
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein structural dynamics in covalent drug design: insights from irreversible and reversible covalent inhibitors
Ruchira
Basu
a and
Steven
Fletcher
 *ab
*ab
aDepartment of Pharmaceutical Sciences, University of Maryland School of Pharmacy, 20 N. Pine St., Baltimore, MD 21201, USA. E-mail: steven.fletcher@rx.umaryland.edu
bUniversity of Maryland Marlene & Stewart Greenebaum Cancer Centre, 20 S. Greene St., Baltimore, MD 21201, USA
First published on 8th January 2026
Abstract
Proteins form complex networks critical to various biological processes; many become involved in disease-related pathologies – only a subset of these proteins are considered to be druggable by conventional, non-covalent small-molecule therapeutics. Covalent drugs, which encompass irreversible inhibitors and reversible covalent inhibitors, are small-molecule modalities that chemically conjugate with their therapeutic targets and have emerged as a strategy to more effectively target these proteins, with structure-based approaches guiding their design, and achieving an improved therapeutic effect, predominantly through sustained inhibitions. In this review, we focus on the impact of covalent bond formation on protein structural dynamics, such as the generation/trapping of cryptic pockets, and how these phenomena may be leveraged in orthosteric and allosteric drug design. Further, while irrreversible inhibitors result in longer residence times with permanent changes of target proteins that will require protein re-synthesis, reversible covalent inhibitors enjoy the benefit of samplng different adducts, wherein one particular conjugate may be favoured through stabilizing structural reogranizations; this may prove significant when a protein presents multiple nucleophilic residues, and selectivity is a concern. Herein, we explore selected case studies that examine the mechanistic consequences of protein–drug conjugations, recommending a more dynamic structural perspective in rational drug development.
1. Introduction
The Human Proteome Project estimates the number of proteins in the human body to be ∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 when correlated with their encoded genes.1 This number increases dramatically based on considerations of associated splice variants (isoforms) or post-translational modifications, such as methylations, acetylations and phosphorylations.2 These proteins interact with ligands and other protein partners to form extensive cellular networks which play critical roles in physiological and pathological processes such as cell proliferation, apoptosis, signal transduction, inflammation, metastasis and directly correlates with our health and well-being.3 Aberrations in the interactome in the form of mutations, amplification or suppression of proteins can contribute to diseases such as cancer, infections, cardiovascular diseases and neurodegenerative disorders, among others.4,5 However, only a subpopulation of this proteome is considered to be druggable (total 854 proteins as described in the Human Protein Atlas) which includes both extracellular and intracellular targets such as receptors, ion channels and enzymes.6 FDA-approved small molecules and biologics (antibodies, peptides, oligonucleotides) are designed to effectively bind to these targets resulting in activation or inhibition of biological pathways, achieving therapeutic effects.7 Among these therapeutic entities, small molecules dominate almost 90% of the pharmaceutical drug market with approximately 50 drugs being approved by the FDA each year.7,8 Depending on the desired physiological response from the target, these small molecule scaffolds are customized to feature special pharmacological attributes and accordingly largely classified, into covalent and non-covalent drugs.9 In addition to druggable proteins, there are also ‘undruggable’ proteins that are challenging to target using conventional drug discovery techniques due to their lack of well-defined binding pockets and high structural plasticity. For both types of drug targets, there has been a significant shift in efforts to modulate their function using covalent drug modalities.10 Fifty years ago, drug discovery efforts typically involved phenotypic library screenings and, accidental breakthroughs, trial-and-error and exhaustive structure–activity relationship studies to build up an idea of the structure of the binding site, all of which can be time-consuming and inefficient. But now, more conventional methods feature rational decision-making coupled with computational modeling, structural biology data and other high-throughput techniques that gradually gained popularity for targeted design of non-covalent drugs.11 More recently, scientific advances have extended this precision to covalent drug design, utilizing structure–activity relationship analyses and detailed biochemical characterization.12
000 when correlated with their encoded genes.1 This number increases dramatically based on considerations of associated splice variants (isoforms) or post-translational modifications, such as methylations, acetylations and phosphorylations.2 These proteins interact with ligands and other protein partners to form extensive cellular networks which play critical roles in physiological and pathological processes such as cell proliferation, apoptosis, signal transduction, inflammation, metastasis and directly correlates with our health and well-being.3 Aberrations in the interactome in the form of mutations, amplification or suppression of proteins can contribute to diseases such as cancer, infections, cardiovascular diseases and neurodegenerative disorders, among others.4,5 However, only a subpopulation of this proteome is considered to be druggable (total 854 proteins as described in the Human Protein Atlas) which includes both extracellular and intracellular targets such as receptors, ion channels and enzymes.6 FDA-approved small molecules and biologics (antibodies, peptides, oligonucleotides) are designed to effectively bind to these targets resulting in activation or inhibition of biological pathways, achieving therapeutic effects.7 Among these therapeutic entities, small molecules dominate almost 90% of the pharmaceutical drug market with approximately 50 drugs being approved by the FDA each year.7,8 Depending on the desired physiological response from the target, these small molecule scaffolds are customized to feature special pharmacological attributes and accordingly largely classified, into covalent and non-covalent drugs.9 In addition to druggable proteins, there are also ‘undruggable’ proteins that are challenging to target using conventional drug discovery techniques due to their lack of well-defined binding pockets and high structural plasticity. For both types of drug targets, there has been a significant shift in efforts to modulate their function using covalent drug modalities.10 Fifty years ago, drug discovery efforts typically involved phenotypic library screenings and, accidental breakthroughs, trial-and-error and exhaustive structure–activity relationship studies to build up an idea of the structure of the binding site, all of which can be time-consuming and inefficient. But now, more conventional methods feature rational decision-making coupled with computational modeling, structural biology data and other high-throughput techniques that gradually gained popularity for targeted design of non-covalent drugs.11 More recently, scientific advances have extended this precision to covalent drug design, utilizing structure–activity relationship analyses and detailed biochemical characterization.12
For most covalent drugs, early-phase drug discovery processes rely heavily on structural studies to elucidate crucial interactions between the target macromolecules and the drug candidates in order to guide drug design.13 Insights from these studies are used in various stages – identification and validation of potential binding sites, predictions of drug–target interactions, understanding mechanisms of action, de novo design and lead optimization.14 Common techniques used to extract such information from protein 3D structures include X-ray crystallography, NMR, hydrogen/deuterium exchange mass spectrometry, small molecule FRET, cryogenic electron microscopy and computer-aided approaches such as molecular dynamic (MD) simulations.15 However, protein macromolecules do not exist in a single rigid form in a physiological context – rather, they are highly adaptable and exist as an ensemble of constantly fluctuating conformational states; more sophisticated CADD software is becoming better able to predict these.16–19 Molecular recognition process involving interactions between these protein states and their native ligands can be triggered through induced fit or conformational selection.20 These events can have important consequences in protein function and are commonly explored in various structural and biological mechanism studies.21,22 Similar to binding their native ligands, conformational alterations in protein structures can be expected to occur as a result of covalent drug binding as well. However, the contribution of these events to drug development studies remains somewhat underrepresented in the literature. Few drug design studies provide a comprehensive analysis of protein structural dynamics or the consequential effects of drug binding, and how these insights could enhance design optimization. While some protein–drug binding events may involve straight-forward structural changes or well-established mechanisms, some conformational shifts in proteins may be fortuitous and hold great potential for drug design efforts. Therefore, a deeper understanding is needed for the effects of drug binding on the global and local structural fluctuations of proteins, and its mechanistic impacts beyond just the placement of the drug motifs in the binding site and geometries of the neighbouring amino acid side chains. To provide a robust foundation for this perspective to the drug development community, this review will discuss selected case studies that have investigated the impact of covalent drug interactions on protein dynamics and how these insights influenced subsequent drug modelling. In order to present the most relevant and up-to-date information, publications exploring clinically-significant therapeutic protein targets in the last decade will be highlighted.
2. Non-covalent versus covalent inhibition
2.1. General overview
Non-covalent inhibitors encompass a class of molecules that rely on intermolecular interactions (such as salt bridges, hydrogen bonding and van der Waals forces) to engage with amino acids at its binding site of target enzymes or receptors. The binding is temporary meaning it involves a rapid steady-state equilibrium process characterized by the thermodynamic equilibrium constant Keq (occupancy-driven pharmacology) (Fig. 1A). In other words, the magnitude of the association (kon) and dissociation (koff) rate constants of the drug molecule quantifies how well it binds to the protein (binding affinity). These values can influence the measured potency (IC50, half-maximal inhibitory concentration) under experimental conditions as described by the Cheng–Prusoff equation. Covalent drugs, on the other hand, possess reactive electrophilic handles called warheads that modulate the target protein via chemical transformation at the complexation site. Their mechanism of action involves a two-step process (Fig. 1B and C) starting with recognition and binding of the molecular scaffold to its complementary site. This resulting complex is reversible in nature, and the on and off kinetics are defined by kon and koff. This is followed by a covalent interaction of the reactive moiety with a neighbouring amino acid on the protein (cysteine, lysine etc.) which is a nucleophilic residue.23,24 An appropriate orientation of the reactive handle and its accessible placement near the side chain is required for a successful adduct formation which can be stable for hours or days. This second step can lead to either a reversible or an irreversible covalent adduct (expanded upon in Section 3.1). In case of irreversible covalent drugs, the second step is represented by the maximal rate of inactivation (kinact) under ligand saturation condition, calculated using concentration-dependent inhibition assays.25 The term [kinact/Kirr] (called inactivation efficiency) is the quantifiable metric generally preferred over IC50 for comparing the potency of these molecules (where Kirr is the global equilibrium constant).26 This is because, unlike non-covalent binding, irreversible covalent inhibition is a mechanism-based pharmacology and therefore, the apparent IC50 value is now no longer time-independent. Both affinity and reactivity needs to be characterized individually so these parameters can be better adjusted for covalent drug design.25 Usually, this is directly measured via covalent labelling using mass spectrometry or indirectly, with time-dependent continuous assays.27 In general, covalent and non-covalent drugs can regulate target proteins via different mechanisms: inactivation of enzymes by out-competing native ligands or blocking catalytic amino acids, disruption of protein–protein interactions, mimicking messenger molecules to activate or inhibit receptors, degradation of misfunctioning proteins or binding to allosteric sites that lead to conformational changes in the protein.28–31 While a non-covalent drug can readily dissociate from its protein binding site, the covalent mode of binding, affording a longer residence time, makes the drug molecule, to all intents and purposes, an integral part of the protein. Due to the stable linkage formed in the second step, the duration of action is prolonged. It follows that any structural impacts on the protein caused by this mechanism may be more pronounced and persistent compared to the non-covalent binding mode. | ||
| Fig. 1 Schematic illustration and overview of the kinetics of three types of mechanisms for: (A) non-covalent inhibitors, (B) irreversible covalent inhibitors, (C) reversible covalent inhibitors. Orange bean: target protein, grey circle: drug molecule, yellow triangle: target residue, red box: warhead. Adapted from ref. 27. | ||
2.2. Significance in drug discovery and historical perspective
The main foci of drug development programs are to strengthen the interaction between the target protein and the drug candidate as well as to reduce potential off-target effects, which influences the drug's pharmacodynamic profile.10,32 Caution should be adopted when comparing binding affinities (Kd) and IC50 values in the context of non-covalent versus covalent inhibitors, because Kd's and IC50's are time-dependent parameters. The preferred parameter for covalent inhibitors is kinact/Kirr, since this is a time-independent parameter.33 Installing a warhead onto a non-covalent inhibitor may enhance the inhibition of a target protein, but it may not. The latter outcome may be due to too short of an incubation time in the assay to permit the covalent bond to form. Or equally possible is that if the covalent bond does form, perhaps, due to an inaccurately positioned warhead, the interactions from the non-covalent portion of the drug have become somewhat compromised. In general, time permitting, an improvement in inhibitory potency would be expected with a congener of a non-covalent inhibitor in which a suitably reactive and appropriately positioned electrophilic warhead has been installed.34 Since the formation of a covalent bond will result in an increased residence time, it is further expected that a covalent inhibitor will yield a sustained inhibition of its therapeutic target.Historically, drug discovery efforts have been predominantly focussed on non-covalent drugs over covalent drugs on account of toxicity concerns, selectivity issues and resistance mechanisms that are often observed for covalent moieties.35 Whether through rational design or high-throughput screening, non-covalent drugs have been long established as effective therapeutic strategies and often used as starting points for covalent drug design.36,37 Even if a non-covalent drug binds non-specifically to proteins other than its target, the low affinity ensures that it gets dislodged before any side-effect can manifest.38 The warheads on covalent drugs, on the other hand, inherently achieving longer residence times, may be susceptible to off-target reactivity that might cause an immune response, other toxic side effects as well as reduce drug availability at the target site.39,40 For example, cysteine-reactive species might bind to high concentrations of intracellular glutathione or other proteins with hyper-reactive cysteines.41 Nevertheless, covalent drug approvals have significantly increased in the last decade with many covalent inhibitors reaching blockbuster status.42,43 Additionally, proteomic screenings during early developmental phases and toxicity screenings during pre-clinical trials can help eliminate compounds with indiscriminatory reactivities.44 Many covalent drugs like aspirin and penicillin were discovered serendipitously over a century ago, where their mechanism was not clinically validated until much later.11 A more targeted approach in medicinal chemistry efforts for covalent drug design (targeted covalent inhibition) became more popular in recent years supported by advancements in structural biology techniques, proteomics studies, bioinformatics and computational biology methods.45–47 Pharmaceutical companies began actively pursuing covalent mechanisms (non-equilibrium binding) of inhibition in the early 2000s, especially for targets like BTK, KRASG12C and EGFR.48 Recent success stories include the development of well-tolerated covalent inhibitors against challenging drug targets such as Ibrutinib (BTK) and Sotorasib (KRASG12C).49,50 The main advantages of covalent drugs are their high potency and prolonged residence time at the target binding site (smaller koff values) which reinforces the modulatory action of the molecule.51 This, in turn, reduces their dosage requirements during formulation and improves patient compliance, which can be a liability for non-covalent drugs.11,52 While non-covalent drugs rely heavily on the affinity component to drive the interaction, covalent drugs need not be very high-affinity ligands as long as the covalent bond formed is selective and rapid, which can ultimately influence the overall efficacy of the drug.53,54 Furthermore, ‘undruggable’ proteins which do not have deep grooves needed for non-covalent drug docking, can still be effectively latched onto by covalent inhibitors via strategically-selected surface-exposed residues.9,55 As of now, there are more than 50 covalent inhibitors either approved or in clinical trials, in different disease states such an cancer, infections, immune, cardiovascular, gastrointestinal or CNS disorders.48,56 This number is predicted to increase as the value and interest in these innovative chemistries rises both in academic circles as well as pharmaceutical companies.52,57
3. Irreversible versus reversible covalent inhibition
3.1. General overview
As previously mentioned, both irreversible and reversible covalent inhibitors exhibit a temporary equilibrium state (Keq) in the initial binding stage followed by the formation of a drug–protein covalent conjugate. The adduct formed is permanent in the case of irreversible covalent binders; the modified protein will need to be re-synthesized. This can be especially useful the in case of undruggable protein targets like KRAS that employ highly competitive endogenous ligands (GDP/GTP) with tight affinities and sub-millimolar cellular concentrations.58 Similarly, this long-standing bond formation can be beneficial for ‘selective’ drugging of specific kinase isoforms via non-catalytic amino acids at their ATP-binding pockets.59 For reversible covalent binders, on the other hand, this covalent bond formed is short-lived as the bond formation can be reversed, effectively releasing/regenerating the protein.60 The rate of this reversal (dissociation) step can be controlled based on the chemistry of the warhead and the protein microenvironment around it.48 For example, introducing a nitrile group at the α-position of an irreversible acrylamide moiety can not only increase its reactivity but also render the warhead's conjugation chemistry reversible.61 Similarly, the orientation of the target amino acid and its protonation state due to the local pH of the binding site, steric factors as well as nature of the other interacting residues can play a significant role on the reaction rate.39 From a kinetics perspective, the difference in the covalent adduct formation step can be explained in terms of the reaction rate constants. For an irreversible binder, the first order rate constant (kinact) describes a full neutralization of the bound target (Fig. 1B) which is incorporated into the equilibrium constant (Kirr). The function of the protein cannot be restored unless it is resynthesized in the cell. In the case of a reversible covalent entity, a secondary equilibrium chemistry is established, represented by the forward and backward reactions (kfr and kbr, respectively) which is included in the global equilibrium constant Krev (Fig. 1C). Significant advances in reversible covalent inhibitors have been made, fuelled by advances in electrophilic warhead chemistries.62 Recent examples for FDA-approved reversible covalent drugs include Paxlovid (nirmatrelvir), a SARS-CoV2 protease inhibitor and Voxelotor, a haemoglobin inhibitor for sickle cell disease.63–65 According to the Drug Hunter's site, Paxlovid was voted 2021 Small Molecule of the Year by the drug development community, largely edging out non-covalent inhibitors like MRTX1133 (KRASG12D) and KB-0742 (CDK9).473.2. Target residues and reactive electrophilic warhead partners
Covalent inhibitor interactions can be grouped under three most common classes based on their mechanistic schemes: nucleophilic addition, addition–elimination and nucleophilic substitution.66 These chemistries can show differing levels of reactivities and specificities based on the electrophilic warhead and nucleophilic target residue partner.67 Different nucleophilic amino acids are selected based on their reactivities and accessibilities on the target proteins (Fig. 2).68 These residues may even be involved in enzymatic catalysis or may be a drug-resistance inducing mutation. Cysteine is the most commonly targeted residue due to the high nucleophilicity of its thiol group, unique redox activity, frequent role in catalysis and/or poorly conserved nature lending selectivity to the target isoform.23 However, due to its low occurrence in drug targets, other residues (such as lysine, serine, threonine, tyrosine, histidine) that show higher prevalence in the proteome are gradually receiving more attention.69 Electrophiles on the warheads can be fine-tuned to preferentially engage specific amino acids and there are a multitude of functional groups to choose from when deciding the type of chemistry required (reversible or irreversible).60 These include acrylamides, haloacetamides, epoxides, sulphur(VI) centres, phosphorous centres, enones, cyanoacrylamides, nitriles, aldehydes or ketones, boronic acids centres like ortho-CHO-phenylboronic acids.62 (Fig. 2). | ||
| Fig. 2 General structures of irreversible and reversible covalent warhead chemistries with their complementary reactive amino acid targets. X = Cl, Br, I. | ||
3.3. Irreversible versus reversible covalent inhibitors
As mentioned in previous sections, irreversible covalent inhibitors have the potential for high selectivities and potencies for their targets due to long-lasting duration of action, ability to bind shallow surface grooves and out-compete high affinity native ligands.56,70 However, this is also accompanied with concerns regarding adverse side-effects on accounts of their indiscriminate reactivities with off-target nucleophiles as well as on-target toxicities.71,72 Reversible covalent drugs, on the other hand, are essentially designed to combine the strengths of covalent (longer residence time) and non-covalent drugs (weaker off-target binding through reversibility).73–75 By controlling the kinetics of the warhead of the inhibitor by introducing modifications on its chemistry, researchers are aiming to strike an optimum balance in the residence time of the binder with the on-target or off-target biomolecule.40,76 Namely, in case of on-target binding, with the initial association of the drug with the protein being cooperatively driven by non-covalent interactions (hydrogen bonding, hydrophobic effects), the association constant kon need not be very fast. The overall uncoupling process between the drug and target, however, should be slow so that there is sufficient time for the molecule to be efficacious in the bound form (increased residence time), based on the turnover rate of the protein.27,76 On the other hand, for weak or unsuitable interactions such as in case of off-target binding, the dissociation should be fast, i.e. the covalent bond should quickly disconnect and the molecule should detach. This ensures that associated toxicity issues are mitigated, thus improving the safety profile of the drug molecule.Overall, the covalent mechanism of action (whether reversible or irreversible) can offer many pharmacological benefits and has been shown to exhibit immense potential to access a wide variety of both druggable and undruggable targets. To further solidify the advantages offered by these modalities, it is imperative to investigate ways in which they modulate target protein conformation – which could impact their binding affinity and selectivity. Therefore, the case studies discussed in the next section will be examining specific covalent inhibitors and their impact on protein structure. Structural changes may include induction of hidden cavities called ‘cryptic pockets’ in the protein body that are not apparent in the apo form – can be an favourable attribute for undruggable targets from a drug discovery perspective.77 This may manifest through side-chain or loop movements, or even domain rearrangements due to interaction of the molecule with classical binding sites. Broadly, in this review, these drugs have been categorised into three sections: active site binders indicating drug molecules that orthosterically compete with natural ligands for their binding site and influence the target protein function; protein–protein interaction (PPI) inhibitors, wherein low-molecular-weight ligands are fashioned to target the PPI interface; and allosteric inhibitors encompassing drug molecules that bind to alternate sites on the protein surface that indirectly modulate its function through conformational adjustments.
4. Methodological approaches: role in structural dynamics and comparison
Whether a protein exhibits local structural rearrangements or allosteric changes due to ligand binding, these alterations can have important functional and biological consequences. Different biophysical methods are employed to evaluate these changes for target validation and drug discovery.78 X-ray crystallography is one technique heavily relied on to obtain a 3D visualization of the protein–ligand bound structure, modeled based on its electron-density maps.79 For covalent drugs, this technique helps provide atomic level details of the exact location and orientation of the covalent bond, whether it is formed or not, and the residues involved in the adduct formation in addition to other structural information.80 Fragment-based covalent drug design often use high throughput X-ray crystal structure information by screening electrophilic fragment libraries to identify and improve on the affinity of the binder through structure-based optimization.81–83 Unexpected structural changes can be observed during these screenings that can leveraged for drug design. A recent success story is the development of Sotorasib (Fig. 10) – where co-crystal structure information of a previously unrecognized cryptic pocket on KRASG12C was exploited in the development of this highly potent inhibitor with favorable ADME properties.84 The Schrödinger SiteMap program is a tool that uses crystal structure information to predict druggability of a target protein binding site based on computational scores.85 A recent study using this program found that ligand-binding initiated conformational changes in some proteins that significantly improved its druggability score compared to its ligand-free state.86 Nonetheless, large sample requirements and the need for high quality crystals for X-ray crystallography can be a significant bottleneck for this process. Additionally, this method does not yield any dynamic information with respect to the binding process which could be crucial for covalent drug development.87 HDX-MS can work around these issues and provide information at peptide-level resolution based on changes in solvent exchange rates upon ligand binding, and associated domain movements.88,89 Fluctuations in the solvent accessibility of the binding site or otherwise, can give a comparative view of the exact regions in the protein involved in ligand interaction or even mechanism of inhibition.88,90Cryptic pocket formation upon ligand binding, as discussed in some of the case studies, were mostly discovered through serendipity.91 However, having prior knowledge of these ‘hidden’ cavities holds great untapped potential in drug discovery.92 While biophysical techniques like X-ray crystallography may not always provide sufficient information about the presence of cryptic sites in the ‘static’ ligand–bound protein structure, MD simulations can serve to account for that protein flexibility under different physiological conditions and be utilized to model these novel binding events along with predicting its kinetics.93–95 Conventional MD simulations, however, may not explore all possible protein conformational changes quickly enough, but enhanced sampling techniques can help accelerate this by searching through multiple metastable states of a protein and identify its relevant druggable conformations.96,97 For a more in-depth overview of current advances in computational modeling methods for exploring cryptic pocket formation in drug targets, the reader is encouraged to read a review by Bemelmans and co-authors.98 In case of allosteric binders, there is no immediate correlation between affinity and efficacy of the drug – thus making it challenging to evaluate and optimize using classical docking approaches. MD simulations have been combined with machine learning to bridge this gap by relating the functional effects of allosteric inhibitors with protein dynamics.99 Principal Component Analysis (PCA) is a dimensionality reduction technique that can be applied to MD simulation datasets to map the trajectory of protein dynamics and discern movement patterns that could be missed through direct observation.100 However, virtual screening depends on high-quality structural information which may not be available for many therapeutic targets. In such cases, AlphaFold (an artificial intelligence program) can be used to provide initial predictions of the protein structure (but may or may not be active state).101 Combining this with MD simulations and deep learning have helped identify previously unknown binding sites for allosteric ligands, in addition to exhibiting positive correlation between the compound's predicted affinities and experimental potencies.102
Finally, cryo-EM is another high resolution technique that can reconstruct the 3D structure of a flash-frozen protein–ligand complex based on images taken at different angles.103 Time-resolved cryo-EM elevates this method by incorporating a dynamic visualization of different intermediate states of a protein captured across different time points in a biochemical process.104 This can potentially provide insights into ligand or drug-induced transitional changes in a protein structure – whether through cryptic site formation or long range allosteric effect, and guide structure-based optimization. However, low throughput nature of cryo-EM, instrument costs along with limited resolution could be a setback to easily incorporating this method into drug discovery pipeline.103
5. Case studies
As introduced in Section 2.1, while it is predicted that the formation of a covalent bond should improve the affinity of the ligand for its target protein, the specific parameter used to confirm that should be kinact/Kirr, which is time-independent, rather than the time-dependent IC50 or Ki data – consideration of the latter can be, at best, ambiguous or, at worst, misleading.54,105 In the case studies of covalent inhibitors that follow, we have provided control data for the non-covalent counterparts, where it was available. Aside from improved affinities, a covalent inhibitor is always expected to yield sustained inhibition. Further, a covalent inhibitor may be able to trap a transient state within a protein's “breathing” motions/structural dynamics wherein an ensemble of populations are sampled, and this may result in the discovery of a cryptic pocket.91,106 The corresponding non-covalent inhibitor would not be capable of trapping such a transient state, and thus would not be expected to bind as tightly, if at all. Moreover, any inhibition would not be sustained.We have surveyed the literature and identified a small number of covalent inhibitors that have accompanying structural data that describes specific changes induced to the proteins; these are summarized in Table 1, and then elaborated below. To the best of our knowledge, there are no reports of inhibitors carrying boronic acid/boronate reversible covalent warheads eliciting changes in protein structure. However, given their prevalence, and their significance in the FDA-approved multiple myeloma drugs bortezomib and ixazomib, we considered this class too significant to be omitted.107 Further, a co-crystal structure has been solved of the adenosine A1 receptor (A1-AR) – a G-protein-coupled receptor (GPCR) that plays vital roles in cardiovascular, renal and neuronal processes – bound to the selective covalent antagonist DU172 whose arylsulfonyl fluoride warhead reacted with Tyr271. However, to the best of our knowledge, this is the only reported high-resolution structure of A1-AR;108 there is no apo crystal structure. Thus it is not clear specifically what impacts the covalent binding interaction had on the structural dynamics of the A1-AR GPCR; given this uncertainty, this example will be presented in the Table only.
| Warhead | Complementary nucleophile | Example target | Example compound | Product | Comments |
|---|---|---|---|---|---|
| Orthosteric inhibitors | |||||

|

|
SARS-CoV2-MPro |

|

|
See case study 1: subtle cryptic pocket formation – opening of S2 and S4 subpockets, by the shifting of Met149, Met165 and Leu167. |

|

|
JAK3 |
|

|
See case study 2: formation of an “arginine pocket” by the shifting of Arg911 and Arg953. |

|

|
BFL-1 |

|

|
See case study 3: hydrophobic cryptic pocket formation – shifting of Phe95. |
| Allosteric inhibitors | |||||

|

|
KRASG12C |

|

|
See case study 4: hydrophobic cryptic pocket formation rotation of the α2-helix, and shifting of Met72. |

|

|
MCL-1 |

|

|
See case study 5: rigidification of BH3 binding groove reduced flexibility of the N-terminus and portions of the α4, α5 and α6 helices. |

|

|
AKT1E17K |

|

|
See case study 6: formation of a neo-zinc chelate-between the phenol oxygen of 20, the Lys17-based salicylaldimine nitrogen, and the sulfurs of Cys296 and Cys310. |
| Additional examples | |||||

|

|
20S proteasome |

|

|
No significant structural changes upon conjugation.107a |

|

|
β-Lactamase (CTX-M-14) |

|

|
No significant structural changes upon conjugation.107b |

|

|
A1-Ar (GPCR) |

|

|
apo-A1-AR crystal structure not reported, so changes upon chemical conjugation unclear.108 |
We begin with covalent and reversible covalent inhibitors inducing the formation of cryptic pockets by reaction with the protein active site (Section 5.1) or the protein surface (Section 5.2) and we discuss how these pockets may be leveraged in drug discovery. The third part of our case studies deals with allosteric inhibitors (Section 5.3) that cause protein structural reorganizations.
5.1. Cryptic pockets: active sites
Cryptic pockets are binding pockets that become apparent only upon small molecule ligand or drug binding, and these can be leveraged in targeting “undruggable” proteins and/or achieving selectivity.98 These phenomena appear less commonly in enzyme active sites, and are more prevalent at protein surfaces.77,109 In the case of the former, the cryptic pocket is more likely to be already present, and is just occluded, while in the latter, it may be completely absent. Structural dynamics of protein active sites are limited, likely owing to the requirement to maintain an ordered well-defined pocket or cavity to ensure catalytic efficiency and selectivity.122,123 We begin with enzyme active sites: SARS-CoV2 MPro, a protease, and JAK3, a kinase. | ||
| Fig. 3 (A) Chemical Structure of the SARS-CoV2 MPro inhibitor 1 (electrophilic warhead highlighted in turquoise); SARS-CoV2 MPro (B) apo protein (PDB ID: 7CAM; (C) co-crystallized with 1 (PDB ID: 8DLB). For clarity, only one protomer is shown. S labels refer to substrate binding pockets. | ||
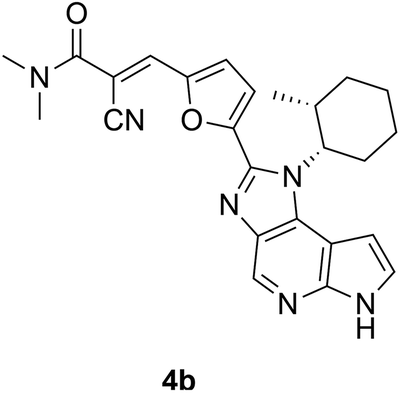
Janus Kinases (JAK) are a family of homologous cytosolic kinases (JAK1/2/3 and TYK2) that function in close association with cytokine receptors and regulate various crucial signal transduction pathways including immune-regulation. Aberrations in JAK-mediated pathways have been implicated in hematologic cancers and immune disorders.116 Compared to its other kinase isoforms, JAK3 is of unique therapeutic interest for selective inhibition, because of its localized expression in the lymphatic system and bone marrow, which can potentially abrogate off-target effects.117 Furthermore, JAK3 structurally differs from JAK1/2 in the availability of a solvent-exposed cysteine (Cys909) near the ATP-binding site. This was leveraged by Förster and colleagues towards the design of selective covalent inhibitors against JAK3 starting with a tricyclic 1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine scaffold like that in compounds 2–7 (Fig. 4).84,118 Their initial designs with irreversible acrylamide-based warheads installed at the imidazole C2 position of parental compound 2 did not yield significantly potent inhibitors, e.g. compound 3: IC50 = 548 nM. Tuning the reactivity of their warhead, they switched to the more electrophilic (but also reversible) α-cyanoacrylamide-based handle,119 arriving at compound 4a (FM-381, Fig. 4), which showed remarkable inhibition of JAK3 kinase activity (IC50 = 127 pM) and excellent JAK3 selectivity (400-, 2700- and 3600-fold over JAK1, JAK2 and TYK2, respectively).84,118 Additionally, kinetic analysis from a reporter displacement assay showed that 4a had a high on-target residence time of 50 min for JAK3 compared to <1.4 min on JAK1, JAK2 and TYK2. When 4a and its analogue 4b were tested against 410 other unrelated kinases, no significant effect was observed on their activities, especially for a set of ten kinases that possessed cysteines at a position equivalent to Cys909 for JAK3. The importance of the ability to form a (reversible) covalent bond was highlighted by inert 5 displaying a moderate IC50 to JAK3 of 181 nM. High-resolution X-ray crystal structures of compounds 4a and 4b complexed with JAK3 (PDB IDs: 5LWM and 5LWN, respectively) revealed formation of a new cryptic ‘arginine pocket’ on the protein surface, decorated with residues Arg911, Asp912 and Arg953 and occupied by the electrophilic warhead.118,120 In each case, the cyclohexyl group was lodged in the ATP-binding site.
 | ||
| Fig. 4 Chemical Structures of JAK3 inhibitors with the electrophilic warheads highlighted in turquoise. | ||
The nitrile component of the 4b warhead formed hydrogen bonds with Arg911 by fully rearranging its side chain orientation, while the Arg953 guanidine group was flipped by almost 180°, relative to the apo protein; when compared with 16 other available crystal structures of JAK3, they concluded that this distinct realignment of the Arg residues to form the cavity was a unique feature caused by the covalent binding event. When comparing the co-crystal structures of JAK3–4b (Fig. 5B (PDB ID: 5LWN), with that of pan-JAK inhibitor 6 (tofacitinib, IC50 = 3.5 nM; Fig. 5A (PDB ID: 3LXK)) and compound 7, a non-acrylamide analogue of 4a (IC50 = 130 nM; Fig. 5C (PDB ID: 6GLB)), the induced pocket in Fig. 5B was seen to be distinctly absent and the two arginines exhibited the same poses for both molecules.120 In all these cases (4b, 5 and 6), the hydrogen bonding interactions of the indole-type scaffold with the hinge residues Glu903 and Leu905 backbones were retained. A clear bond formation between the α-cyanoacrylamide moiety of compound 4a and Cys909 was not captured in the electron density maps.118 This could be due to the rate of reversibility of the covalent bond not being fully represented by the equilibrium binding poses captured by the crystallization process.121 The co-crystal structure of compound 4b, however, exhibited both the covalent and non-covalent modes of binding with Cys909 – thus validating the reversible binding mechanism for these molecules. Either way, based on the observations, it was clear that the induction of this polar cavity is largely driven by the connections introduced by the extended reversible covalent handle. Furthermore, they overlaid the sequence of the arginine pocket of JAK3 with the ten other kinases having a cysteine equivalent to JAK3 Cys909. One of the arginines (at Arg953) was mostly conserved across all the kinases and the Asp912 was replaced by other residues (Glu, Asn and Lys) at their corresponding positions in five of the kinases. Interestingly, it was found that Arg911 is unique to JAK3 – all the other kinases predominantly have leucine at this position, therefore unable to form H-bond contacts with the warhead nitrile group. It could be reasonably inferred that in addition to the cysteine bond formation, the warhead's interaction with Arg911 leading to cavity formation was another factor responsible for the high degree of kinome-wide selectivity demonstrated by compound 4a for JAK3. For these covalent and non-covalent compounds, a comparison of their kinetic parameters such as on-target residence times may have helped further establish a correlation between the covalent bond and the induced pocket formed with their corresponding observed potencies and selectivities.
 | ||
| Fig. 5 JAK3 protein co-crystallized with: (A) the non-covalent inhibitor tofactinib (6; PDB ID: 3LXK); (B) the covalent inhibitor 4b (PDB ID: 5LWN), yellow arrow highlights the cryptic “arginine pocket”; (C) 7, non-acrylamide analogue of 4a (PDB ID: 6GLB). | ||
5.2. Cryptic pockets: protein–protein interactions
While structural dynamics of protein active sites are limited,122,123 PPIs are far more dynamic and malleable, and this flexibility is presumably required to facilitate the recognition of the binding site on one protein by multiple other protein partners,124 as is the case in the BCL-2 family or proteins,125 and because of this cryptic pocket formation in the context of covalent inhibitors may be more profound, as has recently been observed with BFL-1, an anti-apoptotic protein of the BCL-2 family. | ||
| Fig. 6 (A) apo-BFL-1 (PDB ID: 5WHI); (B) BFL1 co-crystallized with D-NA-NOXA SAHB (PDB ID: 5WHH), demonstrating the induction of a hydrophobic, cryptic pocket subjacent to Cys55. | ||
Harvey et al. extended this strategy towards identifying potential small molecule-based alternatives to target the BH3-binding domain.136 They employed a disulfide ‘tethering’ technology to screen a library of disulfide containing compounds against the BFL-1 protein.137 Intact MS and a competitive fluorescence polarization assay were used to verify the most potent binder, 4E14 (compound 8Fig. 7) that derivatized Cys55 irreversibly in BFL-1 and exhibited an IC50 of 1.3 µM. They used hydrogen/deuterium exchange MS (HDX-MS) to study the conformational effects of this covalent derivatization on the protein structure by analyzing the hydrogen–deuterium accessibility/exchangeability in the protein backbone amide Hs. They observed that after 10 s of deuteration upon compound 8 treatment, the regions closer to the cysteine residue in the upper part of the canonical groove (lined by alpha helices α2 and α3) showed maximum protection due to initial interaction and shielding of the binding site. After 10 mins of deuterium exchange, however, this area of protection gradually extended to include the α1–α2 loops and α4, indicating that the covalent engagement led to gradual reduction in structural flexibility of the binding groove and its surrounding connected regions. This experiment was repeated with a C55S-mutated BFL-1 to observe the differences in the absence of the disulfide bond formation with compound 8. Interestingly, a slight deprotection of the canonical groove upon binding was observed, which could potentially indicate an ‘opening motion’ of the α2 helical region initiated due to non-covalent docking of the molecule. This conformational displacement around the groove was previously observed in crystal structure determination for a covalent peptide binder of BFL-1 as well.138 X-ray crystallographic structure of the BFL-1/compound 8 complex (PDB ID: 6VO4) also supported these observations. The indole component of compound 8 docked into the deepest hydrophobic pocket called ‘p2’ in the BH3-binding groove which is normally occupied by a highly conserved leucine in all pro-apoptotic BH3 proteins.139 It was predicted to interact with residues Val48, Leu52, Val74 and Phe95 lining the p2 pocket along with a Glu78, another unique residue that distinguishes BFL-1 from its other pro-survival sister proteins.133 The binding event also allowed the disulfide linker to access a cryptic pocket adjacent to the p2 site formed by Leu52, Leu56, Val74 and Phe95, followed by stabilization of the alpha-helical domains surrounding the binding region. This cryptic pocket is not formed in the case of interaction of a BH3-mimetic peptide with MCL-1, a BFL-1 sister protein, which means this unique rearrangement may be selectively tailored for cysteine-based covalent targeting of BFL-1.138 Structural modifications on the indole component of compound 8 (L- to D-form, addition of a methyl group and replacement to naphthyl) led to reduction or loss in potency in cytochrome c release assay when compared to 8. Docking studies with these analogues also showed less favorable contact with the p2 and cryptic pockets. It would be interesting to further investigate if an inhibitor with more potent interactions with the BFL-1 pocket than compound 8 could lead to further stabilization of the helical domain.
 | ||
| Fig. 7 Chemical structures of small-molecule BFL-1 inhibitors with the electrophilic warheads highlighted in turquoise. | ||
More recently, Lucas and coworkers utilized a rapid mass spectrometry-based screening technology to identify acrylamide-based covalent binders of BFL-1, validated with a FRET-based competition assay to measure potency.140 For one of the hits with poor potency (IC50 ∼ 23 µM), they were unable to acquire a resolvable crystal structure.141 SAR optimization of this molecule improved its potency by 4-fold (IC50 = 5.1 µM (kinact/Kirr = 18 M−1 s−1), compound 9, Fig. 7) which facilitated the acquisition of a high-resolution co-crystal structure (PDB ID: 9FL0); a covalent bond was observed between the acrylamide warhead and Cys55. Significantly, the para-chlorobenzyl group was found in the aforementioned hydrophobic cryptic pocket that had been previously observed by Walensky's team with their stapled peptides.97,134 Further optimization initially focused on attempts to capture a hydrogen bond with Glu78, which yielded, among others, racemic compound 10a (Fig. 7; IC50 = 0.48 µM, kinact/Kirr = 240 M−1 s−1). As predicted from docking studies and the order of magnitude improvement in activity, the co-crystal structure of BFL-1 with compound (±)-10a (PDB ID: 9FKZ) showed that the rac-3-aminocyclopentyl moiety engaged in an H-bond with Glu78 while maintaining the induction of the cryptic hydrophobic pocket –and the covalent bond with Cys55 that were observed with compound 9. Fig. 8 clearly shows this cryptic pocket in the BFL-1-10a complex, whose formation was mainly ascribed to a shift of Phe95 from an “in” position to an “out” position (cyan and grey, respectively, in Fig. 8C). Next, 10a was further optimized by varying the para-chlorobenzyl group: compound (R,R,S)-10b (where the methyl group is the carbon with S stereochemistry) was their most potent with an IC50 of 0.022 µM (kinact/Kirr = 4600 M−1 s−1). Subsequently, they also saw an improvement in cellular potency as judged by activation of caspase 3/7 activity (EC50 = 370 nM for 10b). Interestingly, in the BFL-1 co-crystal structure for compound 10b (PDB ID: 9FKY), in order to accommodate the additional substituents on the benzylic group, the acrylamide warhead was observed to rotate in order to maintain the covalent bond with Cys55 (when compared to the binding pose of 10a). Since the authors’ initial goal was to screen a library of warheads, their quest was focused on the discovery of covalent inhibitors; it does not appear that a non-covalent congener of 9–10b was prepared, so the benefit of the covalent bond formation with this inhibitor scaffold on BFL-1 inhibition is unclear at this time.
 | ||
| Fig. 8 (A) A co-crystal structure of BFL-1 and compound 10a, and (B) a zoomed-in, transverse perspective (PDB ID: 9FKZ); (C) unsurfaced structures of apo BFL-1 (PDB ID: 5WHI; cyan) overlaid with PDB ID: 9FKZ (grey, ligand removed for clarity) highlighting the shift of Phe95 and its role in the hydrophobic cryptic pocket formation. | ||
5.3. Allosteric inhibitors
As with the malleability of protein surfaces at PPIs, it may be envisaged that allosteric binding sites on protein surfaces may likewise be flexible upon covalent ligand binding, generating cryptic pockets and/or structural reoganizations, without pocket formation, but which stabilize the protein–ligand complex. Highly flexible regions of the protein can mold into transient grooves (in specific functional states) and form novel interactions with the drug that have not been discovered before. Therefore, these dynamic regions should be examined more closely for potential movements that can be utilized for allosteric influence on protein activity or protein–protein interactions.Picomolar affinity of GDP/GTP for KRAS renders the design of efficacious (and selective) drugs highly challenging, which is compounded by a scarcity of alternative druggable sites due to a relatively featureless protein surface.145 However, Shokat's group recently discovered new “switch” pockets on the surface of KRASG12C that were found to undergo conformational shifts upon nucleotide binding and regulate effector interactions.147 Ostrem and colleagues found that the mutant cysteine (Cys12) was favorably positioned between the switch regions and was exploited in the screening of a disulfide-tethering compound library using intact protein mass spectrometry.81 One of their initial probes showing the highest degree of dose-dependent modification in the MS assay (compound 11, Fig. 9) was co-crystallized with GDP-bound-KRASG12C (PDB ID: 4LUC). Compound 11 was seen occupying a previously obscured allosteric site called the switch-II pocket (S-IIP), whilst in disulfide conjunction with the target cysteine. This groove was bordered by α2 (switch II loop), α3 helices and central β-sheet. This S-IIP is only accessible in the GDP-bound protein and completely blocked in the active form. Further evaluation of the binding contacts between 11 and switch-II region revealed both hydrophobic and H-bonding contacts as well as the presence of additional sub-sites in the pocket which could be amenable to accommodating structural modifications.
 | ||
| Fig. 9 Chemical structures of KRASG12C inhibitors with the electrophilic warheads highlighted in turquoise. | ||
To generate irreversible inhibitors with extended inhibitory activities and to avoid a sensitivity to cellular redox chemistry, they abandoned the reversible covalent disulfide warhead and shifted to irreversible covalent designs, such as a cysteine-reactive vinylsulfonamide-based warhead (12; Fig. 9), as well as acryalmides.
 | ||
| Fig. 10 KRASG12C, with Cys12 (yellow), Switch I (blue) and Switch II (cyan) regions highlighted; ligands, green (coloured by atom type); GDP, white (coloured by atom type): co-crystallized with (A) GDP (PDB ID: 4L8G); (B) 12 (PDB ID: 4LYF); (C) 13 (PDB ID: 5F2E), Mg2+cation, magenta sphere. | ||
While reversible covalent disulfide 11 exhibited subtle rearrangement of the switch-II region to form S-IIP with minimal influence on the switch-I site, it was shown that irreversible inhibitor 12 displaced residues forming the S-IIP to a greater degree, as observed in the KRASG12C–12 co-crystal structure (PDB ID: 4LYF; Fig. 10B, compare with GDP-bound KRASG12C (Fig. 10A)). This in turn introduced partial disorder in the switch-I region while the electron density for Mg2+ (required for effective GTP binding) was also not detected. Therefore, it is predicted that by indirectly preventing GTP-binding, these compounds not only favored the GDP-bound state of KRAS but also reduced its affinity to effectors such as Ras. This allosteric rearrangement in an otherwise ‘undruggable’ protein surface was not only crucial for inhibitor library screening and the SAR process but also necessary in tilting favor of KRAS towards its inactive state.
Building from Shokat's work, Patricelli et al. designed a potent acrylamide-based covalent inhibitor ARS-853 (compound 13, cellular IC50 1.6 µM, Fig. 9) was selective to GDP-bound KRASG12C but did not interact with the GTP-bound form.148 In the co-crystal structure of GDP-bound KRASG12C–13 (PDB ID: 5F2E; Fig. 10C), this compound occupied the S-IIP, while being covalently bound to Cys12. Rotation of the α-2 helix allowed the hydrophobic S-IIP to harbor the phenyl ring where the chloro- and cyclopropyl groups made van der Waals contacts. The hydroxyl group on the phenyl ring and the warhead carbonyl formed H-bonds with residues Asp69 and a conserved Lys16, respectively. The mechanism of inhibition proposed by these authors further solidified the observations by Ostrem et al.81 Based on a combination of structural and biochemical evaluations, they discovered that the electrophilic warhead moiety of ARS-853 was positioned in the groove that was typically occupied by the terminal phosphate group of GTP, which explained the compound's lack of preference for the GTP-bound active form. They also suggested that there was cooperative interaction between the inhibitor, GDP and Mg2+ ion which stabilized the GDP-bound form of KRASG12C and prevented subsequent GDP-to-GTP exchange. The switch-I and -II loops were also seen to be more conformationally stabilized when compared to their more disordered nature in the active form of KRAS. Lack of structural mobility of the switch regions could, therefore, interfere with effector binding and consequently, deter downstream signaling. Although ARS-853 showed low micromolar potency in cells, it did not show suitable chemical stability in mouse models. Janes and coworkers wanted to further improve the pharmacokinetic properties of ARS-853, namely its metabolic stability and quick kinetic sampling to ‘capture’ the GDP-bound KRAS.149 Replacement of the chlorophenol in the ARS-853 series with a rigid quinazoline scaffold yielded second generation compound, ARS-1620 (compound 14, Fig. 9) with substantial improvement in potency in both biochemical and cell-based assay. Interestingly, only the S-conformational atropisomer form of ARS-1620 showed this high degree of activity, with the R-atropisomer form being completely inactive in biochemical assays. The S-atropisomer exhibited the same mode of engagement in crystal structure (PDB ID: 5V9U; Fig. 10C) as the previous inhibitors – trapping the GDP-bound protein in an inactive state. Upon comprehensive in vivo and in vitro evaluation, ARS-1620 was considered to be the first potent and selective covalent inhibitor of GDP-bound KRASG12C with favorable ADME properties. However, the oncogenic mutations in KRAS stabilize its GTP-bound form, not the GDP-bound state which was being singly targeted by the previous KRAS inhibitors.
Continued optimization of 14 by Canon and colleagues, yielded acrylamide-based lead candidate AMG510 (Sotorasib, 16, Fig. 9) which was the first KRASG12C inhibitor to proceed to clinical trials and was approved in 2021 for the treatment of non-small cell lung cancer.84,150 Last, the inert/saturated, propionamide congeners of 14 and 16, 15 and 17 (Fig. 9), respectively, were unremarkable inhibitors.149 There are multiple other examples of allosteric as well as direct inhibitors of KRAS in literature. The reader is directed to an excellent review article by Chen et al. with comprehensive analysis and extensive structural details of these inhibitors for further information.144
 | ||
| Fig. 11 (A) Chemical Structure of MCL-1 irreversible inhibitor MAIM1 (18), and (B) a cartoon representation of its allosteric mechanism of action. | ||
Benabderrahmane and coworkers used molecular dynamic simulation studies to further explore this hypothesis by introducing a mutation (C286W) in the MCL-1 and observing its behavior in silico.106 The mutant Trp set in motion a coordinated movement across multiple helices and internal loops in their protein simulations. The bulky hydrophobic side chain was seen to transiently pack into a cryptic pocket which altered the secondary structure of the adjacent α3 helix, pushed away the α4 helix and the α3–α4 loop; and reoriented the α3 helix in a ‘standing’ conformation. They also observed the formation of additional salt bridges that were responsible for stabilizing the new alignment of the α3 helix – this deformed the BH3 binding groove and therefore, responsible for inhibiting the BH3 ligand binding. Based on principal component analysis for these dynamics, they described the movements of the protein as a ‘breathing motion’ at the BH3 binding interface along with internal loop motions. They used Metadynamics, which is an enhanced sampling technique in MD simulations, to visualize the free energy landscape (FEL) and profile this ‘breathing’ motion. Their results suggest an equilibrium between two predominant states: a more open conformation that was ‘ready to bind’ to pro-apoptotic partners and a closed conformation that could hinder the anti-apoptotic process. Binding of a ligand or a Trp mutation at the Cys286 allosteric site of MCL-1 seems to favor a shift to the more conformationally rigid closed form of the protein. It would be interesting to further investigate into the cryptic pocket environment induced in MCL-1 by the Trp residue, whether MAIM1 conjugation could cause a similar allostery effect, what side chain interactions are involved, and whether optimization of these contacts could help improve the potency of the molecule.
 | ||
| Fig. 12 Chemical structures of (A) reversible; (B) reversible covalent and (C) irreversible inhibitors of AKT1. Electrophilic warheads highlighted in turquoise. | ||
 | ||
| Fig. 13 Co-crystal structures of (A) AKT1(wt)–ARQ092 (PDB ID: 5KCV); (B) AKT1E17K–20 (PDBID: 8UW9), demonstrating the formation of a covalent salicylaldimine complex with E17K, and a neo-chelate Zn2+ complex that led to structural reorganization of the protein surface. | ||
Another first-in class allosteric inhibitor of AKT1, borussertib (21, Fig. 12), binds to an interfacial pocket, sandwiched between the kinase/catalytic and PH domains (PDB ID: 6HHF).168 The warhead irreversibly engages with Cys296 of the catalytic domain, forms a crucial π–π stacking interaction with Trp80 of the PH domain and other hydrophobic contacts (with Tyr272, Leu210, Leu264, Ile290), stabilizing the interdomain interaction and effectively ‘locking’ AKT1 in an inactive state.169,170 Immunoblot assays with washout experiments were performed to ascertain the duration of action of the covalent drug based on its effect on AKT-mediated signalling. Efficient downregulation was observed up to 24 hrs even after the washout – indicating prolonged inhibitory effect in vivo. The efficacy of borussertib was also confirmed with cellular and xenograft models. Uhlenbrock et al., used this molecule as template to design derivatives for pre-clinical evaluation through SAR analysis.171 One of their most potent and kinase selective candidates, 22 was co-crystallized with AKT1 (PDB ID: 6HHI; Fig. 14). It was also seen to have a similar binding pose as borussertib, with confirmed labelling of Cys296 and additional H-bonding with the backbone amide of Glu85. Biochemical characterization with fluorescence-based assay showed that compared to borussertib (IC50 ∼ 0.8 nM and kinact/Kirr ∼ 0.85 µM−1 s−1), compound 22 showed less favourable values (IC50 ∼ 3.6 nM and kinact/Kirr ∼ 0.2 µM−1 s−1). Nonetheless, this molecule showed potent inhibition across multiple cancer cell lines as well as improved Phase 1 metabolic stability compared to borussertib. This unique class of allosteric inhibitors further validates the importance of leveraging domain-based conformational changes of kinases to alter their activity profiles, using covalent drugs. Considering that non-covalent binders cannot form sufficiently tight complexes with their host proteins (limitation on maximum possible affinity), chemical bonds created by covalent modalities to secure two different regions of a protein together can offer a promising alternative.172 The mechanism of action of 22 (and 21) echoes an intra-protein version of the emerging field of RIPTACs, wherein two ligands for two proteins of interest are coupled together to promote the aggregation, and thereby inhibition, of those two proteins.173,174
 | ||
| Fig. 14 A co-crystal structure of AKT1 and 22 (PDB ID: 6HHI), indicating the small-molecule 22 facilitates the formation of a deactivating intra-domain complex, upon forming a covalent bond with Cys296. | ||
5.4 PROTACs and other degraders
Although outside the scope of this review, given a lack of structural data, covalent and reversible covalent chemistries have recently been implemented in PROTAC design.175,176 PROTACs are bivalent compounds that comprise a ligand for the target protein coupled to a ligand for an E3 ligase, which promotes the ubiquitination of the target protein and subsequent recognition by the proteasome followed by degradation. Importantly, once the degradation is complete, the PROTAC is released, able to promote the degradation of another target protein molecule, and so on. On the one hand, covalent PROTACs may afford a potent and drug-like molecule of lower molecular weight but on the other hand, the recycling characteristic of PROTACs would be compromised, but this may be restored by reversible covalent chemistries.176,177 To the best of our knowledge, there are no reports of impacts on protein structural dynamics of covalent (versus non-covalent) PROTACs, but this information will obviously be of utility in the design of protein specific degraders. One significant concern of PROTACs is that they do not need to be incredibly tight binders, as they operate through event-driven, rather than occupancy-driven, pharmacology, and thus they may inadvertently degrade an unintended protein.175,176 PROTACs built from ligands that induce and bind cryptic pockets may serve to allay these concerns, especially when trying to target a specific isoform within a family of proteins.6. Discussion
Structural changes induced locally in a protein by the interaction with the drug molecule, even with a single residue, such as a cysteine, can be capitalized upon in the design of inhibitors that are selective to the target protein isoform or subtype. Structural biology techniques used to compare the bound form of the protein with the apo form can help unveil these accessible sites. Further, advances in computational chemistry may help identify such pockets, or, at least, guide the medicinal chemist towards a potentially malleable site. These changes can then be further characterized to make more informed decisions about the molecule regarding its improvements in target affinity. Novel sites on a protein surface or its interior can be induced as more pharmacophoric features are added on drug molecule – which warrants a closer look using structural biology characterization in order to make more informed decisions regarding its improvements in target affinity.Applying such strategies may be especially beneficial for the most heavily targeted regulatory proteins such as kinases (as we saw with the JAK3 case study) and G-protein coupled receptors (such as with A1-AR in Table 1), but also with other protein families, such as proteases, e.g., SARS-CoV2-MPro.178 Since kinases tend to have multiple isoforms and highly differential tissue expressions, effective drug candidates that lend selectivity as well as specificity could help reduce potential off-target effects.
In our literature search, we found more examples of cryptic pocket formations in PPIs, rather than in enzyme active sites. As mentioned earlier, this likely stems from the requirement of a protein surface to be inherently more adaptable, permitting the recognition of multiple protein partners, in contrast to more rigid enzyme active sites that have evolved to recognize just one, or a small handful, of ligands. Indeed, the BFL-1 case studies underscores this point, with the formation of a significant lipophilic cavity subjacent to the targeted Cys55. Lucas and co-workers successfully leveraged the discovery of this cryptic pocket to yield a suitable tool compound to further study the effects of BFL-1 inhibition in vivo.
Cryptic pockets have not only been observed at ligand or protein-binding sites, but at allosteric sites, too. For example, inhibitors designed to react with oncogenic mutants of K-RAS, such as KRAS-G12C, exploited the highly ‘malleable’ nature of the protein, facilitating its being locked into a preferred active or inactive state via allosteric modulation due to covalent drug binding. Covalent allosteric inhibitors may also yield structural changes distant to the site of conjugation as demonstrated by the partial inhibition of MCL-1 upon reaction of Cys286 with MAIM1: this somewhat incapacitated the ability of the MCL-1 protein to recognize the BH3 domains of its partner proteins. Interestingly, this effect was emulated by a single point mutation of C286W. Additionally, considering that MCL-1 binds with different pro-apoptotic partners with variable dissociation constants, the extent of ‘rigidification’ of the canonical groove upon allosteric derivatization may vary and inhibit different BH3 protein partners to different extents. This may thus be a source of selectivity, and may also be considered in the context of other PPIs.
Last, while a significant portion of this review has focused on the formation of cryptic pockets, a well-defined cavity is not always formed. An example of this was provided by the development of selective inhibitors of an oncogenic mutation of AKT1, which also showcased the utility of a reversible covalent electrophilic warhead, permitting the sampling of multiple conjugates, until the most stable conjugate (neo zinc chelate) was found.
7. Conclusions
Protein functions are closely associated with their structural dynamics – so is their mode of interaction with other biomolecules including drugs.21 These therapeutic molecules can sample multiple conformational states of proteins and may prefer to bind to, or select for, a specific state, or can induce changes in the structure upon binding. Either way, the case studies discussed in this review article underscore that even minor conformational changes in the target protein domains that manifest upon the chemical association with an inhibitor can reduce or eliminate binding to their native partners. Alternatively, these binders can only partially occupy the spaces proximal to the active sites but have significant competing effect on docking by a native binding partner. There are other examples in the literature describing such protein dynamics for non-covalent inhibitors as well.179–181 In the interest of brevity and relevance to the topic, only covalent inhibitors were highlighted in this article. It would not be unreasonable to expect that most inhibitors currently under research could potentially have different extents of structural impacts on their protein targets – that have simply escaped observation due to lack of enough evidence or the necessary techniques. Acquiring a detailed understanding or complete picture of these structural changes can also be challenging using just one technique. For example, X-ray crystal structures capture only one single conformational state of the protein-inhibitor complex at a time. The snapshots may yield electron density maps that are difficult to model for highly flexible regions, which would be needed to understand the very essence of the dynamic process. Similarly, HDX-MS results can only be used for an indirect inference and not a visual representation of the protein structural changes. For complex protein systems, these experiments can be difficult to perform as well as would require careful analytical processing.Irreversible and reversible covalent inhibitors each present their own benefits. Irreversible inhibitors may induce (presumably permanent) cryptic pockets, as in the BFL-1 case study. Equally, reversible covalent inhibitors may also afford new binding pockets that can be exploited in rational drug design, as in the KRAS case study, although it is predicted these will be transient, and the protein structure will be regenerated upon the ligand's departure. While reversible covalent inhibitors typically exhibit shorter residence times and possibly a reduced therapeutic effect relative to the corresponding irreversible inhibitors, reversibility may mitigate toxicity that might be experienced otherwise. In a landmark publication, Jack Tauton's recent work on the development of selective AKT1-E17K inhibitors using reversible covalent chemistry highlights the inherent advantage of the reversibility and associated structural reorganization that may select for one specific adduct when multiple adducts are possible.
Covalent inhibitors often yield improvements in target protein binding relative to their non-covalent counterparts, although this is not always the case. This may be due to the formation of a single covalent bond in addition to an array of non-covalent interactions that were already present. Alternatively, in the event the warhead was not installed judiciously, crucial non-covalent interactions may be compromised, or even lost, upon the chemical reaction taking place. Of course, sustained inhibition, i.e. an increased period of target occupancy/increased residence time, would always be observed, which can provide a significant therapeutic benefit. In the case studies, where the data were available, we described the relative benefit of the chemical warhead to target binding. Where profound differences in inhibitory activities may be observed is in the event a covalent inhibitor traps a specific transient state of a protein, such as a cryptic pocket, that may then be leveraged in lead optimization: such a pocket may not be accessible to a non-covalent inhibitor, as its existence may be too fleeting to be exploited. In other words, drugs equipped with electrophilic warheads have the inherent ability to harness transient states adopted during a protein's “breathing” motions, providing a source of increased affinity (and maybe selectivity) that would otherwise not seem possible based on static structures.
Author contributions
RB performed the literature search and wrote the manuscript; SF wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
This is not relevant to our review article.Acknowledgements
We thank the University of Maryland Schools of Pharmacy and Medicine for their continued support of our work in this area. Further, this work was supported in part by NIH grant T32 GM158458.Notes and references
- R. Aebersold, J. N. Agar, I. J. Amster, M. S. Baker, C. R. Bertozzi, E. S. Boja, C. E. Costello, B. F. Cravatt, C. Fenselau, B. A. Garcia, Y. Ge, J. Gunawardena, R. C. Hendrickson, P. J. Hergenrother, C. G. Huber, A. R. Ivanov, O. N. Jensen, M. C. Jewett, N. L. Kelleher, L. L. Kiessling, N. J. Krogan, M. R. Larsen, J. A. Loo, R. R. Ogorzalek Loo, E. Lundberg, M. J. Maccoss, P. Mallick, V. K. Mootha, M. Mrksich, T. W. Muir, S. M. Patrie, J. J. Pesavento, S. J. Pitteri, H. Rodriguez, A. Saghatelian, W. Sandoval, H. Schlüter, S. Sechi, S. A. Slavoff, L. M. Smith, M. P. Snyder, P. M. Thomas, M. Uhlén, J. E. Van Eyk, M. Vidal, D. R. Walt, F. M. White, E. R. Williams, T. Wohlschlager, V. H. Wysocki, N. A. Yates, N. L. Young and B. Zhang, Nat. Chem. Biol., 2018, 14, 206–214 Search PubMed.
- L. Jiang, M. Wang, S. Lin, R. Jian, X. Li, J. Chan, G. Dong, H. Fang, A. E. Robinson, F. Aguet, S. Anand, K. G. Ardlie, S. Gabriel, G. Getz, A. Graubert, K. Hadley, R. E. Handsaker, K. H. Huang, S. Kashin, D. G. MacArthur, S. R. Meier, J. L. Nedzel, D. Y. Nguyen, A. V. Segrè, E. Todres, B. Balliu, A. N. Barbeira, A. Battle, R. Bonazzola, A. Brown, C. D. Brown, S. E. Castel, D. Conrad, D. J. Cotter, N. Cox, S. Das, O. M. de Goede, E. T. Dermitzakis, B. E. Engelhardt, E. Eskin, T. Y. Eulalio, N. M. Ferraro, E. Flynn, L. Fresard, E. R. Gamazon, D. Garrido-Martín, N. R. Gay, R. Guigó, A. R. Hamel, Y. He, P. J. Hoffman, F. Hormozdiari, L. Hou, H. K. Im, B. Jo, S. Kasela, M. Kellis, S. Kim-Hellmuth, A. Kwong, T. Lappalainen, X. Li, Y. Liang, S. Mangul, P. Mohammadi, S. B. Montgomery, M. Muñoz-Aguirre, D. C. Nachun, A. B. Nobel, M. Oliva, Y. S. Park, Y. Park, P. Parsana, F. Reverter, J. M. Rouhana, C. Sabatti, A. Saha, A. D. Skol, M. Stephens, B. E. Stranger, B. J. Strober, N. A. Teran, A. Viñuela, G. Wang, X. Wen, F. Wright, V. Wucher, Y. Zou, P. G. Ferreira, G. Li, M. Melé, E. Yeger-Lotem, M. E. Barcus, D. Bradbury, T. Krubit, J. A. McLean, L. Qi, K. Robinson, N. V. Roche, A. M. Smith, L. Sobin, D. E. Tabor, A. Undale, J. Bridge, L. E. Brigham, B. A. Foster, B. M. Gillard, R. Hasz, M. Hunter, C. Johns, M. Johnson, E. Karasik, G. Kopen, W. F. Leinweber, A. McDonald, M. T. Moser, K. Myer, K. D. Ramsey, B. Roe, S. Shad, J. A. Thomas, G. Walters, M. Washington, J. Wheeler, S. D. Jewell, D. C. Rohrer, D. R. Valley, D. A. Davis, D. C. Mash, P. A. Branton, L. K. Barker, H. M. Gardiner, M. Mosavel, L. A. Siminoff, P. Flicek, M. Haeussler, T. Juettemann, W. J. Kent, C. M. Lee, C. C. Powell, K. R. Rosenbloom, M. Ruffier, D. Sheppard, K. Taylor, S. J. Trevanion, D. R. Zerbino, N. S. Abell, J. Akey, L. Chen, K. Demanelis, J. A. Doherty, A. P. Feinberg, K. D. Hansen, P. F. Hickey, F. Jasmine, R. Kaul, M. G. Kibriya, J. B. Li, Q. Li, S. E. Linder, B. L. Pierce, L. F. Rizzardi, K. S. Smith, J. Stamatoyannopoulos, H. Tang, L. J. Carithers, P. Guan, S. E. Koester, A. R. Little, H. M. Moore, C. R. Nierras, A. K. Rao, J. B. Vaught, S. Volpi and M. P. Snyder, Cell, 2020, 183, 269–283.e19 Search PubMed.
- H. Lu, Q. Zhou, J. He, Z. Jiang, C. Peng, R. Tong and J. Shi, Signal Transduction Targeted Ther., 2020, 5, 213 CrossRef PubMed.
- L. Clausen, A. B. Abildgaard, S. K. Gersing, A. Stein, K. Lindorff-Larsen and R. Hartmann-Petersen, Adv. Protein Chem. Struct. Biol., 2019, 114, 61–83 Search PubMed.
- N. Mainali, M. Balasubramaniam, S. Pahal, W. S. T. Griffin, R. J. Shmookler Reis and S. Ayyadevara, Basic Res. Cardiol., 2025, 120, 489–507 CrossRef CAS PubMed.
- M. Gu, S. Sun, Q. You and L. Wang, Molecules, 2023, 28, 1–14 Search PubMed.
- B. G. de la Torre and F. Albericio, Molecules, 2024, 29, 585 Search PubMed.
- M. W. Y. Southey and M. Brunavs, Front. Drug Discovery, 2023, 3, 1–8 Search PubMed.
- T. Tamura, M. Kawano and I. Hamachi, Chem. Rev., 2025, 125, 1191–1253 Search PubMed.
- X. Xie, T. Yu, X. Li, N. Zhang, L. J. Foster, C. Peng, W. Huang and G. He, Signal Transduction Targeted Ther., 2023, 8, 335 CrossRef PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 Search PubMed.
- S. Mozaffari, A. Moen, C. Y. Ng, G. A. F. Nicolaes and K. Wichapong, Res. Pract. Thromb. Haemostasis, 2025, 9, 102691 Search PubMed.
- H. Wei and J. A. McCammon, npj Drug Discovery, 2024, 1, 1–8 CrossRef.
- S. Kalyaanamoorthy and Y. P. P. Chen, Drug Discovery Today, 2011, 16, 831–839 CrossRef CAS PubMed.
- M. Batool, B. Ahmad and S. Choi, Int. J. Mol. Sci., 2019, 20, 2783 CrossRef CAS PubMed.
- X. Du, Y. Li, Y. L. Xia, S. M. Ai, J. Liang, P. Sang, X. L. Ji and S. Q. Liu, Int. J. Mol. Sci., 2016, 17, 1–34 Search PubMed.
- C. Falciani, L. Lozzi, A. Pini and L. Bracci, Chem. Biol., 2005, 12, 417–426 CrossRef CAS PubMed.
- E. P. Raman, W. Yu, O. Guvench and A. D. MacKerell, J. Chem. Inf. Model., 2011, 51, 877–896 CrossRef CAS PubMed.
- M. E. Lanning, W. Yu, J. L. Yap, J. Chauhan, L. Chen, E. Whiting, L. S. Pidugu, T. Atkinson, H. Bailey, W. Li, B. M. Roth, L. Hynicka, K. Chesko, E. A. Toth, P. Shapiro, A. D. MacKerell, P. T. Wilder and S. Fletcher, Eur. J. Med. Chem., 2016, 113, 273–292 Search PubMed.
- D. Li and B. Ji, Med. Nov. Technol. Devices, 2019, 3, 100026 CrossRef.
- D. D. Boehr, R. Nussinov and P. E. Wright, Nat. Chem. Biol., 2009, 5, 789–796 CrossRef CAS PubMed.
- J.-P. Colletier, D. Fournier, H. M. Greenblatt, J. Stojan, J. L. Sussman, G. Zaccai, I. Silman and M. Weik, EMBO J., 2006, 25, 2746–2756 CrossRef CAS PubMed.
- F. Huang, X. Han, X. Xiao and J. Zhou, Molecules, 2022, 27, 7728 CrossRef CAS PubMed.
- L. Zheng, Y. Li, D. Wu, H. Xiao, S. Zheng, G. Wang and Q. Sun, MedComm: Oncol., 2023, 2, 1–54 Search PubMed.
- L. K. Mader and J. W. Keillor, ACS Med. Chem. Lett., 2024, 15, 731–738 CrossRef CAS PubMed.
- B. F. Krippendorff, R. Neuhaus, P. Lienau, A. Reichel and W. Huisinga, J. Biomol. Screen., 2009, 14, 913–923 CrossRef CAS PubMed.
- E. Mons, S. Roet, R. Q. Kim and M. P. C. Mulder, Curr. Protoc., 2022, 2, 1–85 Search PubMed.
- P. C. Sharma, A. Jain, S. Jain, R. Pahwa and M. S. Yar, J. Enzyme Inhib. Med. Chem., 2010, 25, 577–589 CrossRef CAS PubMed.
- Q. Cao, X. Wu, Q. Zhang, J. Gong, Y. Chen, Y. You, J. Shen, Y. Qiang and G. Cao, Front. Pharmacol., 2023, 14, 1–7 Search PubMed.
- M. Jan, A. S. Sperling and B. L. Ebert, Nat. Rev. Clin. Oncol., 2021, 18, 401–417 CrossRef PubMed.
- S. K. De, Med. Chem. Res., 2023, 32, 424–433 CrossRef CAS.
- N. Singh, P. Vayer, S. Tanwar, J. L. Poyet, K. Tsaioun and B. O. Villoutreix, Front. Drug Discovery, 2023, 3, 1–11 Search PubMed.
- A. Thorarensen, P. Balbo, M. E. Banker, R. M. Czerwinski, M. Kuhn, T. S. Maurer, J.-B. Telliez, F. Vincent and A. J. Wittwer, Bioorg. Med. Chem., 2021, 29, 115865 CrossRef CAS PubMed.
- Y. S. Raouf, J. Med. Chem., 2024, 67, 10513–10516 CrossRef CAS PubMed.
- R. A. Bauer, Drug Discovery Today, 2015, 20, 1061–1073 CrossRef CAS PubMed.
- H. Liu, A. Zask, F. Forouhar, S. Iketani, A. Williams, D. R. Vaz, D. Habashi, K. Choi, S. J. Resnick, S. J. Hong, D. H. Lovett, T. Bai, A. Chavez, D. D. Ho and B. R. Stockwell, Nat. Commun., 2025, 16, 1–18 Search PubMed.
- T. A. Baillie, Angew. Chem., Int. Ed., 2016, 55, 13408–13421 Search PubMed.
- A. M. Chan, C. C. Goodis, E. G. Pommier and S. Fletcher, RSC Med. Chem., 2022, 13, 921–928 RSC.
- J. S. Martin, C. J. MacKenzie, D. Fletcher and I. H. Gilbert, Bioorg. Med. Chem., 2019, 27, 2066–2074 CrossRef CAS PubMed.
- Faridoon, R. Ng, G. Zhang and J. J. Li, Med. Chem. Res., 2023, 32, 1039–1062 CrossRef CAS PubMed.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–797 CrossRef CAS PubMed.
- C. A. Smith, A. Ebrahimpour, L. Novikova, D. Farina, A. O. Bailey, W. K. Russell, A. Jain, A. B. Saltzman, A. Malovannaya, B. V. V. Prasad, L. Hu and Y. T. Ghebre, Biochim. Biophys. Acta, Gen. Subj., 2022, 1866, 130149 CrossRef CAS PubMed.
- J. M. Shin and N. Kim, J. Neurogastroenterol. Motil., 2013, 19, 25–35 CrossRef PubMed.
- Y. M. Jhanker, M. F. Kadir, R. I. Khan and R. Hasan, J. Appl. Pharm. Sci., 2012, 2, 1–12 CAS.
- A. Thorarensen, M. E. Dowty, M. E. Banker, B. Juba, J. Jussif, T. Lin, F. Vincent, R. M. Czerwinski, A. Casimiro-Garcia, R. Unwalla, J. I. Trujillo, S. Liang, P. Balbo, Y. Che, A. M. Gilbert, M. F. Brown, M. Hayward, J. Montgomery, L. Leung, X. Yang, S. Soucy, M. Hegen, J. Coe, J. Langille, F. Vajdos, J. Chrencik and J. B. Telliez, J. Med. Chem., 2017, 60, 1971–1993 CrossRef CAS PubMed.
- J. B. Fell, J. P. Fischer, B. R. Baer, J. F. Blake, K. Bouhana, D. M. Briere, K. D. Brown, L. E. Burgess, A. C. Burns, M. R. Burkard, H. Chiang, M. J. Chicarelli, A. W. Cook, J. J. Gaudino, J. Hallin, L. Hanson, D. P. Hartley, E. J. Hicken, G. P. Hingorani, R. J. Hinklin, M. J. Mejia, P. Olson, J. N. Otten, S. P. Rhodes, M. E. Rodriguez, P. Savechenkov, D. J. Smith, N. Sudhakar, F. X. Sullivan, T. P. Tang, G. P. Vigers, L. Wollenberg, J. G. Christensen and M. A. Marx, J. Med. Chem., 2020, 63, 6679–6693 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- M. S. Davids and J. R. Brown, Futur. Oncol., 2014, 10, 957–967 CrossRef CAS PubMed.
- S. S. Zhang and M. Nagasaka, Lung Cancer: Targets Ther., 2021, 12, 115–122 Search PubMed.
- G. Vauquelin, Br. J. Pharmacol., 2016, 2319–2334 CrossRef CAS PubMed.
- H. Kim, Y. S. Hwang, M. Kim and S. B. Park, RSC Med. Chem., 2021, 12, 1037–1045 Search PubMed.
- D. A. Schuetz, W. E. A. de Witte, Y. C. Wong, B. Knasmueller, L. Richter, D. B. Kokh, S. K. Sadiq, R. Bosma, I. Nederpelt, L. H. Heitman, E. Segala, M. Amaral, D. Guo, D. Andres, V. Georgi, L. A. Stoddart, S. Hill, R. M. Cooke, C. De Graaf, R. Leurs, M. Frech, R. C. Wade, E. C. M. de Lange, A. P. IJzerman, A. Müller-Fahrnow and G. F. Ecker, Drug Discovery Today, 2017, 22, 896–911 Search PubMed.
- J. M. Strelow, J. Biomol. Screen., 2017, 22, 3–20 CrossRef CAS PubMed.
- M. Visscher, M. R. Arkin and T. B. Dansen, Curr. Opin. Chem. Biol., 2016, 30, 61–67 CrossRef CAS PubMed.
- D. Schaefer and X. Cheng, Pharmaceuticals, 2023, 16, 1–33 CrossRef PubMed.
- J. Lee and S. Bum Park, Pharmaceuticals, 2022, 15, 1478 CrossRef CAS PubMed.
- G. Kim, R. J. Grams and K.-L. Hsu, Chem. Rev., 2025, 125, 6653–6684 CrossRef CAS PubMed.
- L. Hillebrand and M. Gehringer, Chimia, 2022, 76, 435–447 CrossRef CAS PubMed.
- R. P. Bhole, G. O. Joshi, H. S. Kapare, R. V. Chikhale and S. Chaudhari, Results Chem., 2024, 8, 101615 CrossRef CAS.
- D. Bálint, Á. L. Póti, A. Alexa, P. Sok, K. Albert, L. Torda, D. Földesi-Nagy, D. Csókás, G. Turczel, T. Imre, E. Szarka, F. Fekete, I. Bento, M. Bojtár, R. Palkó, P. Szabó, K. Monostory, I. Pápai, T. Soós and A. Reményi, Nat. Commun., 2024, 15, 8606 CrossRef PubMed.
- Faridoon, J. Zheng, G. Zhang and J. J. Li, Future Med. Chem., 2025, 17, 389–392 CrossRef CAS PubMed.
- S. E. Dalton, O. Di Pietro and E. Hennessy, J. Med. Chem., 2025, 68, 2307–2313 CrossRef CAS PubMed.
- A. Tuley and W. Fast, Biochemistry, 2018, 57, 3326–3337 CrossRef CAS PubMed.
- D. Oksenberg, K. Dufu, M. P. Patel, C. Chuang, Z. Li, Q. Xu, A. Silva-Garcia, C. Zhou, A. Hutchaleelaha, L. Patskovska, Y. Patskovsky, S. C. Almo, U. Sinha, B. W. Metcalf and D. R. Archer, Br. J. Haematol., 2016, 175, 141–153 CrossRef CAS PubMed.
- P. A. Jackson, J. C. Widen, D. A. Harki and K. M. Brummond, J. Med. Chem., 2017, 60, 839–885 CrossRef CAS PubMed.
- P. Ábrányi-Balogh, L. Petri, T. Imre, P. Szijj, A. Scarpino, M. Hrast, A. Mitrović, U. P. Fonovič, K. Németh, H. Barreteau, D. I. Roper, K. Horváti, G. G. Ferenczy, J. Kos, J. Ilaš, S. Gobec and G. M. Keserű, Eur. J. Med. Chem., 2018, 160, 94–107 CrossRef PubMed.
- E. Mons, R. Q. Kim and M. P. C. Mulder, Pharmaceuticals, 2023, 16, 1–46 CrossRef PubMed.
- N. Csorba, P. Ábrányi-Balogh and G. M. Keserű, Trends Pharmacol. Sci., 2023, 44, 802–816 CrossRef CAS PubMed.
- L. Zheng, Y. Li, D. Wu, H. Xiao, S. Zheng, G. Wang and Q. Sun, MedComm: Oncol., 2023, 2, 1–54 Search PubMed.
- D. Basu, A. Richters and D. Rauh, Bioorg. Med. Chem., 2015, 23, 2767–2780 CrossRef CAS PubMed.
- A. Bandyopadhyay and J. Gao, Curr. Opin. Chem. Biol., 2016, 34, 110–116 CrossRef CAS PubMed.
- L. Hillebrand, X. J. Liang, R. A. M. Serafim and M. Gehringer, J. Med. Chem., 2024, 67, 7668–7758 CrossRef CAS PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed.
- D. Patel, Z. E. Huma and D. Duncan, ACS Chem. Biol., 2024, 19, 824–838 CrossRef CAS PubMed.
- J. M. Bradshaw, J. M. McFarland, V. O. Paavilainen, A. Bisconte, D. Tam, V. T. Phan, S. Romanov, D. Finkle, J. Shu, V. Patel, T. Ton, X. Li, D. G. Loughhead, P. A. Nunn, D. E. Karr, M. E. Gerritsen, J. O. Funk, T. D. Owens, E. Verner, K. A. Brameld, R. J. Hill, D. M. Goldstein and J. Taunton, Nat. Chem. Biol., 2015, 11, 525–531 CrossRef CAS PubMed.
- V. Oleinikovas, G. Saladino, B. P. Cossins and F. L. Gervasio, J. Am. Chem. Soc., 2016, 138, 14257–14263 CrossRef CAS PubMed.
- C. Aplin, S. K. Milano, K. A. Zielinski, L. Pollack and R. A. Cerione, J. Phys. Chem. B, 2022, 126, 6599–6607 CrossRef CAS PubMed.
- L. Maveyraud and L. Mourey, Molecules, 2020, 25, 1030 CrossRef CAS PubMed.
- K. Bum-Erdene, D. Liu, G. Gonzalez-Gutierrez, M. K. Ghozayel, D. Xu and S. O. Meroueh, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7131–7139 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. Von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS PubMed.
- K. McAulay, A. Bilsland and M. Bon, Pharmaceuticals, 2022, 15, 1–22 CrossRef PubMed.
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H. L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, J. Med. Chem., 2020, 63, 52–65 CrossRef CAS PubMed.
- T. A. Halgren, J. Chem. Inf. Model., 2009, 49, 377–389 CrossRef CAS PubMed.
- L. Alzyoud, R. A. Bryce, M. Al Sorkhy, N. Atatreh and M. A. Ghattas, Sci. Rep., 2022, 12, 7975 CrossRef CAS PubMed.
- R. Hansen, U. Peters, A. Babbar, Y. Chen, J. Feng, M. R. Janes, L. S. Li, P. Ren, Y. Liu and P. P. Zarrinkar, Nat. Struct. Mol. Biol., 2018, 25, 454–462 CrossRef CAS PubMed.
- D. Deredge, J. Li, K. A. Johnson and P. L. Wintrode, J. Biol. Chem., 2016, 291, 10078–10088 CrossRef CAS PubMed.
- R. Y.-C. Huang and G. Chen, Anal. Bioanal. Chem., 2014, 406, 6541–6558 CrossRef CAS PubMed.
- E. V. Petrotchenko, B. Novy, E. Nagy, K. I. Popov, J. B. Cross, R. Thapar and C. H. Borchers, Comput. Struct. Biotechnol. J., 2025, 27, 3618–3624 CrossRef CAS PubMed.
- V. Oleinikovas, G. Saladino, B. P. Cossins and F. L. Gervasio, J. Am. Chem. Soc., 2016, 138, 14257–14263 CrossRef CAS PubMed.
- A. Kuzmanic, G. R. Bowman, J. Juarez-Jimenez, J. Michel and F. L. Gervasio, Acc. Chem. Res., 2020, 53, 654–661 CrossRef CAS PubMed.
- N. Haloi, E. Karlsson, M. Delarue, R. J. Howard and E. Lindahl, Sci. Adv., 2025, 11, eadr0797 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, X. Li, J. Wu, L. Zhao, W. Li and J. Liu, J. Med. Chem., 2021, 64, 7575–7595 CrossRef CAS PubMed.
- W. Yu, S. Kumar, M. Zhao, D. J. Weber and A. D. J. MacKerell, J. Chem. Theory Comput., 2025, 21, 4964–4978 CrossRef CAS PubMed.
- R. C. Bernardi, M. C. R. Melo and K. Schulten, Biochim. Biophys. Acta, 2015, 1850, 872–877 CrossRef CAS PubMed.
- N. Vithani, S. Zhang, J. P. Thompson, L. A. Patel, A. Demidov, J. Xia, A. Balaeff, A. Mentes, Y. A. Arnautova, A. Kohlmann, J. D. Lawson, A. Nicholls, A. G. Skillman and D. N. LeBard, J. Chem. Inf. Model., 2024, 64, 8258–8273 CrossRef CAS PubMed.
- M. P. Bemelmans, Z. Cournia, K. L. Damm-ganamet, F. L. Gervasio and V. Pande, Curr. Opin. Struct. Biol., 2025, 90, 102975 CrossRef CAS PubMed.
- M. Ferraro, E. Moroni, E. Ippoliti, S. Rinaldi, C. Sanchez-Martin, A. Rasola, L. F. Pavarino and G. Colombo, J. Phys. Chem. B, 2021, 125, 101–114 CrossRef CAS PubMed.
- C. C. David and D. J. Jacobs, Methods Mol. Biol., 2014, 1084, 193–226 CrossRef CAS PubMed.
- R. Nussinov, M. Zhang, Y. Liu and H. Jang, Drug Discovery Today, 2023, 28, 103551 CrossRef CAS PubMed.
- A. Meller, S. De Oliveira, A. Davtyan, T. Abramyan, G. R. Bowman and H. van den Bedem, Front. Mol. Biosci., 2023, 10, 1171143 CrossRef CAS PubMed.
- C. Vénien-Bryan, Z. Li, L. Vuillard and J. A. Boutin, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2017, 73, 174–183 CrossRef PubMed.
- Y. Mao, Curr. Opin. Struct. Biol., 2025, 91, 103001 CrossRef CAS PubMed.
- C. M. Harris, S. E. Foley, E. R. Goedken, M. Michalak, S. Murdock and N. S. Wilson, SLAS Discovery, 2018, 23, 1040–1050 CrossRef CAS PubMed.
- M. Benabderrahmane, R. Bureau, A. S. Voisin-Chiret and J. Sopkova-De Oliveira Santos, J. Chem. Inf. Model., 2020, 60, 3172–3187 CrossRef CAS PubMed.
- (a) M. Groll, C. R. Berkers, H. L. Ploegh and H. Ovaa, Structure, 2006, 14, 451–456 CrossRef CAS PubMed; (b) M. Perbandt, N. Werner, A. Prester, H. Rohde, M. Aepfelbacher, W. Hinrichs and C. Betzel, Sci Rep., 2022, 12, 5510 Search PubMed.
- A. Glukhova, D. M. Thal, A. T. Nguyen, E. A. Vecchio, M. Jörg, P. J. Scammells, L. T. May, P. M. Sexton and A. Christopoulos, Cell, 2017, 168, 867–877.e13 CrossRef CAS PubMed.
- J. R. Porter, K. E. Moeder, C. A. Sibbald, M. I. Zimmerman, K. M. Hart, M. J. Greenberg and G. R. Bowman, Biophys. J., 2019, 116, 818–830 Search PubMed.
- Q. Hu, Y. Xiong, G. H. Zhu, Y. N. Zhang, Y. W. Zhang, P. Huang and G. B. Ge, MedComm, 2022, 3, 1–27 Search PubMed.
- M. Westberg, Y. Su, X. Zou, P. Huang, A. Rustagi, J. Garhyan, P. B. Patel, D. Fernandez, Y. Wu, C. Hao, C. W. Lo, M. Karim, L. Ning, A. Beck, P. Saenkham-Huntsinger, V. Tat, A. Drelich, B. H. Peng, S. Einav, C. T. K. Tseng, C. Blish and M. Z. Lin, Sci. Transl. Med., 2024, 16, 1–14 Search PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- R. Rungruangmaitree, S. Phoochaijaroen, A. Chimprasit, P. Saparpakorn, K. Pootanakit and D. Tanramluk, Sci. Rep., 2023, 13, 1–13 Search PubMed.
- R. P. Joshi, K. J. Schultz, J. W. Wilson, A. Kruel, R. A. Varikoti, C. J. Kombala, D. W. Kneller, S. Galanie, G. Phillips, Q. Zhang, L. Coates, J. Parvathareddy, S. Surendranathan, Y. Kong, A. Clyde, A. Ramanathan, C. B. Jonsson, K. R. Brandvold, M. Zhou, M. S. Head, A. Kovalevsky and N. Kumar, J. Chem. Inf. Model., 2023, 63, 1438–1453 Search PubMed.
- Z. Xia, M. Sacco, Y. Hu, C. Ma, X. Meng, F. Zhang, T. Szeto, Y. Xiang, Y. Chen and J. Wang, ACS Pharmacol. Transl. Sci., 2021, 4, 1408–1421 Search PubMed.
- S. Kwon, Front. Immunol., 2022, 13, 1–9 Search PubMed.
- X. Hu, J. li, M. Fu, X. Zhao and W. Wang, Signal Transduction Targeted Ther., 2021, 6, 402 Search PubMed.
- M. Forster, A. Chaikuad, S. M. Bauer, J. Holstein, M. B. Robers, C. R. Corona, M. Gehringer, E. Pfaffenrot, K. Ghoreschi, S. Knapp and S. A. Laufer, Cell Chem. Biol., 2016, 23, 1335–1340 Search PubMed.
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471–476 Search PubMed.
- M. Forster, A. Chaikuad, T. Dimitrov, E. Döring, J. Holstein, B. T. Berger, M. Gehringer, K. Ghoreschi, S. Müller, S. Knapp and S. A. Laufer, J. Med. Chem., 2018, 61, 5350–5366 CrossRef CAS PubMed.
- H. Lu, J. N. Iuliano and P. J. Tonge, Curr. Opin. Chem. Biol., 2018, 44, 101–109 CrossRef CAS PubMed.
- G. Casadevall, C. Duran and S. Osuna, JACS Au, 2023, 3, 1554–1562 CrossRef CAS PubMed.
- S. Martí, M. Roca, J. Andrés, V. Moliner, E. Silla, I. Tuñón and J. Bertrán, Chem. Soc. Rev., 2004, 33, 98–107 RSC.
- R. Yuan, J. Zhang, J. Zhou and Q. Cong, Mol. Ther., 2025, 33, 2252–2268 Search PubMed.
- J. Kale, E. J. Osterlund and D. W. Andrews, Cell Death Differ., 2018, 25, 65–80 CrossRef CAS PubMed.
- S. Qian, Z. Wei, W. Yang, J. Huang, Y. Yang and J. Wang, Front. Oncol., 2022, 12, 1–16 Search PubMed.
- C. M. Croce, D. Vaux, A. Strasser, J. T. Opferman, P. E. Czabotar and S. W. Fesik, Cell Death Differ., 2025, 32, 1369–1381 CrossRef CAS PubMed.
- J. L. Yap, L. Chen, M. E. Lanning and S. Fletcher, J. Med. Chem., 2017, 60, 821–838 Search PubMed.
- H. Chen, Z. Yan, C. Xiao, Z. Tang and Y. Wan, Eur. J. Med. Chem., 2025, 299, 118079 CrossRef CAS PubMed.
- S. Fletcher, Expert Opin. Ther. Pat., 2019, 29, 909–919 CrossRef CAS PubMed.
- S. Boiko, T. Proia, M. San Martin, G. P. Gregory, M. M. Wu, N. Aryal, M. Hattersley, W. Shao, J. C. Saeh, S. E. Fawell, R. W. Johnstone, L. Drew and J. Cidado, Blood, 2021, 137, 2947–2957 CrossRef CAS PubMed.
- G. Wang, S. T. Diepstraten and M. J. Herold, Biochem. Soc. Trans., 2022, 50, 1119–1128 CrossRef CAS PubMed.
- S. Dutta, T. S. Chen and A. E. Keating, ACS Chem. Biol., 2013, 8, 778–788 CrossRef CAS PubMed.
- A. J. Huhn, R. M. Guerra, E. P. Harvey, G. H. Bird and L. D. Walensky, Cell Chem. Biol., 2016, 23, 1123–1134 CrossRef CAS PubMed.
- A. Friberg, D. Vigil, B. Zhao, R. N. Daniels, J. P. Burke, P. M. Garcia-Barrantes, D. Camper, B. A. Chauder, T. Lee, E. T. Olejniczak and S. W. Fesik, J. Med. Chem., 2013, 56, 15–30 CrossRef CAS PubMed.
- E. P. Harvey, Z. J. Hauseman, D. T. Cohen, T. J. Rettenmaier, S. Lee, A. J. Huhn, T. E. Wales, H. S. Seo, J. Luccarelli, C. E. Newman, R. M. Guerra, G. H. Bird, S. Dhe-Paganon, J. R. Engen, J. A. Wells and L. D. Walensky, Cell Chem. Biol., 2020, 27, 647–656.e6 CrossRef CAS PubMed.
- D. A. Erlanson, A. C. Braisted, D. R. Raphael, M. Randal, R. M. Stroud, E. M. Gordon and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9367–9372 CrossRef CAS PubMed.
- E. P. Harvey, H. S. Seo, R. M. Guerra, G. H. Bird, S. Dhe-Paganon and L. D. Walensky, Structure, 2018, 26, 153–160.e4 Search PubMed.
- S. Acoca, Q. Cui, G. C. Shore and E. O. Purisima, Proteins: Struct., Funct. Bioinf., 2011, 79, 2624–2636 CrossRef CAS PubMed.
- S. C. C. Lucas, A. G. Milbradt, J. H. Blackwell, S. Bonomo, A. Brierley, D. J. Cassar, J. Freeman, T. E. Hadfield, L. A. Morrill, R. Riemens, S. Sarda, S. Schiesser, D. Wiktelius, S. Ahmed, M. J. Bostock, U. Börjesson, C. De Fusco, C. Guerot, D. Hargreaves, S. Hewitt, M. L. Lamb, N. Su, R. Whatling, M. Wheeler and J. G. Kettle, J. Med. Chem., 2024, 67, 11209–11225 CrossRef CAS PubMed.
- S. C. C. Lucas, J. H. Blackwell, U. Börjesson, D. Hargreaves, A. G. Milbradt, M. J. Bostock, S. Ahmed, K. Beaumont, T. Cheung, S. Demanze, A. Gohlke, C. Guerot, A. Haider, V. Kantae, G. W. Kauffman, O. Kinzel, L. Kupcova, M. D. Lainchbury, M. L. Lamb, L. Leon, A. Palisse, C. Sacchetto, R. I. Storer, N. Su, C. Thomson, J. Vales, Y. Chen and X. Hu, J. Med. Chem., 2024, 67, 16455–16479 CrossRef CAS PubMed.
- L. Huang, Z. Guo, F. Wang and L. Fu, Signal Transduction Targeted Ther., 2021, 6, 1–20 CrossRef PubMed.
- A. Singhal, B. T. Li and E. M. O’Reilly, Nat. Med., 2024, 30, 969–983 CrossRef CAS PubMed.
- H. Chen, J. B. Smaill, T. Liu, K. Ding and X. Lu, J. Med. Chem., 2020, 63, 14404–14424 CrossRef CAS PubMed.
- S. Lu, H. Jang, S. Muratcioglu, A. Gursoy, O. Keskin, R. Nussinov and J. Zhang, Chem. Rev., 2016, 116, 6607–6665 CrossRef CAS PubMed.
- L. Qunaj, M. S. May, A. I. Neugut and B. O. Herzberg, Front. Oncol., 2023, 13, 1–8 Search PubMed.
- W. Ning, Z. Yang, G. J. Kocher, P. Dorn and R. W. Peng, Cancers, 2022, 14, 1–12 Search PubMed.
- M. P. Patricelli, M. R. Janes, L. S. Li, R. Hansen, U. Peters, L. V. Kessler, Y. Chen, J. M. Kucharski, J. Feng, T. Ely, J. H. Chen, S. J. Firdaus, A. Babbar, P. Ren and Y. Liu, Cancer Discovery, 2016, 6, 316–329 CrossRef CAS PubMed.
- M. R. Janes, J. Zhang, L. S. Li, R. Hansen, U. Peters, X. Guo, Y. Chen, A. Babbar, S. J. Firdaus, L. Darjania, J. Feng, J. H. Chen, S. Li, S. Li, Y. O. Long, C. Thach, Y. Liu, A. Zarieh, T. Ely, J. M. Kucharski, L. V. Kessler, T. Wu, K. Yu, Y. Wang, Y. Yao, X. Deng, P. P. Zarrinkar, D. Brehmer, D. Dhanak, M. V. Lorenzi, D. Hu-Lowe, M. P. Patricelli, P. Ren and Y. Liu, Cell, 2018, 172, 578–589.e17 Search PubMed.
- J. Canon, K. Rex, A. Y. Saiki, C. Mohr, K. Cooke, D. Bagal, K. Gaida, T. Holt, C. G. Knutson, N. Koppada, B. A. Lanman, J. Werner, A. S. Rapaport, T. San Miguel, R. Ortiz, T. Osgood, J. R. Sun, X. Zhu, J. D. McCarter, L. P. Volak, B. E. Houk, M. G. Fakih, B. H. O’Neil, T. J. Price, G. S. Falchook, J. Desai, J. Kuo, R. Govindan, D. S. Hong, W. Ouyang, H. Henary, T. Arvedson, V. J. Cee and J. R. Lipford, Nature, 2019, 575, 217–223 CrossRef CAS PubMed.
- J. Montero and A. Letai, Cell Death Differ., 2018, 25, 56–64 CrossRef CAS PubMed.
- I. Ploumaki, E. Triantafyllou, I. A. Koumprentziotis, K. Karampinos, K. Drougkas, I. Karavolias, I. Trontzas and E. A. Kotteas, Clin. Transl. Oncol., 2023, 25, 1554–1578 CrossRef CAS PubMed.
- C. L. Day, L. Chen, S. J. Richardson, P. J. Harrison, D. C. S. Huang and M. G. Hinds, J. Biol. Chem., 2005, 280, 4738–4744 CrossRef CAS PubMed.
- F. Ong, K. Kim and M. Y. Konopleva, Cancer Drug Resist., 2022, 5, 380–400 CrossRef CAS PubMed.
- H. Wang, M. Guo, H. Wei and Y. Chen, J. Hematol. Oncol., 2021, 14, 1–18 Search PubMed.
- S. I. Tantawy, N. Timofeeva, A. Sarkar and V. Gandhi, Front. Oncol., 2023, 13, 1–19 Search PubMed.
- S. Lee, T. E. Wales, S. Escudero, D. T. Cohen, J. Luccarelli, C. G. Gallagher, N. A. Cohen, A. J. Huhn, G. H. Bird, J. R. Engen and L. D. Walensky, Nat. Struct. Mol. Biol., 2016, 23, 600–607 CrossRef CAS PubMed.
- N. A. Cohen, M. L. Stewart, E. Gavathiotis, J. L. Tepper, S. R. Bruekner, B. Koss, J. T. Opferman and L. D. Walensky, Chem. Biol., 2012, 19, 1175–1186 CrossRef CAS PubMed.
- E. F. Lee, P. E. Czabotar, H. Yang, B. E. Sleebs, G. Lessene, P. M. Colman, B. J. Smith and W. D. Fairlie, J. Biol. Chem., 2009, 284, 30508–30517 CrossRef CAS PubMed.
- B. D. Manning and A. Toker, Cell, 2017, 169, 381–405 CrossRef CAS PubMed.
- Y. He, M. M. Sun, G. G. Zhang, J. Yang, K. S. Chen, W. W. Xu and B. Li, Signal Transduction Targeted Ther., 2021, 6, 425 CrossRef CAS PubMed.
- J. R. Testa and A. Bellacosa, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10983–10985 CrossRef CAS PubMed.
- M. Sun, G. Wang, J. E. Paciga, R. I. Feldman, Z. Q. Yuan, X. L. Ma, S. A. Shelley, R. Jove, P. N. Tsichlis, S. V. Nicosia and J. Q. Cheng, Am. J. Pathol., 2001, 159, 431–437 CrossRef CAS PubMed.
- H. Hua, H. Zhang, J. Chen, J. Wang, J. Liu and Y. Jiang, J. Hematol. Oncol., 2021, 14, 1–25 Search PubMed.
- T. Adon, S. Bhattacharya, S. R. V. Madhunapantula and H. Y. Kumar, Eur. J. Med. Chem., 2025, 287, 117334 CrossRef CAS PubMed.
- J. D. Carpten, A. L. Faber, C. Horn, G. P. Donoho, S. L. Briggs, C. M. Robbins, G. Hostetter, S. Boguslawski, T. Y. Moses, S. Savage, M. Uhlik, A. Lin, J. Du, Y. W. Qian, D. J. Zeckner, G. Tucker-Kellogg, J. Touchman, K. Patel, S. Mousses, M. Bittner, R. Schevitz, M. H. T. Lai, K. L. Blanchard and J. E. Thomas, Nature, 2007, 448, 439–444 CrossRef CAS PubMed.
- G. B. Craven, H. Chu, J. D. Sun, J. D. Carelli, B. Coyne, H. Chen, Y. Chen, X. Ma, S. Das, W. Kong, A. D. Zajdlik, K. S. Yang, S. H. Reisberg, P. A. Thompson, J. R. Lipford and J. Taunton, Nature, 2025, 637, 205–214 CrossRef CAS PubMed.
- K. A. Pervanidis, G. D. D’Angelo, J. Weisner, S. Brandherm and D. Rauh, J. Med. Chem., 2024, 67, 6052–6063 CrossRef CAS PubMed.
- J. Weisner, R. Gontla, L. Van der Westhuizen, S. Oeck, J. Ketzer, P. Janning, A. Richters, T. Mühlenberg, Z. Fang, A. Taher, V. Jendrossek, S. C. Pelly, S. Bauer, W. A. L. Van Otterlo and D. Rauh, Angew. Chem., Int. Ed., 2015, 54, 10313–10316 CrossRef CAS PubMed.
- J. Weisner, I. Landel, C. Reintjes, N. Uhlenbrock, M. Trajkovic-Arsic, N. Dienstbier, J. Hardick, S. Ladigan, M. Lindemann, S. Smith, L. Quambusch, R. Scheinpflug, L. Depta, R. Gontla, A. Unger, H. Muller, M. Baumann, C. Schultz-Fademrecht, G. Gunther, A. Maghnouj, M. P. Muller, M. Pohl, C. Teschendorf, H. Wolters, R. Viebahn, A. Tannapfel, W. Uhl, J. G. Hengstler, S. A. Hahn, J. T. Siveke and D. Rauh, Cancer Res., 2019, 79, 2367–2378 CrossRef CAS PubMed.
- N. Uhlenbrock, S. Smith, J. Weisner, I. Landel, M. Lindemann, T. A. Le, J. Hardick, R. Gontla, R. Scheinpflug, P. Czodrowski, P. Janning, L. Depta, L. Quambusch, M. P. Müller, B. Engels and D. Rauh, Chem. Sci., 2019, 10, 3573–3585 RSC.
- A. J. T. Smith, X. Zhang, A. G. Leach and K. N. Houk, J. Med. Chem., 2009, 52, 225–233 CrossRef CAS PubMed.
- Z. Ma, A. A. Bolinger and J. Zhou, Drug Discovery Today, 2023, 28, 103774 CrossRef CAS PubMed.
- K. Raina, C. D. Forbes, R. Stronk, J. P. Rappi, K. J. Eastman, N. Zaware, X. Yu, H. Li, A. Bhardwaj, S. W. Gerritz, M. Forgione, A. Hundt, M. P. King, Z. M. Posner, A. D. Correia, A. McGovern, D. E. Puleo, R. Chenard, J. J. Mousseau, J. I. Vergara, E. Garvin, J. Macaluso, M. Martin, K. Bassoli, K. Jones, M. Garcia, K. Howard, M. Yaggi, L. M. Smith, J. M. Chen, A. B. Mayfield, C. A. De Leon, J. Hines, K. J. Kayser-Bricker and C. M. Crews, Cell Chem. Biol., 2024, 31, 1490–1502.e42 CrossRef CAS PubMed.
- D. Lu, X. Yu, H. Lin, R. Cheng, E. Y. Monroy, X. Qi, M. C. Wang and J. Wang, Chem. Soc. Rev., 2022, 51, 9243–9261 RSC.
- W. H. Guo, X. Qi, X. Yu, Y. Liu, C. I. Chung, F. Bai, X. Lin, D. Lu, L. Wang, J. Chen, L. H. Su, K. J. Nomie, F. Li, M. C. Wang, X. Shu, J. N. Onuchic, J. A. Woyach, M. L. Wang and J. Wang, Nat. Commun., 2020, 11, 1–16 Search PubMed.
- M. Yuan, Y. Chu and Y. Duan, Front. Chem., 2021, 9, 1–5 Search PubMed.
- M. Rask-Andersen, S. Masuram and H. B. Schiöth, Annu. Rev. Pharmacol. Toxicol., 2014, 54, 9–26 CrossRef CAS PubMed.
- A. Clyde, S. Galanie, D. W. Kneller, H. Ma, Y. Babuji, B. Blaiszik, A. Brace, T. Brettin, K. Chard, R. Chard, L. Coates, I. Foster, D. Hauner, V. Kertesz, N. Kumar, H. Lee, Z. Li, A. Merzky, J. G. Schmidt, L. Tan, M. Titov, A. Trifan, M. Turilli, H. Van Dam, S. C. Chennubhotla, S. Jha, A. Kovalevsky, A. Ramanathan, M. S. Head and R. Stevens, J. Chem. Inf. Model., 2022, 62, 116–128 CrossRef CAS PubMed.
- N. H. Kazi, N. Klink, K. Gallant, G.-M. Kipka and M. Gersch, Nat. Struct. Mol. Biol., 2025, 32, 1776–1786 CrossRef CAS PubMed.
- A. E. Tron, M. A. Belmonte, A. Adam, B. M. Aquila, L. H. Boise, E. Chiarparin, J. Cidado, K. J. Embrey, E. Gangl, F. D. Gibbons, G. P. Gregory, D. Hargreaves, J. A. Hendricks, J. W. Johannes, R. W. Johnstone, S. L. Kazmirski, J. G. Kettle, M. L. Lamb, S. M. Matulis, A. K. Nooka, M. J. Packer, B. Peng, P. B. Rawlins, D. W. Robbins, A. G. Schuller, N. Su, W. Yang, Q. Ye, X. Zheng, J. P. Secrist, E. A. Clark, D. M. Wilson, S. E. Fawell and A. W. Hird, Nat. Commun., 2018, 9, 5341 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |