DOI:
10.1039/D5YA00059A
(Paper)
Energy Adv., 2025,
4, 1024-1031
ORR durability study on Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB for heavy-duty vehicle operation†
Received
4th March 2025
, Accepted 12th June 2025
First published on 20th June 2025
Abstract
Since durability is one of the key issues for using robust catalysts in fuel cell systems of heavy-duty vehicles (HDV, a durability target of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h) applications, the durability of various core–shell Pt-based alloy catalysts is evaluated. Herein, we present the durability behaviors of Pt3.7Mn/KB (KB: EC600JD), Pt3.9Co/KB and Pt3.7Ni/KB catalysts with similar initial activities for the oxygen reduction reaction (ORR) of fuel cell systems. The durability of the catalysts was evaluated by a square-wave accelerated durability test (ADT) under the HDV operation. Notably, Pt3.9Co/KB largely retained its mass activity (MA) even after 70000 ADT cycles, while Pt3.7Mn/KB and Pt3.7Ni/KB significantly degraded during the tests. Less alteration in MA of Pt3.9Co/KB was consistent with the slight change in the Tafel slope.
000 h) applications, the durability of various core–shell Pt-based alloy catalysts is evaluated. Herein, we present the durability behaviors of Pt3.7Mn/KB (KB: EC600JD), Pt3.9Co/KB and Pt3.7Ni/KB catalysts with similar initial activities for the oxygen reduction reaction (ORR) of fuel cell systems. The durability of the catalysts was evaluated by a square-wave accelerated durability test (ADT) under the HDV operation. Notably, Pt3.9Co/KB largely retained its mass activity (MA) even after 70000 ADT cycles, while Pt3.7Mn/KB and Pt3.7Ni/KB significantly degraded during the tests. Less alteration in MA of Pt3.9Co/KB was consistent with the slight change in the Tafel slope.
Introduction
Highly efficient and non-polluting polymer electrolyte fuel cells (PEFCs) aim to power HDVs which are significant sources of greenhouse gas emissions.1–3 The United States Department of Energy (DOE) has set the 2025 targets for PEFC performance, a total loading of platinum group metal (PGM) and lifetime to achieve >0.3 A cm−2 at 0.8 V, 0.125 mgPt cm−2, after ADT 5000-h for light-duty vehicles (LDVs), and 1.07 A cm−2 at 0.7 V, 0.3 mgPt cm−2, after ADT 25000-h for HDVs such as Class 8 long-haul trucks.1,4 In addition, the Japanese New Energy & Industrial Development Organization (NEDO) has established more aggressive 2035 targets (0.761 V at 2.18 A cm−2, 0.178 mgPt cm−2, after ADT 50000-h) for PEFC systems of HDVs.5 These strict targets require usage of a highly active and robust catalyst for the cathodic reaction of PEFCs. A significant number of disordered and ordered Pt-based alloy catalysts with enhanced ORR activities have been extensively developed so far.6–13 However, the insufficient durability still remains a critical challenge. In connection with the shift of PEFCs from LDV to HDV applications, state-of-the-art Pt-based catalysts have been evaluated for ORR durability under HDV operation conditions. For instance, a commercial Pt catalyst supported on a high surface area carbon (Pt/C) was evaluated and exhibited a high level of degradation.14 Subsequently, the ORR durability of an ordered PtCo intermetallic catalyst (L10-PtCo/C) was examined with 90000 ADT cycles and was about two times more stable than that of Pt/C.15 The catalyst degradation is mainly caused by dissolution of Pt and secondary metals,16,17 agglomeration of nanoparticles,18,19 poisoning by sulfonic acid groups of ionomers20,21 and corrosion of supports.22 These detrimental processes are considerably decreased by protecting catalyst nanoparticles with specific shells. Shell-protection strategies used in catalyst preparation create shells composed of Pt atoms,8,9 carbon cages12,23,24 and oxide clusters25,26 on the surface of alloy nanoparticles. Among them, the Pt-shell has a significant impact on the performance and durability of alloy-core/Pt-shell structured catalysts. This is because the Pt-shell protects secondary metal atoms from dissolution whereas the alloy-core enhances ORR activity by modifying the surface electronic structure of catalyst nanoparticles.27 The core–shell structured alloy catalysts on carbon are often prepared by colloid,28 impregnation,29,30 polyol31 and ball milling32 strategies. However, the widely used colloid, polyol and ball milling methods suffer from incomplete removal of organic contaminants and inhomogeneous dispersion of metal precursors. In contrast, an aqueous solution-based impregnation (ASI) coupled with a rapid heating and quenching (RHQ) treatment is a clean method which can produce even a gram-scale catalyst for a short period of time. In addition, ASI provides homogeneous dispersion of metal precursors without organic contaminants on support, whereas RHQ not only prevents aggregation of nanoparticles by rapid-heating but also preserves defects on nanoparticle surfaces by rapid-cooling.33 On the other hand, a robust catalyst is expected to have a larger average particle size to tolerate HDV PEFC operating conditions while maintaining its high ORR activity.34 Herein, we prepared the core–shell structured alloy catalysts (Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB) with well-controlled particle sizes (4–5 nm) using the ASI-RHQ method. The three alloy catalysts showed comparable initial ORR activities. However, their durability towards ORR was clearly differentiated by ADTs for 70000 (70k) cycles. Notably, MA of Pt3.9Co/KB was less altered by ADT experiments compared to Pt3.7Mn/KB and Pt3.7Ni/KB which significantly declined. The lower alteration in MA of Pt3.9Co/KB correlated with negligible change in the Tafel slope.
Experimental
Chemicals
A ketjen-black carbon support (KB: EC-600JD, SABET: 1270 m2 g−1)35 was purchased from LION Specialty Chemicals. A commercial catalyst (cPt/C, 46.7 wt% Pt, dTEM.: 2.3 nm) and a diaminedinitritoplatinum(II) aqueous solution (Pt 4.558 wt%, [Pt(NH3)2](NO2)2) were purchased from Tanaka Kikinzoku Kogyo (TKK). Cobalt(II) nitrate hexahydrate (CN, Co(NO3)2·6H2O, 98.0%), nickel(II) nitrate hexahydrate (NN, Ni(NO3)2·6H2O, 98.0%), manganese(II) nitrate hexahydrate (MN, Mn(NO3)2·6H2O, 98.0%), HNO3 (60.0%), HClO4 (70.0%), ethanol (99.5%), 1-propanol (infinity pure), a 5 wt% Nafion solution (DE520 CS), and ultrapure water (for LC/MS) were purchased from FUJIFILM Wako Pure Chemical Corporation. All chemicals were used as received.
Catalyst preparation
An aqueous suspension of KB (0.5 g), distilled water (80 mL) and ethanol (1 mL) was sonicated for 5 min and further stirred for 30 min at room temperature. Subsequently, an aqueous solution (10 mL) of diaminedinitritoplatinum(II) solution (Pt: 0.0026 mol) and transition-metal (M) nitrate (M: 0.0012 mol) was added dropwise to the suspension. The resultant mixture was stirred at room temperature for 2 h, vacuum-distilled at 65 °C to remove the solvent, and dried in air at 50 °C overnight. The dry solid was decomposed at 200 °C (a ramping rate of 3 °C min−1) for 2 h under a nitrogen atmosphere and reduced at 100 °C (2.5 °C min−1) and 150 °C (2 °C min−1), respectively, for 30 min under a 5% H2/Ar flow. Subsequently, the powder was exposed to RHQ treatment. In other words, it was rapidly heated to 880 °C within 2 min, maintained at 880 °C for 1 h under the 5% H2/Ar atmosphere and rapidly cooled using a fan to room temperature within 15 min to obtain alloy nanoparticles. Then, the alloy catalyst was treated with 1.0 M HNO3 at 80 °C for 12 h, vacuum-filtered, rinsed with distilled water (1000 mL) and dried in air at room temperature overnight. The obtained core–shell structured alloy catalyst was dried at 80 °C for 10 h under nitrogen, followed by post-heat treatment at 500 °C under the 5% H2/Ar atmosphere for a short period of time. In all cases, the gas flow rate was set at 60 mL min−1.
Characterization
The X-ray diffraction (XRD) patterns were recorded from 10° to 90° 2θ angles at a scanning rate of 2° min−1 and a step of 0.02° on a MiniFlex 600-C diffractometer (Rigaku) using a CuKα radiation (λ = 1.541862 Å) at 40 kV and 15 mA. Transmission electron microscopy (TEM) images were taken at 200 kV with a JEM-2100 electron microscope (JEOL Ltd.). The metal loadings were estimated by calcination in air followed by reduction in 5%H2/Ar on a Seiko SII-EXSTAR TG/TDA-7200 analyser (Nanotechnology Inc.). The metal ratios were determined using a NEX DE X-ray fluorescence (XRF) system (Rigaku). X-ray photoelectron spectroscopy (XPS) scans were performed on a PHI 5000 VersaProbe (ULVAC-PHI, Inc.) using a monochromatic Al Kα X-ray source (1486.6 eV) at 25 W (15 kV, 3 mA). The XPS survey spectra were collected with a pass energy of 187.85 eV in the binding energy (BE) range of 0–1000 eV with a step of 1.0 eV and a dwell time of 20 ms. The core level (narrow) spectra were scanned with a pass energy of 23.5 eV at a step of 0.1 eV and a dwell time of 50 ms. The atomic fraction of M(x) in alloy nanoparticles was determined from XRD data based on Vegard's law (eqn (1)).36| | | aalloy = aMx + aPt(1 − x) | (1) |
where aalloy, aM and aPt are lattice parameters of experimentally prepared alloy catalyst, bulk M and bulk Pt, respectively.
Electrochemical measurements
Electrochemical measurements were performed using a potentiostat (VSP-300, Bio-Logic SAS), an electrode rotator (AFMSRCE, Pine Research Instruments), and a three-electrode electrochemical cell (Pine Research Instruments). A glassy carbon (GC, 0.196 cm2 area, Pine Research Instruments) polished with 0.05 μm alumina slurry was used as a working electrode support. A platinum coil electrode (BAS Inc.) and a reversible hydrogen electrode with a double junction chamber (RHE, BAS Inc.) were employed as a counter electrode (CE) and a reference electrode (RE), respectively. All potential values are referred to RHE and iR-corrected (EiR-free = Emeas. + iRs) after each measurement. A catalyst ink composed of catalyst powder, ultrapure water, and 5 wt% Nafion solution was ultrasonically dispersed in an ice-cooled bath for 1 h, followed by horn sonication (15 W, 60 s).37 The ratios of water:1-propanol and ionomer:carbon (I/C) were 1:3 and 1:2, respectively. The Pt loading was 18 μgPt cm−2. A 10 μL of the catalyst ink was applied on a pre-polished GC tip mounted on an inverted rotator shaft while spinning at 100 rpm and the solvent was dried at 700 rpm in ambient air. The working electrode with a catalyst layer was immersed in 0.1 M HCIO4 solution (∼130 mL). After the electrolyte was purged with nitrogen (300 mL min−1) for 30 min, the working electrode was scanned at 200 mV s−1 from 0.0 to 1.1 V for 100 cycles to obtain stable and clean surface. Last three cyclic voltammograms (CVs) were recorded at 50 mV s−1 from 0.02 to 0.5 V for the calculation of electrochemical surface areas (ECSAs). ECSAs were estimated from the hydrogen adsorption charge (QH) between 0.05 and 0.40 V after double-layer correction (eqn (2)).| | | ESCA (mPt2 g−1) = QH/(2.10 × Ptloading) | (2) |
where 2.10 (C mPt−2) is the electric charge for a monolayer hydrogen on Pt surface and Ptloading (g) is the Pt mass. The solution resistance (Rs = ∼25 Ω) was measured at 0.5 V with an amplitude of 0.015 V in the frequency range of 100000 Hz to 1 Hz. Subsequently, linear sweep voltammograms (LSVs) were measured at 10 mV s−1 from 0.3 V to 1.1 V to record the background currents. After that, the electrolyte was purged with oxygen (300 mL min−1) for 30 min while measuring the open circuit voltage (OCV). LSVs for ORR were scanned at 10 mV s−1 from 0.3 to 1.1 V at rotation speeds of 200, 400, 900, 1600 and 2000 rpm in the O2-saturated electrolyte at room temperature. The kinetic currents (ik) at various potentials were calculated from LSV curves using the Koutecky–Levich formula (eqn (3)).38where im is the measured current, ik is the kinetic current and iL is the diffusion-limited current at 0.4 V. The ik obtained from the intercept of the Koutecky–Levich linear plot at 0.9 V was normalized with Pt loading and ECSA to evaluate the MA and specific activity (SA), respectively, according to eqn (4) and eqn (5).| | | MA (A gPt−1) = ik/Ptloading | (4) |
| | | SA (μA cmPt−2) = ik/ECSA | (5) |
In addition, Tafel slopes were extracted from the plots of EiR-free against log(ik) at low current regions. The square-wave ADTs were performed using constant potential polarization at 0.6 V (3 s) and 0.95 V (3 s) for 70k cycles in the N2-saturated 0.1 M HClO4 at room temperature (25 °C ± 3 °C).39 At the beginning of ADT and after every 10000 (10k) cycles, CVs and LSVs were measured in the N2- and O2-saturated fresh electrolyte, respectively, to evaluate ECSAs and ORR activities. Single cell tests were performed according to a protocol recommended by NEDO.40 The catalyst ink composed of catalyst powder, ultrapure water, ethanol and 5 wt% Nafion solution was ultrasonically dispersed in an ice-bath for 1 h. The ratios of water:ethanol and I/C were 2:3 and 1:1, respectively. The anode/cathode Pt loadings were fixed at 0.17 mgPt cm−2/0.17 mgPt cm−2. The anode (cPt/C, 46.7 wt% Pt, TKK) and cathode (alloy catalyst) electrodes were produced by painting the catalyst inks on carbon papers (MB30, Avcarb). A membrane electrode assembly (MEA, 1 cm2) was fabricated by hot-pressing a proton-exchange membrane (NRE211 Nafion membrane, Chemours) between the anode and cathode electrodes with a force of 2 MPa at 135 °C for 20 min. The MEA was assembled into a single cell and connected to fuel cell testing systems (FC5100 and DB630, Chino, Japan). During I–V measurements, the single cell was operated at 80 °C, and the anode and cathode humidifiers were set at 73 °C. The relative humidity (RH) of inlet gases was maintained at 75%. The hydrogen (418 mL min−1) and air (998 mL min−1) pressurized at 50 kPa-G were supplied to the anode and cathode components, respectively. The I–V polarization curves were recorded starting from the low-current region, at current densities of 2, 4, 6, 10, 20, 50, 75, 100, 200, 400, 600, 800, 1000, 1500, 2000 mA cm−2 while measuring for 10 min at each current density.
Results and discussion
Catalyst preparation
In this study, the core–shell structured alloy catalysts (Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB) were prepared by the ASI-RHQ method according to the scheme shown in Fig. 1. The method consists of five main steps such as impregnation, decomposition-reduction (DR), RHQ, acid-treatment, and post heat-treatment (post-HT), as described in ESI.† The impregnation anchors metal precursors to the KB support while producing their highly homogenous dispersion in an aqueous solution without organic impurities. The DR step sequentially converts the anchored precursors to solid intermediates, which may be partially reduced oxides, via dehydration, decomposition and mild reduction processes. Notably, no oxide phases (Co3O4, CoO, MnO2, MnO, NiO, etc.) were detected in the XRD spectra of solid materials after DR (Fig. S1, ESI†). However, they may exist as amorphous phases. The RHQ step transforms the partially reduced solids into alloy nanoparticles at a high temperature under a reducing condition. The acid-treatment removes M species from the surface of alloy nanoparticles to build the Pt-shell, while the post-HT rearranges Pt atoms at the shell surface. For the alloy catalysts, the Pt:M ratios determined by XRD were essentially identical to the ratios estimated from XRF. This reflected the ASI-RHQ enabling us to obtain well-dissolved solid phases from a highly homogenous dispersion of metal precursors on KB. Previously, the preparation condition of the RHQ method was examined for various Pt–Ru catalysts with the ratio of nPt−Ru/(nPt−Ru + nPt−Pt) which was precisely determined by EXAFS using the BL36XU beamline of SPring-8 (Proposals 2017B7901 and 2018A7901).41 Subsequently, this method was expanded to the preparation of Pt-M catalysts.
 |
| | Fig. 1 A general scheme for the ASI-RHQ preparation method of the core–shell structured alloy catalysts. | |
Characterization
XRD patterns of alloy catalysts (Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB) and cPt/C are shown in Fig. 2. Both Pt3.9Co/KB (PDF#01-072-9178) and Pt3.7Ni/KB similar to cPt/C (Pt: PDF#00-004-0802) provide the characteristic diffraction peaks from a disordered face-centred cubic (fcc) structure with a Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.6,11 On the contrary, the Pt3.7Mn/KB (PDF#01-071-9674) exists as an ordered phase which was identified by the additional peaks at 2θ angles of 22.81°, 32.42°, 52.49°, 57.96°, 72.74° and 77.43°.42 Also, Pt3.7Mn/KB has the fcc structure with a Pmm space group which was confirmed by matching the d-spacings of all the observed peaks.
m space group.6,11 On the contrary, the Pt3.7Mn/KB (PDF#01-071-9674) exists as an ordered phase which was identified by the additional peaks at 2θ angles of 22.81°, 32.42°, 52.49°, 57.96°, 72.74° and 77.43°.42 Also, Pt3.7Mn/KB has the fcc structure with a Pmm space group which was confirmed by matching the d-spacings of all the observed peaks.
 |
| | Fig. 2 The XRD patterns of Pt3.7Mn/KB, Pt3.9Co/KB, Pt3.7Ni/KB and cPt/C. Asterisks indicate the ordered phase of Pt3.7Mn/KB. The reference diffraction peak positions of bulk Mn, Co, Ni and Pt are also shown below. | |
A broad 002 peak at ca. 25° is assigned to the KB support. In addition, the diffraction peaks of the alloy catalysts shifted to wider 2θ angles than those of cPt/C, indicating formation of alloy nanoparticles. The shift was smaller for Pt3.7Mn/KB with larger Mn (137 pm) atoms, while it was larger for Pt3.9Co/KB and Pt3.7Ni/KB containing smaller Co (125 pm) and Ni (125 pm) atoms, respectively.43 On the other hand, XRD patterns after DR, RHQ and post-HT for each alloy catalyst are compared in Fig. S1 (ESI†). Using them, we examined shifts in the diffraction peaks after RHQ and post-HT to confirm the Pt-shell formation. The peaks shifted positively after RHQ whereas they shifted negatively after post-HT following the acid treatment. For example, a smaller shift in the 2θ(220) angle (RHQ → post-HT) was extracted for Pt3.7Mn/KB (68.01° → 67.93°), Pt3.9Co/KB (69.34° → 69.17°) and Pt3.7Ni/KB (69.42° → 69.34°). These shifts indicate that dissolution of the surface M species leads to formation of the Pt-shell. The physical properties such as metal compositions, crystalline structures and particle sizes of the alloy catalysts are summarized in Table 1.
Table 1 Physical properties of the prepared alloy catalysts
| Catalyst (PtnM/KB) |
Composition (wt%) |
Structure |
| Pt3.7Mn/KB |
Pt: 50.1%, Mn: 3.8% |
Ord. fcc |
| Pt3.9Co/KB |
Pt: 48.3%, Co: 3.8% |
Dis. fcc |
| Pt3.7Ni/KB |
Pt: 49.0%, Ni: 4.0% |
Dis. fcc |
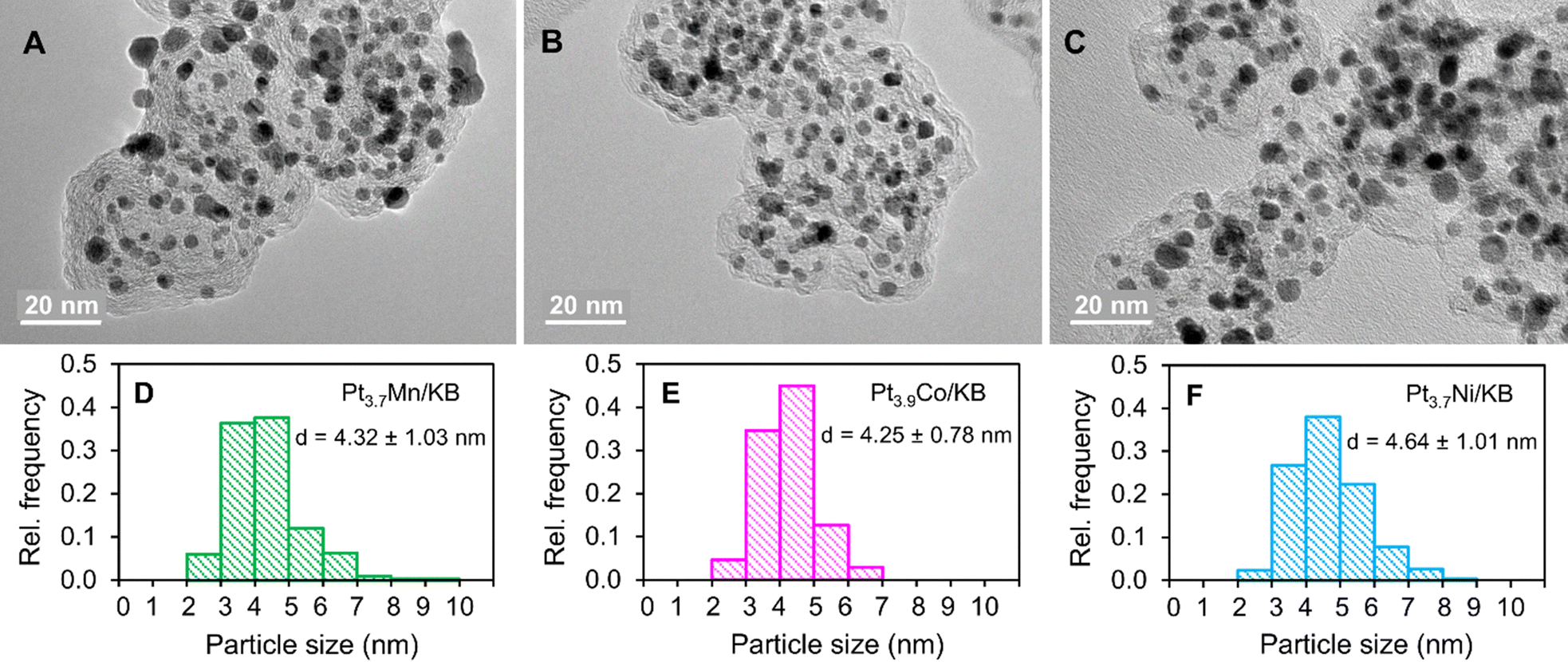
The PtnM (xtotal = 1/(ntotal + 1)) ratios were estimated from XRF results. TG analysis provided a total metal content of 53.9%, 52.1% and 53.0% for Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB, respectively. Based on TG and XRF results, the Pt and M contents in each catalyst were determined, respectively, as given in Table 1. Fig. 3A–C present TEM images of spherically shaped alloy nanoparticles in the prepared catalysts. The average particle sizes (dTEM) estimated from ca. 300 nanoparticles in each catalyst were within the range of 4.2–4.6 nm which are about two times larger than that of cPt/C (2.3 nm). A small number of nanoparticles which were smaller and larger than average also exist in each catalyst.
 |
| | Fig. 3 (A)–(C) TEM images and (D)–(F) particle size distribution histograms of Pt3.7Mn/KB (A) and (D), Pt3.9Co/KB (B) and (E) and Pt3.7Ni/KB (C) and (F) alloy catalysts. | |
We believe that the larger particle sizes of the alloy catalysts have a favourable impact on the lifetime of PEFCs for HDVs. In Fig. 3D–F, the particle size distribution histograms of the alloy catalysts exhibit a narrow distribution, suggesting that the particle sizes were controlled by the RHQ process. Also, TEM images reveal an even dispersion of less agglomerated alloy nanoparticles on the support material, showing potential benefits of the ASI-RHQ preparation method. The XPS analysis was performed to investigate the electronic structure of the surface species of the alloy catalysts before ADT. In the XPS survey spectrum of each catalyst, the signals for Pt, C and O were clearly detected, while the signals for M species were hardly observed which confirms the Pt-shell covers the alloy nanoparticles (Fig. S2, ESI†). In the narrow spectra shown in Fig. 4, the Pt 4f doublets of each catalyst were deconvoluted into the metallic (Pt0) and oxidized (Pt2+ and Pt4+) states.44 The Pt2+ state corresponds to PtO and Pt(OH)2, whereas the Pt4+ state correlates with PtO2. The XPS quantitative analysis showed that the chemical states of Pt were identical on the surfaces of all alloy catalysts. It implies that the surface electronic structure of these alloy catalysts is independent of the M metal species.
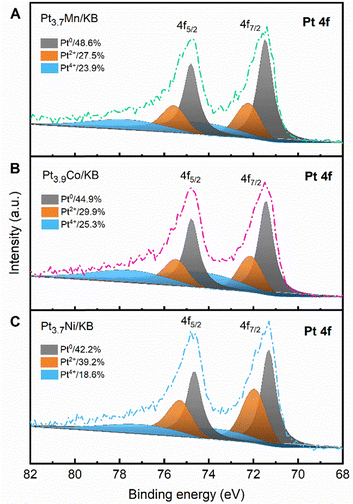 |
| | Fig. 4 The Pt 4f XPS spectra of (A) Pt3.7Mn/KB, (B) Pt3.9Co/KB and (C) Pt3.7Ni/KB catalysts before ADT. | |
Electrocatalytic activity before and after ADT
ORR activities of Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB were evaluated on working electrodes with a catalyst layer (Ptloading: 18 μgPt cm−2 and I/C: 0.5) by a rotating disk electrode (RDE). For comparative studies, the benchmark cPt/C was also studied under the same conditions. The CV curves obtained in N2-saturated 0.1 M HClO4 electrolyte before (initial) and after ADT (70k) are shown in Fig. 5A and B. The initial ECSA values were lower for Pt3.7Mn/KB (56.83 m2 gPt−1), Pt3.9Co/KB (60.27 m2 gPt−1) and Pt3.7Ni/KB (51.08 m2 gPt−1) than cPt/C (78.06 m2 gPt−1) because of their larger particle sizes (4.2–4.6 nm) and modified surface electronic structures. The LSV curves measured at 1600 rpm in O2-saturated 0.1 M HClO4 before and after ADT (70k) are shown in Fig. 5C and D. The initial half-wave potential (E1/2) of the prepared alloy catalysts was more positive by 16–22 mV than that of cPt/C, indicating their high intrinsic ORR activity. The high ORR activity of alloy catalysts stems from the modified electronic structure of surface Pt atoms owing to ligand, strain and ensemble effects induced by M atoms in alloy nanoparticles.45 Kinetic current densities are compared in the Tafel plots at low current density regions. In Fig. 5E, the initial Tafel slopes were 72.39, 70.30, 71.54 and 65.47 mV dec−1 for Pt3.7Mn/KB, Pt3.9Co/KB, Pt3.7Ni/KB, and cPt/C, respectively. As shown in Fig. 5F, the Tafel slopes of the alloy catalysts not only increased but also differentiated from each other after ADT (70k). It shows the changes in their surface chemical states which were initially identical according to the XPS analysis. Among them, Pt3.9Co/KB exhibited the smallest change in its Tafel slope, indicating its stable surface states. The initial SA of Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB catalysts was 708, 978 and 1081 μA cmPt−2, respectively, which are 1.6–2.2 times higher than that of cPt/C (439 μA cmPt−2). In addition, the initial MA of Pt3.9Co/KB (589 A gPt−1), Pt3.7Ni/KB (552 A gPt−1) and Pt3.7Mn/KB (472 A gPt−1) was 1.4–1.7 times higher than that of cPt/C (342 A gPt−1), reflecting their superior ORR activity over that of pure Pt catalysts. The initial activities of the alloy catalysts are comparable, suggesting that catalytic activity is largely independent of the specific alloying metal. This may be attributed to the Pt-shell structures of the alloy catalysts having essentially equivalent surface electronic properties.8 MAs of the alloy catalysts before and after ADT (70k) were compared in Fig. 6A. A relatively low decline in MA was observed for the alloy catalysts tested in this study compared to other studies.12,46 The enhanced durability is attributed not only to the Pt-shell, which inhibits the dissolution of M atoms from the alloy catalysts, but also to the low amount of M atoms present.47–49 In addition to the catalyst characteristics, it might depend on the experimental conditions such as temperature, loading amount and cleanliness of the cell as well as glassware. We thoroughly rinsed the catalyst layer on a GC tip with a fresh electrolyte after every ADT experiment and before each electrochemical measurement. Notably, the lowest loss in MA was observed for the Pt3.9Co/KB (1.7%) compared to Pt3.7Mn/KB (6.4%) and Pt3.7Ni/KB (6.0%) after ADT (70k), indicating its high durability toward ORR. We also examined the practical cell performance of Pt3.9Co/KB under MEA operation conditions to confirm its ORR activity obtained by RDE. The I–V polarization curves were measured on the single cell assembled from MEA (1 cm2), anode (cPt/C, Pt loading: 0.17 mg cm−2) and cathode (Pt3.9Co/KB, Pt loading: 0.17 mg cm−2) electrodes. The measurements were performed at 80 °C and 75% RH, using hydrogen as a fuel and air as an oxidant, under a 50 kPa-G pressure. For comparison, cPt/C was also tested as the cathode catalyst. As shown in Fig. 6B, the Pt3.9Co/KB showed high performances at 0.2 A cm−2 (0.773 V) and at 1.0 A cm−2 (0.697 V) compared to the cPt/C (0.761 V at 0.2 A cm−2 and 0.656 V at 1.0 A cm−2), confirming its high intrinsic activity toward ORR. Additionally, the initial cell performances of Pt3.9Co/KB (ASI-RHQ) and Pt3.6Co/KB (conventional method), prepared using two different approaches, are compared in Fig. S3 (ESI†). The high activity of Pt3.9Co/KB is attributed to ASI-RHQ, which yields homogeneously distributed alloy nanoparticles with relatively uniform sizes. In contrast, the conventional method results in non-uniform particle sizes and greater agglomeration. Briefly, the conventional method consisted of the following steps: (1) formation of Pt nanoparticles on the KB support, (2) precipitation of a cobalt precursor, (3) reduction under a reducing atmosphere, (4) acid-treatment and (5) post-HT. On the other hand, ADT tests on the commercial catalyst (cPt/C) were conducted at 80 °C and 75% RH, using hydrogen as the fuel and air as the oxidant, without back pressure. As shown in Fig. S4 (ESI†), cPt/C exhibited an ECSA loss of nearly 50%, decreasing from 54.6 to 26.1 m2 gPt−1 after 30000 ADT cycles. Based on this result, the durability results observed at 25 °C are considered reasonable.
 |
| | Fig. 5 The CVs (A) and (B), LSVs (C) and (D) and Tafel slopes (E) and (F) of Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB along with cPt/C before (A), (C) and (E) and after (B), (D) and (F) ADT (70k). CVs (at 0 rpm) and LSVs (at 1600 rpm) were measured in the N2- and O2-saturated 0.1 M HClO4 electrolyte, respectively. The Tafel slopes were estimated from the kinetic currents at 0.945–0.983 V. | |
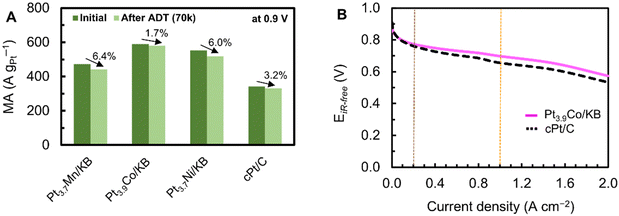 |
| | Fig. 6 (A) MAs at 0.9 V before (initial) and after ADT (70k) of Pt3.7Mn/KB, Pt3.9Co/KB, Pt3.7Ni/KB and cPt/C in O2-saturated 0.1 M HClO4. (B) I–V polarization curves of Pt3.9Co/KB and cPt/C measured at 80 °C, 75% RH, H2/Air 50 kPa-G. | |
Conclusion
In this study, we evaluated the durability of the core–shell structured Pt3.7Mn/KB, Pt3.9Co/KB and Pt3.7Ni/KB catalysts by ADT for 70000 cycles under HDV operation conditions. The alloy catalysts having similar metallic composition, particle size and surface state showed comparable initial activities towards ORR. However, their durability was quite different by ADT evaluation. In comparison to Pt3.7Mn/KB and Pt3.7Ni/KB, the Pt3.9Co/KB well-maintained its MA after ADT (70k), associating with slight variation in the Tafel slope. Therefore, the present study results suggest that the core–shell PtnCo-based alloy catalysts could be key candidates for durable PEFC systems in HDV applications.
Author contributions
G. O. performed synthesis, electrochemical measurement, data curation, formal analysis and manuscript preparation. G. B. and W. N. performed I–V measurements and took TEM images. H. W., K. U. and T. T. provided experimental supervision, data validation and review processes. All authors reviewed the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the ESI.†
Acknowledgements
This work was supported by a grant from New Energy and Industrial Technology Development Organization (NEDO), Japan.
References
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, Nat. Energy, 2021, 6, 462 CrossRef CAS.
- M. De Las, N. Camacho, D. Jurburg and M. Tanco, Int. J. Hydrogen Energy, 2022, 47, 29505 CrossRef.
- S. Li, N. Gjilali, M. A. Rosen, C. Crawford and P.-C. Sui, Int. J. Energy Res., 2022, 46, 11718 CrossRef.
- G. Yang, C.-H. Lee, X. Qiao, S. K. Babu, U. Martinez and J. S. Spendelow, Electrochem. Energy Rev., 2024, 7, 9 CrossRef CAS.
- Fuel Cell and Hydrogen Technology Roadmap: Fuel Cell Roadmap for FCVs/HDVs, NEDO, https://www.nedo.go.jp/content/800020391.pdf.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- C. Lim, A. R. Fairhurst, B. J. Ransom, D. Haering and V. R. Stamenković, ACS Catal., 2023, 13, 14874 CrossRef CAS PubMed.
- V. R. Stamenković, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Marković, J. Am. Chem. Soc., 2006, 128, 8813 CrossRef PubMed.
- C. Wang, M. Chi, D. Li, D. Strmcnik, D. van der Vliet, G. Wang, V. Komanicky, K.-C. Chang, A. P. Paulikas, D. Tripkovic, J. Pearson, K. L. More, N. M. Marković and V. R. Stamenković, J. Am. Chem. Soc., 2011, 133, 14396 CrossRef CAS PubMed.
- V. R. Stamenković, T. J. Schmidt, P. N. Ross and N. M. Marković, J. Phys. Chem. B, 2002, 106, 11970 CrossRef.
- H. Yano, I. Arima, M. Watanabe, A. Iiyama and H. Uchida, J. Electrochem. Soc., 2017, 164, F966 CrossRef CAS.
- Y. Gao, T. Uchiyama, K. Yamamoto, T. Watanabe, S. Tominaka, N. Thakur, R. Sato, T. Teranishi, H. Imai, Y. Sakurai and Y. Uchimoto, ACS Catal., 2023, 13, 10988 CrossRef CAS.
- M. Song, F. Li, Q. Zhang, T. Shen, G. Luo, D. Li and D. Wang, Chem. Eng. J., 2023, 478, 147287 CrossRef CAS.
- X. Wang, L. Hu, K. C. Neyerlin and R. K. Ahluwalia, J. Electrochem. Soc., 2023, 170, 024503 CrossRef CAS.
- R. Ahluwalia, X. Wang, K. Chen, X. Wang and J. S. Spendelow, J. Electrochem. Soc., 2024, 171, 114512 CrossRef CAS.
- S. Cherevko, N. Kulyuk and K. J. J. Mayrhofer, Nano Energy, 2016, 29, 275 CrossRef CAS.
- R. K. Ahluwalia, D. D. Papadias, N. N. Kariuki, J.-K. Peng, X. Wang, Y. Tsai, D. G. Graczyk and D. J. Myers, J. Electrochem. Soc., 2018, 165, F3024 CrossRef CAS.
- B. T. Sneed, D. A. Cullen, R. Mukundan, R. L. Borup and K. L. More, J. Electrochem. Soc., 2018, 165, F3078 CrossRef CAS.
- E. Padgett, V. Yarlagadda, M. E. Holtz, M. Ko, B. D. A. Levin, R. S. Kukreja, J. M. Ziegelbauer, R. N. Andrews, J. Ilavsky and A. Kongkanand, J. Electrochem. Soc., 2019, 166, F198 CrossRef CAS.
- K. Kodama, R. Jinnouchi, T. Suzuki, H. Murata, T. Hatanaka and Y. Morimoto, Electrochem. Commun., 2013, 36, 26 CrossRef CAS.
- K. Kodama, A. Shinohara, N. Hasegawa, K. Shinozaki, R. Jinnouchi, T. Suzuki, T. Hatanaka and Y. Morimoto, J. Electrochem. Soc., 2014, 161, F649 CrossRef CAS.
- N. Macauley, D. D. Papadias, J. Fairweather, D. Spernjak, D. Langlois, R. Ahluwalia, K. L. More, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018, 165, F3148 CrossRef CAS.
- S. G. Ji, H. C. Kwon, T.-H. Kim, U. Sim and C. H. Choi, ACS Catal., 2022, 12, 7317 CrossRef CAS.
- H. Daimon, S. Yamazaki, M. Asahi, T. Ioroi and M. Inaba, ACS Catal., 2022, 12, 8976 CrossRef CAS.
- M. Chisaka, J. Mater. Chem. A, 2024, 12, 18636 RSC.
- R. Nishiizumi, T. Ogawa, K. Sanami, M. Yasutakea, Z. Noda, S. M. Lyth, M. Nishihara, J. Matsuda and K. Sasaki, Int. J. Hydrogen Energy, 2024, 72, 820 CrossRef CAS.
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750 CrossRef CAS.
- K. Okaya, H. Yano, K. Kakinuma, M. Watanabe and H. Uchida, ACS Appl. Mater. Interfaces, 2012, 4, 6982 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yuauth-David, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81 CrossRef CAS PubMed.
- Q. Jia, K. Caldwell, K. Strickland, J. M. Ziegelbauer, Z. Liu, Z. Yu, D. E. Ramaker and S. Mukerjee, ACS Catal., 2015, 5, 176 CrossRef CAS PubMed.
- W. S. Jung and B. N. Popov, ACS Appl. Mater. Interfaces, 2017, 9, 23679 CrossRef CAS PubMed.
- A. Gunnarson, J. D. Bellis, T. Imhof, N. Pfänder, M. Ledendecker and F. Schüth, Chem. Mater., 2023, 35, 2006 CrossRef CAS.
- T. Takeguchi, T. Yamanaka, K. Asakura, E. N. Muhamad, K. Uosaki and W. Ueda, J. Am. Chem. Soc., 2012, 134, 14508 CrossRef CAS PubMed.
- R. Makharia, S. S. Kocha, P. T. Yu, M. A. Sweikart, W. Gu, F. T. Wagner and H. A. Gasteiger, ECS Trans., 2006, 1, 3 CrossRef CAS.
- Y. J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433 CrossRef CAS PubMed.
- S. C. Zignani, E. Antolini and E. R. Gonzalez, J. Power Sources, 2008, 182, 83 CrossRef CAS.
- K. Shinozaki, J. W. Zack, S. Pylypenko, B. S. Pivovar and S. S. Kocha, J. Electrochem. Soc., 2015, 162, F1384 CrossRef CAS.
- S. Treimer, A. Tang and D. C. Johnson, Electroanalysis, 2002, 14, 165 CrossRef CAS.
- T. Nagai, C. Jahn and H. Jia, J. Electrochem. Soc., 2019, 166, F3111 CrossRef CAS.
- PEFC Evaluation Protocol from New Energy and Industrial Technology Development Organization (NEDO), 2023, https://www.nedo.go.jp/content/100963953.pdf and https://www.fc-cubic.or.jp/en/pages/technical-info/.
- D. Kido, M. M. Rahman, T. Takeguchi and K. Asakura, Chem. Lett., 2022, 51, 538 CrossRef CAS.
- M. R. Z. Ghavidel and E. B. Easton, Appl. Catal. B: Environ., 2015, 176–177, 150 CrossRef.
-
P. Atkins, T. Overton, J. Rourke, M. Weller, F. Armstrong and M. Hagerman, Shriver and Atkins' Inorganic Chemistry, 5th edn, 2010, pp. 22–23 Search PubMed.
-
J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data. 1992.
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149, 174705 CrossRef PubMed.
- P. Yu, M. Pemberton and P. Plasse, J. Power Sources, 2005, 144, 11 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS PubMed.
- H. R. Colón-Mercado and B. N. Popov, J. Power Sources, 2006, 155, 253 CrossRef.
- L.-Y. Jiang, A.-J. Wang, X.-S. Li, J. Yuan and J.-J. Feng, ChemElectroChem, 2017, 4, 2909 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Garavdorj
Batnyagt
a,
Waka
Nagano
a,
Hidenobu
Wakita
*ab,
Garavdorj
Batnyagt
a,
Waka
Nagano
a,
Hidenobu
Wakita