
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in the delivery of peptide nucleic acids (PNAs)
Srijani
Sarkar
 and
Niren
Murthy
*
and
Niren
Murthy
*
Department of Bioengineering, Innovative Genomics Institute, University of California Berkeley, Berkeley, CA, USA. E-mail: nmurthy@berkeley.edu
First published on 22nd January 2026
Abstract
Peptide nucleic acid (PNA) oligomers have tremendous potential as therapeutics; however, their delivery is challenging and has limited their development as therapeutics. In recent years, new strategies for delivering water-soluble backbone-modified PNA oligomers into cells for antisense and gene-editing applications have attracted significant attention. This review critically examines earlier delivery approaches and their limitations, highlights recent advances in PNA engineering and nanocarrier design, and discusses the future directions necessary to advance PNA-based therapeutics. By integrating these innovations, PNAs hold the potential to transform biomedical applications and contribute to the next generation of medicine.
1. Introduction
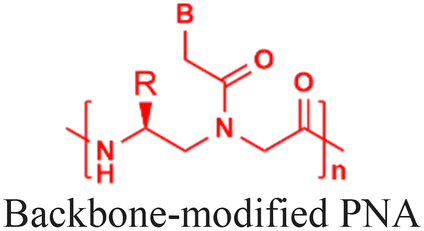
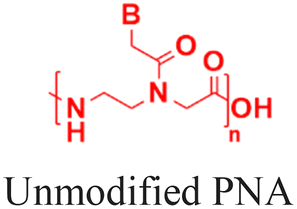
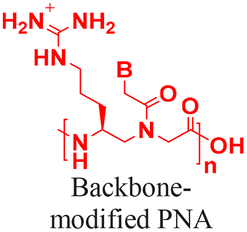
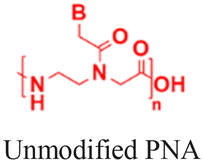
The discovery of antisense oligonucleotides has revolutionized treatment strategies for genetic disorders by inhibiting aberrant protein production of disease-causing messenger RNA (mRNA), noncoding RNA (ncRNA), and microRNA (miRNA).1,2 The antisense oligonucleotides primarily inhibit gene expression by high-affinity and sequence-specific binding to the target gene sequence. Among all the chemical modifications developed to improve oligonucleotide properties (Fig. 1A), peptide nucleic acids, which combine pseudopeptide backbones with nucleobases (Fig. 1B), have emerged as promising constructs and were first reported three decades ago.3 The PNA backbone consists of N-(2-aminoethyl)glycine units linked to nucleobases (adenine or A, thymine or T, guanine or G and cytosine or C) via methylene carbonyl bridges, replacing conventional sugar-phosphate backbones (Fig. 1B).3,4 Since their discovery, PNAs have drawn significant interest because they combine the best chemical features of peptides and nucleic acids and have demonstrated the ability to selectively inhibit gene expression by blocking the interaction of transcription and translation factors, ribosomes, and RNA polymerase with the target sequences (Fig. 1C).5–7 PNA monomer design offers several advantages; key structural benefits are discussed below (Fig. 1D–F). The neutral backbone eliminates electrostatic repulsion between complementary strands, increasing the binding affinity and stability of PNA–DNA and PNA–RNA duplexes (Fig. 1D). The pseudopeptide backbone is resistant to nucleases and proteases and is much more stable in biological fluids than RNA or DNA (Fig. 1E). Finally, PNA monomers are commercially available with Fmoc-protected amines and Boc/Bhoc-protected side chains, making them compatible with standard solid-phase peptide synthesis (Fig. 1F), enabling straightforward oligomer synthesis and easy conjugation with peptides, fluorophores, and lipids to improve properties such as solubility, cellular uptake, and endosomal escape. | ||
| Fig. 1 Structural and functional advantages of peptide nucleic acids (PNAs) compared to other antisense oligonucleotides. (A) Chemical structures of different types of antisense oligonucleotides; (B) chemical structure of the PNA monomer; (C) cartoon representation of inhibiting gene expression by antisense PNAs by acting as a functional blockage for proteins like transcription/translation factors; advantages of PNAs as antisense oligonucleotides for their (D) high-affinity binding ability to target sequences due to neutral backbones, (E) serum stability due to pseudo-peptide nature of backbones, and (F) compatibility with solid phase peptide synthesis. | ||
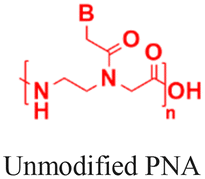
PNAs offer several advantages over traditional antisense oligonucleotides. For example, siRNAs possess a higher molecular weight (4–15 kDa) and negatively charged backbones, whereas PNAs typically have a molecular weight of only 2–4 kDa and neutral and hydrophobic backbones. Again, oligonucleotides with phosphorothioate (PS) or phosphorodiamidate (PMO) backbones need to be larger than PNAs due to their lower binding affinity. Additionally, PS oligonucleotides increase the risk of RNase H-mediated degradation at off-target binding sites. In contrast, PNAs exhibit high binding selectivity and have a lower likelihood of adverse effects from off-target binding.8,9 Numerous studies have established that PNAs can target canonical and non-canonical nucleic acid structures, such as duplexes, quadruplexes, i-motifs, and aptamers.10–15 More importantly, PNAs can invade double-stranded DNA to inhibit transcription and can trigger gene editing. Recent reports on backbone and nucleobase modifications have improved water solubility and binding interactions with target sequences.16–20 Notably, gamma-substituted hydrophilic miniPEG-modified PNAs have demonstrated the ability to invade double-stranded DNA, expanding gene-editing applications across various disease models.8,9,21 Moreover, PNAs have been utilized in the preparation of self-assembled nanostructures for multiple applications, serving as electrochemical biosensors for cancer diagnosis,22 replacement for crosslinked polymers in organic electronic devices,23–27 supramolecular anticoagulants for drug delivery,28 immunogenic triggers,29 and crosslinkers in peptides and polymer hydrogels to increase their stiffness.30–35
Although PNAs demonstrated efficacy in several therapeutic applications under cell-free conditions and in vivo, robust strategies for delivering them are still needed. Currently, thirteen antisense oligonucleotide drugs are approved by the United States Food and Drug Administration (FDA), including nine single-stranded antisense oligonucleotides and four siRNAs.36,37 However, despite the tremendous potential of PNAs as new therapeutics, no PNA-based therapeutics have received FDA approval to date, primarily because delivery remains a significant barrier.38 This review summarizes the historical delivery approaches and their limitations. Additionally, it highlights the latest advances in chemical modifications and nanotechnology-based delivery systems that aim to overcome these obstacles and expand the therapeutic potential of PNAs.
2. Challenges of delivering PNAs into cells
PNA-based therapeutics are limited by poor intracellular delivery and rapid systemic clearance (circulation half-life ≈ 3 minutes), partly due to low membrane permeability and fast renal elimination.39 A broad range of delivery approaches have been explored to address this issue, including physical methods (electroporation, nucleofection, microinjection, and co-transfection), conjugation to cell-penetrating peptides (CPPs), chemical modifications of the backbone, and encapsulation in nanoparticle carriers (Fig. 2). | ||
| Fig. 2 Delivery modalities implemented for PNAs. (A) Common strategies to deliver PNAs into cells: (B) one of the most well-studied strategies is to conjugate PNAs to cell-penetrating peptides (CPPs), which are internalized via endocytosis. | ||
However, many traditional approaches are unsuitable for in vivo applications because of poor PK/PD profiles, toxicity, or limited efficacy. For example, CPP conjugation increases uptake but typically requires high doses because most PNA–peptide conjugates are trapped in endosomes, with only a small fraction reaching the cytosol, which raises concerns about nonspecific interactions and toxicity. Co-delivery with endosome disruptors (e.g., chloroquine or Ca2+) can enhance cytosolic release but may impair cell viability. Again, liposomal delivery often demands additional modifications, such as charged peptides or complementary DNA, to efficiently load PNAs. Recent polymeric nanoparticle approaches offer improved delivery, but they generally lack tissue or cell-type specificity and tend to accumulate in the liver, thereby increasing the risk of hepatotoxicity. Ongoing nanoparticle optimization aims to increase PNA loading and achieve targeted delivery, representing a major advance for the field. Examples of vectors for different PNA oligonucleotides are shown in Table 1 (see Section 3 for a detailed description).
| Serial number | PNA backbone | Delivery platforms | Uptake mechanism | Treated in vivo/in vitro |
|---|---|---|---|---|
| 1 [ref. 16, 19 and 51] |
|
|
Endocytosis | In vitro cell assays |
| 2 [ref. 64, 67, 73 and 74] |

|
|
Endocytosis | In vitro and in vivo assays |
| 3 [ref. 78 and 79] |
|
|
Receptor mediated uptake | In vitro and in vivo assays |
| 4 [ref. 6, 8, 84, 86 and 89] |
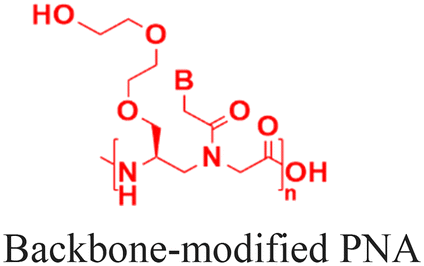
|
|
Endocytosis | In vitro and in vivo assays |
| 5 [ref. 100] |
|

|
Endocytosis | In vitro cell assays |
| 6 [ref. 102] |
|

|
Endocytosis | In vitro cell assays |
3. Classifications of different delivery strategies
In the following section, popularly applied delivery methods for PNAs are discussed separately based on their delivery mechanisms.3.1. Mechanical and electrical transduction
Mechanical or electrical transduction methods can efficiently deliver various biomolecules into cells. These methods disrupt cell membranes and generate transient nanopores that permeabilize the cell to exogenous molecules.40 Hanvey et al. used the microinjection method to deliver PNAs into fibroblast cells and demonstrated their antisense and antigene properties.41 The microinjection method directly injects molecules into living cells by perforating cell membranes with the small, sharp tips of microneedles, which can physically damage cell membranes and cause toxicity. Again, the electroporation method requires an external electric field for membrane permeabilization, and delivery efficacy depends on the electric field parameters, including voltage, electrode type, and other external factors. The electroporation method has been used to deliver PNAs into cells for inducing alternative splicing,42 promoting endogenous gene expression,43 and inhibiting miRNA.44 However, neither microinjection nor electroporation-based methods are suitable for in vivo applications because of three reasons: (1) high toxicity due to irreversible damage to the cell membrane, (2) nonuniform transfection into cells, and (3) the requirement of an external electric field. Although these techniques are incompatible with clinical studies, earlier applications of these delivery methods have helped advancing the PNA field by providing proof of principle of efficacy in cells.3.2. Chemical modifications of the PNA backbone
Several studies have demonstrated that incorporating chemical modification either on the backbone or at the amine or carboxyl terminus can dramatically enhance the delivery of PNAs into cells. For example, conjugation of a streptolysin O moiety to a PNA inhibited the expression of the targeted supFG1 reporter gene 10-fold higher than the control PNA without streptolysin O conjugation.45 Lipophilic moieties such as adamantyl acetic acid (Fig. 3A),46 triphenyl phosphonium cations (Fig. 3B),47 and cholesterol or cholic acid (Fig. 3C)48 attached to the N or C terminal have also successfully delivered PNAs in cells. Gabas and Nielsen conjugated dendrons composed of amino- and guanidino-terminating 2,4-diaminobutanoic acid (Dab) to PNAs and reported successful delivery of dendron-PNAs into HeLa cells via endocytosis and demonstrated their ability to perform splice correction (Fig. 3D).49 Another approach by Hamilton et al. reported lipofection of the PNA/DNA complex to deliver antisense PNAs into cells, which successfully inhibited telomerase activity.50 In the lipofection strategy, the PNA was hybridized with a complementary DNA strand to promote complexation with cationic lipids.50 | ||
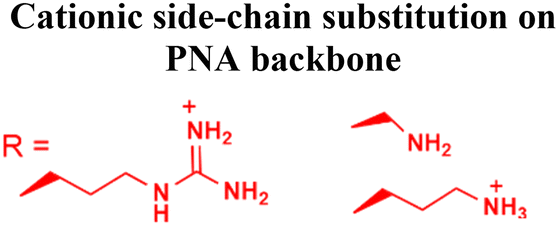
| Fig. 3 Different types of backbone modifications on PNAs to aid delivery into cells. In some cases, lipophilic transporter molecules were attached to the terminal of the PNA backbone like (A) adamantyl acetic acid, (B) triphenylphosphonium cations, (C) diaminobutanoic acid dendrons, and (D) cholic acids. On the other hand, in some cases, different cationic side chains were substituted at the alpha or gamma positions on the PNA backbone, such as (E) aminomethylene, (F) aminopropylene, (G) ethyleneguanidino, (H) arginine side chain, and (I) tetrahydrofuran. | ||
In addition to attaching cationic or hydrophobic groups to the amine or carboxy terminus of the PNA, these groups have also been installed on the side chains of the PNA backbone. Ganesh and coworkers reported a series of cationic amine-substituted backbone-modified PNAs, including aminomethylene-, aminopropylene-, ethyleneamino-, and ethyleneguanidino-PNAs (Fig. 3E-G).19,51,52 Additionally, Ly and coworkers incorporated a guanidinium side chain at the alpha or gamma position of the PNA backbone (Fig. 3H).16,17,53 The gamma-substituted guanidium side chain containing PNAs (GPNAs), reported by Dragulescu-Andrasi et al., showed favorable cellular uptake properties and hybridization to the transcriptional start site of the E-cadherin gene, resulting in sequence-specific antisense effects.54 The GPNAs were less toxic than PNA–polyarginine conjugates, most likely due to the higher amphipathic nature of the PNA–polyarginine conjugates that disrupted cell membranes.54 Another modification introduced by the Appella group replaced the ethylenediamine units on the PNA backbone with cyclic tetrahydrofurans (Fig. 3I).55,56 They also added one lysine at each terminal to increase water solubility. The authors referred to this type of modified PNAs as Lys-thyclotides. The tetrahydrofuran-based backbone led to >50% improvement in cell uptake compared with an unmodified PNA across four cell lines.55 Live cell imaging revealed no colocalization between fluorescein-labeled thyclotide and endosomes, suggesting that thyclotides entered cells via an uptake mechanism different from endocytosis.55 The non-endosomal mechanism of uptake makes this design attractive, but future work will be required to understand the benefits of this backbone modification better.
Although the results obtained with backbone-modified PNAs are exciting, the main drawback of this strategy is the complex chemical synthetic procedures required to make PNA monomers, which could be a significant barrier to scalability and cost-effectiveness in a clinical setting. In addition, positively charged side chains can interact nonspecifically with DNA/RNAs in cells, leading to toxicity.
3.3. PNA–peptide conjugates
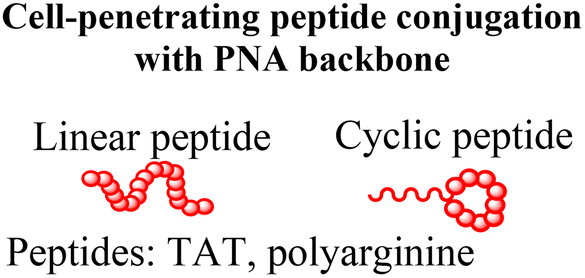
Conjugation of cell-penetrating peptides (CPPs) into PNAs significantly enhances cellular delivery and has been utilized for gene knockdown, miRNA inhibition, splice correction, and antibacterial/antimicrobial/antiviral applications.57–63 Reports include conjugation of cationic peptides (polylysine and polyarginine),64–66 cell-penetrating peptides (TAT, penetratin, and transportan),67–69 nuclear localization peptides,70 and endosomolytic peptides71 into PNAs. A few of the latest reports in this research context are discussed below.A cytosol-localizing internalization peptide (CLIP6) has delivered a splice-switching PNA into glioblastoma cells via a non-endosomal mechanism and modulated splicing in the oncogenic Mnk2 gene.58 Compared to linear cell-penetrating peptides, analogous cyclic peptides show higher cell uptake, miRNA inhibition, and cell apoptosis.59 Additionally, macrocyclic multivalent tetraargininocalix[4]arenes that interact with PNAs noncovalently (via hydrogen bonding and π–π and cation–π interactions) have also been reported to promote cell uptake and inhibit miRNA with efficacy comparable to PNA–polyarginine conjugates.72
Tumor-targeted delivery has been achieved by conjugating a pH-low insertion peptide (pHLIP) into PNAs and backbone-modified PNAs (γPNA) via a disulfide linkage (Fig. 4A). The pHLIP peptide ensures tumor-selective delivery by undergoing a conformational change in the acidic tumor microenvironment, enabling lipid bilayer invasion of tumor cells. Once inside cells, the disulfide linkage is reduced, making the PNA available for silencing miRNAs and inducing apoptosis in cancer cells (Fig. 4A).60,73 Another exciting approach for the cytosolic delivery of PNAs involves constructing a hairpin structure with a cyclized TAT peptide as the loop domain and two short complementary γPNAs (4-mer) as the stem domain (Fig. 4B).74,75 The hairpin conformation imposes a quasi-circular constraint on the TAT peptide, thereby enhancing uptake. Interestingly, cyclized TAT(FAM)-γPNA was reported to escape endosomes more efficiently than the linear TAT(FAM)-γPNA after photoactivation by LED illumination at 490 nm (Fig. 4B). Furthermore, this hairpin construct was used as a carrier molecule by hybridizing the overhang at the stem domain with an additional 13-nucleotide-long antitelomerase γPNA, which effectively inhibited telomerase activity (90% inhibition) by blocking the complexation between telomerase RNA and the TERT protein in cell culture (Fig. 4B).75 Finally, Hakata and Kitamatsu reported delivery of an autophagy-induced peptide (AIP) covalently conjugated with a PNA–PNA duplex, where one of the two PNAs was attached to polyarginine (R8) via a disulfide (S–S) bond.76 The authors demonstrated that the PNA–PNA duplex could induce autophagy more effectively than direct attachment of the AIP to polyarginine.76 Collectively, these approaches demonstrate that peptide conjugation substantially enhances PNA delivery, with some examples overcoming endosomal entrapment and ensuring tumor tissue-specific delivery.
 | ||
| Fig. 4 Latest designs of PNA–peptide conjugates for cell-specific delivery and overcoming endosomal entrapment. (A) Tumor-specific delivery of a pHLIP conjugated PNA that releases the antisense PNA by cleaving the connecting S–S bond in a reducing environment in cells, which can induce cell apoptosis. (B) Cyclization of the TAT peptide by a PNA–PNA duplex that contains an overhang sequence binding to an antisense PNA. The entire molecular construct is internalized into cells via endocytosis, demonstrating photoirradiation-mediated escape from the endosome and inhibition of telomerase activity. | ||
3.4. Receptor-mediated cell delivery
Receptor-mediated uptake enables cell-type selective uptake of PNAs. Bhingardev and Ganesh reported the delivery of N-acetyl galactosamine (GalNac)-conjugated PNAs to mouse livers via the binding of GalNac to the asialoglycoprotein receptor on hepatocytes (Fig. 5A).77 In this study, the authors have designed two different molecular constructs conjugating a 15-mer PNA either to (1) a triantennary GalNAc (GalNAc3) at the N-terminus or (2) three consecutive backbone-modified PNA monomers containing a single GalNAc at their gamma positions (Fig. 5A).77 The GalNAc modification was 40 times more potent at triggering cell internalization than the analogous PNA–(Lys)2 conjugates into HepG2 cells.77 Furthermore, Kumar and Bahal demonstrated that conjugation of a divalent lactobionic acid (diLBA) or GalNac selectively translocated PNAs into hepatocytes after systemic administration in mice.78 They showed that both PNA–diLBA and PNA–GalNAc conjugates predominantly localized (∼90%) in hepatocytes with slightly higher accumulation in the case of GalNAc than diLBA conjugates, irrespective of concentrations, time, and length of the PNA.78 | ||
| Fig. 5 Receptor-mediated uptake of antisense PNAs. (A) PNAs conjugated with N-acetyl galactosamines (GalNAc) bind to the asialoglycoprotein receptor on the cell surface and promote delivery in hepatocytes. (B) The binding of anthrax toxin PA63 to the receptors on the cell surface facilitates the endocytosis of PNA–LFN into cells. Inside endosomes, PA63 promotes conformational rearrangements that form ion-conductive pores, through which PNA–LFN conjugates could translocate into the cytoplasm. | ||
The Pentelute research group has demonstrated PNA delivery using the anthrax-mediated cell uptake mechanism (Fig. 5B).79 This uptake mechanism depends on the interaction between protective antigen (PA) and lethal factor (LFN). In this strategy, the authors conjugated PNAs to the C-terminus of the 263-residue domain of LFN protein via sortase-mediated ligation (PNA–LFN) and incubated cells in the presence of PA to evaluate translocation efficiency. This approach relies on two factors: (1) the binding of anthrax toxin PA83 to its receptors on cell membranes followed by proteolytic cleavage to PA63, which self-assembles to form an octamer and triggers the endocytosis of PNA–LFN into cells, and (2) inside endosomes, PA63 undergoes acid promoted conformational rearrangements to form ion conductive pores, from which PNA–LFN conjugates translocated into the cytoplasm (Fig. 5B).79 This design enables dose-dependent knockdown of the SF3B1 gene at nanomolar concentrations of antisense PNAs across multiple cancer lines.79 Moreover, this technology showed cancer cell-specific gene silencing when PA was engineered to target HER2 (human epidermal growth factor receptor 2), which was overexpressed on cancer cells.79
PNAs have also been delivered into bacteria via targeting membrane transporters. Pienko and Trylska demonstrated that a 14-mer PNA covalently attached to vitamin B12 can enter E. coli cells by hijacking the B12 transport mechanism.80 The interaction between B12 and the B12-specific BtuB receptor on the bacterial outer membrane promoted the cell permeation of PNAs.80 In another study, Pals and Velema used the siderophore-mediated iron uptake pathway to deliver an antisense PNA targeting the acpP gene in E. coli.81 They conjugated an antisense PNA to a tris-catechol siderophore via a click reaction, which was internalized via the iron transport pathway and showed a significant antibacterial effect at a low micromolar concentration.81 The uptake was confirmed to occur exclusively via the iron transport pathway, as E. coli mutants with mutations in iron uptake factors were resistant to the PNA–tris-catechol siderophore conjugate.81 Collectively, targeting transported pathways unique to bacteria is a promising strategy for selective antibacterial applications of PNAs.
3.5. Delivery via nano-assemblies
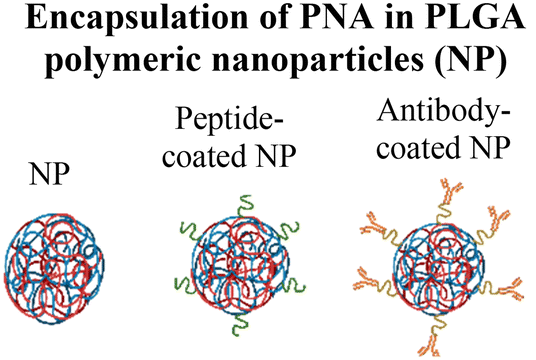
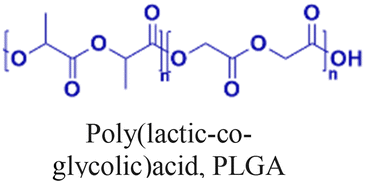
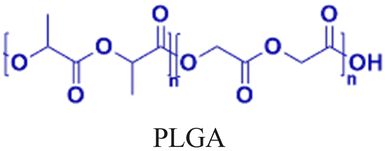
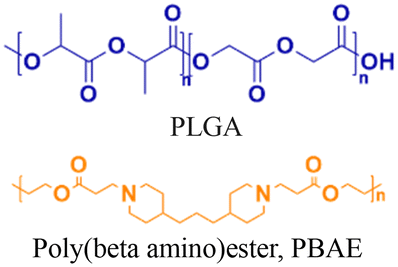
Nanoparticle delivery of both unmodified and backbone-modified PNAs has shown strong potential for in vivo live-cell imaging, targeted gene silencing, and gene editing applications.82,83 The composition of a nanoparticle strongly affects cellular uptake, endosomal escape, and nuclear localization, all of which are key determinants of therapeutic efficacy. To optimize these properties, a variety of structural modifications have been explored. For example, coating nanoparticles with cell-penetrating peptides enhances interactions with the cell membrane and increases uptake (Fig. 6A).84 Hybrid polymeric formulations, such as blends of poly(lactic-co-glycolic acid) (PLGA) with cationic polymers like poly(beta-amino-ester) (PBAE) or poly-histidine, improve endosomal release via the proton sponge effect, thereby increasing intracellular bioavailability and therapeutic potency (Fig. 6B).85 Active targeting via antibody conjugation to nanoparticle surfaces further enables tissue-specific delivery. For example, conjugating antibodies targeting intracellular adhesion molecule-1 (ICAM) improved cellular uptake in bronchial epithelial cells up to 24-fold (Fig. 6C).86 | ||
| Fig. 6 Modifications of the nanoparticle surface to increase delivery efficiency. (A) Coating the nanoparticle surface with cell-penetrating peptides increases cell uptake. (B) The use of a polymer mixture increases the loading and endosomal escape of cargoes. (C) Antibody conjugation enables cell-specific targeting. | ||
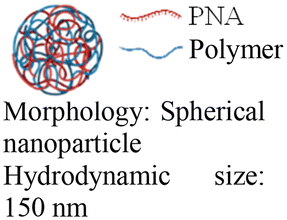
Cheng et al. demonstrated a double-emulsion solvent evaporation method to encapsulate PNAs in PLGA, which generated uniform spherical nanoparticles with a size of ∼150 nm (Table 2, entries 1 and 2).84,87 Surface modification of these PLGA nanoparticles with a cell-penetrating peptide resulted in an ∼4-fold increase in cell uptake compared to the control nanoparticles (Table 2, entry 3).88 Adding poly-L-histidine to PLGA also enhances uptake, and these particles achieved >75% knockdown of miR-155 and a 6-fold reduction in tumor size in mice (Table 2, entry 4).89 Collaborative efforts between the Glazer and Saltzman labs demonstrated site-specific gene editing using triplex-forming PNA and donor DNA delivered via PLGA nanoparticles in different disease models.8,9,90–92 PLGA nanoparticles carrying backbone-modified PNAs (MPγPNA) and donor DNA produced 0.1% editing in the bone marrow in a beta-thalassemia disease model (Table 2, entry 5).8 A similar strategy achieved 7% correction of the CFTR mutation in lung tissue in a mouse model of cystic fibrosis.90,91 Subsequent studies combined PLGA with the cationic polymer poly(beta-amino-esters) (PBAE) and improved the loading efficiency of donor DNA.92 PLGA/PBAE nanoparticles loaded with MPγPNA and donor DNA enabled ∼6% gene editing in the bone marrow and ∼10% gene editing in hematopoietic stem and progenitor cells after a single in utero injection and outperformed treatment efficacy in adult mice (∼4% editing in bone marrow and 7% in hematopoietic stem cells after four doses) (Table 2, entry 6).9 Importantly, none of these studies observed off-target PNA binding or toxicity from PBAE/PLGA nanoparticles even after multiple injections.
| Entry | PNA backbone | Components used to promote nanoassembly | Nanostructure | Tested in vivo/in vitro | Delivery efficiency | Cytotoxicity | Therapeutic effects |
|---|---|---|---|---|---|---|---|
| 1 [ref. 84] |
|
|
|
In vitro cell assays | 99.1% cell uptake | 96–98% cell viability | Deliver PNAs to primary CD34+ human hematopoietic stem cells |
| 2 [ref. 87] |
|
|
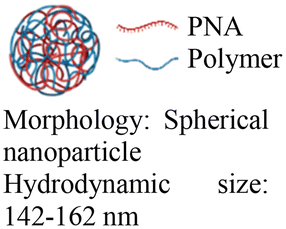
|
In vitro cell assays | Confocal live-cell images confirm cell uptake | No observed toxicity | Block chemokine receptor 5 mRNA expression |
| 3 [ref. 88] |
|
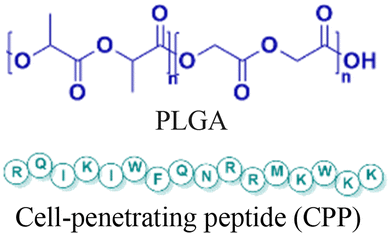
|
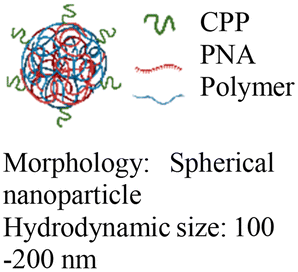
|
In vitro and in vivo mouse model | 3-fold higher cell uptake with surface modification | No observed toxicity | Block micro-mRNA-155 expression and reduce tumor growth |
| 4 [ref. 89] |
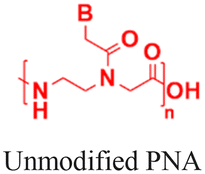
|
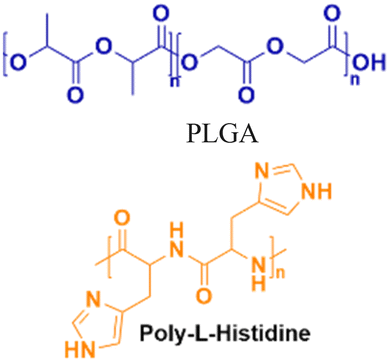
|
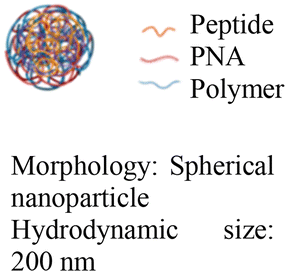
|
In vivo mouse model | Confocal live-cell images confirm cell uptake | No observed toxicity | Block micro-mRNA-155 expression and reduce tumor growth |
| 5 [ref. 8] |
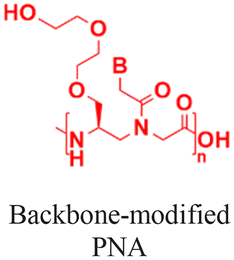
|
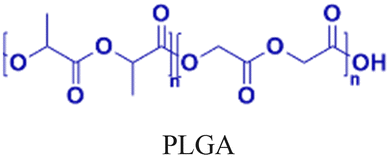
|
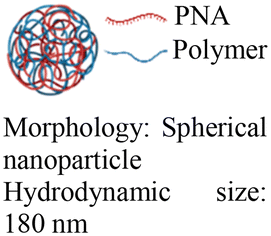
|
In vivo mouse model | Immune fluorescence microscopy confirms delivery into the cell nucleus | No observed toxicity | Editing of the mutated β-globin gene in hematopoietic stem cells |
| 6 [ref. 9 and 92] |
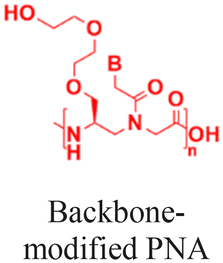
|
|
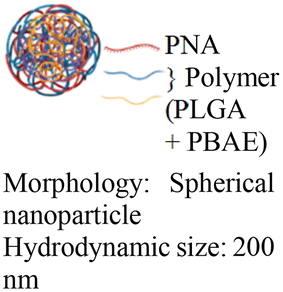
|
In vivo mouse model | Confocal images of fetuses confirm in utero delivery after intra-amniotic administration | No observed toxicity | In utero editing of the mutated β-globin gene |
| 7 [ref. 100] |
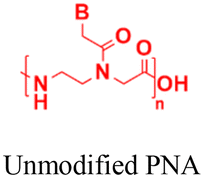
|
|

|
In vitro cell assays | Gal8 recruitment assay shows live-cell endosome-disruptive activity | 50%–90% cell viability | Inhibit miRNA-122 expression |
| 8 [ref. 102] |
|
|
|
In vitro cell assays | 82% cell uptake | ∼100% cell viability | Inhibit miRNA-155 expression and induce cell apoptosis |
In addition to PLGA-based nanoparticles, O’Reilly et al. reported a PNA delivery vehicle composed of polymeric particles made from amphiphilic copolymers that had a hydrophobic polystyrene core and hydrophilic positively charged shells.93 These copolymers encapsulate PNAs via electrostatic interactions or covalent conjugation, and the resulting nanoparticles were used to image iNOS mRNA expression, a marker of inflammation, in live cells.94,95
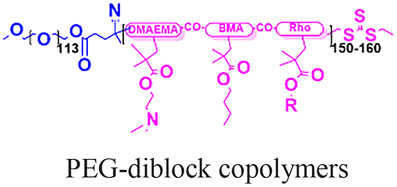
Beyond polymeric nanoparticles, protein and inorganic nanoparticles (zeolite and silica) have been used for PNA delivery.96–100 Kelly et al. combined porous silicon nanoparticles with PEG-based diblock copolymers to create hybrid materials that exhibited improved colloidal stability and facilitated endosomal escape via the proton sponge effect (Table 2, entry 7).100 In this case, the polymer composition determines the nanoparticles’ stability in serum, cytotoxicity, and endosome disruption efficiency.100 Later, Neri et al. implemented a much simpler salt-trapping strategy to enable PNA encapsulation in porous silicon nanoparticles, achieving ∼50% knockdown of a target miRNA in human bronchial epithelial cells.101
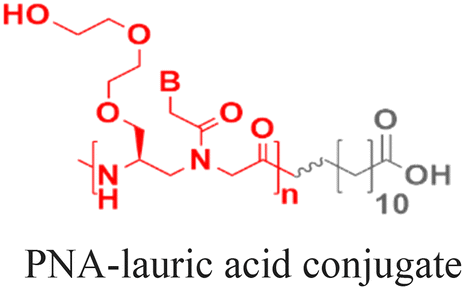
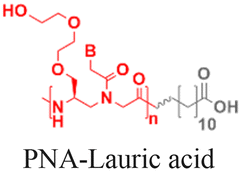
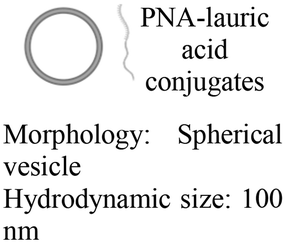
As an alternative to encapsulating PNAs in nanoparticles, one strategy is to design PNA amphiphiles that self-assemble into nanostructures capable of cellular internalization. Malik et al. reported efficient cell uptake of MPγPNA-fatty acid amphiphiles.102 The design had MPγPNA conjugated to a twelve-carbon lauric acid at the N-terminal. The hydrophobic tail drove self-assembly of the amphiphiles into spherical vesicles that were internalized by endocytosis, yielding 82% cellular uptake, 75% miRNA knockdown, and 25% apoptosis (Table 2, entry 8).102
3.6. Clinical translation challenges and opportunities for PNAs
Peptide nucleic acids (PNAs) have attracted intense research interest for three decades. However, their clinical progress lags behind that of other antisense platforms, such as siRNAs and PMOs, which already have multiple FDA-approved drugs.103 PNAs face three interrelated barriers to clinical translation: (1) scalable and cost-effective manufacturing, (2) efficient tissue-selective intracellular delivery, and (3) limited investment. Among these, manufacturing has been the primary impediment. Although native PNA monomers are commercially available, they are substantially more expensive than DNA and RNA monomers because their synthesis is more difficult. In addition, backbone modifications further complicate the synthesis. These backbone modifications are necessary because the original neutral backbone of native PNA monomers reduces water solubility and promotes aggregation, thereby adversely affecting the pharmacokinetic and pharmacodynamic properties of oligonucleotides by shortening the circulation half-life and increasing toxicity. Finally, common delivery platforms, such as cell-penetrating peptides, suffer from endosomal entrapment and insufficient tissue-specific targeting, imposing a delivery barrier that significantly diminishes therapeutic potency.Many of these limitations can be addressed by backbone modifications. Earlier attempts to introduce cationic side chains (amines and guanidine) improved cellular uptake but increased toxicity. In contrast, incorporating neutral hydrophilic groups such as miniPEG and serine side chains into PNA backbones reduces aggregation by improving water solubility. However, the synthesis of these modified monomers is complex and requires challenging purifications, creating manufacturing constraints, high costs, and limited scalability. Overcoming these challenges will require investment in streamlined, high-yield, automatable synthetic routes. Although miniPEG-substituted PNAs offer higher affinity, greater solubility, and improved sequence selectivity, they are not intrinsically cell-permeable. Advanced biocompatible carriers such as ligand-directed systems, pH-responsive polymers, and lipid nanoparticles (LNPs) have demonstrated robust cell-type specificity and efficient endosomal escape for siRNA and mRNA and therefore merit evaluation for PNA delivery. Finally, to justify further investment, PNAs must demonstrate distinct therapeutic advantages over other antisense oligonucleotide platforms. Biophysical characterization and in vitro cell assays have demonstrated the ability of PNAs to invade double-stranded DNA and RNA, whereas other antisense oligonucleotides often fail to bind these targets.13,41,104,105 By invading double-stranded DNA, PNAs can inhibit transcription by arresting RNA polymerase, blocking transcription factor binding, and distorting the helix.41,104,106 Backbone-modified PNAs also selectively bind and disrupt noncanonical secondary structures like guanine quadruplexes (GQ),107 intercalated motifs (i-motifs),108 and hairpins,109–111 which are often inaccessible to other antisense oligonucleotides. These unique capabilities suggest that PNAs could address unmet needs where other oligonucleotides may fail. Fig. 7 summarizes key obstacles to clinical translation of PNAs, the underlying causes, and possible solutions.
 | ||
| Fig. 7 A stepwise representation of the major challenges associated with the clinical translation of PNAs, the root cause for these issues, and potential solutions. | ||
4. Conclusions
Although challenges remain, PNAs offer promising therapeutic opportunities due to their unique biophysical properties. PNAs combine high binding affinity, exceptional sequence selectivity, and chemical stability, making them an attractive platform for both antisense and antigene applications. Importantly, PNAs can invade double-stranded DNA and noncanonical secondary structures, such as guanine quadruplexes, i-motifs, and hairpins, opening therapeutic opportunities that many other oligonucleotides cannot address. Clinical translation will progress through coordinated work across chemistry, nanotechnology, and biology, focusing on optimizing scalable monomer synthesis, improving targeted delivery, and selecting the most appropriate therapeutic targets.Author contributions
Srijani Sarkar: conceptualization, writing – original draft, and writing – review & and editing; Niren Murthy: resources, supervision, and writing – review & and editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.Abbreviations
| PNA | Peptide nucleic acid |
| γPNA | Gamma-substituted backbone-modified PNA |
| CPP | Cell-penetrating peptide |
Data availability
No new experimental data were generated during the preparation of this review. All data are taken from the cited literature. All figures are made from scratch by authors.Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- Y.-K. Kim, Exp. Mol. Med., 2022, 54, 455–465, DOI:10.1038/s12276-022-00757-5.
- Y. Zhu, L. Zhu, X. Wang and H. Jin, Cell Death Dis., 2022, 13, 644, DOI:10.1038/s41419-022-05075-2.
- P. Nielsen, M. Egholm, R. Berg and O. Buchardt, Science, 1991, 254, 1497–1500, DOI:10.1126/science.1962210.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566–568, DOI:10.1038/365566a0.
- R. Brazil, ACS Cent. Sci., 2023, 9, 3–6, DOI:10.1021/acscentsci.3c00016.
- A. Sannigrahi, N. De, D. Bhunia and J. Bhadra, Bioorg. Chem., 2025, 155, 108146, DOI:10.1016/j.bioorg.2025.108146.
- C. Avitabile, M. T. Cerasa, A. D’Aniello, M. Saviano and M. Moccia, Chem. – Eur. J., 2025, 31, e202500469, DOI:10.1002/chem.202500469.
- R. Bahal, N. Ali McNeer, E. Quijano, Y. Liu, P. Sulkowski, A. Turchick, Y.-C. Lu, D. C. Bhunia, A. Manna, D. L. Greiner, M. A. Brehm, C. J. Cheng, F. López-Giráldez, A. Ricciardi, J. Beloor, D. S. Krause, P. Kumar, P. G. Gallagher, D. T. Braddock, W. M. Saltzman, D. H. Ly and P. M. Glazer, Nat. Commun., 2016, 7, 13304, DOI:10.1038/ncomms13304.
- A. S. Ricciardi, R. Bahal, J. S. Farrelly, E. Quijano, A. H. Bianchi, V. L. Luks, R. Putman, F. López-Giráldez, S. Coşkun, E. Song, Y. Liu, W.-C. Hsieh, D. H. Ly, D. H. Stitelman, P. M. Glazer and W. M. Saltzman, Nat. Commun., 2018, 9, 2481, DOI:10.1038/s41467-018-04894-2.
- S. Sarkar and B. A. Armitage, ACS Infect. Dis., 2021, 7, 1445–1456, DOI:10.1021/acsinfecdis.0c00793.
- S. Sarkar, G. Colón-Roura, A. Pearse and B. A. Armitage, Biopolymers, 2023, 114, e23529, DOI:10.1002/bip.23529.
- S. Sarkar, Carnegie Mellon University, 2021.
- S. A. Thadke, V. M. Hridya, J. D. R. Perera, R. R. Gil, A. Mukherjee and D. H. Ly, Commun. Chem., 2018, 1, 79, DOI:10.1038/s42004-018-0080-5.
- P. Bhingardeve, B. R. Madhanagopal and K. N. Ganesh, J. Org. Chem., 2020, 85, 13680–13693, DOI:10.1021/acs.joc.0c01853.
- A. J. Wierzba, E. M. Richards, S. R. Lennon, R. T. Batey and A. E. Palmer, RSC Chem. Biol., 2025, 6, 249–262, 10.1039/D4CB00274A.
- P. Zhou, M. Wang, L. Du, G. W. Fisher, A. Waggoner and D. H. Ly, J. Am. Chem. Soc., 2003, 125, 6878–6879, DOI:10.1021/ja029665m.
- A. Dragulescu-Andrasi, P. Zhou, G. He and D. H. Ly, Chem. Commun., 2005, 244–246, 10.1039/B412522C.
- A. Dragulescu-Andrasi, S. Rapireddy, B. M. Frezza, C. Gayathri, R. R. Gil and D. H. Ly, J. Am. Chem. Soc., 2006, 128, 10258–10267, DOI:10.1021/ja0625576.
- R. Mitra and K. N. Ganesh, Chem. Commun., 2011, 47, 1198–1200, 10.1039/C0CC03988H.
- B. Sahu, I. Sacui, S. Rapireddy, K. J. Zanotti, R. Bahal, B. A. Armitage and D. H. Ly, J. Org. Chem., 2011, 76, 5614–5627, DOI:10.1021/jo200482d.
- S. Malik, J. Lim, F. J. Slack, D. T. Braddock and R. Bahal, J. Controlled Release, 2020, 327, 406–419, DOI:10.1016/j.jconrel.2020.08.026.
- G. Wang, P. Lu, M. Chen, Y. Dong, J. Jiao, Y. Xiang and J. Jiao, Nano Lett., 2025, 25, 8796–8803, DOI:10.1021/acs.nanolett.5c02043.
- O. Berger, L. Adler-Abramovich, M. Levy-Sakin, A. Grunwald, Y. Liebes-Peer, M. Bachar, L. Buzhansky, E. Mossou, V. T. Forsyth, T. Schwartz, Y. Ebenstein, F. Frolow, L. J. W. Shimon, F. Patolsky and E. Gazit, Nat. Nanotechnol., 2015, 10, 353–360, DOI:10.1038/nnano.2015.27.
- O. Berger, E. Yoskovitz, L. Adler-Abramovich and E. Gazit, Adv. Mater., 2016, 28, 2195–2200, DOI:10.1002/adma.201504160.
- V. Basavalingappa, S. Bera, B. Xue, I. Azuri, Y. Tang, K. Tao, L. J. W. Shimon, M. R. Sawaya, S. Kolusheva, D. S. Eisenberg, L. Kronik, Y. Cao, G. Wei and E. Gazit, Nat. Commun., 2019, 10, 5256, DOI:10.1038/s41467-019-13250-x.
- K. Dey, S. R. Chowdhury, E. Dykstra, A. Koronatov, H. P. Lu, R. Shinar, J. Shinar and P. Anzenbacher, J. Mater. Chem. C, 2020, 8, 11988–11996, 10.1039/D0TC02317E.
- K. Dey, S. R. Chowdhury, E. Dykstra, H. P. Lu, R. Shinar, J. Shinar and P. Anzenbacher, ACS Appl. Electron. Mater., 2021, 3, 3365–3371, DOI:10.1021/acsaelm.1c00354.
- M. Dockerill, D. J. Ford, S. Angerani, I. Alwis, L. J. Dowman, J. Ripoll-Rozada, R. E. Smythe, J. S. T. Liu, P. J. B. Pereira, S. P. Jackson, R. J. Payne and N. Winssinger, Nat. Biotechnol., 2025, 43, 186–193, DOI:10.1038/s41587-024-02209-z.
- R. Veneziano, T. J. Moyer, M. B. Stone, E.-C. Wamhoff, B. J. Read, S. Mukherjee, T. R. Shepherd, J. Das, W. R. Schief, D. J. Irvine and M. Bathe, Nat. Nanotechnol., 2020, 15, 716–723, DOI:10.1038/s41565-020-0719-0.
- T.-W. Chu, J. Feng, J. Yang and J. Kopeček, J. Controlled Release, 2015, 220, 608–616, DOI:10.1016/j.jconrel.2015.09.035.
- J. T. M. DiMaio, T. M. Doran, D. M. Ryan, D. M. Raymond and B. L. Nilsson, Biomacromolecules, 2017, 18, 3591–3599, DOI:10.1021/acs.biomac.7b00925.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813, DOI:10.1126/science.aat6141.
- S. Sarkar, C. F. Anderson and J. P. Schneider, Angew. Chem., Int. Ed., 2024, 63, e202313507, DOI:10.1002/anie.202313507.
- S. Sarkar, Biopolymers, 2024, 115, e23567, DOI:10.1002/bip.23567.
- S. Sarkar, C. F. Anderson, Y. Xie, S. J. Lockett and J. P. Schneider, Chem. Mater., 2024, 36, 10216–10226, DOI:10.1021/acs.chemmater.4c01955.
- K. Dhuri, C. Bechtold, E. Quijano, H. Pham, A. Gupta, A. Vikram and R. Bahal, J. Clin. Med., 2020, 9, 2004, DOI:10.3390/jcm9062004.
- S. T. Crooke, B. F. Baker, R. M. Crooke and X. Liang, Nat. Rev. Drug Discovery, 2021, 20, 427–453, DOI:10.1038/s41573-021-00162-z.
- P. E. Nielsen, Q. Rev. Biophys., 2005, 38, 345–350, DOI:10.1017/S0033583506004148.
- B. M. McMahon, D. Mays, J. Lipsky, J. A. Stewart, A. Fauq and E. Richelson, Antisense Nucleic Acid Drug Dev., 2002, 12, 65–70, DOI:10.1089/108729002760070803.
- P. Chakrabarty, P. Gupta, K. Illath, S. Kar, M. Nagai, F.-G. Tseng and T. S. Santra, Mater. Today Bio, 2022, 13, 100193, DOI:10.1016/j.mtbio.2021.100193.
- J. C. Hanvey, N. J. Peffer, J. E. Bisi, S. A. Thomson, R. Cadilla, J. A. Josey, D. J. Ricca, C. F. Hassman, M. A. Bonham and K. G. Au, Science, 1992, 258, 1481–1485, DOI:10.1126/science.1279811.
- S. Z. Hirschman and C. W. Chen, J. Invest. Med., 1996, 44, 347–351 CAS.
- G. Wang, X. Xu, B. Pace, D. A. Dean, P. M. Glazer, P. Chan, S. R. Goodman and I. Shokolenko, Nucleic Acids Res., 1999, 27, 2806–2813, DOI:10.1093/nar/27.13.2806.
- E. Noguchi, N. Shigi and M. Komiyama, Nat. Prod. Commun., 2012, 7, 349–352 CrossRef CAS PubMed.
- A. F. Faruqi, M. Egholm and P. M. Glazer, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1398–1403, DOI:10.1073/pnas.95.4.1398.
- T. Ljungstrøm, H. Knudsen and P. E. Nielsen, Bioconjugate Chem., 1999, 10, 965–972, DOI:10.1021/bc990053.
- A. Muratovska, R. N. Lightowlers, R. W. Taylor, D. M. Turnbull, R. A. Smith, J. A. Wilce, S. W. Martin and M. P. Murphy, Nucleic Acids Res., 2001, 29, 1852–1863, DOI:10.1093/nar/29.9.1852.
- T. Shiraishi and P. E. Nielsen, Bioconjugate Chem., 2012, 15, 196–202, DOI:10.1021/bc200460t.
- I. M. Gabas and P. E. Nielsen, Biomacromolecules, 2020, 21, 472–483, DOI:10.1021/acs.biomac.9b01227.
- S. E. Hamilton, C. G. Simmons, I. S. Kathiriya and D. R. Corey, Chem. Biol., 1999, 6, 343–351, DOI:10.1016/S1074-5521(99)80046-5.
- P. Kumar and D. R. Jain, Tetrahedron, 2015, 71, 3378–3384, DOI:10.1016/j.tet.2015.03.093.
- D. R. Jain, L. Anandi V, M. Lahiri and K. N. Ganesh, J. Org. Chem., 2014, 79, 9567–9577, DOI:10.1021/jo501639m.
- B. Sahu, V. Chenna, K. L. Lathrop, S. M. Thomas, G. Zon, K. J. Livak and D. H. Ly, J. Org. Chem., 2009, 74, 1509–1516, DOI:10.1021/jo802211n.
- A. Dragulescu-Andrasi, S. Rapireddy, G. He, B. Bhattacharya, J. J. Hyldig-Nielsen, G. Zon and D. H. Ly, J. Am. Chem. Soc., 2006, 128, 16104–16112, DOI:10.1021/ja063383v.
- V. Clausse, H. Zheng, H. Amarasekara, M. Kruhlak and D. H. Appella, Nucleic Acids Res., 2022, 50, 10839–10856, DOI:10.1093/nar/gkac864.
- H. Zheng, V. Clausse, H. Amarasekara, S. J. Mazur, I. Botos and D. H. Appella, J. Am. Chem. Soc. Au, 2023, 3, 1952–1964, DOI:10.1021/jacsau.3c00198.
- G. Barkowsky, A.-L. Lemster, R. Pappesch, A. Jacob, S. Krüger, A. Schröder, B. Kreikemeyer and N. Patenge, Mol. Ther.–Nucleic Acids, 2019, 18, 444–454, DOI:10.1016/j.omtn.2019.09.010.
- T. Soudah, M. Mogilevsky, R. Karni and E. Yavin, Bioconjugate Chem., 2017, 28, 3036–3042, DOI:10.1021/acs.bioconjchem.7b00638.
- T. Soudah, S. Khawaled, R. I. Aqeilan and E. Yavin, ACS Omega, 2019, 4, 13954–13961, DOI:10.1021/acsomega.9b01697.
- C. J. Cheng, R. Bahal, I. A. Babar, Z. Pincus, F. Barrera, C. Liu, A. Svoronos, D. T. Braddock, P. M. Glazer, D. M. Engelman, W. M. Saltzman and F. J. Slack, Nature, 2015, 518, 107–110, DOI:10.1038/nature13905.
- U. Tsylents, I. Siekierska and J. Trylska, Eur. Biophys. J., 2023, 52, 533–544, DOI:10.1007/s00249-023-01673-w.
- M. El-Fateh, A. Chatterjee and X. Zhao, Int. J. Antimicro. Agents, 2024, 63, 107083, DOI:10.1016/j.ijantimicag.2024.107083.
- A. Polak, G. Machnik, Ł. Bułdak, W. Wójtowicz, J. Ruczyński, K. Prochera, P. Mucha, P. Rekowski and B. Okopień, Int. J. Pept. Res. Ther., 2025, 31, 114, DOI:10.1007/s10989-025-10776-1.
- U. Koppelhus and P. E. Nielsen, Adv. Drug Delivery Rev., 2003, 55, 267–280, DOI:10.1016/s0169-409x(02)00182-5.
- E. Fabbri, A. Manicardi, T. Tedeschi, S. Sforza, N. Bianchi, E. Brognara, A. Finotti, G. Breveglieri, M. Borgatti, R. Corradini, R. Marchelli and R. Gambari, ChemMedChem, 2011, 6, 2192–2202, DOI:10.1002/cmdc.201100270.
- K. X. Y. Tan and S. Shigenobu, Sci. Rep., 2024, 14, 5378, DOI:10.1038/s41598-024-55179-2.
- M. Pooga, U. Soomets, M. Hällbrink, A. Valkna, K. Saar, K. Rezaei, U. Kahl, J.-X. Hao, X.-J. Xu, Z. Wiesenfeld-Hallin, T. Hökfelt, T. Bartfai and Ü. Langel, Nat. Biotechnol., 1998, 16, 857–861, DOI:10.1038/nbt0998-857.
- S. Abes, J. J. Turner, G. D. Ivanova, D. Owen, D. Williams, A. Arzumanov, P. Clair, M. J. Gait and B. Lebleu, Nucleic Acids Res., 2007, 35, 7396, DOI:10.1093/nar/gkm418.
- B. Chaubey, S. Tripathi and V. N. Pandey, Oligonucleotides, 2008, 18, 9–20, DOI:10.1089/oli.2007.0088.
- S. Cogoi, A. Codognotto, V. Rapozzi and L. E. Xodo, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 971–974, DOI:10.1081/ncn-200059333.
- J. B. Giancola and R. T. Raines, Chem. Commun., 2024, 60, 15019–15022, 10.1039/d4cc05214e.
- J. Gasparello, A. Manicardi, A. Casnati, R. Corradini, R. Gambari, A. Finotti and F. Sansone, Sci. Rep., 2019, 9, 3036, DOI:10.1038/s41598-019-39211-4.
- A. R. Kaplan, H. Pham, Y. Liu, S. Oyaghire, R. Bahal, D. M. Engelman and P. M. Glazer, Mol. Cancer Res., 2020, 18, 873–882, DOI:10.1158/1541-7786.MCR-19-0661.
- X. Tan, M. P. Bruchez and B. A. Armitage, Bioconjugate Chem., 2018, 29, 2892–2898, DOI:10.1021/acs.bioconjchem.8b00495.
- X. Tan, M. P. Bruchez and B. A. Armitage, ChemBioChem, 2019, 20, 727–733, DOI:10.1002/cbic.201800709.
- Y. Hakata, S. Ishikawa, T. Ohtsuki, M. Miyazawa and M. Kitamatsu, Org. Biomol. Chem., 2020, 18, 1978–1986, 10.1039/C9OB02559F.
- P. Bhingardeve, B. R. Madhanagopal, H. Naick, P. Jain, M. Manoharan and K. Ganesh, J. Org. Chem., 2021, 85, 8812–8824, DOI:10.1021/acs.joc.0c00601.
- V. Kumar, A. Wahane, A. Gupta, J. E. Manautou and R. Bahal, Adv. Healthcare Mater., 2023, 12, 2202859, DOI:10.1002/adhm.202202859.
- Z. Lu, B. R. Paolelle, N. L. Truex, A. R. Loftis, X. Liao, A. E. Rabideau, M. S. Brown, J. Busanovich, R. Beroukhim and B. L. Pentelute, ACS Chem. Biol., 2020, 15, 1358–1369, DOI:10.1021/acschembio.9b01027.
- T. Pieńko, J. Czarnecki, M. Równicki, M. Wojciechowska, A. J. Wierzba, D. Gryko, D. Bartosik and J. Trylska, Biophys. J., 2021, 120, 725–737, DOI:10.1016/j.bpj.2021.01.004.
- M. J. Pals, L. Wijnberg, Ç. Yildiz and W. A. Velema, Angew. Chem., Int. Ed., 2024, 63, e202402405, DOI:10.1002/anie.202402405.
- X. Bian, L. Zhou, Z. Luo, G. Liu, Z. Hang, H. Li, F. Li and Y. Wen, ACS Nano, 2025, 19, 4039–4083, DOI:10.1021/acsnano.4c11858.
- W. Chen, Q. Lai, Y. Zhang, L. Mo and Z. Liu, Curr. Med. Chem., 2025, 32, 1378–1390, DOI:10.2174/0109298673266457231123042819.
- C. J. Cheng and W. M. Saltzman, Mol. Pharmaceutics, 2012, 9, 1481–1488, DOI:10.1021/mp300081s.
- R. Bahal, E. Quijano, N. A. McNeer, Y. Liu, D. C. Bhunia, F. Lopez-Giraldez, R. J. Fields, W. M. Saltzman, D. H. Ly and P. M. Glazer, Curr. Gene Ther., 2014, 14, 331–342, DOI:10.2174/1566523214666140825154158.
- V. L. Luks, H. Mandl, J. DiRito, C. Barone, M. R. Freedman-Weiss, A. S. Ricciardi, G. G. Tietjen, M. E. Egan, W. M. Saltzman and D. H. Stitelman, PLoS One, 2022, 17, e0266218, DOI:10.1371/journal.pone.0266218.
- R. Bahal, N. A. McNeer, D. H. Ly, W. M. Saltzman and P. M. Glazer, Artif. DNA: PNA & XNA, 2013, 4, 49–57, DOI:10.4161/adna.25628.
- I. A. Babar, C. J. Cheng, C. J. Booth, X. Liang, J. B. Weidhaas, W. M. Saltzman and F. J. Slack, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1695–E1704, DOI:10.1073/pnas.1201516109.
- A. Wahane, S. Malik, K.-C. Shih, R. R. Gaddam, C. Chen, Y. Liu, M.-P. Nieh, A. Vikram and R. Bahal, ACS Appl. Mater. Interfaces, 2021, 13, 45244–45258, DOI:10.1021/acsami.1c11981.
- N. A. McNeer, K. Anandalingam, R. J. Fields, C. Caputo, S. Kopic, A. Gupta, E. Quijano, L. Polikoff, Y. Kong, R. Bahal, J. P. Geibel, P. M. Glazer, W. M. Saltzman and M. E. Egan, Nat. Commun., 2015, 6, 6952, DOI:10.1038/ncomms7952.
- A. S. Piotrowski-Daspit1, C. Barone, C.-Y. Lin, Y. Deng, D. Wu, T. C. Binns, E. Xu, A. S. Ricciardi, R. Putman, A. Garrison, R. Nguyen, A. Gupta, R. Fan, P. M. Glazer, W. M. Saltzman and M. E. Egan, Sci. Adv., 2022, 8, eabo0522, DOI:10.1126/sciadv.abo0522.
- R. J. Fields, C. J. Cheng, E. Quijano, C. Weller, N. Kristofik, N. Duong, C. Hoimes, M. E. Egan and W. M. Saltzman, J. Controlled Release, 2012, 164, 41–48, DOI:10.1016/j.jconrel.2012.09.020.
- R. K. O’Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083, 10.1039/B514858H.
- Z. Wang, K. Zhang, K. L. Wooley and J.-S. Taylor, J. Nucleic Acids, 2012, 2012, 1–11, DOI:10.1155/2012/962652.
- Z. Wang, K. Zhang, Y. Shen, J. Smith, S. Bloch, S. Achilefu, K. L. Wooley and J.-S. Taylor, Org. Biomol. Chem., 2013, 11, 3159, 10.1039/c3ob26923j.
- K. Langer, C. Coester, C. Weber, H. V. Briesen and J. Kreuter, Eur. J. Pharm. Biopharm., 2000, 49, 303–307, DOI:10.1016/s0939-6411(00)00068-0.
- A. Bertucci, H. Lülf, D. Septiadi, A. Manicardi, R. Corradini and L. De Cola, Adv. Healthcare Mater., 2014, 3, 1812–1817, DOI:10.1002/adhm.201400116.
- X. Ma, G. Devi, Q. Qu, D.-F. K. Toh, G. Chen and Y. Zhao, Bioconjugate Chem., 2014, 25, 1412–1420, DOI:10.1021/bc5002714.
- K. R. Beavers, J. W. Mares, C. M. Swartz, Y. Zhao, S. M. Weiss and C. L. Duvall, Bioconjugate Chem., 2014, 25, 1192–1197, DOI:10.1021/bc5001092.
- I. B. Kelly III, R. B. Fletcher, J. R. McBride, S. M. Weiss and C. L. Duvall, ACS Appl. Mater. Interfaces, 2020, 12, 39602–39611, DOI:10.1021/acsami.0c05827.
- M. Neri, J. Kang, J. M. Zuidema, J. Gasparello, A. Finotti, R. Gambari, M. J. Sailor, A. Bertucci and R. Corradini, ACS Biomater. Sci. Eng., 2022, 8, 4123–4131, DOI:10.1021/acsbiomaterials.1c00431.
- S. Malik, V. Kumar, C. Liu, K. Shih, S. Krueger, M. Nieh and R. Bahal, Adv. Funct. Mater., 2022, 32, 2109552, DOI:10.1002/adfm.202109552.
- M. D. Pérez-Carrión, I. Posadas and V. Ceña, Pharmacol. Res., 2024, 201, 107102, DOI:10.1016/j.phrs.2024.107102.
- B. A. Janowski, K. Kaihatsu, K. E. Huffman, J. C. Schwartz, R. Ram, D. Hardy, C. R. Mendelson and D. R. Corey, Nat. Chem. Biol., 2005, 1, 210–215, DOI:10.1038/nchembio724.
- S. A. Thadke, J. D. R. Perera, V. M. Hridya, K. Bhatt, A. Y. Shaikh, W.-C. Hsieh, M. Chen, C. Gayathri, R. R. Gil, G. S. Rule, A. Mukherjee, C. A. Thornton and D. H. Ly, Biochemistry, 2018, 57, 2094–2108, DOI:10.1021/acs.biochem.8b00062.
- H. J. Larsen and P. E. Nielsen, Nucleic Acids Res., 1996, 24, 458–463, DOI:10.1093/nar/24.3.458.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474, DOI:10.1038/s41580-020-0236-x.
- S. Tao, Y. Run, D. Monchaud and W. Zhang, Trends Genet., 2024, 40, 853–867, DOI:10.1016/j.tig.2024.05.011.
- W. J. Krzyzosiak, K. Sobczak, M. Wojciechowska, A. Fiszer, A. Mykowska and P. Kozlowski, Nucleic Acids Res., 2012, 40, 11–26, DOI:10.1093/nar/gkr729.
- C. A. Ryan, M. M. Rahman, V. Kumar and E. Rozners, J. Am. Chem. Soc., 2023, 145, 10497–10504, DOI:10.1021/jacs.2c12488.
- S. F. Saei, V. Baskevics, M. Katkevics and E. Rozners, ACS Chem. Biol., 2025, 20, 179–185, DOI:10.1021/acschembio.4c00662.
| This journal is © The Royal Society of Chemistry 2026 |