
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulation of intramolecular hydrogen bonds in conjugated pseudoladder polymer for semiconductivity and solution-processability†
Octavio
Miranda
a,
Paola
Mantegazza
b,
Vikki N.
Shinde
a,
Zhiqiang
Cao
c,
Steve
Shelton
d,
Stefania
Moro
b,
Caden
Deverter
a,
Yi
Liu
 d,
Xiaodan
Gu
*c,
Giovanni
Costantini
*b and
Lei
Fang
*a
d,
Xiaodan
Gu
*c,
Giovanni
Costantini
*b and
Lei
Fang
*a
aDepartment of Chemistry, Texas A&M University, 3255 TAMU, College Station, Texas 77843, USA. E-mail: fang@chem.tamu.edu
bSchool of Chemistry, University of Birmingham, Birmingham B15 2TT, UK. E-mail: g.costantini@bham.ac.uk
cSchool of Polymer Science and Engineering, The University of Southern Mississippi, Hattiesburg, Mississippi 39406, USA. E-mail: xiaodan.gu@usm.edu
dThe Molecular Foundry, Lawrence Berkeley National Laboratory, One Cyclotron Road, Berkeley, California 94720, USA
First published on 7th July 2025
Abstract
The conformational coplanarity and local rigidity of π-conjugated backbones are critical for the semiconducting performance of organic electronic materials. While fusing the aromatic system into a ladder-type structure effectively enhances these properties, it also often results in poor solution processability and hence limits their transition to device application. To address this challenge, an intramolecularly hydrogen-bonded pseudoladder polymer (HPLP) system based on alternating hydrogen bond donating benzobisimidazole (BBI) and hydrogen bond accepting benzodifuran (BDF) units, is designed and synthesized. A Boc-protected precursor of HPLP allows for feasible solution processing of the polymer into thin films. Subsequently, in situ thermal Boc-deprotection generates the HPLP polymer, in which intramolecular hydrogen bonds form between each pair of neighboring repeating units, inducing coplanarity and rigidity throughout the entire backbone. This process is accompanied by a significant red-shift of the absorption spectrum, reduced bandgap, and enhanced rigidity, as confirmed by NMR, UV-Vis, and density functional theory analyses. HPLP films exhibit a three-order-of-magnitude enhancement in charge carrier mobility compared to the Boc-protected precursor and demonstrate excellent solvent resistance in organic thin-film transistors.
Introduction
The conformational coplanarity and local rigidity of pi-conjugated backbones have been identified as two critical factors for the semiconducting performances of organic electronic materials.1–3 A coplanar and rigid backbone often facilitates charge transport through enhanced intramolecular pi-conjugation, stronger intermolecular electronic coupling, and lower reorganization energy during charge transfer. A common strategy to induce rigid coplanar conformation is to employ a fused-ring, ladder-type backbone constitution, which restricts torsional rotation between polymer subunits. This strategy is utilized, for example, by installing a second strand of covalent bonds along the backbone of conjugated polymers,4–8 affording conjugated ladder polymers as in the case of BBL9 and BTI10 (Fig. 1(a)). However, the application of these rigid, coplanar ladder polymers often presents challenges, primarily due to their poor solution processability, which hinders the fabrication of high-quality thin films for device applications. This represents a major dilemma in developing and optimizing semiconducting polymers that possess both excellent electronic performance and processability.11–13 | ||
| Fig. 1 Structural formulas of semiconducting polymers with coplanarity induced by (a) covalent ring-fusing, (b) hydrogen-bonds, and (c) weak non-covalent interactions. | ||
A widely used approach to address the aforementioned challenge involves the incorporation of weak intramolecular interactions in conjugated polymers to produce high performance semiconductors,14 such as DPPTT-TVTOEt, PTTz, and F-P3HT (Fig. 1(c)).15 These weak interactions include non-classical hydrogen bonds (CH–N, CH–O, CH–F) that are in the range of 1–2 kcal mol−1 and heteroatom interactions (S–S, S–O, S–N, S–F) falling below 1 kcal mol−1.16,17 This strategy induces a coplanar conformation without forming an overly rigid, poorly soluble ladder-type backbone, and has enabled the development of high-performance, solution-processable polymers for electronic and photovoltaic applications.18–21 Meanwhile, conventional hydrogen bonds have been proposed as an ideal way to achieve stronger noncovalent interactions (2–10 kcal mol−1) in the pursuit of a pseudoladder polymer structure that possesses a more well-defined coplanar conformation.22–25 In addition, hydrogen bonds can be manipulated through masking and unmasking by simple chemical transformations, such as protection with a tert-butyloxycarbonyl (Boc) group and corresponding high-temperature deprotection.26,27 These features make it possible to achieve highly rigid, coplanar, and solvent-resistant pseudoladder polymers without sacrificing solution processability, achieved through a precursor in which hydrogen bonds are masked. Meijer and coworkers pioneered the incorporation of intramolecular hydrogen bonds in conjugated macromolecules by constructing pseudoladder structures consisting of alternating 1,4-phenylenediamide and 1,4-pyrazine repeating units,22 as well as oligomers consisting of benzothiadiazole and pyrrole repeating units (Fig. 1(b), BTP).23 More recently, the Zhao28 and Fang29 groups have each explored this approach on acetylene-linked conjugated backbones (Fig. 1(b), ImA). It has been observed that fusing hydrogen bond-donating and accepting units into the backbone is essential for establishing robust intramolecular hydrogen bonding, due to the more favorable entropy contribution in such systems. Although the semiconducting performance of these materials has not been reported, the early work of Meijer notably shows a high conductivity of 1 S cm−1 achieved in the iodine-doped thin films of BTP oligomers.23 Recent efforts to implement this molecular design strategy into semiconducting systems have demonstrated promising performance in small molecules30 and non-ladder conjugated polymers.24,25 However, a semiconducting pseudoladder polymer featuring its backbone entirely locked by intramolecular hydrogen bonds has yet to be reported. Herein, we present such a hydrogen-bonded pseudoladder polymer (HPLP) system, which is solution-processable through the manipulation of latent intramolecular hydrogen bonds. This system exhibits desired semiconducting electronic properties and solvent resistance in device tests, owing to the rigid and coplanar backbone induced by these intramolecular hydrogen bonds.
Results and discussion
The hydrogen-bonded pseudoladder polymer HPLP (Scheme 1) is composed of alternating benzobisimidazole (BBI) units as the hydrogen bond donors and benzodifuran (BDF) units as the hydrogen bond acceptors. This design allows the formation of a completely ‘pseudoladder’ constitution through a sequence of uninterrupted intramolecular hydrogen bonds between all neighboring conjugated units along the polymer backbone. The BBI unit was selected for this work for multiple reasons. First, the hydrogen-bond donating imidazole moieties are fused to the aromatic backbone, so that the resulting hydrogen bonds can form with a relatively low entropy penalty, rendering it strong and robust even at elevated temperatures. Second, BBI is feasible to synthesize via a simple condensation reaction and versatile for the attachment of various functional groups and solubilizing side chains.31–33 Third, the BBI unit can be easily masked pre-polymerization using Boc protecting groups and unmasked post-polymerization through heat-induced Boc-deprotection. Finally, BBI has been extensively utilized in the construction of conjugated small molecules and polymers,34–43 as well as ladder polymers10,11 which demonstrate unique optoelectronic properties and outstanding stability. To couple with BBI, a benzofuran-derived, hydrogen-bond accepting comonomer was chosen on account of its synthetic feasibility, and previously reported good performance in organic photovoltaics.44 It is worth mentioning that the use of comonomers containing nitrogen centers as the hydrogen bond accepting sites, such as pyridine or pyrazine-fused derivatives, did not yield the desired synthetic outcome due to their negative impact on metal-catalyzed cross-coupling reactions and their tendency to promote undesired Boc-deprotection of neighboring hydrogen bond donors. | ||
| Scheme 1 Synthesis of benzobisimidazole-derived monomers 3a and 3b, small molecular models 4 and 5, polymer precursors BocP-a and BocP-b, and pseudoladder polymers HPLP-a and HPLP-b. | ||
In order to optimize the synthetic conditions and to instruct the characterization of the desired polymeric product, a small molecular model compound 5 was designed and synthesized prior to the synthesis of HPLP (Scheme 1). First, benzobisimidazole 1a and 1b are prepared in over 94% yields through simple condensation reactions of 1,2,4,5-tetraminobenzene hydrochloride salt with a carboxylic acid of choice (2-butylhexanoic acid for 1a and 3-octyltridecanoic acid for 1b). Bromination of 1a and 1b afforded the corresponding intermediates 2a and 2b. A mixed solvent of chloroform and acetic acid is needed here to fully dissolve 1a and 1b due to their poor solubility in most organic solvents. Although 2a and 2b seem suitable for cross-coupling, their poor solubility and the presence of N–H hydrogens prevented efficient conversion during our initial attempts of direct cross-coupling reactions. Therefore, 2a and 2b were subjected to Boc-protection, which proceeds quickly at 50 °C and converts the insoluble starting material suspended in tetrahydrofuran into a homogenous solution of the desired product. Note that two isomers are possible as the result of syn- and anti-protection of the two imidazole moieties. The more favorable anti-protected product 3a or 3b was isolated in good yields (55% and 51%, respectively).
Subsequently, trimethyltin-functionalized benzofuran was used as the coupling partner to react with 3avia palladium catalyzed Stille reaction to afford small molecule model 4, in which the hydrogen bonds are masked by Boc groups. In parallel, to synthesize the target polymer, Stille coupling reactions of 3a or 3b with a benzodifuran comonomer functionalized with two trimethylstannyl groups was conducted to give polymer BocP-a or BocP-b, respectively, in high efficiency. It is noteworthy that the Boc groups were partially removed during the Stille reaction, although both the resulting products BocP-a and BocP-b remained soluble in common organic solvents. The number average molar mass (Mn) and polydispersity (Đ) of both polymers were measured on an analytical gel permeation chromatography that was calibrated by polystyrene standards (Fig. S17 and Table S1, ESI†). BocP-a showed a measured Mn of 11.5 kg mol−1 and Đ = 2.15; while BocP-b had a higher Mn of 28.6 kg mol−1 and Đ = 1.98. Note that BocP-a is decorated with much smaller 1-butylpentyl side chains compared to the 2-octyldodecyl side chains of BocP-b, resulting in the molar mass obtained for BocP-a being considerably lower than that of BocP-b due to solubility limitations for longer polymer chains during the reaction. BocP polymers were purified and separated into fractions of different molar masses (Table S1, ESI†) on a preparative gel permeation chromatography to be used for various analyses such as nuclear magnetic resonance (NMR) and UV-Vis spectroscopy, cyclic voltammetry (CV), and scanning tunneling microscopy (STM). This was done to ensure lower polydispersity, and to accommodate the different solubility requirements for each mode of analysis.
Thermogravimetric analysis of compound 4 and BocP are marked by a significant loss in weight percent between 180–200 °C corresponding to Boc cleavage with no more significant decomposition occurring until the temperature exceeded 350 °C (Fig. S18 and S19, ESI†). These observations assure that thermal treatment at 180 °C under an inert atmosphere does not result in significant decomposition of the molecular backbone other than Boc cleavage. Indeed, synthetically, the Boc groups in 4 can be removed simply by heating at 180 °C for 1 h under a nitrogen atmosphere to afford the unmasked product 5 in quantitative yield. Quantitative conversion of BocP to the desired hydrogen-bonded pseudo ladder polymer HPLP was also achieved under the same conditions. The 1H NMR (Fig. 2(a)) and IR (Fig. S15 and S16, ESI†) spectra clearly illustrate the presence of Boc groups in 4 and BocP-a, and the cleavage of Boc groups in 5 and HPLP-a. Due to the partial cleavage of Boc groups in BocP-a during Stille polymerization, there is a weak peak at ∼10.7 ppm in the 1H NMR spectrum and a weak peak at 3417 cm−1 in the IR spectrum, respectively, which correspond to the unmasked N–H group. After thermal Boc cleavage, a higher intensity peak of the unmasked intramolecular hydrogen-bonded N–H group is observed in both 1H NMR and IR spectra.
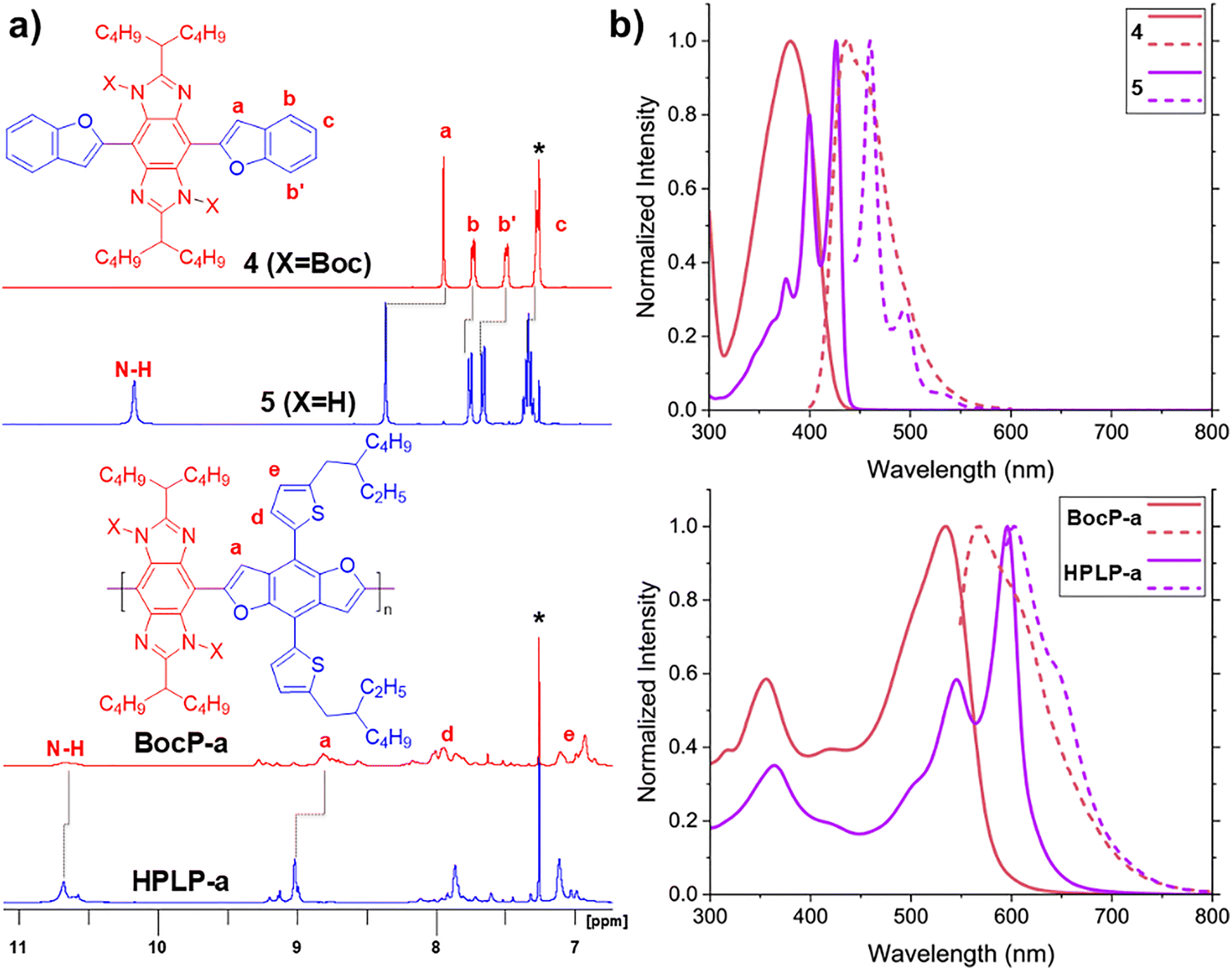 | ||
| Fig. 2 (a) 1H NMR spectra and of the Boc-masked species 4 and BocP-a (Mn = 7.3 kg mol−1, Đ = 1.83) and (b) UV-Vis (solid line) and fluorescence (dashed line) spectra of 4 and BocP-a (Mn = 16.1 kg mol−1, Đ = 1.42) in comparison with the corresponding unmasked derivatives. | ||
Overall, Boc-protected polymers BocP-a and BocP-b exhibited good solubility in common organic solvents such as toluene, chlorobenzene, chloroform, and tetrahydrofuran. Upon Boc-deprotection, however, the solubility of HPLP polymers was greatly decreased (Table S2, ESI†). It should be noted that the well resolved 1H NMR peaks observed for HPLP-a in Fig. 2(a) were obtained using a sample of low molar mass (BocP-a: Mn = 7.3 kg mol−1, Đ = 1.83) due to the poorer solubility of higher polymer chains. For HPLP-a and -b, this low solubility was taken as an advantage to fabricate solvent-resistant thin films, by solution-processing the soluble precursor BocP-a or -b followed by in situ thermal Boc-cleavage, yielding thin films of HPLP exhibiting excellent resistance against a variety of organic solvents (Table S2 and Fig. S21, ESI†). This unique property renders this system highly promising for fabricating solvent-resistant functional polymer thin films for devices that are durable under strong organic solvents and suitable for multi-layer solution processing techniques.
Boc-cleavage also results in significant changes to the optical properties and electronic structures of the designed conjugated systems (Table 1). Conversion of 4 to 5 resulted in a 45 nm redshift in λmax accompanied by an astounding change in absorption features from one broad peak in 4 to well-defined vibrational peaks for 5 (Fig. 2(b)). The redshift, i.e. decrease of optical bandgap, indicates better π-conjugation between the repeating units due to the more coplanar conformation, while the narrowed vibrational peak indicates the local rigidity of the π-system has increased drastically. Similar changes in the UV-Vis and fluorescence spectra peak patterns between BocP-a and HPLP-a were also observed along with an increased redshift of 62 nm and a decreasing stoke shift (Δ) from 35 to 6 nm, respectively (Fig. 2(b) and Table 1). Although boc-protected precursors show similar absorption onsets to their hydrogen-bonded counterparts indicating similar band gaps, cyclic voltammetry showed significant changes to HOMO energy levels and small changes to LUMO levels. The similar absorption onset is likely a result of relatively coplanar conformations in boc-protected precursors bring present in low concentrations, whereas their hydrogen bonded counterparts exist in mostly coplanar conformations as indicated by their sharp vibration peak pattern and λmax lying nearer to their respective onset absorptions. To obtain accurate CV measurements in solution, a lower molar mass sample of 2-octyldodecyl-decorated BocP-b (Mn = 13.5 kg mol−1, Đ = 1.30) and HPLP-b were investigated as the model polymers due to the improved solubility of HPLP-b in tetrahydrofuran compared to that of HPLP-a. CV measurements (Fig. S29 and S30, ESI†) demonstrate a significantly decreased bandgap of 5 (1.59 eV) and HPLP-b (1.38 eV) compared to 4 (1.82 eV) and BocP-b (2.17 eV), respectively (Table 1). The decreased bandgaps in both 5 and HPLP is primarily attributed to the increase of the HOMO level after removal of the electron-withdrawing Boc groups and the coplanar conformation induced by the intramolecular hydrogen bonds, with little change to the LUMO being observed.
To gauge the strength of these intramolecular hydrogen bonds and the coplanarity of the designed conjugated system, torsional analysis was performed using density functional theory (DFT) on a BBI-benzofuran model molecule by rotating about the bridging single bond in 15° intervals and recording free energy at B3LYP/6-31G(d) level of theory (Fig. 3(a)). Results showed rotational barriers of 9.6 kcal mol−1 and a calculated coplanarity parameter45 of 〈cos2(φ)〉 = 0.93. This result suggested that intramolecular hydrogen bonding interactions lead to energetically favorable coplanar conformations in the gas phase. Single crystals of compounds 4 and 5 were grown for X-ray diffraction analysis (Fig. 3(b) and (c)). Despite its sterically demanding Boc groups, 4 demonstrates a fairly small dihedral angle of 21° between the BBI unit and the benzofuran unit (Fig. 3(a), bottom). This dihedral angle is likely decreased by the weak C–H⋯N hydrogen bonding interactions between 2-position of the furan ring and the basic nitrogen center on the BBI unit, evidenced by the short C⋯N distance of 3.15 Å falling within traditional hydrogen bonding distance. In the unmasked molecule 5, a smaller dihedral angle of 13° was observed between the BBI unit and the benzofuran. The short distances of O–N (2.85 Å) and C–N (3.04 Å) confirm the formation of two hydrogen bonds N–H⋯O and C–H⋯N on both sides of the covalent C–C bond. It is interesting to observe that 5 exhibits a non-zero dihedral angle in the solid-state structure, while gas-phase DFT calculations suggest that the dihedral angle should be zero. This discrepancy is likely due to strain induced by intermolecular interactions in the solid state, primarily the intermolecular interaction between the furan oxygen and the BBI's N–H unit that competes against intramolecular hydrogen bonding. This interaction is evident from the relatively short intermolecular O–N distance of 3.3 Å in the crystal structure (Fig. 3(d)). Further evidence for this interaction is provided by an endothermic peak observed at ∼200 °C during differential scanning calorimetry (DSC) of single crystals of 5 (Fig. S19b, ESI†). This peak does not appear in the second scan of the same sample following a melt-solidify cycle or in a powder sample of 5 (Fig. S19a, ESI†). Considering the solution-phase conformation of 5 and the polymers HPLP, it is believed that such intermolecular interactions are not dominant, allowing the intramolecularly hydrogen-bonded molecule to adopt a near-zero dihedral angle in solution as suggested by DFT calculations.
 | ||
| Fig. 3 (a) Relative energy (ΔG) and probability density (P) obtained through torsional analysis by rotating 15° across the bridging C–C bond of a benzobisimidazole–benzofuran system using density functional theory at B3LYP/6-31G(d) level of theory; (b)–(d) front view (top) and side view (bottom) of single molecule crystal structures of 4 and 5, as well as crystal packing of 5 showing intermolecular hydrogen-bonding interactions. | ||
Supporting the DFT calculations, variable temperature-NMR spectra of HPLP-a show little change from 20 to 100 °C (Fig. 4(a)), and variable temperature UV-Vis spectra of its thin films show only a slight blue shift when taken to 250 °C (Fig. 4(b)). Similar behavior was observed in the solution-phase UV-Vis analyses of HPLP-a and HPLP-b (Fig. S27 and S28, ESI†), as well as NMR and UV-vis spectra of small molecule analog 5a (Fig. S13 and S26, ESI†). The high-temperature robustness of intramolecular hydrogen bonds in 5 and HPLP-a can be attributed to the unfavorable entropy change for breaking these hydrogen bonds, due to the additional solvation of the hydrogen-bond donor and acceptor sites once they are dissociated. These results suggest that, although not as strong as the covalent bonds used in traditional ladder polymers, these intramolecular hydrogen bonds can still be used for the same purpose of locking conjugated polymers into a highly robust coplanar rigid conformation.
 | ||
| Fig. 4 (a) Variable-temperature NMR of HPLP-a (derived from BocP-a: Mn = 7.3 kg mol−1, Đ = 1.83) in toluene-d6, and (b) variable-temperature UV-Vis spectra of HPLP-a (from BocP-a: Mn = 16.1 kg mol−1, Đ = 1.42) in thin film. | ||
Both BocP-b (Mn = 12.8, Đ = 1.5) and HPLP-b polymers were analyzed by ultrahigh vacuum STM after having been deposited as monolayer films in vacuum by electrospray deposition onto an atomically clean, flat Au(111) surface (Fig. S32 and S33, ESI†).46–48 In both cases, the polymers appear as extended linear structures with few bends. No substantial difference could be observed between BocP-b and HPLP-b, likely due to the planarizing effect of face-on surface adsorption on the backbone conformation. In areas of higher coverage (Fig. 5), the polymers tend to self-assemble into locally compact arrangements by aligning their backbones parallel to each other. The average distance between polymers, d = 3.5 ± 0.5 nm, is compatible with a packing model in which the side chains of adjacent polymers interlock with a minimal interdigitation (Fig. S33, ESI†), similar to what has been observed in other conjugated polymers with a high density of side chains attached to the backbones.46 Polymer crossings are occasionally observed at near-perpendicular angles (Fig. 5 and Fig. S32a, ESI†), a feature that was recently highlighted as beneficial for promoting a highly interconnected charge carrier transport network, thereby enabling high charge transport mobility.47,48 In addition, the molar mass of the BocP-b sample was evaluated by measuring the lengths of 607 individual chains identified in the STM images. The statistical average molar mass and its distribution closely align with the results obtained from GPC (Fig. S34, ESI†). This finding demonstrates the powerful capability of STM in accurately measuring and calibrating the molar mass of rigid conjugated polymers.
 | ||
| Fig. 5 (a) STM image of a high coverage area of BocP-b (Mn = 19.3 kg mol−1, Đ = 1.50) deposited by electrospray deposition on Au(111). (b) Zoomed-in image showing sections of the polymers align parallel to each other, with a distance d between them. | ||
To test the effect of hydrogen bond-induced coplanarity on electronic performance, organic thin-film transistors composed of BocP-a (Mn = 16.1 kg mol−1, Đ = 1.42) and HPLP-a generated in situ from BocP-a are tested (Fig. 6). A bottom-gate/top-contact device architecture is employed. To fabricate the devices, BocP-a polymers were spin-coated onto n-octadecyltrichlorosilane (OTS)-treated SiO2/Si substrates, while gold was deposited as the source and drain electrodes, detailed in the ESI.† The as fabricated device of BocP-a shows a low hole mobility of 5.2 × 10−5 cm2 V−1 s−1. Such device was directly heated in N2 atmosphere at 180 °C for 1 h to convert BocP-a in the active layer into HPLP-ain situ. The resulting devices show a hole mobility of 4.6 × 10−2 cm2 V−1 s−1, marking a significant increase for three orders of magnitude. An improvement was also observed when comparing BocP-b to HPLP-b with films exhibiting hole mobilities of 8.5 × 10−9 cm2 V−1 s−1 and 3.4 × 10−6 cm2 V−1 s−1, respectively (Fig. S36, ESI†). The drastic enhancement in charge carrier mobility can be attributed to change in electronic band gap, lower reorganization energies, and improved packing of the pseudoladder polymer chains containing intramolecular hydrogen-bonding interactions. To investigate this, atomic force microscopy (AFM) was performed on hydrogen-bonded polymers and their Boc-protected precursors (Fig. S37, ESI†). AFM revealed a reduction in film thickness from BocP-a (40 nm) to HPLP-a (26 nm), while the root-mean-square (rms) roughness remained similar (0.5 nm). A less pronounced thickness change was observed between BocP-b (42 nm) and HPLP-b (35 nm), with the rms roughness again unchanged (1.7 nm). The decreased film thickness in the hydrogen-bonded polymers correlates with their enhanced charge transport, while the higher rms roughness of HPLP-b compared to HPLP-a may contribute to its lower charge-carrier mobilities.
 | ||
| Fig. 6 (top) Schematic illustration of the thermal treatment process on the OFET device of BocP-a, enabling the in situ transformation of the active layer from BocP-a to HPLP-a; (bottom) transfer characteristics of the OFET devices before (BocP-a) and after (HPLP-a) thermal treatment at VDS = 60 V. | ||
Along with the extraordinary improvement in electronic performance upon hydrogen bond-induced coplanarity, HPLP-a lies among some of the highest mobilities achieved for benzodifuran based conjugated polymers.49–55 As benzobisimidazole is unexplored in the field of organic semiconducting materials, its effect on coplanarity and rigidity of polymer chains may serve to maximize performance of other high-performing monomers as it has done with benzodifuran.
Conclusion
In conclusion, we demonstrate a feasible approach to achieve conjugated pseudoladder polymers locked into a coplanar conformation by intramolecular hydrogen bonds, and the unique changes that result in the properties of the polymers. The incorporation of Boc protecting groups also allows for processability of the polymer precursors for solution-based device fabrication, while allowing access to hydrogen bond-induced coplanar conformation through simple in situ thermal treatment. The transition from a flexible, noncoplanar polymer chain to a rigid coplanar conformation was accompanied by a significant decrease in solubility and in the bandgap. The rigidity of the hydrogen bonded polymers was confirmed by the sharp vibrational peaks in the UV-vis spectra, and demonstrated to be highly robust as evidenced by a series of variable temperature analyses. OFET devices were fabricated by solution processing the soluble Boc-protected precursor followed by in situ Boc-deprotection. The device performance of the hydrogen bonded pseudoladder polymer, HPLP-a, showed an improvement in hole mobility by almost 3 orders of magnitude when compared to its Boc-protected counterpart. Solvent resistance of the semiconducting HPLP films also shows potential for multilayer film processing in organic electronic devices. This work demonstrates the potential of hydrogen-bond manipulation as a versatile approach for developing solution-processable, high-performance semiconducting polymers, effectively addressing the trade-off between electronic performance and processability in organic electronic materials.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compound 4 has been deposited at the CCDC under 2422384 and can be obtained from https://www.ccdc.cam.ac.uk/structures/Search?Ccdcid=2422384&DatabaseToSearch=Published. Crystallographic data for compound 5 has been deposited at the CCDC under 2422385 and can be obtained from https://www.ccdc.cam.ac.uk/structures/Search?Ccdcid=2422385&DatabaseToSearch=Published.Acknowledgements
O. M. acknowledges the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-2139772. The authors acknowledge the Robert A. Welch Foundation (A-1898) for financial support of this work. Portions of this research were conducted with the advanced computing resources provided by Texas A&M High Performance Research Computing. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.References
- A. R. Murphy and J. M. J. Fréchet, Organic Semiconducting Oligomers for Use in Thin Film Transistors, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Charge Transport in Organic Semiconductors, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- H. Sirringhaus, 25th Anniversary Article: Organic Field-Effect Transistors: The Path Beyond Amorphous Silicon, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- J. Lee, B. B. Rajeeva, T. Yuan, Z. Guo, Y. Lin, M. Al-Hashimi, Y. Zheng and L. Fang, Thermodynamic synthesis of solution processable ladder polymers, Chem. Sci., 2016, 7, 881–889 RSC.
- Y. Zou, X. Ji, J. Cai, T. Yuan, D. J. Stanton, Y. Lin, M. Naraghi and L. Fang, Synthesis and Solution Processing of a Hydrogen-Bonded Ladder Polymer, Chem, 2017, 2, 139–152 CAS.
- J. Lee, H. Li, A. J. Kalin, T. Yuan, C. Wang, T. Olson, H. Li and L. Fang, Extended Ladder-Type Benzo[k]tetraphene-Derived Oligomers, Angew. Chem., Int. Ed., 2017, 56, 13727 CrossRef CAS PubMed.
- X. Ji, M. Leng, H. Xie, C. Wang, K. R. Dunbar, Y. Zou and L. Fang, Extraordinary electrochemical stability and extended polaron delocalization of ladder-type polyaniline-analogous polymers, Chem. Sci., 2020, 11, 12737–12745 RSC.
- X. Wu, Q. He, Z. Zhou, T. L. D. Tam, C. Tang, M. Lin, M. Moser, S. Griggs, A. Marks, S. Chen, J. Xu, I. McCulloch and W. L. Leong, Stable n-Type Perylene Derivative Ladder Polymer with Antiambipolarity for Electrically Reconfigurable Organic Logic Gates, Adv. Mater., 2024, 36, 2308823 CrossRef CAS PubMed.
- A. Harbuzaru, I. Arrechea-Marcos, A. D. Scaccabarozzi, Y. Wang, X. Guo, M. Caironi, J. T. López Navarrete, M. C. Ruiz Delgado and R. Ponce Ortiz, Ladder-type bithiophene imide-based organic semiconductors: understanding charge transport mechanisms in organic field effect transistors, J. Mater. Chem. C, 2020, 8(44), 15759–15770 RSC.
- R. L. Van Deusen, Benzimidazo-benzophenanthroline polymers, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 211–214 CrossRef CAS.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, Integrated Materials Design of Organic Semiconductors for Field-Effect Transistors, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- J. Lee, A. J. Kalin, T. Yuan, M. Al-Hashimi and L. Fang, Fully conjugated ladder polymers, Chem. Sci., 2017, 8, 2503–2521 RSC.
- L. Ding, Z. Yu, X. Wang, Z. Yao, Y. Lu, C. Yang, J. Wang and J. Pei, Polymer Semiconductors: Synthesis, Processing, and Applications, Chem. Rev., 2023, 123, 7421–7497 CrossRef CAS PubMed.
- K. Yang, Z. Chen, Y. Wang and X. Guo, Alkoxy-Functionalized Bithiophene/thiazoles: Versatile Building Blocks for High-Performance Organic and Polymeric Semiconductors, Acc. Mater. Res., 2023, 237–250 CrossRef CAS.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Organic and Polymeric Semiconductors Enhanced by Noncovalent Conformational Locks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, M. Olvera de la Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, Controlling Conformations of Conjugated Polymers and Small Molecules: The Role of Nonbonding Interactions, J. Am. Chem. Soc., 2013, 135, 10475–10483 CrossRef CAS.
- M. Liu, X. Han, H. Chen, Q. Peng and H. Huang, A molecular descriptor of intramolecular noncovalent interaction for regulating optoelectronic properties of organic semiconductors, Nat. Commun., 2023, 14, 2500 CrossRef CAS PubMed.
- D. Ding, G. Kim, Y. Kim, F. D. Eisner, E. Gutiérrez-Fernández, J. Martín, M. Yoon and M. Heeney, Influence of Backbone Curvature on the Organic Electrochemical Transistor Performance of Glycolated Donor–Acceptor Conjugated Polymers, Angew. Chem., Int. Ed., 2021, 60, 19679 CrossRef PubMed.
- J. Chen, S. Cong, L. Wang, Y. Wang, L. Lan, C. Chen, Y. Zhou, Z. Li, I. McCulloch and W. Yue, Backbone coplanarity manipulation via hydrogen bonding to boost the n-type performance of polymeric mixed conductors operating in aqueous electrolyte, Mater. Horiz., 2023, 10, 607–618 RSC.
- X. Zhang, X. Gu and H. Huang, Low-Cost Nonfused-Ring Electron Acceptors Enabled by Noncovalent Conformational Locks, Acc. Chem. Res., 2024, 57, 981–991 CrossRef CAS PubMed.
- Y. Wu, Y. Yuan, D. Sorbelli, C. Cheng, L. Michalek, H. Cheng, V. Jindal, S. Zhang, G. LeCroy, E. D. Gomez, S. T. Milner, A. Salleo, G. Galli, J. B. Asbury, M. F. Toney and Z. Bao, Tuning polymer-backbone coplanarity and conformational order to achieve high-performance printed all-polymer solar cells, Nat. Commun., 2024, 15, 2170 CrossRef CAS PubMed.
- D. A. P. Delnoye, R. P. Sijbesma, J. A. J. M. Vekemans and E. W. Meijer, π-Conjugated Oligomers and Polymers with a Self-Assembled Ladder-like Structure, J. Am. Chem. Soc., 1996, 118, 8717–8718 CrossRef CAS.
- H. A. M. Van Mullekom, J. A. J. M. Vekemans and E. W. Meijer, Alternating copolymer of pyrrole and 2,1,3-benzothiadiazole, Chem. Commun., 1996, 2163–2164 RSC.
- B. Liu, J. Li, W. Zeng, W. Yang, H. Yan, D. Li, Y. Zhou, X. Gao and Q. Zhang, High-Performance Organic Semiconducting Polymers by a Resonance-Assisted Hydrogen Bonding Approach, Chem. Mater., 2021, 33, 580–588 CrossRef CAS.
- M. U. Ocheje, R. B. Goodman, K. Lu, Y. Wang, L. A. Galuska, L. Soullard, Z. Cao, S. Zhang, M. Yadiki, X. Gu, Y. Chiu and S. Rondeau-Gagné, Precise Control of Noncovalent Interactions in Semiconducting Polymers for High-Performance Organic Field-Effect Transistors, Chem. Mater., 2021, 33, 8267–8277 CrossRef CAS.
- B. Li, R. Li, P. Dorff, J. C. McWilliams, R. M. Guinn, S. M. Guinness, L. Han, K. Wang and S. Yu, Deprotection of N-Boc Groups under Continuous-Flow High-Temperature Conditions, J. Org. Chem., 2019, 84, 4846–4855 CrossRef CAS PubMed.
- M. Ryan, D. Lynch, S. G. Collins and A. R. Maguire, Selective Thermal Deprotection of N-Boc Protected Amines in Continuous Flow, Org. Process Res. Dev., 2024, 28, 1946–1963 CrossRef CAS PubMed.
- W. Hu, N. Zhu, W. Tang and D. Zhao, Oligo(p-phenyleneethynylene)s with Hydrogen-Bonded Coplanar Conformation, Org. Lett., 2008, 10, 2669–2672 CrossRef CAS PubMed.
- C. Zhu, A. U. Mu, C. Wang, X. Ji and L. Fang, Synthesis and Solution Processing of a Rigid Polymer Enabled by Active Manipulation of Intramolecular Hydrogen Bonds, ACS Macro Lett., 2018, 7, 801–806 CrossRef CAS PubMed.
- A. U. Mu, Y. Kim, M. Miranda, M. Vazquez, J. Strzalka, J. Xu and L. Fang, Hydrogen-Bond-Promoted Planar Conformation, Crystallinity, and Charge Transport in Semiconducting Diazaisoindigo Derivatives, ACS Mater. Lett., 2022, 4, 1270–1278 CrossRef CAS.
- H. T. M. Le, N. S. El-Hamdi and O. S. Miljanić, Benzobisimidazole Cruciform Fluorophores. The, J. Org. Chem., 2015, 80, 5210–5217 CrossRef CAS PubMed.
- A. Bhuwalka, J. F. Mike, M. He, J. J. Intemann, T. Nelson, M. D. Ewan, R. A. Roggers, Z. Lin and M. Jeffries-EL, Quaterthiophene–Benzobisazole Copolymers for Photovoltaic Cells: Effect of Heteroatom Placement and Substitution on the Optical and Electronic Properties, Macromolecules, 2011, 44, 9611–9617 CrossRef CAS.
- S. Tannir, R. Chavez III, G. Molina III, A. Tomlinson and M. Jeffries-EL, Evaluating the Effect of Extended Conjugation and Regioisomerism on the Optoelectronic Properties and Device Efficiencies of Blue Light-Emitting Benzobisoxazoles, Chem. Mater., 2024, 36, 4945–4954 CrossRef CAS.
- A. J. Boydston, P. D. Vu, O. L. Dykhno, V. Chang, A. R. Wyatt, A. S. Stockett, E. T. Ritschdbrff, J. B. Shear and C. W. Bielawski, Modular Fluorescent Benzobis(imidazolium) Salts: Syntheses, Photophysical Analyses, and Applications, J. Am. Chem. Soc., 2008, 130, 3143–3156 CrossRef CAS PubMed.
- Z. Guo, N. R. Song, J. H. Moon, M. Kim, E. J. Jun, J. Choi, J. Y. Lee, C. Bielawski, J. L. Sessler and J. Yoon, A Benzobisimidazolium-Based Fluorescent and Colorimetric Chemosensor for CO2, J. Am. Chem. Soc., 2012, 134, 17846–17849 CrossRef CAS PubMed.
- C. J. Lefaux, B. Kim, N. Venkat and P. T. Mather, Molecular Composite Coatings on Nafion Using Layer-by-Layer Self-Assembly, ACS Appl. Mater. Interfaces, 2015, 7, 10365–10373 CrossRef CAS PubMed.
- W. Seo, K. L. Carpenter, J. A. Gaugler, W. Shao, K. A. Werling, P. M. Fournier, D. S. Lambrecht and A. Star, Polybenzobisimidazole-derived two-dimensional supramolecular polymer, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1095–1101 CrossRef CAS.
- X. Wang, M. Shan, X. Liu, M. Wang, C. M. Doherty, D. Osadchii and F. Kapteijn, High-Performance Polybenzimidazole Membranes for Helium Extraction from Natural Gas, ACS Appl. Mater. Interfaces, 2019, 11, 20098–20103 CrossRef CAS PubMed.
- Y. Zhao, X. Shen, S. He, X. Han and Z. Ni, Relationship between cis–trans isomerism and optical and electrical properties based on benzidiimidazole-thiophene copolymer, Synth. Met., 2018, 245, 175–181 CrossRef CAS.
- C. Yu, Q. Zheng, L. Wang, T. Wang, X. Zheng and G. Gao, A prototype of benzobis(imidazolium)-embedded conjugated polyelectrolyte: Synthesis by direct C–H arylation and fluorescent responses to anions, Chin. Chem. Lett., 2022, 33, 2425–2428 CrossRef CAS.
- A. J. Boydston, K. A. Williams and C. W. Bielawski, A Modular Approach to Main-Chain Organometallic Polymers, J. Am. Chem. Soc., 2005, 127, 12496–12497 CrossRef CAS PubMed.
- Z. Sun, Y. Liu, J. Chen, C. Huang and T. Tu, Robust Iridium Coordination Polymers: Highly Selective, Efficient, and Recyclable Catalysts for Oxidative Conversion of Glycerol to Potassium Lactate with Dihydrogen Liberation, ACS Catal., 2015, 5, 6573–6578 CrossRef CAS.
- J. Chen, J. Wu and T. Tu, Sustainable and Selective Monomethylation of Anilines by Methanol with Solid Molecular NHC-Ir Catalysts, ACS Sustainable Chem. Eng., 2017, 5, 11744–11751 CrossRef CAS.
- X. Li, Y. Li, Y. Zhang and Y. Sun, Recent Progress of Benzodifuran-Based Polymer Donors for High-Performance Organic Photovoltaics, Small Sci., 2022, 2, 2200006 CrossRef CAS PubMed.
- Y. Che and D. F. Perepichka, Quantifying Planarity in the Design of Organic Electronic Materials, Angew. Chem., Int. Ed., 2021, 60, 1364–1373 CrossRef CAS PubMed.
- D. A. Warr, L. M. A. Perdigão, H. Pinfold, J. Blohm, D. Stringer, A. Leventis, H. Bronstein, A. Troisi and G. Costantini, Sequencing conjugated polymers by eye, Sci. Adv., 2018, 4, eaas9543 CrossRef PubMed.
- S. Moro, N. Siemons, O. Drury, D. A. Warr, T. A. Moriarty, L. M. A. Perdigão, D. Pearce, M. Moser, R. K. Hallani, J. Parker, I. McCulloch, J. M. Frost, J. Nelson and G. Costantini, The Effect of Glycol Side Chains on the Assembly and Microstructure of Conjugated Polymers, ACS Nano, 2022, 16, 21303–21314 CrossRef CAS PubMed.
- S. Moro, S. E. F. Spencer, D. W. Lester, F. Nübling, M. Sommer and G. Costantini, Molecular-Scale Imaging Enables Direct Visualization of Molecular Defects and Chain Structure of Conjugated Polymers, ACS Nano, 2024, 18, 11655–11664 CrossRef CAS PubMed.
- M. Xiao, B. Kang, S. B. Lee, L. M. A. Perdigão, A. Luci, D. A. Warr, S. P. Senanayak, M. Nikolka, M. Statz, Y. Wu, A. Sadhanala, S. Schott, R. Carey, Q. Wang, M. Lee, C. Kim, A. Onwubiko, C. Jellett, H. Liao, W. Yue, K. Cho, G. Costantini, I. McCulloch and H. Sirringhaus, Anisotropy of Charge Transport in a Uniaxially Aligned Fused Electron-Deficient Polymer Processed by Solution Shear Coating, Adv. Mater., 2020, 32, 2000063 CrossRef CAS PubMed.
- H. Makki, C. A. Burke and A. Troisi, Microstructural Model of Indacenodithiophene-co-benzothiadiazole Polymer: π-Crossing Interactions and Their Potential Impact on Charge Transport, J. Phys. Chem. Lett., 2023, 14, 8867–8873 CrossRef CAS PubMed.
- J. F. Coker, S. Moro, A. S. Gertsen, X. Shi, D. Pearce, M. P. Schelling, Y. Xu, W. Zhang, J. W. Andreasen, C. R. Snyder, L. J. Richter, M. J. Bird, I. McCulloch, G. Costantini, J. M. Frost and J. Nelson, Perpendicular crossing chains enable high mobility in a noncrystalline conjugated polymer, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2403879121 CrossRef PubMed.
- L. Huo, Y. Huang, B. Fan, X. Guo, Y. Jing, M. Zhang, Y. Li and J. Huo, Synthesis of a 4,8-dialkoxy-benzo[1,2-b:4,5-b′]difuran unit and its application in photovoltaic polymer, Chem. Commun., 2012, 48, 3318–3320 RSC.
- L. Huo, H. Ye, Y. Wu, Z. Li, X. Guo, M. Zhang, S. Zhang and J. Huo, Conjugated and Nonconjugated Substitution Effect on Photovoltaic Properties of Benzodifuran-Based Photovoltaic Polymers, Macromolecules, 2012, 45, 6923–6929 CrossRef CAS.
- B. Ebenhoch, S. A. J. Thomson, K. Genevičius, G. Juška and I. D. W. Samuel, Charge carrier mobility of the organic photovoltaic materials PTB7 and PC71BM and its influence on device performance, Org. Electron., 2015, 22, 62–68 CrossRef CAS.
- X. Li, K. Weng, H. W. Ryu, J. Guo, X. Zhang, T. Xia, H. Fu, D. Wei, J. Min, Y. Zhang, H. Y. Woo and Y. Sun, Non-Fullerene Organic Solar Cells Based on Benzo[1,2-b:4,5-b′]difuran-Conjugated Polymer with 14% Efficiency, Adv. Funct. Mater., 2020, 30, 1906809 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2422384 and 2422385. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc01365h |
| This journal is © The Royal Society of Chemistry 2025 |