
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the UV luminescence of Bi-doped LiYGeO4: a journey from first principles to temperature-dependent photoluminescence
Inês
Proença
*a,
José D.
Gouveia
 b,
Ana V.
Girão
c,
Marco
Peres
def,
Rodrigo
Mateus
df,
Luís C.
Alves
fg,
Duarte M.
Esteves
de,
Ana L.
Rodrigues
fg,
Maria I.
Dias
fg,
Katharina
Lorenz
def,
Luís
Rino
a,
Florinda M.
Costa
a,
Teresa
Monteiro
a and
Joana
Rodrigues
*a
b,
Ana V.
Girão
c,
Marco
Peres
def,
Rodrigo
Mateus
df,
Luís C.
Alves
fg,
Duarte M.
Esteves
de,
Ana L.
Rodrigues
fg,
Maria I.
Dias
fg,
Katharina
Lorenz
def,
Luís
Rino
a,
Florinda M.
Costa
a,
Teresa
Monteiro
a and
Joana
Rodrigues
*a
aDepartamento de Física e i3N, Universidade de Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: inesbrazproenca@ua.pt; joana.catarina@ua.pt
bDepartamento de Física e CICECO – Instituto de Materiais de Aveiro, Universidade de Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal
cDepartamento de Engenharia de Materiais e Cerâmica e CICECO – Instituto de Materiais de Aveiro, Universidade de Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal
dIPFN, Instituto Superior Técnico, Campus Tecnológico e Nuclear, 2695-066 Bobadela, Portugal
eINESC-MN, Instituto de Engenharia de Sistemas de Computadores – Microssistemas e Nanotecnologia, 1000-029 Lisboa, Portugal
fDECN, Instituto Superior Técnico (IST), Campus Tecnológico e Nuclear, 2695-066 Bobadela, Portugal
gC2TN, Instituto Superior Técnico, Campus Tecnológico e Nuclear, 2695-066 Bobadela, Portugal
First published on 28th May 2025
Abstract
Materials exhibiting persistent luminescence (PersL) have garnered attention due to their unique ability to emit light for extended periods after the excitation stops. LiYGeO4 has arisen as a promising host for PersL due to its notable defect abundance. When doped with Bi, it can provide a long-lasting ultraviolet emission, which may find interesting applications in areas such as photodynamic therapy or self-sustained photocatalysis. In the present paper, undoped and Bi-doped LiYGeO4 (0.5 mol%) samples were synthesized by solid-state reaction. X-ray diffraction confirmed the presence of the LiYGeO4 phase, alongside residual yttrium germanate phases. Additionally, we calculated its previously undocumented band structure using hybrid density functional theory to provide new theoretical insights. These calculations indicate that LiYGeO4 has a direct bandgap at the Γ point, further supported by the absorption data. Elemental analyses allowed quantification of the samples, identifying the loss of Li during the synthesis. Room temperature (RT) photoluminescence (PL) showed a strong emission band peaked around 350–360 nm. Despite the extensive research conducted on LiYGeO4:Bi, the mechanisms underlying the PersL phenomenon remain unclear. To address this, we conducted temperature-dependent PL from 17 K to RT, using a 325 nm photon excitation. We propose that, at RT, the observed emission arises from the overlap of the 3P(1)1 → 1S0 and 3P(2)1 → 1S0 Bi3+ intraionic transitions, sublevels of the 3P1 state, which is completely split under the local crystal field generated by the ion environment. However, at cryogenic temperatures, the 3P0 → 1S0 transition is the dominant one. Moreover, PersL emission was achieved for nearly 7 h, with 250 nm photon excitation for 10 min, arising from a contribution of the overlapped 3P(1)1 → 1S0 and 3P(2)1 → 1S0 transitions.
1. Introduction
In recent years, materials exhibiting the optical phenomenon known as persistent luminescence (PersL) have gained significant attention due to their remarkable properties. The main driving force for such interest is the versatility that the unique ability of these materials to emit light for extended periods without continuous excitation can bring to PersL-based innovative applications. Indeed, PersL phosphors have been explored in multiple applications across different stages of maturity, some more well-established, such as emergency signage, safety indications, luminous paint, or watch dials, others still in the research and development phase, e.g. anti-counterfeiting, photocatalysis, data storage, security, sensing, and bio-imaging, among others.1–7Among the numerous PersL emitters explored, considerable research has been undertaken regarding the visible and near-infrared (NIR) spectral regions, however, there is a notable gap in the reported ultraviolet (UV) PersL emitters, despite their potential in several applications such as photodynamic therapy, 3D-printing technology, disinfection, and persistent photocatalysis.4,8,9 So far, in the UVA range (315–400 nm), lithium yttrium germanate (LiYGeO4) appears as a promising host candidate, mainly due to its abundance of intrinsic defects, such as vacancies, antisites, and interstitials that can act as trap defects to mediate PersL emission.10,11 LiYGeO4 has gained significant attention since it is able to provide PersL in different spectral regions depending on the dopant introduced as the emitting centre.11–17 This matrix is particularly important to provide PersL in the UVA spectral region, especially when doped with trivalent bismuth (Bi3+). Indeed, to the best of the authors’ knowledge, LiYGeO4 is the matrix that led to the longest PersL time in this spectral region (nearly 300 h, including photostimulation).18 Moreover, the selection of this specific host is highlighted by the fact that changing its elements (e.g. Li and Ge by Na and Si, respectively) leads to detrimental results either by an undesirable redshift of the emission or by creating traps that are too deep to participate in the PersL mechanism.10,19 Nevertheless, there are still a great number of open questions regarding the fundamental properties of this material. For instance, Qiao et al.10 and Zhou et al.11 estimated its bandgap through hybrid density functional theory (DFT), having obtained values of 6.84 eV and 6.75 eV, respectively. However, these values are considerably different from the experimental ones. Particularly, Shao et al.16 and Shi et al.18 reported values of 4.51 eV and 4.6 eV, respectively, for the bandgap, obtained through diffuse reflectance and absorption analysis. Such differences are likely related to the methods used to analyse the experimental data, which often do not consider the influence of the defect states that are known to be present near the band edges (the so-called tail states) and that have a significant effect on the bandgap energy determination.
In the particular case of UVA PersL, ions with the s2 ground state electronic configuration, as is the case with the Bi3+, have been selected as promising emitting centres. This ion has been studied as a luminescent activator for several decades owing to its versatility of emitting in a wide spectral range, from the UV to the IR, depending on the host in which it is incorporated.20–23 Such characteristics derive from the strong influence that the surrounding environment, i.e., the local crystalline field, has on its energy levels.24–26 Bismuth is a nontoxic heavy metal that presents valence states varying from −3 to +5. While bismuth anions (Bi3−, Bi2−, Bi−) are unsuitable for luminescence applications since they do not show emission, the cations (Bi2+, Bi3+, Bi5+) are viable for such purposes.24 Among those, Bi3+ is the most stable when incorporated in inorganic hosts,25 resulting in PersL emission peaking at ∼350 nm when introduced in LiYGeO4, which can last for several hours after stopping the initial photon excitation, typically at 250 nm.4,10,16,18,27 So far, and based on the findings reported by Shi et al.,18 the optimal doping concentration for achieving the most effective PersL in this material appears to be 0.5 mol% Bi. In fact, Shi et al.18 and Shao et al.16 reported PersL durations of ∼72 h (without any additional stimulation) using this doping concentration. In all the reported cases, the intraionic 3P1 → 1S0 transition of Bi3+ has been pointed out as the one dominating the emission at room temperature (RT) and responsible for PersL.4,10,18
LiYGeO4, an olivine-type material, crystallizes in an orthorhombic phase within the D2h16 space group.16,28,29 Its structure consists of a corner- and edge-sharing of [GeO4] tetrahedra, [LiO6] octahedra, and [YO6] octahedra, connected by oxygen atoms (see Fig. 1). Its cations (Ge4+, Li+, and Y3+) possess, respectively, ionic radii and coordination numbers of rGe4+ = 0.53 Å, CN = 4; rLi+ = 0.76 Å, CN = 6 and rY+ = 0.9 Å, CN = 6.16,18 The Li-ion occupies the 4a site (Wyckoff position), characterized by a Ci symmetry with an inversion centre (centrosymmetric), while the Y-ion is located at a 4c site, possessing a Cs symmetry, without an inversion centre.30 According to Qiao et al.,10 when Bi3+ (rBi3+ = 1.03 Å4,10,16,31) is introduced into the LiYGeO4 matrix, it is mostly located at the Y3+ site due to the similar radii, with the incorporation into the Ge4+ or Li+ sites being negligible, as also expected taking into account the charge mismatch. Additionally, in the same work,10 the authors performed a detailed DFT study regarding possible intrinsic point defects, including vacancies (VLi, VY, VGe, VO1,O2,O3), antisites (LiY, LiGe, YLi, YGe, GeLi, GeRE), and interstitials (Lii, Yi, and Gei). The interstitial defects Yi and Gei were discarded from the discussion since no stable geometries were found for them. Regarding the remaining point defects, the most stable spin configuration was obtained. The formation energies of the defects were evaluated considering different positions of the Fermi level (EF) within the bandgap, but since the materials were synthesized in an oxidizing atmosphere, which corresponds to an EF slightly lower than the middle of the bandgap, the value of 0.5 eV below the middle position was the main focus. They concluded that, in the case of the LiYGeO4, the most stable point defects (and their respective formation energies) are Y2+Li (−0.9 eV) > Ge+Y (−0.57 eV) ≈ LiiY (−0.57 eV) > Ge3+Li (0.32 eV) > V−Li (0.66 eV).10
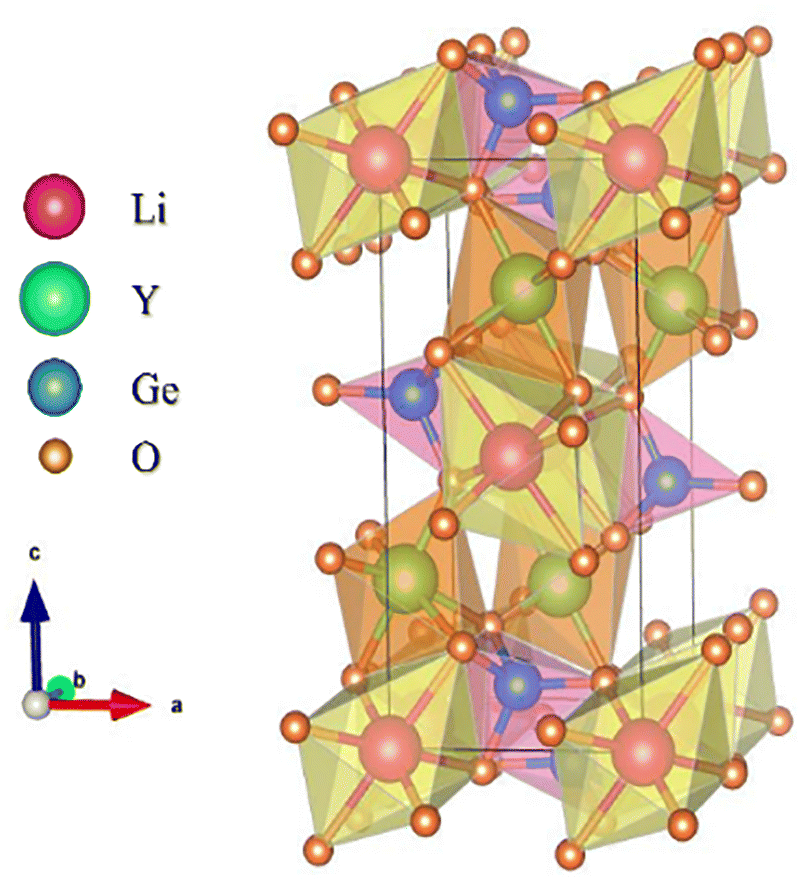 | ||
| Fig. 1 Schematic representation of the LiYGeO4 unit cell. The model was created using the free program VESTA and the data were adapted from the CIF file of mp-504948 using the information from the 04-002-3479 card. | ||
In its trivalent charge state, Bi3+ presents the electronic configuration [Xe] 4f14 5d10 6s2, with the ground level corresponding to 1S0. When excited, the s1p1 electron configuration leads to the formation of an upper 1P spin-singlet state and a lower 3P spin-triplet state, with the latter further split by the spin–orbit interaction, resulting in 3P0, 3P1, and 3P2 states in increasing energy. The non-degenerate 1P level results in the 1P1 state.4,18,20,21 Upon UV photon excitation, the 1S0 → 3P0 transition is strictly forbidden due to spin and parity selection rules, which frequently makes it too weak to be observed, except at cryogenic temperatures (T < 80 K).4,25 However, the 1S0 → 1P1 transition is spin-allowed, while the 1S0 → 3P1 and 1S0 → 3P2 transitions become partially allowed due to spin–orbit coupling and admixture of the wave functions by phonon coupling.4
Although already evaluated in other hosts such as Y2O3 or La2O3 for more than three decades,24,32–34 to date, no temperature-dependent luminescence studies have been reported on Bi-doped LiYGeO4, which is considered a fairly new material that is gaining momentum lately, essentially due to its PersL properties. Hence, in this study, nominally undoped LiYGeO4 (LYG) and 0.5 mol% Bi-doped LiYGeO4 (LYGBi) samples were produced by solid-state reaction and the thermal, structural, elemental, and optical properties were investigated by thermogravimetry (TGA), X-ray diffraction (XRD), nuclear reaction analysis (NRA), particle-induced X-ray emission (PIXE), Rutherford backscattering spectroscopy (RBS), elastic backscattering spectrometry (EBS), diffuse reflectance (DR), RT steady-state photoluminescence (PL) and PL excitation (PLE), temperature-dependent PL (from 17 K to RT), thermoluminescence (TL) as well as afterglow measurements. Valuable insights were obtained with the temperature-dependent PL, allowing us to identify that the main transition observed at RT arises from the mixture of the transitions from the 3P1 sublevels, which are completely split by the crystal field, to the 1S0 ground state, while at cryogenic temperatures the 3P0 → 1S0 transition is the dominant one. These results highlight the importance of a detailed temperature-dependent PL study on the correct assignment of the observed optical transitions, which can have important implications in material optimization and its further applications. Regarding PersL, it was seen that this phenomenon could be attained when the samples were excited with 250 nm (4.96 eV) photons for 10 min, while longer wavelengths were seen to be ineffective for PersL activation. The observed emission can be ascribed to a combination of the 3P(1)1 → 1S0 and 3P(2)1 → 1S0 transitions. Additionally, hybrid DFT calculations were performed, providing the band structure determination with bandgap energy that closely matches the value obtained experimentally via diffuse reflectance measurements.
2. Experimental details
2.1 Materials preparation
In the present paper, nominally undoped and 0.5 mol% Bi-doped LiYGeO4 samples were prepared by a high-temperature solid-state reaction. The quantity of Bi doping was selected based on previous studies performed by the authors in which the nominal Bi amount varied (0.1, 0.5, 1 and 5 mol%, not shown), as well as on formerly reported research,18 which concluded that the maximum PersL emission was achieved at 0.5 mol% of Bi.18 The raw materials Li2CO3 (99%, Panreac), Y2O3 (99.99%, Thermo Scientific Chemicals), GeO2 (99.998%, Thermo Scientific Chemicals) and Bi2O3 (99.8%, Alfa Aesar) were weighed according to the stoichiometric ratio to obtain LiYGeO4 and LiYGeO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bi (0.5 mol%) samples. The precursors were mixed in an agate ball mill with ethanol for 4 h. Then, the mixtures were left to dry on a hot plate at 90 °C overnight to evaporate all the ethanol. Afterwards, the mixtures were transferred to alumina crucibles and fired at 800 °C for 2 h in an oven (heating rate of 5 °C min−1). After this step, the powders were ground and homogenized in an agate mortar and pressed into pellets with a 13 mm diameter (5-ton uniaxial press, 2 min). These pellets were subjected to a thermal annealing treatment in air at 1000 °C for 40 h in an oven (heating rate of 5 °C min−1). Multiple samples were prepared for each batch, and these batches were repeated in the same conditions to assess the homogeneity and reproducibility of the materials. The analysed properties were found to be consistent, and the data presented in this paper are representative of all the samples.
:Bi (0.5 mol%) samples. The precursors were mixed in an agate ball mill with ethanol for 4 h. Then, the mixtures were left to dry on a hot plate at 90 °C overnight to evaporate all the ethanol. Afterwards, the mixtures were transferred to alumina crucibles and fired at 800 °C for 2 h in an oven (heating rate of 5 °C min−1). After this step, the powders were ground and homogenized in an agate mortar and pressed into pellets with a 13 mm diameter (5-ton uniaxial press, 2 min). These pellets were subjected to a thermal annealing treatment in air at 1000 °C for 40 h in an oven (heating rate of 5 °C min−1). Multiple samples were prepared for each batch, and these batches were repeated in the same conditions to assess the homogeneity and reproducibility of the materials. The analysed properties were found to be consistent, and the data presented in this paper are representative of all the samples.
2.2 Materials characterization
Thermal characterization of the materials was performed in a Hitachi Nexta STA300, placing 15.692 mg in an alumina crucible, under a mix of O2 and N2 (air composition) and using a ramp temperature program mode between 24–1180 °C, with a rate of 10 °C min−1.The crystalline structure of the samples was assessed by XRD, conducted under a PANalytical X'Pert PRO diffractometer operating in the Bragg–Brentano configuration and with CuKα radiation (λ = 1.54056 Å). DR was performed in a UVVIS JASCO V-780, with an integrating sphere JASCO ISN-901i, in the wavelength range 190–1600 nm with a bandwidth of 5 nm and a speed of 100 nm min−1.
The elemental and stoichiometric composition of the samples was characterized through ion beam analysis techniques. All the measurements were conducted with the help of the 2.5 MV Van de Graaff accelerator installed at the Laboratory of Accelerators and Radiation Technologies (LATR) of Instituto Superior Técnico (IST) using MeV H+ or He+ beams.35 Distinct events are simultaneously observed from the particle spectra. The contents of light elements such as O or Li are commonly evaluated through the corresponding elastic backscattering yields via EBS. The contents of heavier elements such as Y, Ge or Bi are followed through Rutherford backscattering yields via RBS. Simultaneously, the depth profiling and contents of Li may be obtained via NRA by following the yield of the 7Li(p,α)4He nuclear reaction.36,37 This reaction has a high Q value (17.35 MeV), avoiding the superposition of the NRA yield with those induced by the EBS and RBS events.38 The collection of particle spectra was performed using a dedicated chamber equipped with two unsealed Si PIN diode detectors (Hamamatsu; depletion layer thickness of 300 μm) with an energy resolution of 14 keV. For NRA, the detector was positioned at a scattering angle of −165°, and the nuclear reaction 7Li(p,α)4He was used to determine the Li concentration along the sample's depth, while for EBS and RBS, the detector was placed at +165°; in this case, a higher signal amplification of the detection system was used to enhance the individual analysis of the EBS and RBS yields. The particle spectra were acquired simultaneously, with a total integrated beam charge of 5 μC using a proton beam collimated to a dimension of 0.5 × 0.5 mm2, with an energy of 2.1 MeV, at which the cross-section of the 7Li(p,α)4He nuclear reaction reaches a maximum of approximately 5 mb sr−1.36,37 Using the Oxford Microbeams nuclear microprobe beamline for focusing a 2 MeV proton beam down to 5 × 5 μm2 and scanning a sample area up to 2640 × 2640 μm2, PIXE spectra were collected with a 30 mm2 SDD detector. The deconvolution and quantitative analysis with the particle and PIXE spectra were carried out using the NDF39,40 and GUPIXWIN41 software, respectively.
Steady-state PL and PLE spectroscopy measurements were conducted at RT using Fluorolog-3 Horiba Scientific setup, equipped with a 450 W Xe lamp coupled to a monochromator as the excitation source, a double additive grating Gemini 180 monochromator (1200 grooves mm−1 and 2 × 180 mm) in the excitation side and a triple grating iHR550 spectrometer in the emission (1200 grooves mm−1 and 550 mm). The intensity of the PersL with time (afterglow) was carried out in the same equipment, using a kinetics mode. The samples were excited with the same Xe lamp for 10 minutes with 250 nm photons, after which the excitation was stopped and the decay curves were acquired. Additionally, temperature-dependent PL measurements were recorded on a selected sample using as excitation the 325 nm (∼3.81 eV) line of a continuous helium–cadmium (He–Cd) laser (I0 < 0.6 W cm−2). In this case, the samples were placed on a cold finger He cryostat and the luminescence was dispersed by a SPEX 1704 monochromator (1 m, 1200 grooves mm−1), being detected with a water-cooled Hamamatsu RCA C31034 photomultiplier. The temperature was varied from 17 K up to RT. These experiments ensured optical alignment at a 90-degree geometry between sample irradiation and signal detection. The presented spectra were corrected to the spectral response of the measurement systems. In addition, the Jacobian transformation was considered for the quantitative analysis of the temperature-dependent PL data.
The thermoluminescence (TL) measurements were performed using a Risø thermoluminescence/optically-stimulated luminescence reader (TL/OSL-DA-20), manufactured by DTU Physics, at the Luminescence Dating Laboratory of IST. A 90Sr/90Y β-source (with a nominal activity of 825 MBq and a dose rate in quartz of 0.062 ± 0.001 Gy s−1) was used to excite the samples for 200 s. The TM–TSTOP method was employed to identify the number of traps present in the samples, by monitoring the temperature position of the first peak (TM) in the glow curve as a function of the pre-heating temperature TSTOP, which was varied between 323 and 523 K.42 After each irradiation and pre-heating, a full TL scan was acquired from RT up to 673 K at a constant heating rate of 2 K s−1.
2.3 Modelling and computational details
The spin-polarized first-principles calculations were performed using the VASP code43 at the HSE06 level of theory,44 with the fraction of exact exchange in the hybrid functional set to 0.30 to allow the calculated bandgap (6.01 eV) to match the experimental one. The bandgaps calculated with different fractions of exact exchange (α) in the hybrid functional, which were tested in preliminary calculations, are 5.59 eV (α = 0.25) and 6.42 eV (α = 0.35). The range-separated hybrid functional HSE06 was used for all the calculations to maximise the agreement with the experimental results. This functional is known to produce essentially the same results as PBE0,45 as we verified by calculating the band diagram of LiYGeO4 and finding a band structure that qualitatively resembles the ones given in the literature,10,11 which capture the most important band features but show an underestimated bandgap. This way, we obtain a more reliable band structure of this material,46,47 which confirms the standard-DFT results in the literature, and was essential for determining the positions of defect-related levels in the bandgap of LiYGeO4.The simulations were run on a 1 × 2 × 2 LiYGeO4 supercell, with periodic boundary conditions in all Cartesian directions. The explicitly treated electrons were 2s1 for Li, 4s2 4p6 5s2 4d1 for Y, 4s2 4p2 for Ge, 2s2 2p4 for O, and 5d10 6s2 6p3 for Bi, while the remaining core electrons were tackled using the projector augmented-wave method.48 For all calculations except those of the band structure, the Brillouin zone was sampled using only the k-point Γ, given the extremely high computational cost of hybrid DFT calculations when using the plane-wave formalism.49 The cutoff value for the plane-wave basis set was 415 eV. The force and energy convergence criteria for structural and electronic relaxation were set to 0.01 eV Å−1 and 10−5 eV, respectively. The lattice parameters of bulk LiYGeO4 were obtained by optimizing atomic positions, cell shape and cell volume of a unit cell of the material, using the residual minimization method with direct inversion in the iterative subspace, which is ideal for systems with more than a few tens of degrees of freedom that are already close to their ground state structure. In turn, electronic orbitals were optimized with the blocked-Davidson scheme, which is the default in VASP, and their occupancies were determined using a Gaussian smearing with a width of 0.01 eV.
3. Results and discussion
3.1 Band structure calculations
The lattice parameters calculated for LiYGeO4 are a = 11.19 Å, b = 6.34 Å, and c = 5.07 Å. To the extent of our knowledge, the band structure of LiYGeO4 has been calculated with standard GGA functionals only,10 which are famously known to underestimate the bandgap of materials. Here, using the HSE06 functional, we intend to obtain a more reliable band structure of this material, which is shown in Fig. 2. The plot is remarkably similar to the one found by Qiao et al.,10 albeit with a significantly wider bandgap, 6.01 eV, which is almost twice as much as the one calculated at the PBE level. Qualitatively, both methods predict the bandgap to be a direct one at the Γ point, and the top of the valence band to be rather flat, especially along the Γ–Z direction of k-points. | ||
| Fig. 2 Calculated band structure of LiYGeO4 using the HSE06 hybrid functional. The indicated energy is the calculated bandgap value, the blue/red lines indicate the occupied/unoccupied electronic states, and the horizontal dashed line lies at the Fermi energy. | ||
The most favourable location of Bi atoms in the LiYGeO4 structure, as well as its most stable charge state, are at a Y site and Bi3+, respectively.10 The calculations of Qiao et al.10 further showed that the formation energy of BiY is at least 1.0 eV lower than that of the stoichiometric isomer BiLi–LiY. Here, we verified this fact and obtained a difference of 1.24 eV between the formation energies of these two defects. Furthermore, we went a step further and compared BiY with BiGe–GeY, having concluded that the latter is thermodynamically disadvantageous by 2.90 eV. Thus, we completely covered the three possibilities of a Bi atom replacing any cation in LiYGeO4.
Upon substitution of a single Y atom by a Bi one, the surrounding structure undergoes nearly no relaxation. In fact, the relaxation energy (the energetic difference between the BiY model after and before atomic position optimization) is a mere 0.25 eV. Furthermore, the average Y–O distance in LiYGeO4 was estimated at 2.27 Å, while the average BiY–O one is 2.35 Å, i.e., only 3% larger. Given the low Bi content of the LYGBi synthesized here (0.5 mol%), this substitution should have a negligible effect on the experimentally determined lattice parameters. Note that, although a BiY defect alone has Cs symmetry (the same as the Y sites in the pristine material), the presence of neighbouring defects that are easily found on LiYGeO4, such as YLi antisites, or Li vacancies or interstitials, further reduces the local symmetry to C1. Nonetheless, this should have the same impact on the splitting of energy levels as a Cs environment.
We calculated the band energies at the Γ point of bulk LiYGeO4 and with a BiY defect, which are displayed in Fig. 3(a). Upon replacement of a Y atom by a Bi one in the simulation supercell, a localized state, associated with Bi3+ s orbitals, appears within the bandgap of the material, 4.91 eV below the conduction band minimum (CBM). In other words, the transfer of an electron from the ground state level of Bi3+ to the CBM should be associated with an energy of around 5 eV.
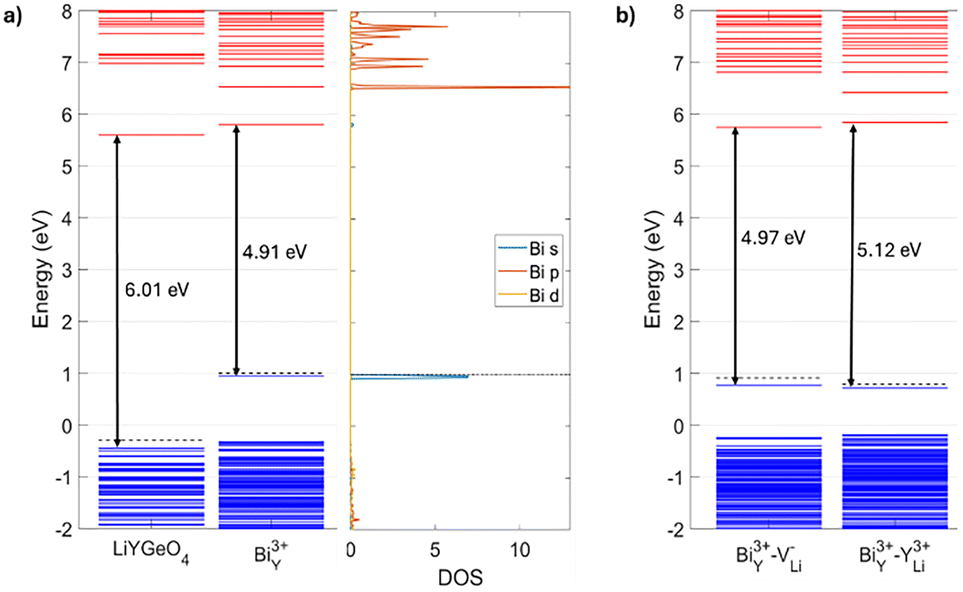 | ||
| Fig. 3 (a) Band energies at the Γ point of bulk LiYGeO4, or with a Bi3+Y defect. The density of states of the defective model, projected onto the Bi atom, is also shown. (b) Band energies at the Γ point of models in which a Bi3+Y has a neighbouring V−Li or a Y3+Li. In the band energy diagrams, blue (red) lines denote occupied (unoccupied) levels. The dashed line is drawn at the Fermi energy. | ||
Given the high probability of occurrence of Li vacancies (VLi) and YLi antisites, it should be common to find a BiY near such a defect. In fact, given that (i) Li vacancies are very common, (ii) the most stable position of a Bi atom is at a Y site, and (iii) in pristine LiYGeO4 the defect with the lowest formation energy is YLi, it stands to reason that a combination of these three defects, yielding BiY–YLi, should be observable. Such a combination can be regarded as a region of the material in which a dopant Bi atom occupies a Y site, having pushed the Y atom towards a Li-vacant site. For this reason, we performed calculations using models with a combination of BiY and a neighbouring VLi or YLi. According to the results, both are stable – the reconstruction of the lattice around the defective area is minimal and accompanied by a small relaxation energy, of a few tenths of an eV, as in the case of a simple BiY. In addition, the formation energies of BiY–VLi and BiY–YLi are lower than that of BiY. In particular, the formation energy of BiY near a Li vacancy is lower by around 0.8 eV than in the absence of such a vacancy. This implies that BiY is thermodynamically easier to form when the material has Li vacancies or, equivalently, that BiY are attracted to regions of the material where there are Li vacancies. The most stable charge states of the two composite defects are Bi3+Y–V−Li and Bi3+Y–V3+Li. The respective band energies at the Γ point are illustrated in Fig. 3(b) and appear to be very similar to those of BiY. Finally, according to our theoretical calculations, the energetic difference between having two BiY replacing two nearest Y atoms and having two infinitely separated BiY is a mere 1 meV. This implies that BiY defects practically do not interact (i.e., they do not attract or repel each other), and therefore can be distributed in any way in the LiYGeO4 lattice.
3.2 Thermal, structural and elemental analysis
Thermal analysis was used to assess the behaviour of the precursor powders in the mixture with the desired stoichiometry with temperature variation, as the TG and DTG curves shown in Fig. 4. | ||
| Fig. 4 Thermogravimetric and corresponding derivative curves for the LiYGeO4 synthesis, starting from the precursor's powders in the used stoichiometry. | ||
The TG curve clearly shows four major weight loss steps within areas A, B, D and E, and very small weight gains are identified within the temperature range of area C. The first weight loss of around 0.4% (area A) can be associated with the evaporation of residual water as well as CO2 release.50 The second major weight loss of 0.3% (area B) is related to the decomposition of Li2CO3 and conversion to LiO2, followed by further rearrangement between the precursors/products (area C) since weight gain is observed within area C.51 During this reorganization of the overall crystalline host, evaporation of lithium and/or of the remaining cations may take place to some extent.51,52 The variation in weight observed around 1000 °C results from the crystallization of the final LiYGeO4 (area D), culminating in a small weight loss (area E), possibly due to the further sublimation of lithium.52,53
XRD was performed to assess the phase composition of the prepared samples, and the results are depicted in Fig. 5(a). Table 1 presents the phase composition of the LYG and LYGBi samples. As can be seen by inspecting both the figure and the table, the produced samples exhibit a polyphasic nature. Nevertheless, the LiYGeO4 (ICDD card 04-002-3479) crystalline phase is the predominant one (∼88%), and the estimated lattice parameters are a = 11.1359 Å, b = 6.3305 Å and c = 5.0628 Å for LYG, and a = 11.1346 Å, b = 6.3315 Å and c = 5.0622 Å for LYGBi, well in line with previously reported ones,11,53 and notably close to those resulting from our calculations. The tetragonal Y2Ge2O7 (ICDD card 04-001-6776) phase is also present in small amounts in both samples. Besides, sample LYG also exhibits small percentages of Y2GeO5 (ICDD card 00-023-1484) and Li2GeO3 (ICDD card 00-029-0806) phases, while sample LYGBi shows evidence of the Bi2GeO5 (ICDD card 04-011-8707) phase, whose presence is most likely the result of an inhomogeneity in the distribution of Bi during the mixture of the precursors. Note that the Y2Ge2O7, Y2GeO5 and Li2GeO3 phases are related to the LiYGeO4 instability and the breakdown of the stoichiometry at high temperatures, especially because of the sublimation of lithium mentioned above.14,30 The Wyckoff positions of the ions in the LiYGeO4 phase were also confirmed using XRD data, and these positions are compatible with the previous description, supporting that Li is located at the 4a site and Y occupies the 4c site. By comparing the peak position of the graphs in Fig. 5(a), a slight shift (∼0.0079°) is observed. This shift is likely attributed to the incorporation of Bi into the lattice, as this ion is expected to occupy the position of the Y ion and has a slightly larger radius.
 | ||
| Fig. 5 (a) X-ray diffraction patterns of the produced samples. The data are vertically shifted for clarity. (b) PIXE data for both LYG and LYGBi samples. For the LYG sample, the data demonstrate the existence of Bi contamination. The data are shifted vertically for clarity. (c) Particle spectra collected with a 2.1 MeV H+ beam. The data correspond to the EBS with the NRA spectra obtained with the detector located at −165° and their respective fits (red and orange lines). In addition to identifying the different contributions, the input associated with the signal resulting from the overlap of two backscattered particles hitting the detector simultaneously, referred to as pile-up, is also identified. (d) EBS spectra, and the different contributions estimated from the fit using the NDF software. The latter two spectra were obtained with a similar detector positioned at 165° and with higher signal amplification to achieve higher resolution in this region, thereby enabling more precise identification of the different barriers. | ||
| Sample | Composition | Crystal system | % |
|---|---|---|---|
| LYG | LiYGeO4 | Orthorhombic | 88.6 |
| Y2Ge2O7 | Tetragonal | 6.5 | |
| Y2GeO5 | Monoclinic | 3.2 | |
| Li2GeO3 | Orthorhombic | 1.7 | |
| LYGBi | LiYGeO4 | Orthorhombic | 88.4 |
| Y2Ge2O7 | Tetragonal | 5.1 | |
| Bi2GeO5 | Orthorhombic | 6.5 |
The stoichiometric analysis of the LYG and LYGBi samples was carried out using the simultaneous analysis of the particle yields. Since the samples are composed of light and heavy elements, different approaches were carried out. The quantification of the individual contributions of the heavier elements, namely Y, Ge and Bi, was assessed by a PIXE analysis, while the quantification of light elements was assessed through the combination of EBS and NRA. As shown in Fig. 5(b), from the PIXE spectra, it is possible to confirm the presence of Y, Ge, and Bi in both samples. Using the GUPIXWIN software code,41 the concentration of these elements was accurately estimated (see Table 2). Besides the host and dopant elements, small quantities of other undesired elements (Fe and Zn) were identified, with percentages lower than 0.01%, so their information was not included in Table 2 but can be seen in Fig. 5(b). It is worth noting that the presence of Bi in the undoped sample is related to trace contamination of the raw Y2O3 and Li2CO3 precursors, as reported in the corresponding Certificate of Analysis provided by the supplier (analysis method: mass spectrometry). The same is valid for the remaining impurities that were identified, most of them originating from trace contaminants of the used raw materials. The relative concentrations of Y, Ge and Bi estimated from the fit of PIXE spectra were subsequently used to define the fitting model for EBS and NRA spectra in order to estimate the concentration of all elements using the NDF software. In defining the fitting model, contaminations of Cr, Fe, K, and Ca, identified in the PIXE spectra, were not considered due to low concentrations. It is also noteworthy that the individual EBS and NRA spectra fittings, represented by the red and orange lines in Fig. 5(c) and (d), were performed simultaneously. These fits, in addition to allowing the evaluation of stoichiometry, clearly suggest that the samples exhibit high homogeneity in depth. EBS events enable identifying and quantifying the light elements by analysing the barriers associated with corresponding backscattered particles.37,54,55 In addition, RBS yields enable a similar analysis involving heavier elements.56 However, as can be seen in Fig. 5(c), in the case of the complex matrices involving different light and heavy elements, it is challenging to accurately identify and quantify the elemental concentration of each element due to the overlap of the barriers, as in the case of Bi, Y and Ge, or due to their low signal, as in the case Li. In particular, for the Li, to address this limitation, complementary NRA analysis of the 7Li(p,α)4He nuclear reaction was performed. As can be seen in Fig. 5(c), the high Q-value of the reaction38 enables the clear separation of the induced alpha particle yield from those associated with the proton yields elastically backscattered by heavier elements such as Y, Ge, Bi, and by light elements such as O and Li. Despite the much lower sensitivity of EBS for Li quantification, a correspondence is obtained from the analysis of both Li-EBS and Li-NRA yields.
| Sample | Elements | wt% | Nominal wt% |
|---|---|---|---|
| LYG | Li | 1.63 ± 0.46 | 2.97 |
| Y | 38.75 ± 0.88 | 38.08 | |
| Ge | 32.91 ± 0.75 | 31.10 | |
| O | 26.64 ± 0.76 | 27.40 | |
| Bi | 0.066 ± 0.002 | — | |
| LYGBi | Li | 2.40 ± 0.19 | 2.97 |
| Y | 37.75 ± 0.37 | 38.08 | |
| Ge | 32.00 ± 0.31 | 31.10 | |
| O | 27.65 ± 0.32 | 27.40 | |
| Bi | 0.213 ± 0.002 | 0.45 | |
Furthermore, Fig. 5(b) suggests that the Bi content in the intentionally doped sample differs from the nominal one (0.213 wt% vs. ∼0.447 wt%). The lower content is likely related to the loss of this element during the synthesis process.57 As can be seen, a significant amount of Li is also lost during the synthesis process, especially in the case of sample LYG, resulting in Li vacancies in the structure, as already anticipated by other works, although no final quantification was performed.14,58 The remaining cations display wt% close to the nominal ones, with negligible differences that can be accounted for by the error of the measurement. Nevertheless, as pointed out by the thermal analysis, reorganization of the crystalline network takes place during the synthesis, and some losses may have occurred during the process.
Please note that the values presented in Table 2 represent average values obtained from the probed samples. Consistent with the XRD data, which confirmed the presence of the Bi2GeO5 phase, the Bi distribution in the LYGBi sample is non-uniform. Analysis of various random spots revealed significant variations in Bi content, while the Ge and Y concentrations remained relatively constant throughout the sample.
3.3 Optical assessment
 | ||
| Fig. 6 (a) Absorption (grey filled lines), photoluminescence excitation (blue and red full-filled lines, monitored at 358 nm (∼3.46 eV)) and photoluminescence (solid lines, acquired under 250 nm (4.96 eV) and 300 nm (∼4.13 eV) photon excitation) spectra obtained for the studied samples. The spectra were shifted vertically for the sake of clarity. (b) Bandgap energy estimation from the Tauc plot method. The linear part of the plot is extrapolated to the energy axis. (c) Energy level diagram of the Bi3+ ion, the vertical arrow represents the 3P1 → 1S0 transition of the Bi3+ ion. Note that the presence of the Bi3+-related transitions in the LYG sample is related to the presence of Bi contaminants in the raw precursors. | ||
Besides this absorption, both samples exhibit a lower intensity absorption band at ∼212 nm (∼5.85 eV), followed by a strong asymmetric absorption band peaked around 250 nm (∼4.96 eV). The latter was seen to widen and increase its intensity with increasing dopant concentration, as confirmed here by the high intensity of this band in the LYGBi sample, which suggests a correlation with the presence of the Bi3+ ion. By comparing the present data with the energy positions estimated by Qiao et al.10 for the most common defects present in this compound, the low-intensity band at ∼5.85 eV (∼0.68 eV below the CBM) is compatible with the energy position estimated for GeLi antisites, while the strong absorption band is most likely related to the 1S0 → 1P1 transition of the Bi3+ ions.24 As stated above, when considering the Russell–Saunders (LS) coupling scheme, only the transitions from the 1S0 ground state to the 1P1 excited state are allowed by the electric dipole. However, the strong spin–orbit coupling leads to interaction between the 3P1 and 1P1 levels, resulting in mixed states. Thus, the ΔS = 0 rule is relaxed, and the 1S0 → 3P1 becomes partially allowed. Likewise, the interaction with the lattice phonons also makes the 1S0 → 3P2 transition possible.24 As so, and according to the diagram in Fig. 6(c), the two strongly asymmetric bands centred at ∼310 nm and 296 nm (∼4 eV and ∼4.19 eV, respectively) observed in the PLE spectra are probably associated with the 1S0 → 3P1 and 1S0 → 3P2 transitions, exhibiting an energy separation around ∼100–200 meV (considering the peak position of the maximum of the excitation bands and their similar full widths at half maximum). The broadening of the bands can be associated with the vibronic levels that originate from the interaction with the lattice phonons.24 Moreover, one must take into account that, when introduced in the Y sites, the Bi ions will be placed at a Cs or C1 symmetry, as mentioned in Section 3.1, in which the three-fold degenerate 3P1 and 1P1 and the five-fold degenerate 3P2J-levels will be completely split.24
Additionally, a closer inspection of the PLE spectra also reveals the presence of an absorption band at 245 nm (∼5.06 eV), which can be assigned to a metal-to-metal charge transfer (MMCT) transition. Indeed, in the case of Bi ions, besides the 6s2 → 6s16p1 interconfigurational transitions, other luminescence signals are commonly identified in different matrices, as is the case of this MMCT transition.24,25,61 As reported by other authors, above the 1S0 → 1P1 band, but below the absorption of the host, a further band is generally encountered and attributed to an MMCT state. This band generally occurs at fairly low energy and may overlap or replace the absorption bands associated with the Bi3+ ion intraionic transitions.24 The MMCT state results from the interaction between the Bi3+ levels and the electronic levels of host metal cations having d0 or d10 configuration, which, in the present case, corresponds to Ge4+ ([Ar] 3d10).25,61 Boutinaud61 reported a quantitative model for estimating the position of the MMCT energies of Bi3+ ions in different oxide hosts based on the electronegativity of the metal cations (Mn+), the coordination number of the Bi3+ and the host Mn+, as well as the shortest interatomic distances between them.24,61 Note that such a model is only indicative for oxides. Therefore, the electronegativities of the host cations strongly influence the energies of charge transfer excitations.24,61 Considering Ge4+ as the metal cation involved in this transition, this could be represented as Bi3+(6s2)/Ge4+(d10) → Bi4+ (6s1)/Ge3+(s1).61 Qiao et al.10 reported that in the case of LiYGeO4, this band should occur around 225–234 nm, which fairly matches the value obtained for the PLE band at 245 nm (∼5.06 eV). Such an MMCT transition corresponds to the transfer of an electron from the ground level (1S0) of the Bi3+ ion to the CBM of the host.24,61 Assuming that this is indeed the present case, this band allows us to locate the 1S0 level within the bandgap of the material reported here. The value of 5.06 eV is compatible with the diagrams in Fig. 3 and with the energy estimated by Qiao et al.,10 which reported that the Bi3+ ground level was ∼5 eV below the CBM. According to Boutinaud's models,61,62 when this transition is present at energies higher than that of the 1S0 → 3P1 transition, excitation through this band can be followed by emission either from the MMCT or via the 3P1,0 → 1S0, after transfer of the absorbed energy. Such findings may also explain why the PersL reported here (see Section 3.3.4) is mostly promoted when the samples are excited with photons with wavelengths ∼230–250 nm and not when excited directly to the ion excited levels, as was also reported in previous works,18 indicating that the PersL mechanism must be mediated by the conduction band of the host.
In the case of the PL acquired at both 250 nm and 300 nm photon excitation, the emission spectra display a maximum at ∼358 nm in both cases, with only a slight shift in the peak position, which will be further discussed in the next section. This value is in agreement with the previous reports in the literature regarding Bi-doped LiYGeO4 samples, also presenting a similar spectral shape.16,18 As already mentioned in the Introduction section, such a band has been attributed to the 3P1 → 1S0 transition, with no further discussion. Nevertheless, the possibility of different Bi centres contributing to the observed transitions cannot be ruled out, as it is known that the sample contains other phases. Indeed, these additional phases may also present luminescence bands associated with the incorporation of Bi3+ ions. However, as noted by previous authors, the observed UV emission is unlikely to arise from the inclusion of Bi3+ in the secondary phases Y2Ge2O4 or Bi2GeO5, in which this ion is known to emit in the green63 or infrared64 spectral regions. Conversely, for the Y2GeO5 phase, Bi3+-related emission is expected to occur in the UV spectral region, peaking at approximately 370 nm.63,65 Therefore, a minor contribution from this phase cannot be completely ruled out. The same applies to the Li2GeO3 phase. To the best of the authors’ knowledge, there are currently no reports on Bi incorporation as a dopant in this matrix, thus, no comparison could be made that allowed us to discard such influence fully. Notwithstanding, as the Y2GeO5 and Li2GeO3 phases are not present in the LYGBi sample, and the obtained spectra for the two samples are very similar, if this contribution is indeed present, it should be negligible.
 | ||
| Fig. 7 (a) Temperature-dependent PL spectra of the LYG sample acquired with 325 nm photon excitation. (b) Spectra obtained at 17 K, 110 K and 290 K presenting the respective Gaussian deconvolution; here the data and the respective Gaussian fits were vertically shifted for clarity. The pink, blue and purple Gaussian fits correspond to the 3P0 → 1S0, 3P(1)1 → 1S0 and 3P(2)1 → 1S0 transitions, respectively, as can be seen in the diagrams alongside. The green lines correspond to the cumulative fit. (c) Temperature dependence of the integrated intensity for each transition, normalized to the maximum value of each transition. The dashed lines correspond to the fit of each transition behaviour according to eqn (1) and (2). (d) Schematic representation of the Bi3+ configurational coordinate diagram model for the ground and the first three excited states (3P0, 3P(1)1 and 3P(2)1). | ||
To further analyse the observed transitions, the spectra were deconvoluted into Gaussian functions, as displayed in Fig. 7(b). At 17 K, the emission maxima are shifted to longer wavelengths, peaked at ∼380 nm (∼3.26 eV), when compared to the RT spectrum. It can also be seen that the band is strongly asymmetric, pointing to the overlap of more than one electronic transition. Moreover, at 17 K, the emission is much broader than in the case of RT. The spectral deconvolution allowed us to conclude that the emission is dominated by a component peaked at ∼3.13 eV (pink line in Fig. 7(b)), with a small contribution from another band at ∼3.38 eV (blue line in Fig. 7(b)). For temperatures higher than 70 K, the low-energy component is no longer observed. Note that up to 70 K, the overall PL intensity decreases, while from 90 K to RT, an increase in the intensity was progressively observed (Fig. 7(a)), accompanied by a high-energy shift up to RT. At 110 K, the main emission shifts to high energy, compared to emission at 17 K, with the emission band now peaked at 363 nm (∼3.42 eV). By deconvolution of this band, the appearance of a higher-energy component (at ∼3.5 eV, violet line) becomes clear, which is seen to contribute to a shift in the overall emission towards higher energies, in line with what was displayed in Fig. 6(a) for the RT data. When raising the temperature to RT the main maximum is peaked at 358 nm (∼3.46 eV) and composed of two components, which were attributed to the splitting of the 3P1 level due to the crystal field, since the ion is placed in a Cs/C1 symmetry, thus explaining the slight shift in the peak of the emission band depending on the excitation wavelength observed in Fig. 6(a).
Although the transition from the 3P0 level to the ground state is not expected to contribute to absorption due to its forbidden nature, previous works on other matrices24,33,34,66 have reported that at lower temperatures (<80 K), and as a result of mixing the 3P0 and 3P1 states induced by the second derivatives of strains caused by nearby point defects, the 3P0 → 1S0 transition can be observed. Indeed, several authors have reported this transition as the dominant one at low temperatures.24,66 Therefore, in the present case, the low-energy component is suggested to correspond to such a transition. According to Boulon et al.,33 despite being considered metastable, the 3P0 level does make an important contribution to the total emission at very low temperatures, in which the 3P1 → 1S0 transition has a low intensity due to the lower population of the 3P1 level at such temperatures. As the temperature increases, the thermal population from the 3P0 to the 3P1 levels takes place, resulting in the increased intensity of the 3P1 → 1S0 transition, which is accompanied by a blueshift of the overall PL emission. The temperature dependence of the PL intensity of each transition is depicted in Fig. 7(c), highlighting the mentioned behaviour. Moreover, it also shows that both high-energy transitions increase their intensity up to RT, presenting nearly the same intensity dependence, which is a strong indication that both are related to the 3P1 excited level, being assigned to the lower energy 3P(1)1 sub-level and the intermediate 3P(2)1 at higher energies, respectively. This is further corroborated by their energy separation (∼140 meV). A schematic representation of these levels is shown in Fig. 7(d). Hence, the emission observed at RT is composed of an overlap of the 3P(1)1 → 1S0 and 3P(2)1 → 1S0 transitions, explaining the broad and asymmetric band that is typically observed in this material.16,18,27
However, it is important to note that the above interpretation represents the explanation that seems most likely to the authors, taking into account the data presented here. Nevertheless, the possibility that the emission observed at RT arises from an overlap of the 3P2 and 3P1 states cannot be entirely excluded. Even so, the fact that the temperature behaviour is the same for the two higher energy components strongly supports the present assumption.
In general, and as shown in Fig. 7(c), by exciting the sample with 325 nm photons, the intensity of the 3P0 → 1S0 decreases over the entire temperature range in which this transition is observed (17 K to 70 K), while, as already mentioned, the 3P(1)1 → 1S0 and 3P(2)1 → 1S0 increase up to RT. As mentioned above, this behaviour indicates the presence of a thermal population mechanism among the energy levels of the Bi3+ ion. As so, the effect of thermal depopulation of the 3P0 state on its luminescence intensity, i.e., the non-radiative competitive processes involved in its temperature dependence, can be expressed by the classical model,1,67,68
 | (1) |
 | (2) |
| Transition | E pop (meV) | E depop (meV) |
|---|---|---|
| 3P0 → 1S0 | — | 9.3 ± 3.5 |
| 3P(1)1 → 1S0 | 36.0 ± 1.6 | — |
| 3P(2)1 → 1S0 | 25.1 ± 15.6 | — |
 | ||
| Fig. 8 Thermoluminescence study of the two produced samples (LYG and LYGBi) by the TM–TSTOP method (panels a and b) and by glow curve computational fitting (panels c and d). The insets in the upper panels show the evolution of the temperature position of the first, TM, as a function of the pre-heating temperature, TSTOP, used to gradually depopulate the traps. | ||
Bearing this in mind, the curve was fitted to the sum of four general-order69 TL peaks using the TL-Fitter software, which is freely available on GitHub,70 according to the following model:
 | (3) |
Considering two of the common acceptance criteria for TL fitting, namely the TL figure of merit below 3% and the acceptability parameter below 0.40, the fitting quality is quite good.71,72 The obtained activation energies Ei are shown in Fig. 8(c) and (d) next to the corresponding peaks. Comparing both samples, the estimated energy values differ at most by 0.12 eV, which further suggests that the traps are indeed similar in these samples. The differences observed in the glow curves are likely related to differences in the trap concentrations and in the frequency parameters, which depend on the structural properties of the samples.
It should be noted that the extraction of unique parameters from glow curve fitting is notoriously difficult due to the ambiguity in the role of different parameters, considering the number of free variables. However, the energies obtained from the fits are in close agreement with those reported in the literature for this material. For instance, Lyu and Dorenbos73 report a trap in LiYGeO4 with an activation energy of 0.76 eV, which is attributed to Bi2+ and compares very well with trap 1 in the present work. On the other hand, the results are also quite close to several traps estimated by Qiao et al.10 using DFT. In particular, trap 1 can tentatively be assigned to the +/0 level of GeLi, (predicted at an energy of 0.71 eV), trap 2 to the +/0 level of Lii (predicted at an energy of 0.84 eV) and traps 3 and 4 to the +/− and 2 +/+ levels of YLi (predicted at energies of 1.08 and 1.25 eV, respectively). Notably, the latter is expected in LYGBi, as the incorporation of Bi into the lattice may displace Y atoms, which in turn may occupy Li sites.16,18 Moreover, the obtained activation energy for trap 1 is also close to the absorption band measured in these samples (absorption band identified at ∼212 nm/∼5.85 eV, which corresponds to ∼0.68 eV below the CBM), as discussed above, which further stresses its role in the PersL phenomenon.
 | ||
| Fig. 9 Decay profiles acquired for 7 h after exciting the samples with 250 nm photons for 10 min. The illustration of the pellets depicts a representation of the reduction in emission over time. The percentage intensity values shown in the figure are relative to the initial intensity measured at t = 0 s (immediately after the excitation was turned off). | ||
Analysing the decay profiles reveals a rapid intensity drop within the initial minutes, with a ∼97% reduction after 1 hour, after which the decrease of the PersL intensity becomes more gradual over time. To investigate the mechanism behind this behaviour, namely the defect trap level distribution involved in capturing and de-trapping of the photoexcited carriers, the measured data were analysed. The best fit of the decay curves was obtained using a combination of stretched exponential functions (see Fig. 9), following the equation,
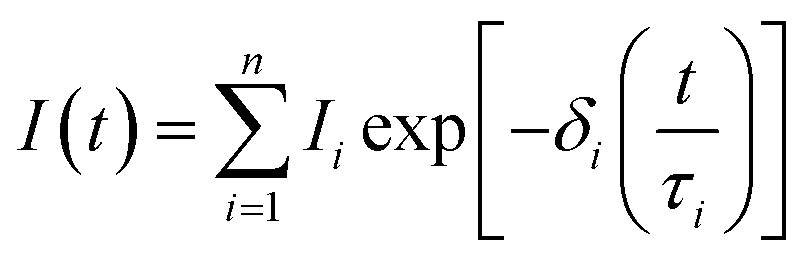 | (4) |
| Sample | τ (s) | δ |
|---|---|---|
| LYG | τ 1 = 3.0 ± 0.5 | δ 1 = 0.733 ± 0.159 |
| τ 2 = 136.8 ± 4.1 | δ 2 = 0.198 ± 0.005 | |
| τ 3 = 342.5 ± 7.7 | δ 3 = 1.000 ± 0.047 | |
| τ 4 = 892.2 ± 62.2 | δ 4 = 0.750 ± 0.015 | |
| LYGBi | τ 1 = 137.9 ± 4.0 | δ 1 = 0.74 ± 0.03 |
| τ 2 = 310.1 ± 4.3 | δ 2 = 0.32 ± 0.45 | |
| τ 3 = 312.9 ± 90.7 | δ 3 = 0.77 ± 0.04 | |
| τ 4 = 343.1 ± 1.6 | δ 4 = 0.78 ± 0.07 | |
A simplified scheme of the process described can be seen in Fig. 10. Since the PersL is only observed when using 250 nm excitation light, which, as indicated by the PLE results, corresponds to the excitation of the MMCT transition, it is reasonable to consider that the electrons must be excited from the ground level of the Bi3+ (1S0) state to the CBM (process labelled as 1 in Fig. 10). After which the electron should be captured by the intrinsic point defects located below the CBM (process 2). Once the excitation is removed, thermal energy from the surrounding environment stimulates the release of the trapped electron, starting the de-trapping process. The shallower the depths of the trap, the faster the detrapping process, corresponding to shorter decay times. This process will then populate the excited states of Bi3+, either through the conduction band or directly from the defect levels (process 3). The electrons then return to the ground state of the emitting levels, emitting photons, and resulting in the observed UVA PersL (process 4).
 | ||
| Fig. 10 Schematic diagram illustrating the PersL phenomenon in LiYGeO4: Bi upon excitation in the MMCT band. The depths of the defect traps were labelled according to the absorption and TL results. The PersL phenomenon involves exciting electrons from the ground state of Bi3+ (1S0) to the CBM (process 1), followed by their capture by defect traps below the CBM (process 2). Once the excitation ceases, thermal energy releases the trapped electrons to the CB again or directly to excited states of Bi3+, leading to the emission of UVA PersL as the electrons return to the ground state (processes 3 and 4). | ||
4. Conclusions
Lithium yttrium germanate samples, nominally undoped and intentionally doped with 0.5 mol% of Bi, were prepared by high-temperature solid-state reaction, at 1000 °C for 40 hours. The band structure of the material was calculated using the HSE06 hybrid functional, which predicts a room temperature direct bandgap at the Γ point with a value of 6.01 eV. The calculations also confirm that the most favourable position for the Bi atoms in the LiYGeO4 structure is at the Y site, in line with previous reports of other authors. Additionally, Bi located at the Y site appears to be thermodynamically easier to form when there are Li vacancies, both by replacing Y atoms and by pushing Y atoms to Li vacancies, as it tends to be attracted to regions where these vacancies are present.The produced samples are predominantly composed of the LiYGeO4 crystalline phase, although small amounts of other phases were also present, resulting from the instability of the LiYGeO4 at high temperatures, which may result in the breakdown of the desired stoichiometry due to lithium sublimation.
Elemental analysis revealed a Bi contamination in the undoped sample, introduced due to trace contaminations in the percursor powders. Furthermore, the Bi content in the intentionally doped sample differs from the nominal one, likely due to its evaporation during the synthesis process, which was highlighted in our thermal analysis. The loss of Li was confirmed by the elemental analysis, leading to the formation of Li vacancies.
The optical analysis showed very similar results for both samples. Absorption measurements indicate a steep slope band at shorter wavelengths, suggesting a direct bandgap for the material with a value of ∼6.05 eV, consistent with our hybrid DFT results. Under 250 nm and 300 nm photon excitation, the PL spectra revealed a strong UVA emission band peaked at ∼358 nm. Further assessment of the temperature-dependent PL provided valuable insights into the origin of the observed luminescence. Under 325 nm photon excitation, which corresponds to direct excitation of the Bi ion's excited levels, when the sample is cooled to cryogenic temperatures, a redshift in the peak position (regarding RT) is observed, accompanied by a broadening of the band. By analysing the behaviour of the PL spectra with rising temperature, we propose that at 17 K the emission arises from an overlap of the 3P0 → 1S0 and 3P(1)1 → 1S0 transitions, with the 3P0 → 1S0 transition being the dominant one. As the temperature increases, a thermal population from the 3P0 to the 3P1 levels occurs, leading to increased intensity of the 3P(1)1 → 1S0 transition, accompanied by a blueshift of the overall PL emission. At temperatures above 70 K, the 3P0 component is no longer observed, and the development of another component, from 110 K, was detected, which we associate with the 3P(2)1 → 1S0 transition. Thus, at RT, the PL emission peaked at 358 nm is attributed to the splitting of the 3P1 level and is composed of the overlap of the 3P(1)1 → 1S0 and 3P(2)1 → 1S0 transitions.
It was also inferred that PersL is only observed by direct excitation into the MMCT state, which corresponds to the excitation of an electron from the Bi3+ ground state to the minimum of the host conduction band, as this phenomenon could only be activated when photons with 250 nm were used to illuminate the samples, matching the estimated energy for this transition. Longer wavelengths were not effective for the observation of this phenomenon. As so, attaining PersL requires the excitation of an electron from the 1S0 ground state of the Bi3+ to the CBM, which is then captured by the trap levels. Afterglow measurements revealed that the intensity of the PersL could be detected for about 7 hours after turning off the excitation. The fact that 4 different decay times were needed to describe the afterglow profile is well in line with the 4 trap levels identified by thermoluminescence, suggesting that those are indeed the traps involved in the trapping/detrapping process that mediates the observed PersL. Thermoluminescence measurement allowed us to conclude that at least four traps were present in the analysed samples, with activation energies of 0.71–0.77 eV, 0.86–0.89 eV, 1.05–1.08 eV and 1.11–1.23 eV. The fact that the estimated energy values exhibit only a slight difference (less than 0.12 eV), suggests that similar traps are involved in the two samples, however with distinct defect concentrations, with different trapping/detrapping kinetics. Those traps were tentatively assigned to the +/0 level of GeLi, the +/0 level of Lii and the +/− and 2 +/+ levels of YLi, as previously reported by DFT calculation.
Author contributions
Inês Proença: formal analysis, investigation, writing – original draft, writing – review & editing. José D. Gouveia: formal analysis, investigation, writing: review & Editing Ana V. Girão: investigation, writing: review & editing. Marco Peres: formal analysis, investigation, writing: review & editing. Rodrigo Mateus: investigation, writing: review & editing. Luis C. Alves: investigation, writing: review & editing. Duarte M. Esteves: formal analysis, investigation, writing: review & editing. Ana L. Rodrigues: investigation. Maria I. Dias: formal analysis, investigation. Katharina Lorenz: formal analysis, writing: review & editing. Luís Rino: data curation, formal analysis, writing: review & editing. Florinda M. Costa: conceptualization, formal analysis, writing: review & editing. Teresa Monteiro: conceptualization, formal analysis, writing: review & editing. Joana Rodrigues: conceptualization, methodology, formal analysis, funding acquisition, investigation, supervision, writing: original draft, review & editing.Data availability
Data for this article are available at Mendeley Data at https://data.mendeley.com/datasets/zgw9v3h4nw/1.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by national funds through the FCT – Portuguese Foundation for Science and Technology under the i3N project UIDB/50025/2020 & UIDP/50025/2020 & LA/P/0037/2020 and project 2023.00054.RESTART. This work was also developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI 10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI 10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI 10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). J. Rodrigues acknowledges FCT for Programme Stimulus of Scientific Employment Support, grants 2022.00010.CEECIND and CEECINSTLA/00005/2022, J. D. Gouveia acknowledges the FCT grant 2023.06511.CEECIND, in the scope of the Individual Call to Scientific Employment Stimulus – 6th Edition, and D. M. Esteves acknowledges FCT for the PhD grant 2022.09585.BD (DOI: 10.54499/2022.09585.BD). The authors thank MSc Celeste Azevedo and M. R. Soares (CICECO – Aveiro Institute of Materials) for the diffuse reflectance and XRD experiments, respectively. Acknowledgements are also due to the project 101217446 POEMS, which is supported by the Chips Joint Undertaking and its members, including top-up funding by FCT.Notes and references
- M. S. Batista, J. Rodrigues, M. S. Relvas, J. Zanoni, A. V. Girão, A. Pimentel, F. M. Costa, S. O. Pereira and T. Monteiro, Optical Studies in Red/NIR Persistent Luminescent Cr-Doped Zinc Gallogermanate (ZGGO:Cr), Appl. Sci., 2022, 12, 2104 Search PubMed.
- D. Poelman, D. Van Der Heggen, J. Du, E. Cosaert and P. F. Smet, Persistent phosphors for the future: fit for the right application, J. Appl. Phys., 2020, 128, 240903 CAS.
- H. Shi and Z. An, Ultraviolet afterglow, Nat. Photonics, 2019, 13, 74–75 CAS.
- X. Wang and Y. Mao, Emerging Ultraviolet Persistent Luminescent Materials, Adv. Opt. Mater., 2022, 10, 2201466 Search PubMed.
- S. Wu, Y. Li, W. Ding, L. Xu, Y. Ma and L. Zhang, Recent Advances of Persistent Luminescence Nanoparticles in Bioapplications, Nano-Micro Lett., 2020, 12, 1–26 CrossRef PubMed.
- J. Xu and S. Tanabe, Persistent luminescence instead of phosphorescence: history, mechanism, and perspective, J. Lumin., 2019, 205, 581–620 CrossRef CAS.
- C. Liao, F. Liu, H. Wu, H. Wu, L. Zhang, G. Hui Pan, Z. Hao, X. Jun Wang and J. Zhang, Red-to-ultraviolet light chargeable Sm2+-activated deep-red persistent phosphor for simultaneous bioimaging and bio-temperature sensing, Acta Mater., 2024, 279, 120322 Search PubMed.
- W. Wang, Z. Sun, X. He, Y. Wei, Z. Zou, J. Zhang, Z. Wang, Z. Zhang and Y. Wang, How to design ultraviolet emitting persistent materials for potential multifunctional applications: a living example of a NaLuGeO4:Bi3+,Eu3+ phosphor, J. Mater. Chem. C, 2017, 5, 4310–4318 RSC.
- P. Xiong and M. Peng, Recent advances in ultraviolet persistent phosphors, Opt. Mater. X, 2019, 2, 100022 Search PubMed.
- Z. Qiao, X. Wang, C. Heng, W. Jin and L. Ning, Exploring Intrinsic Electron-Trapping Centers for Persistent Luminescence in Bi3+-Doped LiREGeO4 (RE = Y, Sc, Lu): Mechanistic Origin from First-Principles Calculations, Inorg. Chem., 2021, 60, 16604–16613 Search PubMed.
- S. Zhou, B. Lou, C. G. Ma and M. Yin, First-principles study on persistent luminescence mechanism of LiYGeO4:Eu3+, J. Rare Earths, 2023, 41, 1519–1524 CrossRef CAS.
- T. Dai, G. Ju, Y. Lv, Y. Jin, H. Wu and Y. Hu, Luminescence properties of novel dual-emission (UV/red) long afterglow phosphor LiYGeO4:Eu3+, J. Lumin., 2021, 237, 118193 Search PubMed.
- H. Cui, Y. Cao, Y. Zhang, L. Li, G. Li, S. Xu, Y. Wang, J. Zhang and B. Chen, Upconversion luminescence thermal enhancement and emission color modulation of LiYGeO4:Er3+/Yb3+ phosphors, J. Alloys Compd., 2022, 927, 167107 CrossRef CAS.
- M. Misevicius, E. Griniuk and R. Ramanauskas, Solid-state synthesis of LiYGeO4 and luminescence properties of Dy-doped samples, Mater. Chem. Phys., 2023, 306, 128082 Search PubMed.
- T. Zhang, H. Guo, Q. Shi, J. Qiao, C. Cui, P. Huang and L. Wang, Investigation of a novel Pr3+-activated LiYGeO4 phosphor with red long-persistent luminescence, J. Lumin., 2024, 267, 120382 CrossRef CAS.
- P. Shao, P. Xiong, Y. Xiao, Q. Chen, Y. Sun, N. Yan, D. Chen and Z. Yang, Novel spectral band: ultraviolet A mechanoluminescence from Bi3+-doped LiYGeO4, J. Mater. Chem. C, 2022, 102, 16670–16678 Search PubMed.
- P. Shao, P. Xiong, D. Jiang, Z. Chen, Y. Xiao, Y. Sun, D. Chen and Z. Yang, Tunable and enhanced mechanoluminescence in LiYGeO4:Tb3+ via Bi3+ → Tb3+ energy transfer, J. Mater. Chem. C, 2022, 11, 2120–2128 Search PubMed.
- J. Shi, X. Sun, S. Zheng, X. Fu, Y. Yang, J. Wang and H. Zhang, Super-Long Persistent Luminescence in the Ultraviolet A Region from a Bi3+-Doped LiYGeO4 Phosphor, Adv. Opt. Mater., 2019, 7, 1900526 Search PubMed.
- X. Wang, C. Heng, Z. Qiao and L. Ning, First-principles study on luminescent properties of Bi3+-doped ALuGeO4 (A = Li, Na): insights into effects of host cation on emission wavelength, J. Lumin., 2022, 244, 118700 CrossRef CAS.
- Q. Sun, T. Sakthivel, B. Devakumar, S. Wang, L. Sun, J. Liang, S. J. Dhoble and X. Huang, Realizing bright blue-red color-tunable emissions from Gd2GeO5:Bi3+,Eu3+ phosphors through energy transfer toward light-emitting diodes, J. Lumin., 2020, 222, 117127 Search PubMed.
- G. Annadurai, L. Sun, H. Guo and X. Huang, Bright tunable white-light emissions from Bi3+/Eu3+ co-doped Ba2Y5B5O17 phosphors via energy transfer for UV-excited white light-emitting diodes, J. Lumin., 2020, 226, 117474 CrossRef CAS.
- W. Zhao, S. Qi, J. Liu and B. Fan, Luminescence properties and energy transfer of a color-tunable Y2GeO5:Bi3+,Eu3+ phosphor, J. Alloys Compd., 2019, 787, 469–475 CrossRef CAS.
- A. Krasnikov, E. Mihokova, M. Nikl, S. Zazubovich and Y. Zhydachevskyy, Luminescence spectroscopy and origin of luminescence centers in Bi-doped materials, Crystals, 2020, 10, 208 CrossRef CAS.
- H. C. Swart and R. E. Kroon, Ultraviolet and visible luminescence from bismuth doped materials, Opt. Mater. X, 2019, 2, 100025 CAS.
- Y. Tang, M. Deng, M. Wang, X. Liu, Z. Zhou, J. Wang and Q. Liu, Bismuth-Activated Persistent Phosphors, Adv. Opt. Mater., 2023, 11, 2201827 CrossRef CAS.
- P. Dang, D. Liu, G. Li, A. A. Al Kheraif and J. Lin, Recent Advances in Bismuth Ion-Doped Phosphor Materials: Structure Design, Tunable Photoluminescence Properties, and Application in White LEDs, Adv. Opt. Mater., 2020, 8, 1901993 CrossRef CAS.
- W. J. Zhang, L. Zheng, S. C. Qi, J. X. Li, D. M. Xue, X. Q. Liu and L. B. Sun, Maintaining the configuration of a light-responsive metal-organic framework: LiYGeO4:Bi3+-incorporation-induced long-term bending through short-time light irradiation, J. Mater. Chem. A, 2023, 11, 17484–17490 RSC.
- X. Wang, Y. Chen, P. A. Kner and Z. Pan, Gd3+-activated narrowband ultraviolet-B persistent luminescence through persistent energy transfer, Dalton Trans., 2021, 50, 3499–3505 RSC.
- T. Lyu, P. Dorenbos, C. Li, S. Li, J. Xu and Z. Wei, Unraveling electron liberation from Bi2+ for designing Bi3+-based afterglow phosphor for anti-counterfeiting and flexible X-ray imaging, Chem. Eng. J., 2022, 435, 135038 CrossRef CAS.
- B. Fan, W. Zhou, S. Qi and W. Zhao, Eu3+-doped NaYGeO4: a novel red-emitting phosphors for ultraviolet or blue chips excited white LEDs, J. Solid State Chem., 2020, 283, 121158 CrossRef CAS.
- M. Esquivel-Gaon, S. Anguissola, D. Garry, A. D. C. Gallegos-Melgar, J. M. Saldaña, K. A. Dawson, A. De Vizcaya-Ruiz and L. M. Del Razo, Bismuth-based nanoparticles as the environmentally friendly replacement for lead-based piezoelectrics, RSC Adv., 2015, 5, 27295–27304 RSC.
- A. M. Van De Craats and G. Blasse, The quenching of bismuth(III) luminescence in yttrium oxide (Y2O3), Chem. Phys. Lett., 1995, 243, 559–563 CrossRef CAS.
- G. Boulon, C. Pedrini, M. Guidoni and C. Pannel, Étude de la cinétique des centres luminogènes Bi3+ dans les cristaux, J. Phys., 1975, 36, 267–278 CrossRef CAS.
- G. Boulon, Processus de photoluminescence dans les oxydes et les orthovanadates de terres rares polycristallins activés par L’ion Bi3+, Le J. Phys., 1971, 32, 333–347 CAS.
- E. Alves, K. Lorenz, N. Catarino, M. Peres, M. Dias, R. Mateus, L. C. Alves, V. Corregidor, N. P. Barradas, M. Fonseca, J. Cruz and A. Jesus, An insider view of the Portuguese ion beam laboratory, Eur. Phys. J. Plus, 2021, 136, 684 CrossRef.
- V. Paneta, A. Kafkarkou, M. Kokkoris and A. Lagoyannis, Differential cross-section measurements for the 7Li(p,p0) 7Li, 7Li(p,p1) 7Li, 7Li(p,α0)4He, 19F(p,p0) 19F, 19F(p,α0)16O and 19F(p,α1,2)16O reactions, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 288, 53–59 CrossRef CAS.
- I. A. E. A. Nuclear Data Section, Ion Beam Analysis Nuclear Data Library.
- D. R. Tilley, J. H. Kelley, J. L. Godwin, D. J. Millener, J. E. Purcell, C. G. Sheu and H. R. Weller, Energy levels of light nuclei A = 8, 9, 10, Nucl. Phys. A, 2004, 745, 155–362 CrossRef.
- N. P. Barradas and C. Jeynes, Advanced physics and algorithms in the IBA DataFurnace, Nucl. Instrum. Methods Phys. Res., Sect. B, 2008, 266, 1875–1879 CrossRef CAS.
- N. P. Barradas, C. Jeynes and R. P. Webb, Simulated annealing analysis of Rutherford backscattering data, Appl. Phys. Lett., 1997, 71, 291–293 CrossRef CAS.
- J. L. Campbell, N. I. Boyd, N. Grassi, P. Bonnick and J. A. Maxwell, The Guelph PIXE software package IV, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 3356–3363 CrossRef CAS.
- S. W. S. Mckeever, Analysis of Complex Thermoluminescence Glow-Curves: Resolution into individual peaks, physica status solidi (a), 1980, 62, 331–340 CrossRef CAS.
- G. Kresse and J. Furthmü, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- T. M. Henderson, A. F. Izmaylov, G. Scalmani and G. E. Scuseria, Can short-range hybrids describe long-range-dependent properties?, J. Chem. Phys., 2009, 131, 044108 CrossRef PubMed.
- J. D. Gouveia and J. R. B. Gomes, Effect of surface composition on the stability of Ti- and V-based oxycarbide and oxynitride MXenes, Mater. Today Phys., 2024, 46, 101481 CrossRef CAS.
- I. Proença, J. D. Gouveia, M. R. Soares, L. Rino, F. M. Costa, T. Monteiro and J. Rodrigues, Next-Generation Multilevel Anti-Counterfeiting Technologies: Deciphering the UV/Violet Luminescence of Bi-Doped Yttrium Germanates, Phys. Status Solidi RRL, 2025, 2500102 CrossRef.
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. D. Gouveia and J. Coutinho, Can we rely on hybrid-DFT energies of solid-state problems with local-DFT geometries?, Electron. Struct., 2019, 1, 015008 CrossRef CAS.
- L.-M. Loembe, Z. Fu, W. Wang and H. Wang, Sol–Gel Synthesis and Characterization of Lithium Yttrium Oxide, Int. J. Res. Eng. Technol., 2015, 2321–7308 Search PubMed.
- P. Pasierb, R. Gajerski, S. Komornicki and M. Rekas, Structural properties and thermal behavior of Li2CO3–BaCO3 system by DTA, TG and XRD measurements, J. Therm. Anal. Calorim., 2001, 65, 457–466 CrossRef CAS.
- I. Koseva, V. Nikolov, N. Petrova, P. Tzvetkov and M. Marychev, Thermal behavior of germanates with olivine structure, Thermochim. Acta, 2016, 646, 1–7 CrossRef CAS.
- K. Cheng, C. Li, H. Xiang, Y. Sun and L. Fang, LiYGeO4: Novel low-permittivity microwave dielectric ceramics with intrinsic low sintering temperature, Mater. Lett., 2018, 228, 96–99 CrossRef CAS.
- S. Bashkint and H. T. Richards, Proton Bombardment of the Lithirrm Isotopes*, Phys. Rev., 1951, 84, 1124–1129 Search PubMed.
- A. F. Gurbich, SigmaCalc recent development and present status of the evaluated cross-sections for IBA, Nucl. Instrum. Methods Phys. Res., Sect. B, 2016, 371, 27–32 Search PubMed.
- R. Mateus, M. B. Costa, L. C. Alves, N. Catarino, R. C. da Silva, M. Dias, M. Guedes, A. C. Ferro and E. Alves, Ion beam analysis of Li-Sn alloys for fusion applications, Nucl. Instrum. Methods Phys. Res., Sect. B, 2021, 486, 55–62 CrossRef CAS.
- B. Kindler, B. Lommel, E. Celik Ayik, A. Huebner, J. Steiner and V. Yakusheva, Comparison of Bi2O3-targets produced by thermal evaporation and RF magnetron sputtering, EPJ Web Conf., 2023, 285, 10001 CrossRef CAS.
- T. Lyu and P. Dorenbos, Towards information storage by designing both electron and hole detrapping processes in bismuth and lanthanide-doped LiRE(Si,Ge)O4 (RE = Y, Lu) with high charge carrier storage capacity, Chem. Eng. J., 2020, 400, 124776 CrossRef CAS.
- P. Kubelk and F. Mulk, A contribution to the optics of pigments, Z. Technol. Phys, 1931, 12, 593 Search PubMed.
- P. Makuła, M. Pacia and W. Macyk, How To Correctly Determine the Band Gap Energy of Modified Semiconductor Photocatalysts Based on UV-Vis Spectra, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef.
- P. Boutinaud, Revisiting the spectroscopy of the Bi3+ ion in oxide compounds, Inorg. Chem., 2013, 52, 6028–6038 CrossRef CAS PubMed.
- P. Boutinaud and E. Cavalli, Predicting metal-to-metal charge transfer in closed-shell transition metal oxides doped with Bi3+ or Pb2+, Chem. Phys. Lett., 2011, 503, 239–243 CrossRef CAS.
- F. Zhao, P. Guo, G. Li, F. Liao, S. Tian and X. Jing, Luminescent properties of Eu3+, Tb3+ or Bi3+ activated yttrium germanates, Mater. Res. Bull., 2003, 38, 931–940 CrossRef CAS.
- X. Li, W. Zhang, W. Cui, J. Li, Y. Sun, G. Jiang, H. Huang, Y. Zhang and F. Dong, Reactant activation and photocatalysis mechanisms on Bi-metal@Bi2GeO5 with oxygen vacancies: a combined experimental and theoretical investigation, Chem. Eng. J., 2019, 370, 1366–1375 CrossRef CAS.
- B. Fan, J. Liu, W. Zhao and S. Qi, Luminescence and energy transfer of a single-phased phosphor Y2GeO5:Bi3+,Tm3+,Tb3+,Eu3+ for white UV LEDs, Opt. Mater., 2019, 90, 33–39 CrossRef CAS.
- A. E. Hughes and G. P. Pxms, The Luminescence Spectra of Bi3+ Ions in MgO and CaO, Phys. Status Solidi, 1975, 71, 707–718 Search PubMed.
- C. Nico, R. Fernandes, M. P. F. Graça, M. Elisa, B. A. Sava, R. C. C. Monteiro, L. Rino and T. Monteiro, Eu3+ luminescence in aluminophosphate glasses, J. Lumin., 2014, 145, 582–587 CrossRef CAS.
- Y. Tokida and S. Adachi, Photoluminescence spectroscopy and energy-level analysis of metal–organic-deposited Ga2O3:Cr3+ films, J. Appl. Phys., 2012, 112, 063522 Search PubMed.
- A. J. J. Bos, Theory of thermoluminescence, Radiat. Meas., 2006, 41(1), S45–S56 CrossRef.
- D. Esteves, TL-Fitter, 2025, https://github.com/DuarteME/TL-Fitter.
- J. M. Kalita and M. L. Chithambo, Comprehensive kinetic analysis of thermoluminescence peaks of α-Al2O3:C,Mg, J. Lumin., 2017, 185, 72–82 Search PubMed.
- G. Kitis, TL glow-curve deconvolution functions for various kinetic orders and continuous trap distribution: Acceptance criteria for E and s values, 2001, vol. 247 Search PubMed.
- T. Lyu and P. Dorenbos, Liquid nitrogen temperature to 700 K Bi3+ thermoluminescence: toward wide-temperature-range light dosimeters for versatile anti-counterfeiting, information storage, and X-ray imaging, Appl. Phys. Rev., 2024, 11, 041417 CAS.
- I. Pelant and J. Valenta, Luminescence Spectroscopy of Semiconductors, 2012 Search PubMed.
- D. M. Esteves, M. S. Batista, J. Rodrigues, A. V. Girão, L. C. Alves, A. L. Rodrigues, M. I. Dias, F. M. Costa, K. Lorenz, S. O. Pereira, T. Monteiro and M. Peres, Photoluminescence, persistent luminescence and thermoluminescence studies of Cr-doped zinc gallogermanate (ZGGO:Cr), J. Mater. Chem. C, 2024, 12, 19359–19370 RSC.
| This journal is © The Royal Society of Chemistry 2025 |