
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a spectrophotometric assay for high-throughput screening and mechanistic characterization of glucose-1-phosphate thymidylyltransferase inhibitors
Bronwyn E.
Rowland
 a,
Jesse C.
Fuller
ab,
Chigozie L.
Okolie
b and
David L.
Jakeman
*ab
a,
Jesse C.
Fuller
ab,
Chigozie L.
Okolie
b and
David L.
Jakeman
*ab
aDepartment of Chemistry, Dalhousie University, Halifax, Nova Scotia B3H 4R2, Canada, david.jakeman@dal.ca
bCollege of Pharmacy, Dalhousie University, Halifax, Nova Scotia B3H 4R2, Canada
First published on 24th April 2026
Abstract
The sugar β-L-rhamnose is often a conserved component of bacterial polysaccharide capsules, an extracellular structure featured in many drug-resistant, pathogenic bacteria. The β-L-rhamnose biosynthetic enzymes (RmlA-D) have therefore become key targets in antibiotic development. However, inhibitor discovery for these enzymes, particularly α-D-glucose-1-phosphate thymidylyltransferase, has been hindered by a lack of robust activity assays. To address this challenge, we report a continuous, coupled-enzyme assay for screening α-D-glucose-1-phosphate thymidylyltransferase inhibitors, using Streptococcus pneumoniae Cps2L as a model enzyme. This platform was used to characterize ligand binding to Cps2L, identifying uridine diphosphate-α-D-glucose and pyrophosphate as substrates of the reverse pyrophosphorylase reaction, and to explore inhibition by thymidine diphosphate-β-L-rhamnose (TDP-Rha). A rapidly reversible binding mechanism at the allosteric site was identified for TDP-Rha (IC50 = 45.1 ± 2.3 µM) using progress curve shape analysis and an engineered E253D Cps2L variant. Offering high-throughput capabilities and detailed kinetic resolution, the developed assay is well-suited for inhibitor discovery and mechanistic characterization.
Introduction
Multi-drug resistant bacteria remain a threat to global health as acquired resistance continues to outpace new iterations of modern antibiotics. The declining utility of traditional antibacterials, which predominantly inhibit peptidoglycan synthesis, has prompted interest in targeting the polysaccharide capsule instead. This extracellular structure surrounds the peptidoglycan and is commonly featured in highly pathogenic, drug-resistant bacteria, such as Acinetobacter baumannii,1Mycobacterium tuberculosis,2Klebsiella pneumoniae,3Staphylococcus aureus,4 and Streptococcus pneumoniae.5Capsular polysaccharides are composed of repeating oligosaccharide units, in which β-L-rhamnose, a 6-deoxyhexose sugar, frequently appears as a conserved building block.6,7 The production of β-L-rhamnose in bacteria proceeds through a single pathway involving four enzymes (Scheme 1).8 In this pathway, thymidine triphosphate (TTP) and α-D-glucose-1-phosphate (Glc-1-P) are transformed into thymidine diphosphate-β-L-rhamnose (TDP-Rha), facilitated in sequence by an α-D-glucose-1-phosphate thymidylyltransferase (RmlA), a 4,6-dehydratase (RmlB), a 3,5-epimerase (RmlC), and a 4-keto reductase (RmlD). TDP-Rha ultimately serves as the glycosyl donor for rhamnosyltransferases in capsular polysaccharide synthesis, as well as a feedback inhibitor of RmlA (Fig. 1A).9,10
 | ||
Scheme 1 Bacterial TDP-β-L-rhamnose biosynthetic pathway. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Some RmlB variants require Mg2+. bR = bacterial glycan acceptor. Some RmlB variants require Mg2+. bR = bacterial glycan acceptor. | ||
 | ||
| Fig. 1 Known inhibitors of α-D-glucose-1-phosphate thymidylyltransferases. (A) Endogenous feedback inhibitor, TDP-Rha. (B) P. aeruginosa RmlA synthetic inhibitors. (C) and (D) S. pneumoniae Cps2L synthetic inhibitors. | ||
Rhamnose-rich capsular polysaccharides are most commonly found in Gram-positive bacteria of the genera Streptococcus and Enterococcus.11 However, many mycobacteria also utilize β-L-rhamnose in their capsule, such as M. tuberculosis, where a rhamnose linkage anchors arabinogalactan polysaccharides to the peptidoglycan.12,13 Similar to capsular polysaccharides, lipopolysaccharides on the surface of Gram-negative bacteria often possess rhamnan components as well, such as in Pseudomonas aeruginosa14 and Salmonella enterica.15 Functional studies across species show that a knockout of any rml gene strongly correlates to severely attenuated bacterial growth and impaired biofilm formation.13,16–23 Notably, ΔrmlA mutants in Mycobacterium smegmatis (the model strain for M. tuberculosis) and P. aeruginosa show increased susceptibility to macrophage-mediated killing and reduced colonization capacity in mouse models.22,23 The critical role of β-L-rhamnose in bacterial fitness and virulence, coupled with its complete absence in humans,6 underscores the pathway as an attractive target for antibiotic development.
As the first committed step of TDP-Rha biosynthesis, we are primarily interested in the inhibition of α-D-glucose-1-phosphate thymidylyltransferase. The strongest known inhibitor of this enzyme was discovered by Naismith, Westwood, and co-workers.24 This group initially discovered a thymine-like lead from a commercial library with a potent IC50 of 73 ± 10 nM against P. aeruginosa RmlA (Fig. 1B, compound 1). Subsequent structural optimization efforts mainly hindered inhibition, except for the addition of bromine on the benzyl substituent (Fig. 1B, compound 2, IC50 = 34 ± 2 nM).24,25 Despite nanomolar potency, only modest whole-cell activity in M. tuberculosis was found for compound 2 (MIC100 > 25 µg mL−1), conceivably due to poor membrane permeability. Conversely, our group has previously applied a rational design approach for inhibitors targeting the RmlA homolog in S. pneumoniae, known as Cps2L. A series of sugar phosphonates were designed as non-hydrolyzable analogues of β-L-rhamnose pathway metabolites, although exhibited weaker inhibition (Fig. 1C, IC50s > 5 mM, Kis ≥ 70 µM).26,27 Similar inhibition trends were observed for our tri- and tetraphosphate analogues of TDP-Glc (Fig. 1D), synthesized to resemble the transition state stabilized by Cps2L.28 Although P. aeruginosa RmlA and S. pneumoniae Cps2L have a relatively high sequence similarity (68%), Naismith and Westwood's compound 1 showed only minor inhibition (<10% at 25 mM) against Cps2L. This discrepancy likely occurs because 1 binds at the RmlA allosteric site, a region that is less conserved in the Cps2L homolog.26,29,30 With limited alternative leads, a therapeutically useful RmlA/Cps2L inhibitor remains elusive.
A major obstacle in the discovery of RmlA/Cps2L inhibitors has been establishing a robust screening assay. Techniques such as HPLC,31–33 capillary electrophoresis,34 or ESI-MS35 have been used to measure thymidylyltransferase activity through direct detection of the TDP-Glc product. However, these methods are low throughput and often require high substrate or enzyme concentrations. As the alternative to direct detection, coupled-enzyme assays use auxiliary enzymes to transform the product into an analyte more amenable to quantification. Some coupled-enzyme assays are discontinuous, whereby samples of a reaction are quenched at various times (e.g. with heat or acid) and the total product at each time point is measured. An example is the malachite green assay, the most common high-throughput method for measuring RmlA/Cps2L activity. Its main drawback is the need to individually collect, quench, and measure samples at each reaction time point. Continuous coupled-enzyme assays overcome this limitation through real-time reaction monitoring. For example, Elling and co-workers developed a continuous assay coupled to the oxidation of NADH,36 but it requires the use of four coupling enzymes. We previously developed an RmlA/Cps2L activity assay coupled to uric acid production,27 but one of the coupling enzymes (purine nucleoside phosphorylase) can bind thymidine-based inhibitors as pseudo-substrates, complicating their evaluation.
In this study, we present a continuous, coupled-enzyme assay that circumvents limitations of earlier RmlA/Cps2L activity assays. This method exploits the bidirectional catalysis of α-D-glucose-1-phosphate thymidylyltransferases, measuring activity via the reverse pyrophosphorylase reaction. Building on the work of Kornfeld, Melo, and Glaser with P. aeruginosa RmlA,10,37 we adapted their approach into a robust microtiter plate format, which was used to kinetically characterize UDP-Glc and PPi as S. pneumoniae Cps2L substrates. To further validate the assay, a known α-D-glucose-1-phosphate thymidylyltransferase inhibitor, TDP-Rha, was tested against Cps2L. Our findings support a rapidly reversible binding mechanism for TDP-Rha at the enzyme's allosteric site. Together, this work provides a platform for high-throughput identification and mechanistic characterization of novel α-D-glucose-1-phosphate thymidylyltransferase inhibitors.
Materials and methods
General methods and instrumentation
Primers were obtained from Integrated DNA Technologies and restriction enzymes from New England Biolabs (NEB). Digests were performed according to manufacturer guidelines. Plasmid DNA was isolated using the NEB Monarch Spin Plasmid Miniprep Kit, according to manufacturer instructions, and quantified with a Denovix DS-7 microvolume spectrophotometer. Sanger sequencing was performed by the London Regional Genomics Center (Western University) using universal T7 Promoter and T7 Terminator primers. All chemicals were obtained from Sigma-Aldrich and used without further purification unless otherwise stated. Plasmid bearing N-terminal His6-tagged Cps2L (pET-28a(+)-wtCps2L, EC 2.7.7.24) was cloned and expressed in Escherichia coli BL21(DE3) as previously described.31 α-Phosphoglucomutase (α-PGM) from rabbit muscle (CAS No. 9001-81-4, EC 5.4.2.2), α-D-glucose-6-phosphate dehydrogenase (G6PD) from Leuconostoc mesenteroides (Product No. 10165875001, EC 1.1.1.363), and inorganic pyrophosphatase (IPP) from E. coli (CAS No. 9024-82-2, EC 3.6.1.1) were obtained commercially. Enzyme stocks were prepared in assay buffer containing 25% glycerol, aliquoted (50–200 µL), and stored at −20 °C. 3-(N-Morpholino)propanesulfonic acid (MOPS, 40 mM) was used as assay buffer, prepared in Millipore water and adjusted to pH 7.5. Fresh Cps2L working solutions (50 µM) were prepared daily in cold assay buffer and stored on ice. Enzyme assay reagent stocks were prepared in assay buffer (unless otherwise stated), aliquoted (200 µL), and stored at −20 °C, except for MgCl2 and Pi which were stored at 4 °C. Solutions of malachite green oxalate, ammonium molybdate (in 7 N HCl), and Tween-20 were prepared in Millipore water and stored at room temperature. Absorbance and pathlength were measured on a SpectraMax Plus 384 UV-vis microplate reader using the PathCheck feature and SoftMaxPro 4.6 software. Enzyme assays were performed in duplicate in 96-well flat-bottom microtiter plates (Corning Costar). GraphPad Prism 10.1.1 was used for nonlinear regression analyses and plotting.Site-directed mutagenesis
The Cps2L variant E253D was generated by site-directed mutagenesis using the NEB Q5 Site-Directed Mutagenesis Kit, performed as instructed by the manufacturer. For efficient subcloning, pET-28a(+)-wtCps2L31 was first transformed into NEB 5-alpha competent E. coli. The plasmid pET-28a(+)-wtCps2L, isolated from NEB 5-alpha cells, served as the template for site-directed mutagenesis. Polymerase chain reactions were conducted on a Biometra TProfessional Basic 96 Thermocycler using the forward and reverse primers shown in Table S1. The forward primer introduced the E253D mutation and the reverse primer added a silent mutation at N245 to create an AclI recognition site. AclI digests of pET-28a(+)-wtCps2L and plasmid isolated from site-directed mutagenesis transformants were compared and used to identify colonies bearing the mutated sequence (Fig. S1A). Sanger sequencing was performed to ensure no other alterations to the nucleotide sequence were introduced. For protein overexpression, the mutant plasmid DNA (pET-28a(+)-mtCps2L) was transformed into NEB BL21(DE3) competent E. coli and stored in 50% glycerol at −70 °C.Protein overexpression and purification
Protein overexpression and purification was performed as previously described with minor modifications.38 Freshly streaked LB agar plates (50 µg mL−1 kanamycin) were grown overnight at 37 °C. LB media (20 mL, 50 µg mL−1 kanamycin) was inoculated with a single colony and grown overnight (37 °C, 250 RPM). After 15–17 hours, 1 L of LB media (4 × 250 mL, 50 µg mL−1 kanamycin) was inoculated by adding 2.5 mL of starter culture to each 250 mL of media. Cultures were grown to an OD600 of 0.6–0.9 (37 °C, 250 RPM), after which they were subjected to a cold-shock at 4 °C for 30–60 min. Cultures were then induced with IPTG (0.4 mM) and grown overnight (room temperature, 250 RPM). Cells were harvested by centrifugation (Sorvall Legend RT, 4000 × g, 10 min) and stored at −70 °C until purification. To purify, cell pellets were thawed on ice and resuspended with 20 mL of lysis buffer, made up of 3 mL glycerol, 1 mL 10% Triton X-100, 10 mg lysozyme, 20 µg DNase I, and 16 mL Wash Buffer (40 mM MOPS pH 7.5, 300 mM NaCl, 30 mM imidazole). The cell suspension was stirred for 1 hour at 4 °C in an ice bath and then sonicated (Cole Parmer GEX 750, 50% amplitude, 5 × 5 s pulses with a 30 s rest between pulses). The cellular debris was cleared by centrifugation (Labnet Prism R, 17000 × g, 10 min, 4 °C). The supernatant was diluted 1:1 with Wash Buffer and then immediately applied to a 5 mL nickel-charged column (Cytiva, HisTrap High Performance column) pre-equilibrated with 25 mLs of Wash Buffer. Flow-through was collected as Fraction 1 (F1), followed by column washing with 3 × 25 mLs of Wash Buffer (F2–4). Elution was initiated with 2 × 25 mLs of a 1:4 dilution of Wash Buffer and Elution Buffer (40 mM MOPS pH 7.5, 300 mM NaCl, 250 mM imidazole) (F5 and F6). Elution was completed by applying 3 mL Elution Buffer, incubating the column at 4 °C for 20 min, followed by 3 × 10 mLs Elution Buffer (F7–9). Wash and Elution buffers were stored at 4 °C and applied to the column via syringe. All fractions were collected on ice and then stored at 4 °C. SDS-PAGE was performed to identify fractions containing pure protein (usually F5–8), which were then concentrated and buffer exchanged into the assay buffer using a centrifugal filter device (10 kDa molecular weight cut-off). Purified protein was visualized by SDS-PAGE (Fig. S1B) and concentrations were determined by absorbance at 280 nm using the calculated extinction coefficient of 29340 M−1 cm−1.31 We found that Cps2L E253D degraded more rapidly in storage (assay buffer with 25% glycerol, −20 °C) compared to the wild-type (wt).
Malachite green colorimetric assay
Cps2L activity was measured using the malachite green assay as previously described with some modifications.39 Malachite green reagent was prepared fresh daily by combining 0.3% w/v malachite green oxalate, 2% w/v ammonium molybdate (in 7 N HCl), 1% w/v Tween-20, and Millipore water in a 1:1:1:7 ratio. This solution was incubated at room temperature for at least 30 min upon which the colour turned from dark green to gold. Incubated malachite green reagent was filtered (hydrophilic PVDF membrane filter) to remove any precipitate. Enzyme reactions (1×) contained TTP (100 µM), α-D-glucose-1-phosphate (Glc-1-P, 100 µM), MgCl2 (5 mM), IPP (0.8 enzyme units (EU) mL−1), and Cps2L (10 nM) in a final volume of 30 µL. To prepare these reactions, a 1.2× mixture of all reagents except Cps2L was made up in assay buffer and then aliquoted (25 µL) into 0.2 mL microcentrifuge tubes. Reactions were initiated using a multichannel pipette with 6× Cps2L (5 µL, final concentration of 10 nM) prepared immediately before. A negative control was included alongside each reaction, substituting Cps2L with buffer. At 4 minutes post-initiation, reactions were quenched with formic acid (0.1%, 30 µL) and centrifuged to pellet precipitated protein (Labnet Prism R, 17000 × g, 5 min, 4 °C). The supernatant of each quenched reaction (50 µL) was transferred into microtiter plate wells, alongside two sets of monophosphate standards prepared in assay buffer (50 µL, 1.5-fold serial dilution from 70 µM to 6.15 µM Pi, and a buffer control). A multichannel pipette was used to dispense the previously prepared malachite green reagent into all wells (50 µL). After incubation of the plate (37 °C, 25 min), absorbance at 630 nm was measured. The absorbances of monophosphate standard replicates were averaged, normalized to the buffer control, and then fit by linear regression to generate a standard curve. Total Pi concentration of each enzyme reaction was calculated using this standard curve and corrected by subtracting the background Pi concentration of the corresponding negative control.
NADPH-coupled spectrophotometric assay
NADPH-coupled assay enzyme reactions (1×) contained uridine diphosphate-α-D-glucose (UDP-Glc, 300 µM), PPi (100 µM), NADP+ (500 µM), α-D-fructose bisphosphate (F-1,6-BP, 100 µM), MgCl2 (8 mM), α-PGM (1.5 EU mL−1), G6PD (1.5 EU mL−1), and Cps2L (50 nM) in a final volume of 200 µL. To prepare these reactions, a 1.11× mixture of all reagents except Cps2L was made up in assay buffer and then aliquoted (180 µL) down a column of microtiter plate wells. Reactions were initiated using a multichannel pipette with 10× Cps2L (20 µL, final concentration of 50 nM) prepared immediately before. Absorbance at 340 nm was measured every 20 s at 25 °C for 10–30 min. The Beer–Lambert Law was used to calculate the concentration of NADPH produced, given the measured absorbance and pathlength of each well and the known extinction coefficient of NADPH (6.22 mM−1 cm−1 at 340 nm). A standard curve can also be used to determine product concentrations. NADPH concentrations were normalized by subtracting the background concentration at t = 0 min. Initial velocities (vi) were determined by linear regression of the first 2 min of linear product formation, as determined by the plotted progress curves.Z′-factor calculations
Z′-factors for the NADPH-coupled and malachite green assays were calculated from 16 positive controls and 16 negative controls, prepared according to the described assay-specific conditions. Uninhibited Cps2L reactions served as positive controls, whereas buffer vehicle was used in place of Cps2L in the negative controls. Z′-factor was calculated according to eqn (1),40 where µ+ and µ– refer to the mean primary readout of the positive and negative controls, respectively, and σ+ and σ– refer to the standard deviation of these respective readouts. The primary readout of the NADPH-coupled assay is the vi of absorbance measurements, whereas the primary readout of the malachite green assay is the fixed absorbance measured at 4 minutes post-initiation. For visual comparison, the readouts of both assays were individually scaled from 0–100% and then graphed on a shared axis as the relative enzyme activity across each replicate. | (1) |
Cofactor effects on NADPH-coupled assay
To evaluate the effect of Mg2+ and F-1,6-BP concentrations on the developed NADPH-coupled assay, a 1.25× reaction mixture containing all reagents except Cps2L and the selected cofactor (Mg2+ or F-1,6-BP) was prepared. The cofactor was serially diluted 2-fold in assay buffer to produce working solutions at 10× the desired final concentration, alongside a buffer control. After aliquoting the reaction mixture (160 µL) down a column of microtiter plate wells, a multichannel pipette was used to add the serial dilutions to the column (20 µL). Reactions were initiated with Cps2L, measured, and processed as described (vide supra). Relative activity was calculated as the percent ratio of each measured vi to that of the maximum observed vi.Cps2L kinetic parameters
To determine the apparent kinetic parameters of Cps2L using the developed NADPH-coupled assay, a 1.25× reaction mixture containing all reagents except Cps2L and the selected substrate (UDP-Glc or PPi) was prepared. In these experiments, MgCl2 was reduced to 5 mM to minimize Mg2+-PPi precipitation, Cps2L was reduced to 25 nM, and NADP+ was increased to 1 mM to avoid limiting the rate, given the concentrated substrate conditions. For the analysis of UDP-Glc, PPi was fixed at 0.5 mM, while UDP-Glc was serially diluted 2-fold in assay buffer to produce working solutions at 10× the desired final concentrations, alongside a buffer control. After aliquoting the reaction mixture (160 µL) down a column of microtiter plate wells, a multichannel pipette was used to add the substrate serial dilutions to the column (20 µL). Reactions were initiated with Cps2L, measured, and processed as described (vide supra). An analogous procedure was used for the analysis of PPi, where UDP-Glc was fixed at 0.5 mM and PPi was serially diluted in the described fashion. Michaelis–Menton curves generated from duplicate experiments were analyzed by nonlinear regression, according to eqn (2), where vi is initial velocity in µM min−1, [Et] is the total enzyme concentration of 0.025 µM, and [S] is the substrate concentration in µM. This analysis yielded Kappm, Vappmax, and kappcat for each substrate, along with the SEM for each parameter. These values were used to calculate catalytic efficiency (kappcat/Kappm).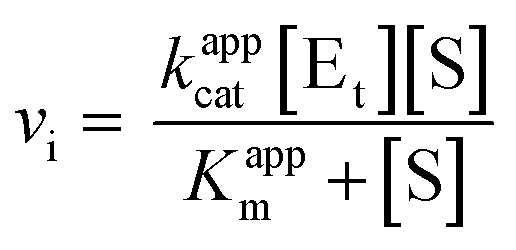 | (2) |
TDP-Rha dose–response curves
To generate a TDP-Rha dose–response curve using the described NADPH-coupled assay, a 1.25× reaction mixture of all reagents except Cps2L and TDP-Rha (obtained from Biosynth) was prepared. The inhibitor was serially diluted 3-fold in assay buffer to produce working solutions at 10× the desired final concentration, alongside a buffer control. After aliquoting the reaction mixture (160 µL) down a column of microtiter plate wells, a multichannel pipette was used to add the inhibitor serial dilutions to the column (20 µL). The absorbance at 340 and 280 nm was monitored every 30 s for 5 min to ensure no reaction occurred without addition of Cps2L. Reactions were initiated with Cps2L, measured, and processed as described (vide supra).To generate the dose–response curve using the malachite green assay, a 1.5× reaction mixture of all reagents except Cps2L and TDP-Rha was prepared. The inhibitor was diluted in assay buffer to produce working solutions at 10× the desired final concentration, alongside a buffer control. After aliquoting the reaction mixture (20 µL) into 0.2 mL microcentrifuge tubes, a multichannel pipette was used to add the inhibitor serial dilutions to the tubes (5 µL). Reactions were initiated with Cps2L, quenched, measured, and processed as described (vide supra).
Enzyme activity was calculated as the percent ratio of each measured vi (NADPH-coupled assay) or end-point Pi concentration (malachite green assay) to that of the buffer control. Dose–response curves generated from duplicate experiments were analyzed by nonlinear regression, according to eqn (3), where Y is percent enzyme activity, [I] is the log of the inhibitor concentration, h is the Hill slope, Max is the upper limit of the curve (constrained to ≤100%), and Min is the lower limit (constrained to ≥0%). This analysis yielded the IC50 of each inhibitor, as well as the Hill slope and range of the dose–response curve, along with the SEM for each parameter.
 | (3) |
Cu2+ effect on NADPH-coupled assay
To evaluate the effect of Cu2+ on the developed NADPH-coupled assay, a 1.25× reaction mixture of all reagents except Cps2L and CuSO4 was prepared. CuSO4 was serially diluted 1.75-fold in water to produce working solutions at 10× the desired final concentration, alongside a water control. After aliquoting the reaction mixture (160 µL) down a column of microtiter plate wells, a multichannel pipette was used to add the CuSO4 serial dilutions to the column (20 µL). Reactions were initiated with Cps2L, measured, and processed as described (vide supra). Upon reaction completion, the pH of each well was measured (Sentron SI series MicroFET pH/T probe). IC50 was calculated as previously described for the NADPH-coupled assay (vide supra).Mg2+ effect on G6PD activity
A stock solution of Mg2+ was serially diluted 2-fold in assay buffer to produce working solutions at 10× the desired final concentration. Assay buffer (150 µL), a 20× mixture of G6P and NADP+ (10 µL), and the serial dilutions (20 µL) were mixed in a microtiter plate. A control that did not contain Mg2+ was included in the experiment. Reactions were initiated with 10× G6PD (20 µL) and monitored at 340 nm every 20 s (25 °C, 30 min total). Final concentrations were 0.01 EU mL−1 G6PD, 100 µM NADP+, 500 µM G6P, and 0–10 mM Mg2+. The concentration of NADPH at each time point was calculated as described (vide supra). Relative activity was calculated as the percent ratio of each measured vi to that of the maximum observed vi.Molecular modelling
AlphaFold3 was used to model the tertiary structure of S. pneumoniae Cps2L (pTM = 0.91). The crystal structure of P. aeruginosa co-crystallized with TDP-Rha was sourced from RCSB PDB (PDB code 1G3L). Protein structures were visualized in MolSoft ICM-Browser.Results and discussion
NADPH-coupled assay vs. malachite green assay
In the developed assay, Cps2L activity is measured via the appearance of NADPH using α-phosphoglucomutase (α-PGM) and α-D-glucose-6-phosphate dehydrogenase (G6PD) as coupling enzymes (Scheme 2, orange). Selection of these coupling enzymes was guided by early cuvette-based assays performed by Kornfeld, Melo, and Glaser with P. aeruginosa RmlA.10,37 This technique is flexible, accommodating either TDP-Glc or the noncanonical substrate UDP-Glc. However, we developed the method with UDP-Glc due to its wider commercial availability. Upon α-D-glucose-1-phosphate (Glc-1-P) production by Cps2L in the pyrophosphorylase direction, α-PGM catalyzes conversion to α-D-glucose-6-phosphate (Glc-6-P). α-D-Fructose-1,6-bisphosphate (F-1,6-BP) or α-D-glucose-1,6-bisphosphate (G-1,6-BP) is included as a necessary phosphoryl donor for activation of the α-PGM catalytic serine.41,42 Glc-6-P is subsequently taken up by G6PD to produce a gluconolactone via the reduction of NADP+. The resulting formation of NADPH enables reactions to be monitored in a microplate reader at 340 nm. Initial velocities could be obtained within 10 minutes of reaction initiation. In comparison, the malachite green assay measures Cps2L activity via the appearance of Pi using inorganic pyrophosphatase (IPP) as a coupling enzyme (Scheme 2, green). In general, the developing reagent (malachite green, ammonium molybdate, and a surfactant) is added to quenched and centrifuged samples, which are then incubated for colour development (usually 37 °C for 30 min). The total Pi concentration of each sample, and thus the total TDP-Glc produced by Cps2L, is quantified by absorbance at 600–660 nm and a monophosphate standard curve (Fig. S2).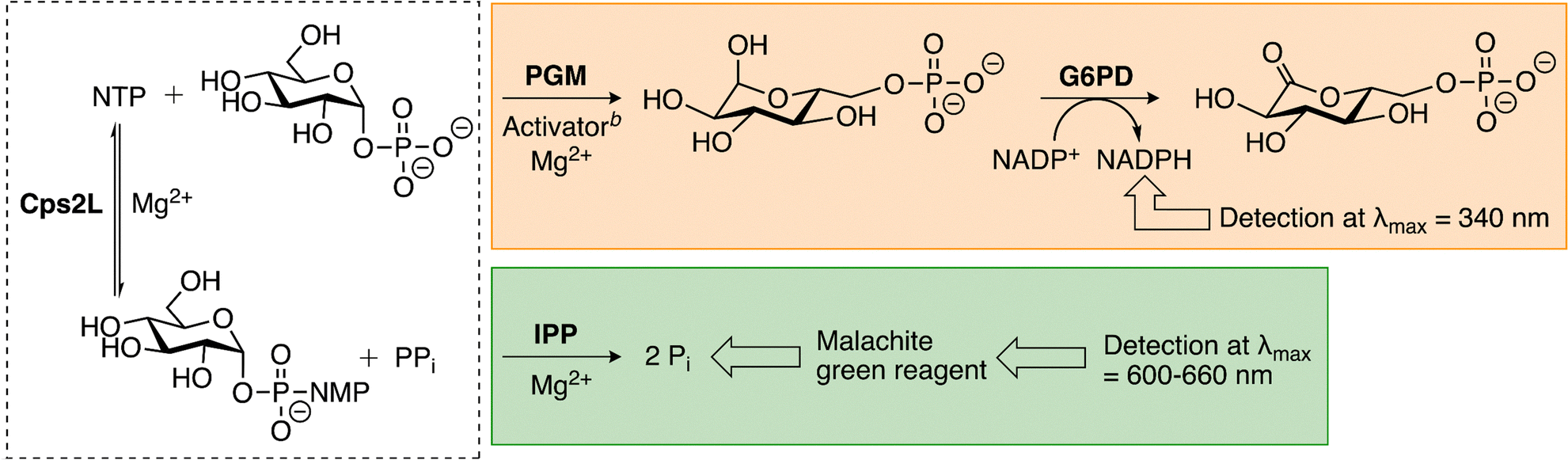 | ||
| Scheme 2 NADPH-coupled assay and malachite green assay for measuring Cps2L activity.a aDashed box is Cps2L reaction (NTP = TTP or UTP, NMP = TMP or UMP). Orange box is NADPH-coupled assay. Green box is malachite green assay. bActivator is F-1,6-BP or G-1,6-BP. | ||
Z′-factor was used to assess the quality of the assays for high-throughput inhibitor screening, with scores between 0.5 and 1 indicating excellent assay performance.40 The NADPH-coupled assay achieved a Z′-factor of 0.90, while the malachite green assay scored 0.75, consistent with literature (0.70).43 The lower score of the malachite green assay likely reflects the variability among positive control replicates (Fig. 2), which is typical for a discontinuous format. Both methods are suitable for high-throughput screening, but the NADPH-coupled assay provides greater precision.
 | ||
| Fig. 2 Quality assessment of the NADPH-coupled assay and malachite green assay for high-throughput inhibitor screening. A series of positive controls (PC) and negative controls (NC) were prepared for each assay (n = 16). NCs substituted Cps2L with an equal volume of buffer. Cps2L activity was measured as an initial rate (NADPH-coupled assay) or an end-point absorbance (malachite green assay). Assay readouts were independently scaled from 0–100% and then graphed on a shared axis, revealing the relatively high precision of the NADPH-coupled assay. The unscaled data were used to calculate Z′-factor. | ||
Characterization of Cps2L reaction kinetics
The NADPH-coupled assay was used to study Cps2L kinetics. Enzyme velocity was calculated from the first 2 min of steady-state, linear product formation, with rate doubling confirmed up to 100 nM Cps2L (Fig. 3A and B). At higher enzyme levels, a brief lag phase was observed at the beginning of the reaction, most likely due to rate limitation by the auxiliary enzymes. All coupled-enzyme assays will display a lag period when the rate of the first enzymatic reaction is greater than the rate of the subsequent reactions. This lag must be minimized with optimization of enzyme concentrations.44,45 To avoid the lag period observed in our NADPH-coupled assay, all experiments were carried out at 50 nM Cps2L or lower. We did not observe a lag phase under the experimental conditions used in the malachite green assay (Fig. S2B). | ||
| Fig. 3 (A) NADPH-coupled assay progress curves at varying Cps2L concentrations. Prominent lag period observed above 50 nM Cps2L. (B) Reaction velocity as a function of Cps2L concentration. Velocity calculated from a linear regression of the first 2 min of linear product formation, as determined by the plotted progress curves. Rate doubling observed up to 100 nM Cps2L. Effect of (C) Mg2+ and (D) F-1,6-BP on the developed NADPH-coupled assay. Relative activity represents the percent ratio of each measured vi to that of the maximum observed vi in the dataset. Optimal concentrations of 8 mM MgCl2 and 100 µM F-1,6-BP were determined. Data shown are mean ± SD (n = 2). Some error bars are smaller than the data points. | ||
Optimal concentrations of cofactors Mg2+ and F-1,6-BP were also determined using the developed NADPH-coupled assay. Mg2+ is an essential cofactor of both rabbit muscle α-PGM46 and α-D-glucose-1-phosphate thymidylyltransferases,37,47–49 and a reported non-essential activator of G6PD from certain species.50–52 Accordingly, Mg2+ was required for activity, with 8 mM selected as the optimal assay concentration (Fig. 3C). Excess Mg2+ and PPi will result in insoluble Mg2+-PPi precipitates.53 Although G-1,6-BP is typically used to activate α-PGM, we used the fructose analogue due to commercial backorders. This phosphoryl donor is also effective, likely by α-PGM-mediated conversion of F-1,6-BP and Glc-1-P or Glc-6-P to G-1,6-BP, producing α-D-fructose-6-phosphate as a by-product (Scheme S1).41,54,55 A concentration of 100 µM F-1,6-BP was determined to be optimal (Fig. 3D).
Under the selected assay conditions, kinetic parameters for S. pneumoniae Cps2L were determined using UDP-Glc and PPi as substrates. Michaelis–Menten plots for each substrate (Fig. 4 and Fig. S3) yielded the apparent Km, Vmax, and kcat values (Table 1). The Kappm values of UDP-Glc and PPi were similar to those found for P. aeruginosa RmlA in a study from 1964 (0.6 mM and 0.66 mM, respectively).10 This early study remains the only other kinetic analysis of UDP-Glc and PPi as co-substrates of a α-D-glucose-1-phosphate thymidylyltransferase as other works have focused on the canonical substrate, TDP-Glc, as the sugar donor instead. While the Km associated with UDP-Glc has not been quantified outside of S. pneumoniae and P. aeruginosa, enzymatic turnover has been observed in Cps2L homologs from S. enterica LT2,33E. coli,56,57 and Sulfolobus tokodaii.58 More recently, E. coli RmlA has been shown to produce UDP-Glc as a nutrient-responsive signaling molecule that stalls polysaccharide biosynthesis and cell division, indicating a physiological role for α-D-glucose-1-phosphate thymidylyltransferase substrate promiscuity.57 As the preferred substrate of α-D-glucose-1-phosphate thymidylyltransferase, reported Kappm values for TDP-Glc are consistently lower than what is observed here for UDP-Glc.10,33,56,58 Consequently, a relatively high amount of UDP-Glc is required to achieve a useful reaction rate, which may cause competitive inhibitors to appear less potent due to increased substrate competition.59 Structural differences between uridine and thymidine could also influence active site or allosteric site conformations, although such effects are expected to be minimal. The crystal structure of P. aeruginosa RmlA indicates that the thymine methyl group, which is absent in uracil, does not form intermolecular interactions in the active site.29 Moreover, a crystal structure of Aneurinibacillus thermoaerophilus RmlA complexed with UDP-glucose shows that only one additional binding interaction is formed by uridine's C2 hydroxyl group compared to thymidine.60
 | ||
| Fig. 4 Michaelis–Menten curves for (A) UDP-Glc and (B) PPi as Cps2L substrates. Cps2L activity was measured using the developed NADPH-coupled assay at various concentrations of the substrate of interest while the other substrate was fixed at 0.5 mM. Calculated vis were plotted against the concentration of the varied substrate and then fit to eqn (2). Data shown are mean ± SD (n = 2). Some error bars are smaller than the data points. | ||
| Substrate |
K
appma (mM) |
V
appmaxa (µmol mg−1 s−1) |
k
appcata (s−1) |
k
appcat/Kappmb (mM−1 s−1) |
|---|---|---|---|---|
| a Data shown are mean ± SEM (n = 2). b Data shown are mean ± propagated uncertainty from kappcat and Kappm SEMs (n = 2). | ||||
| UDP-Glc | 0.52 (± 0.04) | 0.46 (± 0.02) | 15.6 (± 0.5) | 30.3 (± 3.2) |
| PPi | 0.61 (± 0.04) | 0.51 (± 0.02) | 17.5 (± 0.6) | 28.9 (± 3.1) |
TDP-rhamnose inhibition of Cps2L
We anticipate this assay will be valuable for screening novel inhibitor candidates of α-D-glucose-1-phosphate thymidylyltransferases. To demonstrate its utility, we examined TDP-Rha inhibition of Cps2L using the developed NADPH-coupled assay and compared results to the malachite green assay. TDP-Rha was chosen as its mechanism of inhibition is well-characterized in other α-D-glucose-1-phosphate thymidylyltransferases, allowing for additional assay validation. The continuous nature of the NADPH-coupled assay enables Cps2L activity to be derived from initial velocities obtained from linear regression of the progress curves. For the malachite green assay, Cps2L activity was estimated from the total product concentration after a 4-minute reaction. IC50 was chosen as a simple measure of potency, acknowledging that its precise value can vary with assay conditions. Despite this limitation, both methods produced similar dose–response curves (Fig. 5A). The NADPH-coupled assay yielded an IC50 of 45.1 ± 2.3 µM and a Hill slope of −1.8 ± 0.2. A Hill slope >|1| is indicative of cooperative binding.24,39 No change in background absorbance (340 or 280 nm) was detected in the absence of Cps2L (Fig. S4). The malachite green assay yielded similar potency and cooperativity trends, with an IC50 of 34.5 ± 3.1 µM and a Hill slope of −1.3 ± 0.1. The results across both methods are consistent with values reported for other α-D-glucose-1-phosphate thymidylyltransferases.39 | ||
| Fig. 5 TDP-Rha inhibition of S. pneumoniae Cps2L. (A) Similar TDP-Rha dose–response curves were generated using either the NADPH-coupled assay or the malachite green assay. Percent Cps2L activity represents the ratio of each measured vi (NADPH-coupled assay) or end-point Pi concentration (malachite green assay) to that of a buffer control (0 µM TDP-Rha). (B) Progress curves collected using the NADPH-coupled assay under varying concentrations of TDP-Rha. Inset highlights the volume of data gathered within the first 5 minutes of the reaction. (C) Concentration of PPi produced after 4 minutes, measured using the malachite green assay under varying concentrations of TDP-Rha. 15× fewer time points are acquired within the first 5 minutes of the reaction compared to the NADPH-coupled assay. Data shown are mean ± SD (n = 2). Some error bars are smaller than the data points. | ||
The NADPH-coupled assay is efficient, generating a 30-minute progress curve within the set-up time required for a single malachite green end-point measurement. This continuous assay also provides high kinetic resolution, yielding 15× more data points than the malachite green method within a 5-minute reaction window (Fig. 5B and C). This detail is advantageous in high-throughput screening as progress curve shape can provide preliminary insight into inhibitor mechanism. Within the first ∼10% of substrate consumption, uninhibited enzyme reactions produce a linear progress curve, whereas inhibited reactions will appear either linear or biphasic, depending on the mechanism involved.61–63 Linear progress curves suggest a rapid equilibrium is reached between enzyme and inhibitor, consistent with a non-covalent binding mechanism. We observed this curve shape for TDP-Rha (Fig. 5B inset), which agrees with the competitive and non-competitive inhibition modes reported in other α-D-glucose-1-phosphate thymidylyltransferases.9,10 By contrast, biphasic progress curves are indicative of a time-dependent binding mechanism. As such, the initial rate of the inhibited reaction is described by its vi as well as a subsequent steady-state velocity (vs). Time-dependent binding is characteristic of irreversible-covalent inhibitors (vs = 0) as well as reversible-covalent or slow-binding non-covalent inhibitors (vs ≠ 0, vs < vi).64 Non-linear regression of these biphasic progress curves can be used to rapidly derive covalent inhibition constants from a single experiment.65–67
TDP-Rha activity against Cps2L was probed using an allosteric mutant of the enzyme to provide further mechanistic characterization and assay validation. Recent work has shown that a E256D/D104N variant of S. enterica LT2 RmlA experiences purely competitive inhibition by TDP-Rha rather than the mixed inhibition observed in wt enzyme.39 With E256D targeting the allosteric site and D104N deemed a serendipitous stabilizing mutation,68 it was concluded that allosteric regulation by TDP-Rha was eliminated in the mutant. We engineered the analogous E253D variant of Cps2L and evaluated its sensitivity to TDP-Rha inhibition. Despite the minor structural difference between glutamate and aspartate, Cps2L E253D exhibits nearly 2× and 10× greater activity than wt Cps2L at 50 µM and 500 µM TDP-Rha, respectively, indicating a similar attenuation of allosteric control (Fig. 6A). Zheng et al. suggest that their E256D/D104N mutant of S. enterica LT2 RmlA is resistant to TDP-Rha inhibition due to the loss of a hydrogen bond between E256 and the C4 carbonyl of the pyrimidine ring.68 Based on the crystal structure of P. aeruginosa RmlA, it is also possible that E253 hydrogen bonds with a nearby arginine residue facilitating a key cation-π interaction with the thymidine pyrimidine ring (Fig. 6B and Fig. S5). The critical role of R217 (S. pneumoniae Cps2L) is supported by its strict conservation across α-D-glucose-1-phosphate thymidylyltransferases, and the attenuation of allosteric control observed in a R220A variant of S. enterica LT2 RmlA, where R220 is equivalent to Cps2L R217.68
 | ||
| Fig. 6 (A) Inhibition of wt Cps2L and the E253D variant by TDP-Rha (50 µM and 500 µM), measured using the developed NADPH-coupled assay. Percent Cps2L activity represents the ratio of each measured vi to that of a buffer control (0 µM TDP-Rha). Attenuation of allosteric control observed in Cps2L E253D. Data shown are the mean ± SD (n = 2). (B) The tertiary structure of Cps2L E253D was predicted using AlphaFold3 (green) and aligned to P. aeruginosa RmlA (grey, PDB: 1G3L).29 The E253D mutation in S. pneumoniae Cps2L (residue marked in green) potentially disrupts a cation-π interaction formed between an arginine residue and the thymine ring of TDP-Rha. Hydrogen bonding between E255 and R219 in P. aeruginosa RmlA is shown (equivalent residues in S. pneumoniae Cps2L are E253 and R217). | ||
Effect of Cu2+ on the NADPH-coupled assay
The effect of Cu2+ on the developed assay was investigated as rabbit muscle α-PGM is known to be inactivated by trace CuSO4.69–71 Under our experimental conditions, reactions were highly sensitive to CuSO4, with inhibition detected at concentrations as low as 1 µM and an IC50 of 2.5 ± 0.1 µM on the overall system (Fig. 7). A micro pH probe was used to confirm that reaction pH remained stable across CuSO4 concentrations and thus did not contribute to the observed inhibition. As a coupled-enzyme assay, it remains a challenge to resolve the effect of Cu2+ on each enzyme. However, it is likely that Cu2+ primarily affects α-PGM, as this ion has been shown to exhibit strong competitive and non-competitive inhibition of the enzyme.70 Consequently, the assay is poorly suited for the screening of inhibitors synthesized using a copper catalyst unless residual Cu2+ is rigorously removed to below 1 µM (∼64 ppb). This also highlights the need for a deep understanding of a coupled-enzyme system used for inhibitor screening, as any significant inhibition of coupling enzymes can yield misleading results. | ||
| Fig. 7 Effect of Cu2+ on the developed NADPH-coupled assay. Percent activity represents the ratio of each measured vi to that of a buffer control (0 µM CuSO4). Data points were fitted to eqn (3). Rate inhibition detected at CuSO4 concentrations exceeding 1 uM. Data shown are mean ± SD (n = 2). Some error bars are smaller than the data points. | ||
Since the activity of Cps2L and α-PGM is dependent on Mg2+, divalent cations in addition to Cu2+ are expected to affect the assay and should be avoided. Indeed, rabbit muscle α-PGM is also inhibited by Zn2+, Hg2+, and Be2+.70–72 Exceptions are Mn2+ and Co2+ which can substitute for Mg2+ and activate Cps2L or α-PGM.71,73 Unlike some G6PD variants, we found that L. mesenteroides G6PD activity is minimally dependent on Mg2+ (Fig. S6). As such, it is less likely that L. mesenteroides G6PD will be inhibited by divalent cations, which is supported by its resistance to Zn2+-mediated inhibition.74
Conclusions
In this work, we established a robust, NADPH-coupled assay compatible with high-throughput screening and characterization of α-D-glucose-1-phosphate thymidylyltransferase inhibitors. Using this assay, UDP-Glc was identified as a substrate for S. pneumoniae Cps2L, and the binding affinities for both UDP-Glc and PPi were determined. Furthermore, TDP-Rha inhibition of Cps2L was studied, revealing an IC50 near 40 µM and a rapidly reversible binding mechanism at the allosteric site. The engineered Cps2L E253D variant facilitates the identification of inhibitors that bind the allosteric site. In future work, a Cps2L variant bearing an active site mutation would enable definitive identification of active site inhibitors as well.The NADPH-coupled assay has potential to be particularly useful in the identification and characterization of covalent inhibitors, both reversible and irreversible, targeting Cps2L or other α-D-glucose-1-phosphate thymidylyltransferases. The high kinetic resolution of this assay lends itself seamlessly to the determination of kinact/KI or  through non-linear regression analysis of the generated progress curves.65–67 Furthermore, as opposed to discontinuous assays, the continuous format readily supports jump-dilution experiments and ‘substrate spike tests’, enabling assessment of inhibitor reversibility and drug-target residence times.66,67
through non-linear regression analysis of the generated progress curves.65–67 Furthermore, as opposed to discontinuous assays, the continuous format readily supports jump-dilution experiments and ‘substrate spike tests’, enabling assessment of inhibitor reversibility and drug-target residence times.66,67
As with all coupled-enzyme assays, any potent inhibitors identified against Cps2L should be screened for off-target activity against the auxiliary enzymes, α-PGM and G6PD, particularly covalent inhibitors bearing a highly reactive warhead or a non-specific scaffold. Counter-screening can be achieved by spiking the reaction mixture with the substrate of a coupling enzyme and monitoring for an apparent decrease in inhibitor potency. Alternatively, Cps2L and its substrates can be omitted entirely from the reaction mixture and replaced with the α-PGM substrate, Glc-1-P, to directly assess inhibition of the coupling enzymes. Verified Cps2L inhibitors may be further evaluated against other α-D-glucose-1-phosphate thymidylyltransferases associated with bacterial pathogenicity to assess broad-spectrum activity. Future studies employing the developed NADPH-coupled assay to investigate small-molecule inhibition of α-D-glucose-1-phosphate thymidylyltransferases will be reported in due course.
Author contributions
Bronwyn Rowland: investigation, formal analysis, methodology, visualization, validation, data curation, conceptualization, writing – original draft, writing – review & editing. Jesse Fuller and Chigozie Okolie: methodology, validation, conceptualization. David Jakeman: supervision, project administration, funding acquisition, conceptualization, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Abbreviations
| α-PGM | α-Phosphoglucomutase |
| EU | Enzyme units |
| F-1,6-BP | α-D-Fructose-1,6-bisphosphate |
| G-1,6,-BP | α-D-Glucose-1,6-bisphosphate |
| Glc-1-P | α-D-Glucose-1-phosphate |
| Glc-6-P | α-D-Glucose-6-phosphate |
| G6PD | α-D-Glucose-6-phosphate dehydrogenase |
| IPP | Inorganic pyrophosphatase |
| MOPS | 3-(N-Morpholino)propanesulfonic acid |
| NC | Negative control |
| PC | Positive control |
| TDP-Rha | Thymidine diphosphate-β-L-rhamnose |
| TTP | Thymidine triphosphate |
| UDP-Glc | Uridine diphosphate-α-D-glucose |
| v i | Initial velocity |
| v s | Steady-state velocity |
| wt | Wild-type |
Data availability
The materials, methods and data supporting this article have been included as part of the supplementary information (SI). Supplementary information includes verification of Cps2L E253D mutant, additional kinetic spectra for malachite green and NADPH-coupled assays, and the predicted structure of S. pneumoniae Cps2L. See DOI: https://doi.org/10.1039/d6cb00002a.Accession codes: Cps2L α-D-glucose-1-phosphate thymidylyltransferase, UniProt ID: Q8CWT6.
Acknowledgements
We thank members of the Jakeman group (E. Ospanow, and A. Alkaş) for helpful discussions and suggestions. This research was financially supported by the Natural Sciences and Engineering Research Council (NSERC), the Canadian Institute of Health Research (CIHR), and a Killam Predoctoral Scholarship (B. Rowland).References
- J. K. Singh, F. G. Adams and M. H. Brown, Front. Microbiol., 2019, 9, 1–8 CrossRef CAS PubMed.
- R. Kalscheuer, A. Palacios, I. Anso, J. Cifuente, J. Anguita, W. R. Jacobs, M. E. Guerin and R. Prados-Rosales, Biochem. J., 2019, 476, 1995–2016 CrossRef CAS PubMed.
- C. Opoku-Temeng, S. D. Kobayashi and F. R. DeLeo, Comput. Struct. Biotechnol. J., 2019, 17, 1360–1366 CrossRef CAS PubMed.
- N. Mohamed, Y. Timofeyeva, D. Jamrozy, E. Rojas, L. Hao, N. C. S. de Monerri, J. Hawkins, G. Singh, B. Cai, P. Liberator, S. Sebastian, R. G. K. Donald, I. L. Scully, C. H. Jones, C. B. Creech, I. Thomsen, J. Parkhill, S. J. Peacock, K. U. Jansen, M. T. G. Holden and A. S. Anderson, PLoS One, 2019, 14, 1–33 Search PubMed.
- J. C. Paton and C. Trappetti, Microbiol. Spectrum, 2019, 7, 1–15 CrossRef PubMed.
- A. Adibekian, P. Stallforth, M.-L. Hecht, D. B. Werz, P. Gagneux and P. H. Seeberger, Chem. Sci., 2011, 2, 337–344 RSC.
- S. Gao, W. Jin, Y. Quan, Y. Li, Y. Shen, S. Yuan, L. Yi, Y. Wang and Y. Wang, Npj Biofilms Microbiomes, 2024, 10, 1–15 Search PubMed.
- M.-F. Giraud and J. H. Naismith, Curr. Opin. Struct. Biol., 2000, 10, 687–696 CrossRef CAS PubMed.
- R. L. Bernstein and P. W. Robbins, J. Biol. Chem., 1965, 240, 391–397 CrossRef CAS PubMed.
- A. Melo and L. Glaser, J. Biol. Chem., 1965, 240, 398–405 CrossRef CAS PubMed.
- M.-Y. Mistou, I. C. Sutcliffe and N. M. van Sorge, FEMS Microbiol. Rev., 2016, 40, 464–479 CrossRef CAS PubMed.
- M. McNeil, M. Daffe and P. J. Brennan, J. Biol. Chem., 1990, 265, 18200–18206 CrossRef CAS PubMed.
- Y. Ma, F. Pan and M. McNeil, J. Bacteriol., 2002, 184, 3392–3395 CrossRef CAS PubMed.
- R. Rahim, L. L. Burrows, M. A. Monteiro, M. B. Perry and J. S. Lam, Microbiology, 2000, 146, 2803–2814 CrossRef CAS PubMed.
- X.-M. Jiang, B. Neal, F. Santiago, S. J. Lee, L. K. Romana and P. R. Reeves, Mol. Microbiol., 1991, 5, 695–713 CrossRef CAS PubMed.
- H. Qu, Y. Xin, X. Dong and Y. Ma, FEMS Microbiol. Lett., 2007, 275, 237–243 CrossRef CAS PubMed.
- W. Li, Y. Xin, M. R. McNeil and Y. Ma, Biochem. Biophys. Res. Commun., 2006, 342, 170–178 CrossRef CAS PubMed.
- Y. Yamashita, K. Tomihisa, Y. Nakano, Y. Shimazaki, T. Oho and T. Koga, Infect. Immun., 1999, 67, 3693–3697 CrossRef CAS PubMed.
- S. L. van der Beek, A. Zorzoli, E. Çanak, R. N. Chapman, K. Lucas, B. H. Meyer, D. Evangelopoulos, L. P. S. de Carvalho, G. Boons, H. C. Dorfmueller and N. M. van Sorge, Mol. Microbiol., 2019, 111, 951–964 CrossRef CAS PubMed.
- A. P. Bischer, C. J. Kovacs, R. C. Faustoferri and R. G. Quivey Jr., J. Bacteriol., 2020, 202, 1–14 CrossRef PubMed.
- R. G. Quivey Jr., E. J. Grayhack, R. C. Faustoferri, C. J. Hubbard, J. D. Baldeck, A. S. Wolf, M. E. MacGilvray, P. L. Rosalen, K. Scott-Anne, B. Santiago, S. Gopal, J. Payne and R. E. Marquis, Mol. Oral Microbiol., 2015, 30, 474–495 CrossRef CAS PubMed.
- D. Qu, X. Zhao, Y. Sun, F.-L. Wu and S.-C. Tao, Front. Microbiol., 2021, 12, 1–15 Search PubMed.
- A. A. Elamin, S. Steinicke, W. Oehlmann, Y. Braun, H. Wanas, E. A. Shuralev, C. Huck, M. Maringer, M. Rohde and M. Singh, PLoS One, 2017, 12, 1–18 CrossRef PubMed.
- M. S. Alphey, L. Pirrie, L. S. Torrie, W. A. Boulkeroua, M. Gardiner, A. Sarkar, M. Maringer, W. Oehlmann, R. Brenk, M. S. Scherman, M. McNeil, M. Rejzek, R. A. Field, M. Singh, D. Gray, N. J. Westwood and J. H. Naismith, ACS Chem. Biol., 2013, 8, 387–396 CrossRef CAS PubMed.
- G. Xiao, M. S. Alphey, F. Tran, L. Pirrie, P. Milbeo, Y. Zhou, J. K. Bickel, O. Kempf, K. Kempf, J. H. Naismith and N. J. Westwood, Bioorg. Med. Chem., 2021, 50, 1–8 CrossRef PubMed.
- M. W. Loranger, S. M. Forget, N. E. McCormick, R. T. Syvitski and D. L. Jakeman, J. Org. Chem., 2013, 78, 9822–9833 CrossRef CAS PubMed.
- S. M. Forget, A. Jee, D. A. Smithen, R. Jagdhane, S. Anjum, S. A. Beaton, D. R. J. Palmer, R. T. Syvitski and D. L. Jakeman, Org. Biomol. Chem., 2015, 13, 866–875 RSC.
- D. A. Smithen, S. M. Forget, N. E. McCormick, R. T. Syvitski and D. L. Jakeman, Org. Biomol. Chem., 2015, 13, 3347–3350 RSC.
- W. Blankenfeldt, M. Asuncion, J. S. Lam and J. H. Naismith, EMBO J., 2000, 19, 6652–6663 CrossRef CAS PubMed.
- J. Hoskins, W. E. Alborn, J. Arnold, L. C. Blaszczak, S. Burgett, B. S. DeHoff, S. T. Estrem, L. Fritz, D.-J. Fu, W. Fuller, C. Geringer, R. Gilmour, J. S. Glass, H. Khoja, A. R. Kraft, R. E. Lagace, D. J. LeBlanc, L. N. Lee, E. J. Lefkowitz, J. Lu, P. Matsushima, S. M. McAhren, M. McHenney, K. McLeaster, C. W. Mundy, T. I. Nicas, F. H. Norris, M. O’Gara, R. B. Peery, G. T. Robertson, P. Rockey, P.-M. Sun, M. E. Winkler, Y. Yang, M. Young-Bellido, G. Zhao, C. A. Zook, R. H. Baltz, S. R. Jaskunas, P. R. Rosteck, P. L. Skatrud and J. I. Glass, J. Bacteriol., 2001, 183, 5709–5717 CrossRef CAS PubMed.
- S. C. Timmons, R. H. Mosher, S. A. Knowles and D. L. Jakeman, Org. Lett., 2007, 9, 857–860 CrossRef CAS PubMed.
- S. A. Beaton, M. P. Huestis, A. Sadeghi-Khomami, N. R. Thomas and D. L. Jakeman, Chem. Commun., 2009, 238–240 RSC.
- L. Lindquist, R. Kaiser, P. R. Reeves and A. A. Lindberg, Eur. J. Biochem., 1993, 211, 763–770 CrossRef CAS PubMed.
- Y. Wang, Y. Xu, A. V. Perepelov, Y. Qi, Y. A. Knirel, L. Wang and L. Feng, J. Bacteriol., 2007, 189, 8626–8635 CrossRef CAS PubMed.
- C. J. Zea and N. L. Pohl, Anal. Biochem., 2004, 328, 196–202 CrossRef CAS PubMed.
- J. E. Ritter, C. Berlin and L. Elling, Anal. Biochem., 1996, 234, 74–82 CrossRef CAS PubMed.
- S. Kornfeld and L. Glaser, J. Biol. Chem., 1961, 236, 1791–1794 CrossRef CAS PubMed.
- S. M. Forget, J.-S. Zhu, B. E. Rowland and D. L. Jakeman, ACS Catal., 2025, 15, 13898–13903 CrossRef CAS.
- M. Zheng, M. C. Zheng, H. Kim and T. J. Lupoli, J. Am. Chem. Soc., 2023, 145, 15632–15638 CrossRef CAS PubMed.
- J.-H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- J.-S. Zhu, K. M. Stiers, S. M. Winter, A. D. Garcia, A. F. Versini, L. J. Beamer and D. L. Jakeman, ACS Omega, 2019, 4, 7029–7037 CrossRef CAS PubMed.
- N. Neumann, S. Friz and K. Forchhammer, mBio, 2022, 13, 1–15 CrossRef CAS PubMed.
- S. Sha, Y. Zhou, Y. Xin and Y. Ma, J. Biomol. Screening, 2012, 17, 252–257 CrossRef CAS PubMed.
- W. Van Roy, G. Woronoff, A. M. Jimenez Valencia, T. Stakenborg and W. A. Clarke, Biochem. Eng. J., 2020, 161, 1–13 CrossRef.
- F. B. Rudolph, B. W. Baugher and R. S. Beissner, in Methods in Enzymology, Academic Press, London, 1979, vol. 63, pp. 22–42 Search PubMed.
- C. Milstein, Biochem. J., 1961, 79, 574–584 CrossRef CAS PubMed.
- W. A. Barton, J. Lesniak, J. B. Biggins, P. D. Jeffrey, J. Jiang, K. R. Rajashankar, J. S. Thorson and D. B. Nikolov, Nat. Struct. Biol., 2001, 8, 545–551 CrossRef CAS PubMed.
- H. A. Brown, J. B. Thoden, P. A. Tipton and H. M. Holden, Protein Sci., 2018, 27, 441–450 CrossRef CAS PubMed.
- J. Sivaraman, V. Sauvé, A. Matte and M. Cygler, J. Biol. Chem., 2002, 277, 44214–44219 CrossRef CAS PubMed.
- G. E. Glock and P. McLean, Biochem. J., 1953, 55, 400–408 CrossRef CAS PubMed.
- K. J. Lendzian, Planta, 1978, 141, 105–110 CrossRef CAS PubMed.
- L. P. K. Lee and G. W. Kosicki, Can. J. Biochem., 1967, 45, 1279–1282 CrossRef CAS PubMed.
- N. M. Stover, K. Ganko and R. D. Braatz, Biotechnol. Bioeng., 2024, 121, 2636–2647 CrossRef CAS PubMed.
- J. V. Passonneau, O. H. Lowry, D. W. Schulz and J. G. Brown, J. Biol. Chem., 1969, 244, 902–909 CrossRef CAS PubMed.
- M. Hirose, E. Sugimoto and H. Chiba, Agric. Biol. Chem., 1972, 36, 2157–2162 CAS.
- S. Zuccotti, D. Zanardi, C. Rosano, L. Sturla, M. Tonetti and M. Bolognesi, J. Mol. Biol., 2001, 313, 831–843 CrossRef CAS PubMed.
- M. C. Zheng, J. C. Bryant, A. Koid, M. Sheshova, H. Kim, M. A. Jorgenson and T. J. Lupoli, Proc. Natl. Acad. Sci., 2026, 123, 1–12 Search PubMed.
- Z. Zhang, M. Tsujimura, J. Akutsu, M. Sasaki, H. Tajima and Y. Kawarabayasi, J. Biol. Chem., 2005, 280, 9698–9705 CrossRef CAS PubMed.
- J. Yang, R. A. Copeland and Z. Lai, SLAS Discovery, 2009, 14, 111–120 CrossRef CAS PubMed.
- T. J. Chen, W. T. Chien, C. C. Lin and W. C. Wang, 4HO8: Crystal structure of glucose 1-phosphate thymidylyltransferase from Aneurinibacillus thermoaerophilus complexed with UDP-glucose and thymidine, RCSB Protein Data Bank, https://www.rcsb.org/structure/4HO8, (accessed 6 April 2026).
- R. Copeland, in Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, John Wiley & Sons, Ltd, Hoboken, 2nd edn, 2013, pp. 203–244 Search PubMed.
- C. Zimmer, J. Brauer, D. Ferenc, J. Meyr, P. Müller, H.-J. Räder, B. Engels, T. Opatz and T. Schirmeister, Molecules, 2024, 29, 1–16 CrossRef PubMed.
- B. E. Rowland, K. D. Roy, A. Alkaş, T. Kuot, M. Barsoum, J. Fuller, C. A. J. Naudet, E. Ospanow and D. L. Jakeman, RSC Chem. Biol., 2026 10.1039/D6CB00070C.
- P. Klein, F. Barthels, P. Johe, A. Wagner, S. Tenzer, U. Distler, T. A. Le, P. Schmid, V. Engel, B. Engels, U. A. Hellmich, T. Opatz and T. Schirmeister, Molecules, 2020, 25, 2064 Search PubMed.
- L. K. Mader and J. W. Keillor, RSC Med. Chem., 2025, 16, 2517–2531 Search PubMed.
- L. K. Mader, J. E. Borean and J. W. Keillor, RSC Med. Chem., 2025, 16, 63–76 Search PubMed.
- B.-F. Krippendorff, R. Neuhaus, P. Lienau, A. Reichel and W. Huisinga, SLAS Discovery, 2009, 14, 913–923 CrossRef CAS PubMed.
- M. Zheng, M. Zheng and T. J. Lupoli, ACS Infect. Dis., 2022, 8, 2035–2044 CrossRef CAS PubMed.
- J. W. Long and W. J. Ray, Biochim. Biophys. Acta BBA – Protein Struct., 1970, 221, 522–528 Search PubMed.
- C. Milstein, Biochem. J., 1961, 79, 591–596 CrossRef CAS PubMed.
- V. Jagannathan and J. M. Luck, J. Biol. Chem., 1949, 179, 561–578 CrossRef CAS PubMed.
- T. Hashimoto, J. G. Joshi, C. del Rio and P. Handler, J. Biol. Chem., 1967, 242, 1671–1679 Search PubMed.
- S. A. Beaton, P. M. Jiang, J. C. Melong, M. W. Loranger, S. Mohamady, T. I. Veinot and D. L. Jakeman, Org. Biomol. Chem., 2013, 11, 5473–5480 Search PubMed.
- W. Jiang and W. G. Niehaus, Exp. Mycol., 1986, 10, 289–293 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |