptor oligothiophene structure.28,29 Our successful conditions are described in Fig. 3. Use of a modern catalyst well-known to be effective for Suzuki coupling (i.e. SPhos-Pd-G430) was compatible both with the parent substrate 7 and 6-N-substituted 13, delivering the double-coupling products 19 and 20. When the heteroaryl chloride was hindered (16), such conditions only return hydrolysed starting material (15). Using anhydrous conditions was therefore one necessary modification. The second was the use of copper(I) additives, which has been shown to greatly improve the coupling of CF3-substituted pyridines,31 and proved to be critical here. In the referenced report, the pyridine is the nucleophile of the reaction unlike in our case—so we do not suggest that the impact of Cu(I) on the mechanism of the reaction is necessarily identical. Regardless of the underlying reason, these conditions were uniquely able to afford 21.
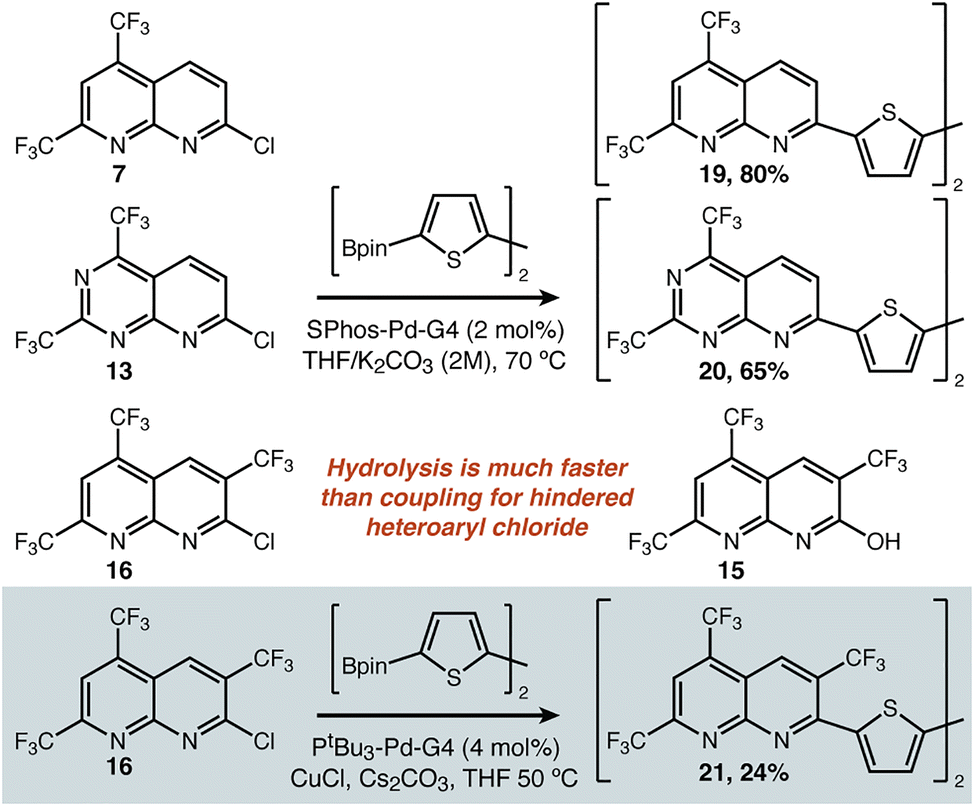 | ||
| Fig. 3 Conditions for C–C bond formation via cross coupling for both sterically accessible and sterically hindered substrates. | ||
Estimation of electron withdrawing ability: with confirmation that these building blocks could be elaborated by cross-coupling, we sought to demonstrate their electron withdrawing ability. Since the breakthrough report of Adachi in 2012,32 it has been realized that organic small molecules with linked electron-rich donors and electron-poor acceptors—often constructed so there is a significant twist between their donor/acceptor π-systems—commonly display thermally-activated delayed fluorescence (TADF) behaviour and have LUMO energy levels determined mostly by the structure of the acceptor.33–37 Density functional theory calculations were carried out on a suite of model compounds (Fig. 4) using PBE0 functional38 and 6-31G(d,p) basis set.39. We also synthesized three of these compounds (22–24) to ensure the fluorinated heterocycle could be used as shown (see SI). DFT results show that the LUMO is centred on the fluorinated acceptor, and thus the energy of this molecular orbital reveals their highly electron-deficient nature. Many of these compounds have LUMO levels that are lower in energy than triazine (25), phthalimide (26), dicyanopyridine (27), or heptazine (28). Our compounds in general are predicted to be around heptazine, or between heptazine and a very electron deficient pyrazine acceptor (29) that has emission in the near infrared when combined with a diphenyl amine donor.40
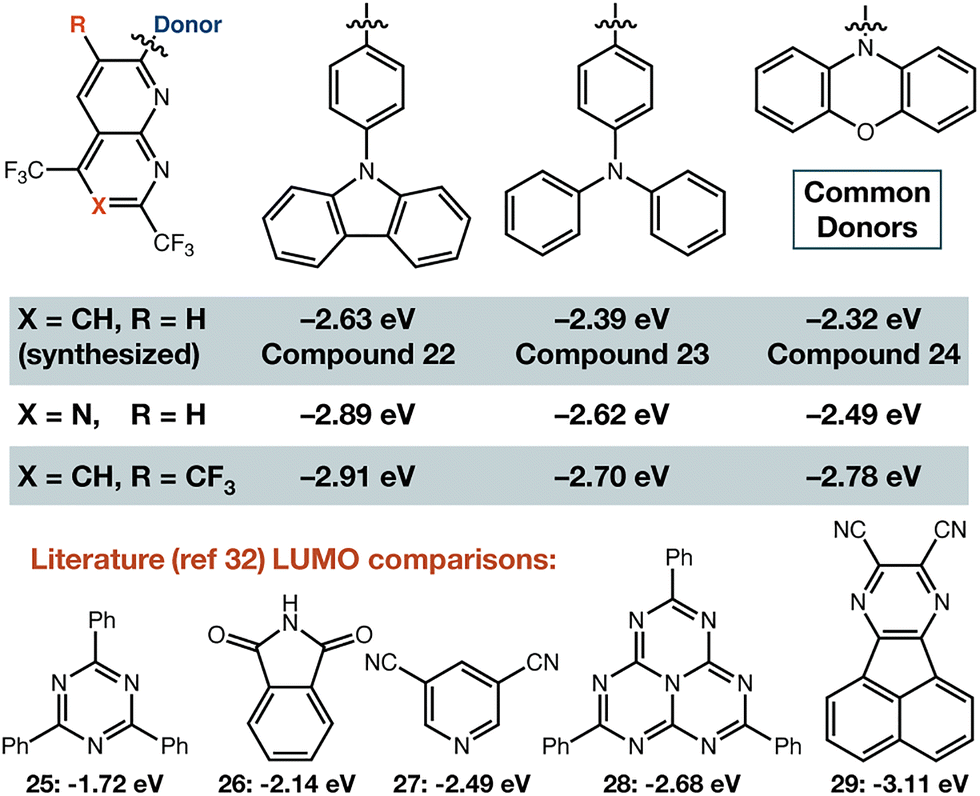 | ||
| Fig. 4 LUMO energies for representative molecules. DFT was performed with PBE0 functional and 6-31G(d,p) basis set in both this work and ref. 37. | ||
One of our laboratories recently reported the fabrication of host-free OLED devices under air.41 That report used an organic polymer (i.e. “Super Yellow”42) as the emissive layer, so we sought to incorporate 7 into an appropriate polymer. Creating building blocks for polymerization (Fig. 5, 31 and 32) also allowed us to further demonstrate the chemistry of these systems (see SI). We selected the backbone polymer described by Bryce43— which uses fluorene 33 and sulfone 34 as the major components—to incorporate our building blocks by Suzuki polymerization (Fig. 5). Incorporation of 31 and 32 in both low (2 mol%) and high (10 mol%) amounts led to excellent yields of polymer, with emitter loading having a significant effect on emissive properties. Molecular weight analysis of the polymers (at both heterocycle loadings) displayed reasonable MN (32–62 kDa), MW (50–89 kDa), and PDI (1.39–1.56). Our recently described OLED fabrication procedure (with no additional optimization) delivered devices for initial analysis.41 Polymers derived from 31 and 32 show green-yellow and orange-red electroluminescence, respectively (see SI). When compared to previous reports using heptazine acceptors in combination with carbazole44 and phenoxazine45 donors, the observed emission maxima further support the electron deficient nature of these systems.
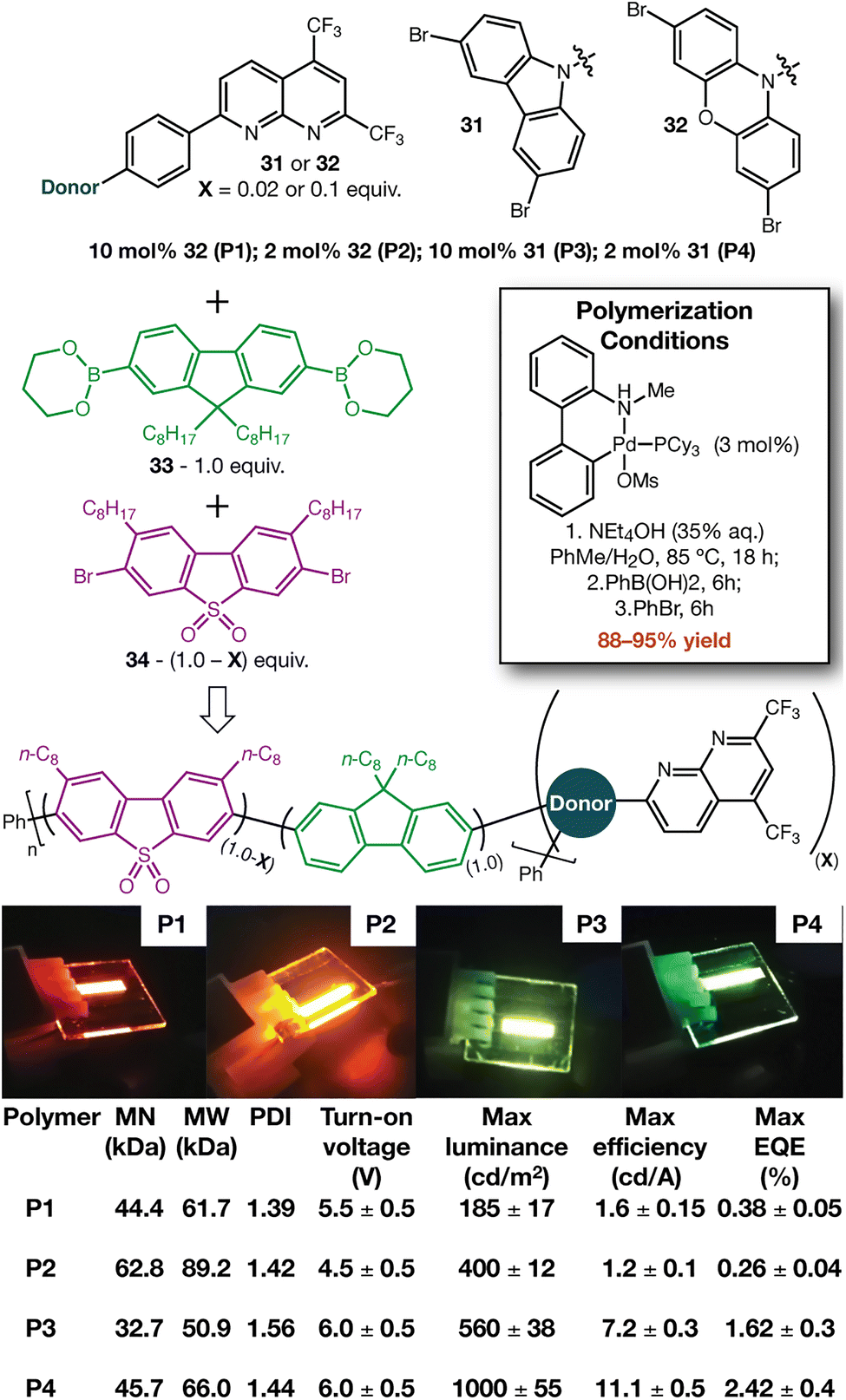 | ||
| Fig. 5 Synthesis of light-emitting polymers with electron deficient heterocycles incorporated. OLED devices were fabricated and tested as in ref. 41. | ||
We set out to increase the electron deficient nature of the naphthalene ring system without using π-electron withdrawing groups. Azaheterocycles that feature five of the seven possible C–H positions modified to N or CF3 were produced. Catalytic and stoichiometric chemistry shows that these building blocks can be incorporated into more elaborate organic molecules. DFT calculations and the observed colours of the OLEDs produced speak to the highly electron withdrawn nature of these systems.
SSB synthesized small molecules and polymers. DRT synthesized small molecules and performed preliminary calculations. SP conducted the theoretical study and produced illustrations. AC fabricated and tested polymer OLEDs. EZ-C analysed results. GCW co-conceptualized project and analysed results. JVH co-conceptualized project, analysed results, and drafted manuscript. All authors edited the draft manuscript.
This project is funded by European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska Curie grant agreements No. 891606 (TADFNIR). S. P. thanks IIT Tirupati for computational resources. The St Andrews team thanks the EPSRC (EP/Z535291/1, EP/W007517/1, and EP/W015137/1) for financial support. The UCalgary team was supported by, and thanks, Alberta Innovates (Strategic Projects Grant G2018000901) and the Canada First Research Excellence Fund.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, spectral information, OLED characteristics, and detailed DFT outputs. See DOI: https://doi.org/10.1039/d5cc04446d.Notes and references
- E. Eichler, C. S. Rooney and H. W. R. Williams, J. Heterocycl. Chem., 1976, 13, 41–42 CrossRef CAS.
- H. W. R. Williams and C. S. Rooney, US Pat., 4031103, 1977 Search PubMed.
- H. W. R. Williams and C. S. Rooney, US Pat., 3962262, 1976 Search PubMed.
- H. W. R. Williams and C. S. Rooney, US Pat., 3843663, 1974 Search PubMed.
- S. Matsumoto, T. Umeno, N. Suzuki, K. Usui, M. Kawahata and S. Karasawa, Chem. Commun., 2022, 58, 12435–12438 RSC.
- Y.-Y. Wu, Y. Chen, G.-Z. Gou, W.-H. Mu, X.-J. Lv, M.-L. Du and W.-F. Fu, Org. Lett., 2012, 14, 5226–5229 CrossRef CAS PubMed.
- T. Umeno, L. Muroi, Y. Kayama, K. Usui, K. Hamada, A. Mizutani and S. Karasawa, Bioconjugate Chem., 2023, 34, 1439–1446 CrossRef CAS PubMed.
- N. Harada, Y. Abe, S. Karasawa and N. Koga, Org. Lett., 2012, 14, 6282–6285 CrossRef CAS PubMed.
- Z. Yin, Z. Zhang, J. F. Kadow, N. A. Meanwell and T. Wang, J. Org. Chem., 2004, 69, 1364–1367 CrossRef CAS PubMed.
- R. A. Lowe, D. Taylor, K. Chibale, A. Nelson and S. P. Marsden, Bioorg. Med. Chem., 2020, 28, 115442 CrossRef CAS.
- K. Yang, Q. Dang, P.-J. Cai, Y. Gao, Z.-X. Yu and X. Bai, J. Org. Chem., 2017, 82, 2336–2344 CrossRef CAS PubMed.
- K. Yang, Q. Dang and X. Bai, Eur. J. Org. Chem., 2017, 1922–1925 CrossRef CAS.
- K. Yang, Z. Yang, Q. Dang and X. Bai, Eur. J. Org. Chem., 2015, 4344–4347 CrossRef CAS.
- M. De Rosa, D. Arnold and D. Hartline, J. Org. Chem., 2013, 78, 8614–8623 CrossRef CAS.
- V. O. Iaroshenko, A. Maalik, D. Ostrovskyi, A. Villinger, A. Spannenberg and P. Langer, Tetrahedron, 2011, 67, 8321–8330 CrossRef CAS.
- V. O. Iaroshenko, A. Bunescu, A. Spannenberg and P. Langer, Chem. – Eur. J., 2011, 17, 7188–7192 CrossRef CAS PubMed.
- Q. Dang and Y. Liu, Tetrahedron Lett., 2009, 50, 6758–6760 CrossRef CAS.
- V. O. Iaroshenko, Synthesis, 2009, 3967–3974 CrossRef CAS.
- G. Xu, L. Zheng, S. Wang, Q. Dang and X. Bai, Synlett, 2009, 3206–3210 CAS.
- Q. Dang, E. Carruli, F. Tian, F. W. Dang, T. Gibson, W. Li, H. Bai, M. Chung and S. J. Hecker, Tetrahedron Lett., 2009, 50, 2874–2876 CrossRef CAS.
- V. O. Iaroshenko, Y. Wang, D. V. Sevenard and D. M. Volochnyuk, Synthesis, 2009, 1851–1857 CrossRef CAS.
- V. O. Iaroshenko, D. V. Sevenard, A. Kotljarov, D. M. Volochnyuk, A. O. Tolmachev and V. Y. Sosnovskikh, Synthesis, 2009, 731–740 CrossRef CAS.
- V. O. Iaroshenko, D. M. Volochnyuk, W. Yan, M. V. Vovk, V. J. Boiko, E. B. Rusanov, U. M. Groth and A. A. Tolmachev, Synthesis, 2007, 3309–3318 CAS.
- M. De Rosa, D. Arnold, E. Blythe, M. S. Farrell, T. Seals, K. Wills and M. Medved, Heterocycl. Commun., 2007, 13, 97–100 CrossRef CAS.
- Q. Dang, Y. Liu and Z. Sun, Tetrahedron Lett., 2001, 42, 8419–8422 CrossRef CAS.
- X. Dai, et al.. WIPO-PCT Pat., WO2022/049521A1, 2022 Search PubMed.
- Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411–14415 CrossRef CAS PubMed.
- A. S. Tanwar and F. Meyer, CrystEngComm, 2025, 27, 736–748 RSC.
- L. Zhang, N. S. Colella, B. P. Cherniawski, S. C. B. Mannsfeld and A. L. Briseno, ACS Appl. Mater. Interfaces, 2014, 6, 5327–5343 CrossRef CAS PubMed.
- N. C. Bruno, N. Niljianskul and S. L. Buchwald, J. Org. Chem., 2014, 79, 4161–4166 CrossRef CAS PubMed.
- J. Z. Deng, D. V. Paone, A. T. Ginnetti, H. Kurihara, S. D. Dreher, S. A. Weissman, S. R. Stauffer and C. S. Burgey, Org. Lett., 2009, 11, 345–347 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- J. M. Dos Santos, et al. , Chem. Rev., 2024, 124, 13736–14110 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Jensen, WIREs Compl. Mol. Sci., 2013, 3, 273–295 CrossRef CAS.
- Y. Yuan, Y. Hu, Y.-X. Zhang, J.-D. Lin, Y.-K. Wang, Z.-Q. Jiang, L.-S. Liao and S.-T. Lee, Adv. Funct. Mater., 2017, 27, 1700986 CrossRef.
- A. C, D. K. Dubey, M. Pahlevani and G. C. Welch, Adv. Mater. Technol., 2021, 6, 2100264 CrossRef CAS.
- S. Gambino, A. K. Bansal and I. D. W. Samuel, Org. Electron., 2013, 14, 1980–1987 CrossRef CAS.
- K. T. Kamtekar, H. L. Vaughan, B. P. Lyons, A. P. Monkman, S. U. Pandya and M. R. Bryce, Macromolecules, 2010, 43, 4481–4488 CrossRef CAS.
- D. M. Mayder, R. Hojo, W. L. Primrose, C. M. Tonge and Z. M. Hudson, Adv. Funct. Mater., 2022, 40, 2204087 CrossRef.
- Y. Kang, L. Zhao and J. Leng, Chem. Phys. Lett., 2018, 698, 187–194 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |