
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron rich N-heterotriangulenes as host materials for OLEDs†
Stefan
Warrington
 a,
Joseph
Cameron
a,
Claire
Wilson
a,
Dmytro
Volyniuk
b,
Dylan
Wilkinson
a,
Juozas Vidas
Grazulevicius
b,
Peter J.
Skabara
*a and
Graeme
Cooke
*a
a,
Joseph
Cameron
a,
Claire
Wilson
a,
Dmytro
Volyniuk
b,
Dylan
Wilkinson
a,
Juozas Vidas
Grazulevicius
b,
Peter J.
Skabara
*a and
Graeme
Cooke
*a
aSchool of Chemistry, University of Glasgow, University Place, Glasgow, G12 8QQ, UK. E-mail: Peter.Skabara@glasgow.ac.uk; Graeme.Cooke@glasgow.ac.uk
bDepartment of Polymer Chemistry and Technology, Kaunas University of Technology, Barsausko 59, LT-51423, Kaunas, Lithuania
First published on 19th June 2025
Abstract
Triphenylamine-based materials have been extensively studied as hole-transporting materials for organic semiconductor devices due to their high hole mobility and thermal stability. For example, they may be used as either hole-transporting layers in solar cells or p-type host materials in the emissive layer of OLEDs. Here, we present the development of a series of fused triarylamine derivatives in the form of N-heterotriangulene-based systems featuring electron-rich pendent groups and have investigated their structural properties using X-ray crystallography, and their optical and redox properties. Space-charge-limited current measurements were carried out to assess their potential to be used as p-type materials and the materials were applied as hosts in OLEDs with a solution-processed 4CzIPN-doped emissive layer. The best-performing devices exhibited a maximum external quantum efficiency of 6.9%. We have shown that the host can disperse the emitter molecules effectively and avoid dimer formation thereby enhancing OLED performance.
Introduction
Nitrogen-containing polycyclic aromatic materials have been of interest as redox-active organic compounds in a range of organic electronic applications. For example, triphenylamine (TPA, Fig. 1a) has been actively employed as a unit in hole-transporting, hole-injection, and electron-blocking materials, as well as a donor motif for push–pull materials due to the electron-rich nature of the material. TPAs have been subject to several reviews over the last decade, focusing on their useful application in several organic and hybrid electronic devices.1–4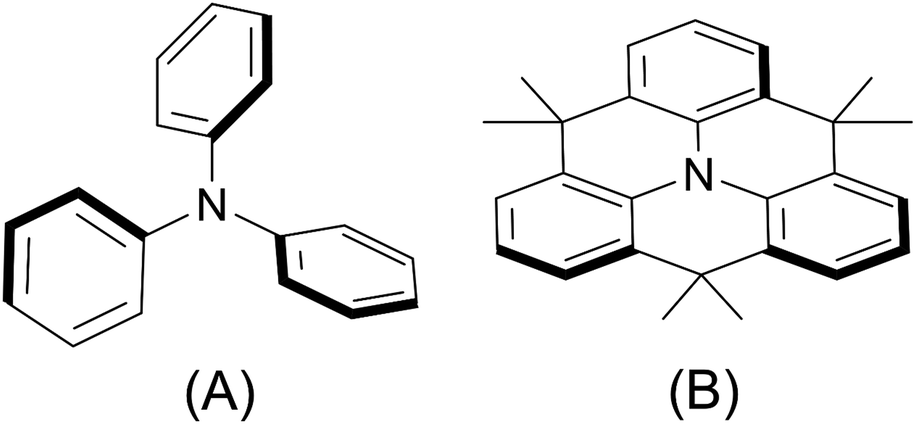 | ||
| Fig. 1 (A) TPA and (B) N-heterotriangulene structure. | ||
The molecular configuration of the TPA moiety has a significant impact on its charge transporting and intramolecular properties, including aggregation suppression. TPA typically possesses a trigonal planar geometry influenced by the resonance delocalisation of the appended aromatic groups and the sp3-hybridised central nitrogen atom. This configuration results in a “propeller”-like structure which is directed by the steric repulsion of the neighbouring phenyl rings manifesting as a dihedral angle ranging from 37–50° between the ring and the central C–N bond5,6 (Fig. 1a). The tilting of the aromatic residues reduces orbital overlap occurring between their π-orbitals and the nitrogen lone pair. Twisting of the molecular structure in this manner can deter formation of tightly packed molecular aggregates and enhance solubility. However, these materials tend to possess low thermal and morphological stability. Additionally, the relatively short π-conjugation required to effectively delocalise and generate radical cations leads to the somewhat short lifetime of charge-generated states.7 One way to overcome these shortfalls is to structurally constrain the TPA which can be an effective method to retain the strengths of TPAs while overcoming their drawbacks (Fig. 1b).8 This planarisation of the TPA has multiple beneficial influences such as increasing the interaction of the nitrogen lone pair with the π-orbitals of the appended phenyl rings which effectively extends conjugation and affects the photophysical properties including a bathochromic shift of the absorbance and emission bands.7 Moreover, the charge transport properties are also enhanced as the increased planarity of the materials leads to a greater degree of π–π stacking, and thus orbital overlap between molecules.
This locking of the molecular conformation of a triphenylamine into a planar structure can be achieved by bridging the phenyl residues to afford structures known as an N-heterotriangulenes (N-HTs) (Fig. 1b). This interesting class of materials was first synthesised with a bridging carbonyl group by Hellwinkel and Melan.9 The work was followed by the synthesis of the methylene-bridged derivative by the same authors.10 The methylene-bridged N-HT systems are the most relevant to this manuscript having proved a useful building block for dye sensitised solar cells,11 hole transport materials for hybrid perovskite solar cells,12 thermally activated delayed fluorescence (TADF) emissive materials, host and emissive materials for organic light-emitting diodes (OLEDs),13,14 and as a non-fullerene acceptor (NFA) for organic photovoltaics.15 These materials have been the subject of several review articles regarding their synthesis and properties.8,16 As the bridging methylene groups between the aromatic rings of the N-HT improve morphological and thermal stability of their corresponding devices, further steric bulk could be used to command an even greater degree of film stability.
Jiang et al. have reported that a para-tolyl bridged N-HT derivative was an effective host for the triplet emitter Ir(ppy)3 showing a maximum current efficiency of 83.5 cd A−1 and a maximum power efficiency of 71.4 Lm W−1 for green electrophosphorescence.17 This work has inspired several derivatives that employ the para-tolyl N-HT core and further functionalisation of the TPA phenyl unit (through the para position with respect to the central nitrogen), thereby extending the conjugation of the π-system. To date materials with TPA and carbazole,18 fluorene,19 TPA-OMe20 and thiophene21 have all been successfully applied in organic electronic devices. However, functionalisation at the bridging positions with units other than para-tolyl groups has been comparatively less well studied. For example, Zhao et al. have investigated the role that functionalisation has on the properties of a push–pull dye for dye-sensitised solar cells compared to its TPA analogue.22 In another example, Jin et al. have developed an amphiphilic partially-bridged N-HT derivative which proved to be an effective HTM in perovskite and organic solar cells.23 More recently, Kivala and co-workers synthesised a fluorene-bridged N-HT in which the steric bulk around the central nitrogen facilitates the formation of long-lived stable radical cations that possess interesting optoelectronic properties.24,25 Monkman, Bryce and co-workers have investigated the role differing bridging moieties have on the delayed fluorescence (via rISC with upper-triplet states), of N-HT derivatives. Interestingly, it was suggested that the control of packing imparted by the host on the emitter plays an important role on whether delayed emission is observed or not.26
Here, we describe a series of four N-HT derivatives featuring electron-rich moieties attached to the bridging positions. Importantly, the materials have been designed so that the electron-rich units are not directly conjugated to the TPA core and provide 3D structures where the triarylamine core unit is sterically and electronically isolated by the bridging units. In addition, we describe the optical, electronic, redox and solid-state structures of these materials and investigate their ability to act as host materials in OLEDs fabricated using the emitter 4CzIPN.
Results and discussion
Synthesis
The synthesis of FTPA-OMe, FTPA-Thio, FTPA-NMe2 and FTPA-TPA is outlined in Scheme 1. Intermediate 1 was prepared following a modified version of a previously reported synthesis.27 The compounds were obtained in two-step synthetic protocols involving lithium-halogen exchange with the respective aryl halide followed by the sixfold nucleophilic addition to the ester 1. This was followed by acid-catalysed (AcOH/HCl, CH3SO3H, or AcOH/H2SO4), ring-closing dehydration reactions to afford the N-HT derivatives. All the target materials were characterised by 1H NMR, 13C NMR, mass spectrometry, and single crystal X-ray crystallography (see the ESI†). The thermal properties were also studied using thermogravimetric analysis and differential scanning calorimetry (Fig. S9, ESI†), and all investigated materials exhibited high thermal stability over 400 °C, with FTPA-TPA retaining 95% of its mass until 519 °C. | ||
| Scheme 1 Synthetic route to FTPA-OMe, Thio, NMe2 and TPA. | ||
X-ray crystallography
The structures of FTPA-OMe, FTPA-Thio, FTPA-NMe2 and FTPA-TPA were determined using single crystal X-ray crystallography (Fig. 2), and the details are provided in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ‡ In all four structures it can be seen that there is significant flattening of the TPA core compared to the parent TPA structure. The dihedral angle between the mean planes of the core phenyl units and the plane of the central three carbon atoms ranged from 11–26°, which are considerably less than typical TPA dihedral angles of 37–50° (Fig. 2).6 For FTPA-OMe, FTPA-Thio and FTPA-TPA the appended aryl units are situated pseudo axial (p-ax) from the bridging sp3 hybridised carbon and three are situated pseudo equatorial (p-eq), forming a sterically hindered side of TPA and a less hindered open side. However, the FTPA-NMe2 has two p-ax and one p-eq aryl groups on one face of the core TPA and the inverse on the other forming a kinked structure.
‡ In all four structures it can be seen that there is significant flattening of the TPA core compared to the parent TPA structure. The dihedral angle between the mean planes of the core phenyl units and the plane of the central three carbon atoms ranged from 11–26°, which are considerably less than typical TPA dihedral angles of 37–50° (Fig. 2).6 For FTPA-OMe, FTPA-Thio and FTPA-TPA the appended aryl units are situated pseudo axial (p-ax) from the bridging sp3 hybridised carbon and three are situated pseudo equatorial (p-eq), forming a sterically hindered side of TPA and a less hindered open side. However, the FTPA-NMe2 has two p-ax and one p-eq aryl groups on one face of the core TPA and the inverse on the other forming a kinked structure.
 | ||
| Fig. 2 Molecular structures for the N-HT series determined by X-ray diffraction studies, displayed as wireframes and hydrogens omitted for clarity. (A) FTPA-OMe with minor disorder components omitted, (B) FTPA-Thio with lattice DCM solvent molecules omitted, (C) FTPA-NMe2 (a region of poorly defined lattice solvent was accounted for using SQUEEZE), and (D) FTPA-TPA. | ||
Theoretical calculations
The electronic and structural properties of the N-HTs were investigated using density functional theory calculations utilising the Gaussian 09 W software package. The HOMO and LUMO distributions on the optimised structures are shown in Fig. 3. The predicted HOMOs were OMe (−4.79 eV), Thio (−5.21 eV), NMe2 (−4.24 eV) and TPA (−4.77 eV) and the LUMOs were OMe (−0.42 eV), Thio (−0.94 eV), NMe2 (−0.08 eV) and TPA (−0.57 eV). The theoretical HOMO levels indicate that the materials may be suitable as hole-transporting materials.28,29 The FMOs show HOMOs located on the central TPA while the LUMOs are more diffuse, overlapping both central TPA and appended aromatic groups.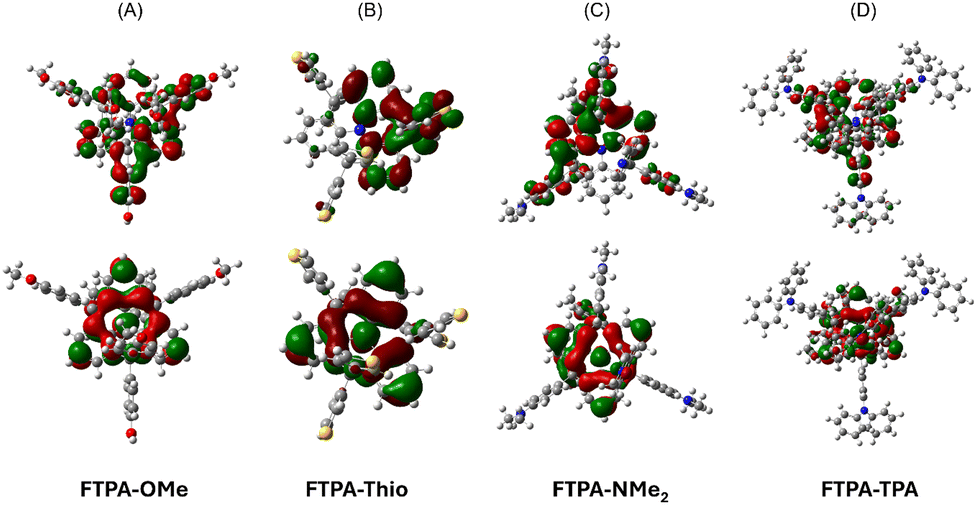 | ||
| Fig. 3 Optimised geometry of the FTPA series, highlighting the HOMO (top) and LUMO (bottom) (A) FTPA-OMe, (B) FTPA-Thio, (C) FTPA-NMe2 and (D) FTPA-TPA. | ||
Optical properties
The optical properties of the materials were investigated using UV-vis and fluorescence spectroscopy (Fig. 4). FTPA-OMe displayed a λmax at 312 nm from the π → π* of the central TPA core with two small bands around 280 nm and 240 nm attributed to the peripheral phenyl residues. FTPA-Thio displays similar spectra where there is a weaker λmax at 305 nm, attributed to a π → π* of the core, and a larger band at 251 nm attributed to a π → π* transition in the appended thiophenes. The FTPA-NMe2 derivative shows a peak at 310 nm which can be attributed to π → π* transition of the TPA core while the λmax is at 268 nm. This can be attributed to the π → π* transition of the appended dimethylaniline groups. Finally, the FTPA-TPA derivative has slightly different band structure with only one strong band found at 309 nm which can be attributed to a π → π* for the FTPA core and the appended TPAs which leads to a broadening of the absorbance band. The fluorescence properties of the N-HT derivatives displayed emission wavelengths ranging from 368 to 374 nm, with most of the emission occurring on the edge of the UV and the visible regions. The Stokes shift for all the materials range from 60 to 64 nm (1.67 × 105–1.56 × 105 cm−1) with FTPA-NMe2 possessing the largest Stokes shift.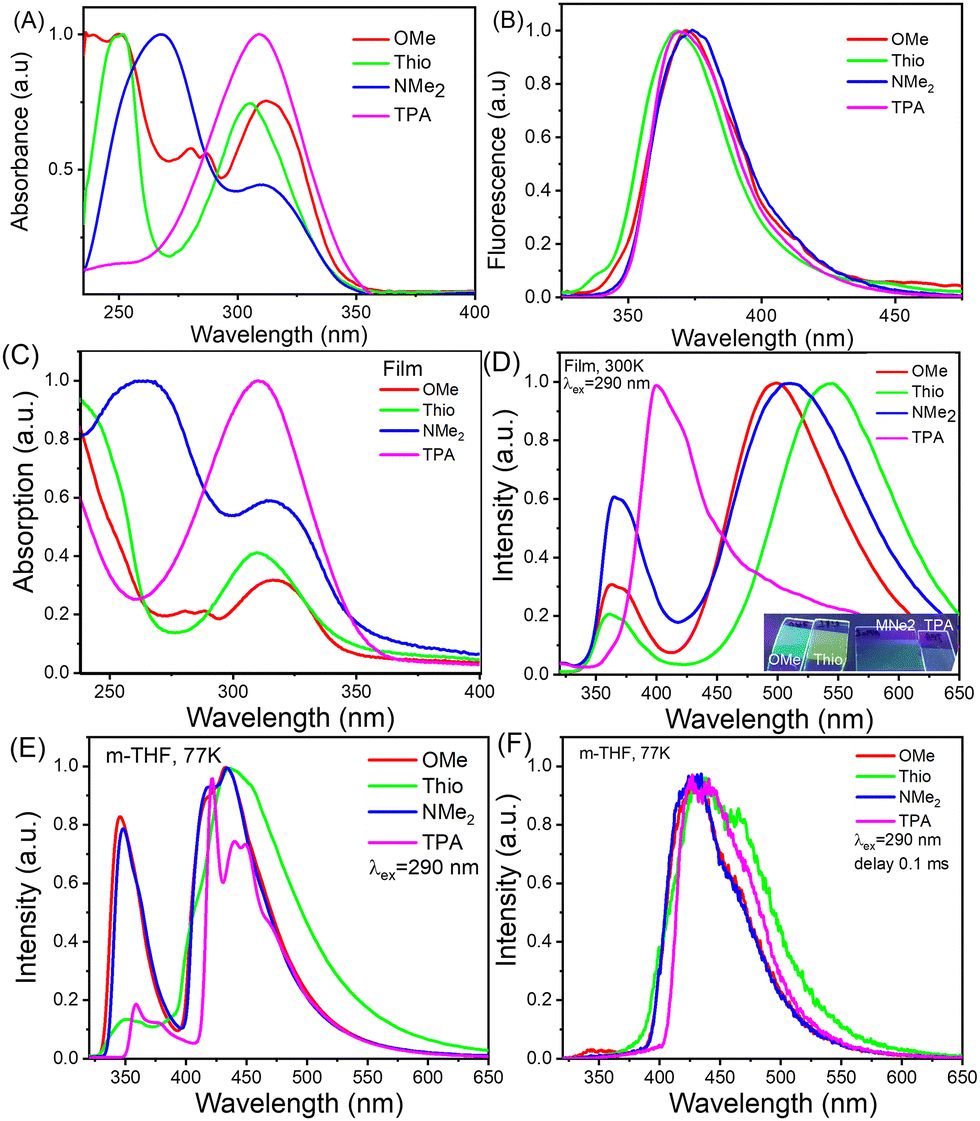 | ||
| Fig. 4 Solution and film optical properties: (A) and (C) UV/Vis absorbance spectra for all N-HTs, performed at a concentration of 5 × 10−6 M in CH2Cl2. (B) and (D) Fluorescence spectra of the solutions (B) and films (D) of the compounds. The spectra of the solutions were recorded at a concentration of 1 × 10−6 M in CH2Cl2. The films were obtained by thermal evaporation. The inset in D shows an image of the films under UV excitation. Emission spectra at 77K PL (E) and associated phosphorescence spectra (F) of the solutions of the compounds in 2-methyltetrahydrofuran (2-MeTHF) (1 × 10−5 M). Phosphorescence spectra were recorded using a delay time of 0.1 ms after excitation. | ||
In the case of the vacuum-deposited films of the N-HT derivatives (Fig. 4C), very similar absorption spectra were obtained to those recorded in solution. In contrast, the fluorescence spectra were found to be different (Fig. 4D). High-energy bands which resulted from recombination of locally excited (LE) states of the core were similar to those of the solutions. The key differences are related to the low-energy broad unstructured bands that are charge transfer (CT) in nature (Table 1). The CT bands are most likely related to intramolecular twisted CT but not to intermolecular CT (excimer) formation due to the strongly non-planar structures of these compounds.
| Media | OMe | Thio | NMe2 | TPA | |
|---|---|---|---|---|---|
| a Electron Affinity (EA) was outside of the solvent window and was estimated from the optical HOMO–LUMO gap and the ionisation energy (IE), using EA = IE − Eopt. Singlet (ES1) and triplet (ET1) energies respectively were taken from the onsets of fluorescence and phosphorescence spectra of the solutions of compounds in 2-MeTHF recorded at 77 K (Fig. 7E and F). | |||||
| HOMO DFT (eV) | — | −4.79 | −5.21 | −4.24 | −4.77 |
| LUMO (aEA) (eV) | — | −0.42 (−1.76) | −0.94 (−1.76) | −0.08 (−1.54) | −0.57 (−1.86) |
| λ onset (nm) | CH2Cl2 | 350 | 341 | 348 | 350 |
| E opt (eV) | 3.54 | 3.64 | 3.56 | 3.54 | |
| E S1 (eV) | 2-MeTHF, 77 K | 3.75 | 3.75 | 3.71 | 3.52 |
| E T1 (eV) | 3.16 | 3.19 | 3.16 | 3.05 | |
| λ PL (nm) | Film | 365, 509 | 360, 542 | 366, 510 | 400 |
| Φ PL | Toluene | 0.10 | 0.10 | 0.02 | 0.09 |
| Φ PL | Film | 0.20 | 0.12 | 0.10 | 0.09 |
| IE (eV) | TBA(PF6) | −5.3 | −5.4 | −5.1 | −5.4 |
| μ SCLCh (cm2 V−1 s−1) | Film | 1.0 × 10−4 | 3.0 × 10−5 | 2.4 × 10−4 | 2.0 × 10−4 |
| μ TOFh (cm2 V−1 s−1) | Film | — | 7.4 × 10−4, 7.4 × 10−5 | — | — |
The emission spectra at 77 K are shown in Fig. 4E, with phosphorescence spectra in Fig. 4F. The low temperature emission spectra are similar in shape to those measured at room temperature. An exception is the emission of FTPA-TPA which shows vibronic structure due to the restricted movement of this larger molecule at low temperature. The calculated T1 energies (Table 1) do not show much variation, between 3.05–3.19 eV, suggesting that the materials should be similarly suitable to act as a host material in OLEDs.
The photoluminescence quantum yields (PLQY) of the N-HT series were measured in both solution and thin films (Fig. S12 and S13, ESI†). In solution, all the materials exhibited low PLQYs: FTPA-OMe, FTPA-Thio and FTPA-TPA showed values around ΦPL ≈ 0.1, while FTPA-NMe2 was significantly lower at ΦPL = 0.02. Notably, the PLQYs increased when measured in the neat thin films. FTPA-OMe doubled to ΦPL = 0.2, FTPA-Thio increasing moderately to ΦPL = 0.12 and FTPA-NMe2 showed a five-fold increase to ΦPL = 0.10. However, FTPA-TPA exhibited no change, remaining at ΦPL = 0.09.
The low observed PLQY values are consistent with the intended function of the N-HT series as hole-transporting host materials, where efficient emission is not a design priority. However, the increase in PLQY from solution to film, particularly for FTPA-OMe and FTPA-NMe2, was unexpected and suggests the presence of intra- or intermolecular interactions in the solid state that enhance the radiative pathways. This prompted further investigation into the aggregation-induced effect, which we examined by correlating emission behaviour with crystallographic data.
Aggregation study
To further investigate the solid-state emission behaviour of these materials, the steady-state emission spectra of the aggregates were collected (Fig. 5). Aggregation was induced by gradually increasing the water fraction (fw) in THF/water mixtures while maintaining a constant analyte concentration. The appended aromatic groups are expected to rotate freely in solution; upon aggregation, this rotational freedom is restricted, potentially enhancing emission intensity—a feature of aggregation-induced emission (AIE). The photophysical response of each material upon aggregation was complex, and none of the compounds displayed classical AIE behaviour. FTPA-OMe showed minimal changes in emission with increasing fw until 80%, at which point the emission intensity decreased significantly, likely due to precipitation. FTPA-Thio and FTPA-NMe2 displayed similar behaviour: as fw increased, a red-shifted emission band emerged at 525 nm and 476 nm respectively while the original blue emission bands (366 nm and 367 nm, respectively) diminished. These changes occurred up to 80% fw for FTPA-Thio and 90% fw for FTPA-NMe2. FTPA-TPA also exhibited the emergence of a red-shifted band at 499 nm; however, it remained relatively weak compared to the original blue emission in neat THF, and no overall enhancement in emission intensity was observed. | ||
| Fig. 5 Fluorescence intensity of FTPA-OMe (A), FTPA-Thio (B), FTPA-NMe2 (C), and FTPA-TPA (D) in THF/water mixtures with increasing water fractions (fw) from 0% to 90%, performed at constant concentration of 1 × 10−5 M (λex = 310 nm). | ||
To further explore the relationship between emission behaviour and structural differences among the N-HT materials, we examined their crystal packing in more detail. Among the four single-crystal X-ray structures obtained, only FTPA-Thio and FTPA-NMe2 contain solvent molecules in the lattice (∼10% occupancy), suggesting less efficient packing compared to FTPA-OMe (0% solvent) and FTPA-TPA (1.4% solvent). This trend may partially explain the more modest changes in emission observed upon aggregation for FTPA-OMe and FTPA-TPA, which exhibit more efficient molecular packing in the solid state.
Beyond solvent inclusion, there is limited additional evidence of close molecular packing in the solid state (see ESI† for further details on crystal packing). The only notable short contact is found in FTPA-Thio, where the sulfur atoms S4 and S7 from two oppositely oriented molecules which are separated by 3.321 Å, shorter than the sum of their van der Waals radii (3.630 Å), as shown in Fig. 6. This interaction may contribute to the distinct emission features observed for FTPA-Thio in the aggregated state.
 | ||
| Fig. 6 Crystal packing of FTPA-Thio, highlighting the intermolecular S⋯S contact between atoms S4 and S7 of adjacent, oppositely oriented molecules. Displayed as ellipsoids with hydrogens and solvent molecules omitted for clarity. | ||
Taken together, these observations suggest increase in PLQY from solution to neat films for FTPA-OMe and FTPA-NMe2 likely arises from the restriction of non-radiative decay pathways in the solid state, possibly via suppression of intramolecular rotations. This is consistent with the well-established restriction of intramolecular motion mechanism, often invoked in AIE-active systems. However, the lack of classical AIE behaviour and the emergence of red-shifted emission bands in THF/water mixtures suggest the formation of weakly emissive excimer-like states. Notably, FTPA-TPA did not show PLQY enhancement in films, possibly due to its already constrained structure in solution limiting further restriction upon aggregation.
Electrochemical properties
The electrochemical properties of the materials were investigated using solution-state cyclic voltammetry (CV) and square-wave voltammetry (SWV) (Fig. 7). FTPA-OMe displayed a quasi-reversible oxidation wave at 0.53 V vs. Fc/Fc+ giving an ionisation energy (IE) of −5.3 eV, whereas FTPA-Thio displayed a quasi-reversible oxidation wave at 0.56 V vs. Fc/Fc+ (IE −5.4 eV). FTPA-TPA and FTPA-NMe2 on the other hand displayed complex multi-electron processes suggesting that the peripheral redox-active groups exist in different electrochemical environments. The SWV of FTPA-TPA displayed two quasi-reversible redox events at 0.62 and 0.95 V vs. Fc/Fc+, the first of which at 0.6 V equating to an IE of −5.4 eV. Interestingly, the CV of FTPA-NMe2 displayed a single irreversible oxidation wave as well as two irreversible reduction waves. The SWV was similar to that of FTPA-TPA, with a complex oxidation process occurring, the first oxidation occurring at 0.29 V vs. Fc/Fc+ equating to an IE of −5.1 eV. No clear reduction redox events were observed due to the limitations of the solvent and the electrolyte chosen. Therefore, an approximation of the electron affinity (EA) and LUMO can be estimated using the optical HOMO–LUMO gap (Eopt) and subtracting this from the experimentally observed ionisation energy (IE). (Table 1) This neglects the binding energy but can be used as an approximation. FTPA-OMe has an Eopt of 3.54 eV which approximates to a LUMO of −1.76 eV. The FTPA-Thio derivative gives an Eopt of 3.64 eV (LUMO = −1.76 eV), FTPA-NMe2 has an Eopt of 3.56 eV (LUMO = −1.54 eV), and FTPA-TPA has an Eopt of 3.54 eV (LUMO = −1.86 eV). | ||
| Fig. 7 Electrochemical analysis of all compounds (SWV left, CV right). All measurements were conducted at 5 × 10−4 M−1 in CH2Cl2 with 0.1 M TBAPF6 as the supporting electrolyte and ferrocene as an external standard. | ||
The observed ionisation energies are very similar to that reported for TPA (5.23 eV) and that of the related N-HT featuring para-tolyl groups (5.22 eV).17 As would be expected, the more electron-rich para-anisole derivative has a slightly lower IE and the even more electron-donating diphenylamine even lower yet. The thiophene derivative also has substantial influence on the IE, lying around the same as the TPA derivative. However, the NMe2 derivative displayed the highest IE, around 0.1 eV higher than that of TPA.
Hole mobility (space-charge-limited current, SCLC)
The hole mobility (p-type) of the materials was determined using hole-only devices with a device architecture ITO/PEDOT:PSS/Host/Al. The N-HT material was deposited by spin-coating and the thickness of the films was determined to be around 100 nm. The current–voltage curves were recorded (Fig. 8A), and the hole mobility was calculated using the Mott–Gurney law.30FTPA-OMe, FTPA-NMe2 and FTPA-TPA had the highest hole mobilities (FTPA-OMe: μh = 1.0 × 10−4 cm2 V−1 s−1; FTPA-NMe2: μh = 2.4 × 10−4 cm2 V−1 s−1; FTPA-TPA: μh = 2.0 × 10−4 cm2 V−1 s−1), an order of magnitude higher than FTPA-Thio (μh = 3.0 × 10−5 cm2 V−1 s−1).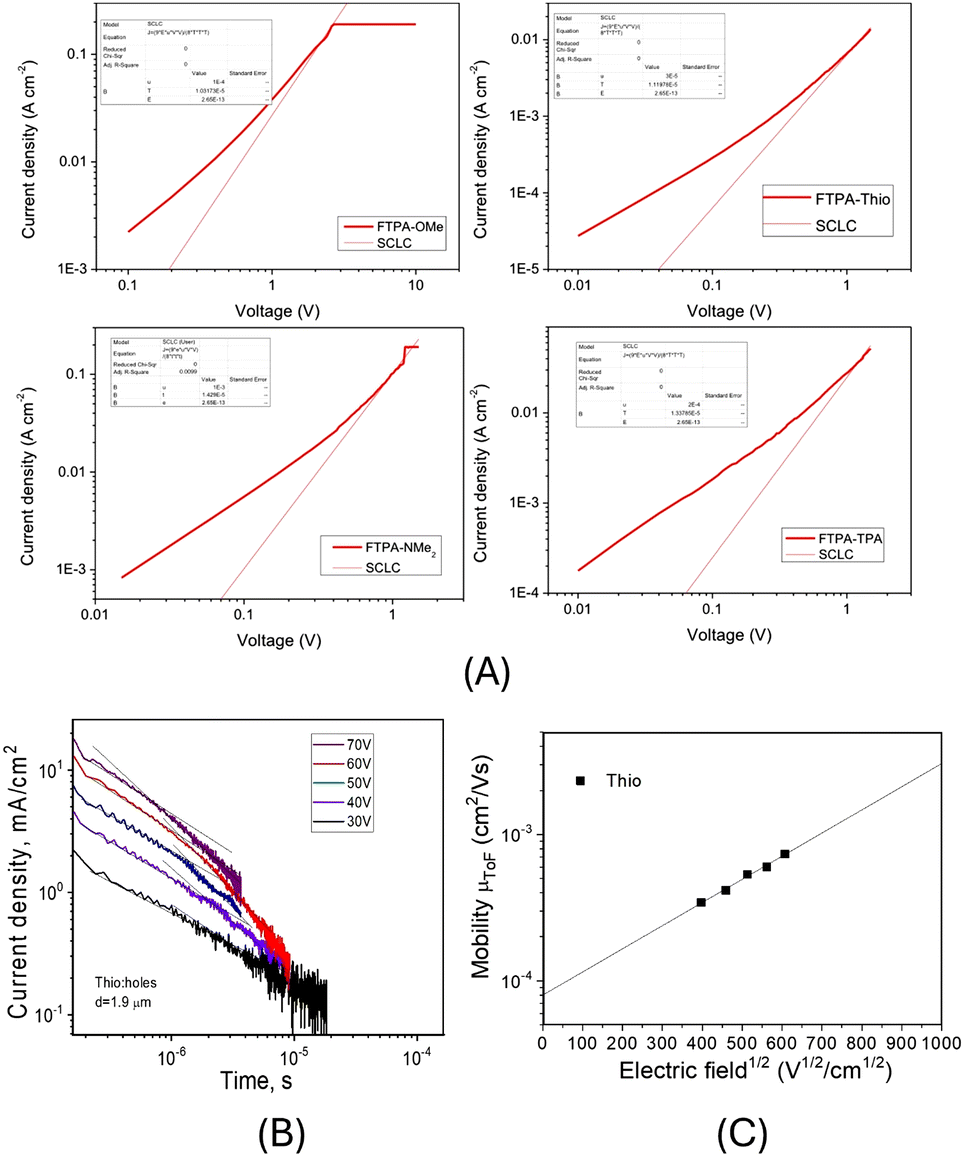 | ||
| Fig. 8 (A) J–V curves with space-charge-limited current mobility fitting. (B) TOF data and (C) electric field dependence of hole mobility in vacuum-deposited film of FTPA-Thio. | ||
When the time-of-flight (TOF) method was used for investigation of charge-transporting properties of vacuum-deposited films, the transit times were observed only for the compound FTPA-Thio (Fig. 8B). Nonetheless, hole-transporting properties of FTPA-OMe, FTPA-NMe2, and FTPA-TPA are confirmed by the results of the charge-transport measurements by charge carrier extraction by linearly increasing voltage (CELIV) method (see Fig. S10 and S11 for further details, ESI†). Using the transit time value, a hole mobility of 7.4 × 10−4 cm2 V−1 s−1 at an electric field of 3.6 × 105 V cm−1 was determined. By fitting of the experimental data by the Poole–Frenkel type mobility (μh = μ0 × exp(βE(0.5))), a zero-field mobility μ0 of 7.4 × 10−5 cm2 V−1 s−1 was obtained which is just slightly higher than that obtained by the space-charge-limited current measurements. It should be noted that compound FTPA-Thio (which showed TOF mobility) also demonstrated the best device efficiency as discussed below.
OLED device results
Due to having acceptable hole mobilities, the molecules were applied as p-type host materials for OLED devices. The hole transport and emissive layers were solution-processed, whereas the electron transport layer (TPBi, 35 nm), electron injection layer (LiF, 1 nm) and cathode (Al, 100 nm) were deposited by thermal evaporation. FTPA-NMe2 was poorly soluble at the required concentration for deposition and therefore was not studied further as a host material for OLEDs with solution-processed emissive layers. However, although it has not been studied here, we believe that the molecules could in principle also be suitable for deposition by thermal evaporation.The thermally activated delayed fluorescence emitter, 4CzIPN, was used as the emissive material with the N-HT host materials in OLEDs. 4CzIPN was chosen as it has previously been used in highly efficient OLEDs, with reported external quantum efficiencies >25% for evaporated devices. Furthermore, it can be prepared easily from commercial reagents (4CzIPN is also commercially available), and unlike some TADF emitters it is also applicable for solution-processing. Suzuki et al. reported that a solution-processed emissive layer of 6 wt% 4CzIPN in CBP led to OLEDs with maximum EQE of 18.5%.31 Although these devices had a short luminance half-life of just over 2 hours, this was improved to over 180 hours when a novel host, CBPC, and optimum electron transport layers were introduced, but led to a reduction of maximum EQE to 9.9%, highlighting the challenge of achieving high performance and lifetime.31 Komatsu and co-workers reported a spin-coated emissive layer of 4CzIPN in a CBP host and various electron transport layers.32 At an emitter concentration of 5 wt%, OLEDs containing TPBi gave a maximum EQE of 7.0%, while B3PyPB and B4PyMBM-containing devices gave 15% and 12%, respectively.32
The device performance of 4CzIPN-based OLEDs with the N-HT hosts is summarised below in Table 2, while plots of EQE vs. luminance are shown in Fig. 9. Of the OLEDs tested, it is clear that FTPA-Thio performs best as a host material for the 4CzIPN-based OLEDs, achieving a maximum EQE of 6.9% and maximum luminance of 1319 cd m−2. This is comparable to devices prepared by Komatsu et al. where a 5% 4CzIPN concentration led to an EQE of 7.0%.32 The performance is significantly reduced when FTPA-OMe (1.7%, 740 cd m−2) or FTPA-TPA (1.1%, 603 cd m−2) are used as hosts, suggesting that FTPA-Thio is a better candidate, despite having a lower hole mobility, as measured using the SCLC model.
| EML | Turn on voltage [V] | Maximum luminance [cd m−2] | Maximum current efficiency [cd A−1] | Maximum power efficiency [lm W−1] | Maximum EQE [%] | CIE 1931 coordinates |
|---|---|---|---|---|---|---|
| 10% 4CzIPN/FTPA-OMe | 3.1 | 740 | 4.8 | 3.5 | 1.7 | 0.40, 0.58 |
| 10% 4CzIPN/FTPA-Thio | 3.5 | 1319 | 22.1 | 15.6 | 6.9 | 0.39, 0.60 |
| 10% 4CzIPN/FTPA-TPA | 3.5 | 604 | 3.0 | 1.9 | 1.1 | 0.40, 0.58 |
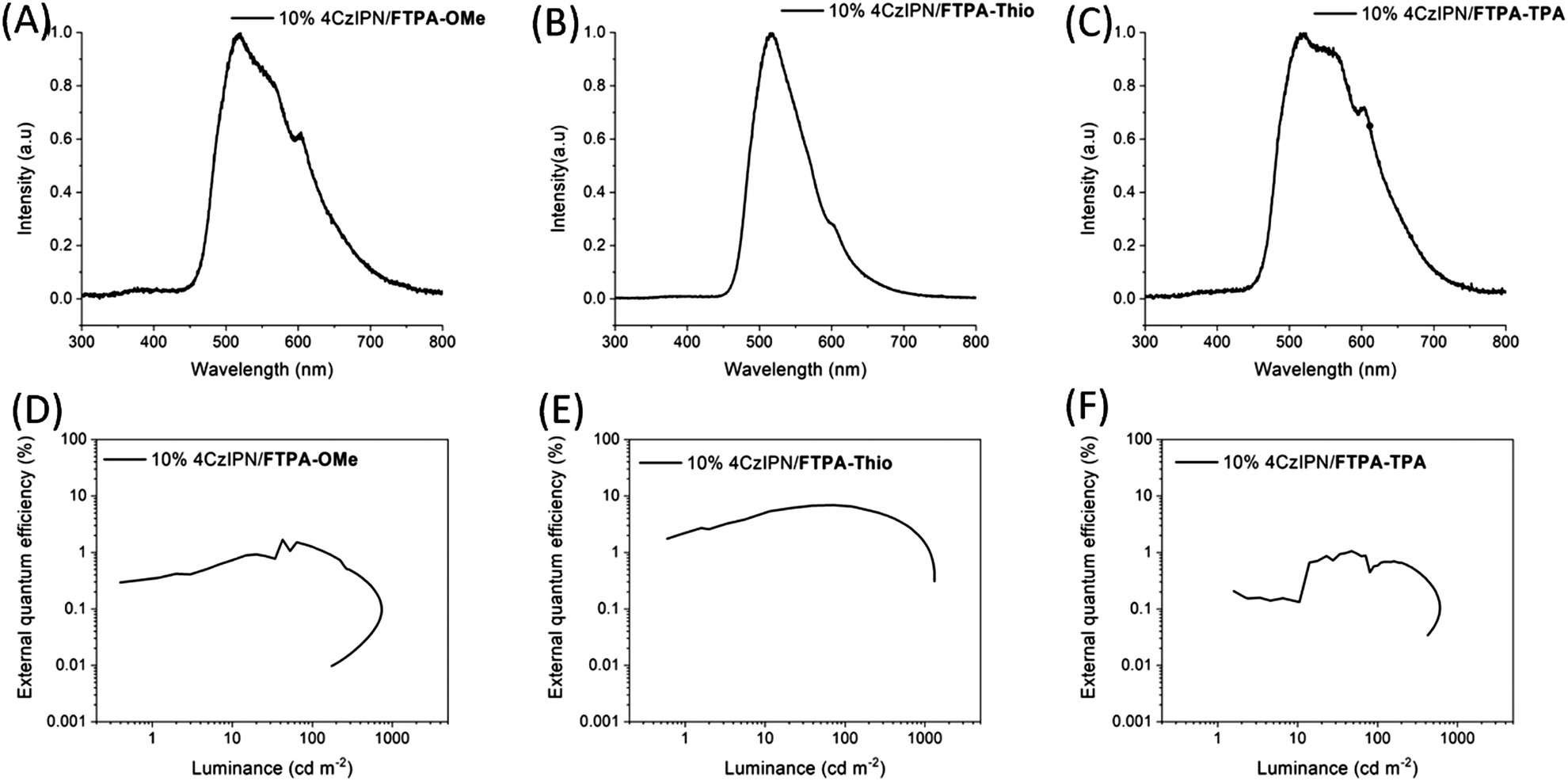 | ||
| Fig. 9 Electroluminescence spectra of OLEDs with (A) FTPA-OMe, (B) FTPA-Thio and (C) FTPA-TPA host materials. Plots of external quantum efficiency vs. luminance for OLEDs containing 4CzIPN emitter with (D) FTPA-OMe, (E) FTPA-Thio and (F) FTPA-TPA host. | ||
The electroluminescence (EL) spectra of the devices give an indication as to why there is a difference in performance between the host materials. The EL spectrum of 4CzIPN/FTPA-Thio has a peak at 518 nm, with only a small shoulder at 605 nm. However, the spectrum for 4CzIPN/FTPA-OMe is broader. Its main peak is at 520 nm but it also has a large shoulder emerging at 567 nm and another large peak at 604 nm, and its relative intensity to the main peak is much larger than the shoulder in the EL spectrum for the OLED with a 4CzIPN/FTPA-Thio emissive layer. The spectrum for 4CzIPN/FTPA-TPA emissive layer also has its most intense peak at 519 nm, but the shoulder at 560 nm is only slightly less intense than the main peak. The peak at 602 nm is even more intense relative to the main peak compared to the spectra for the other two hosts. The peak at ∼560 nm can be attributed to the 4CzIPN dimer.33 This has a low PLQY,33 therefore, large quantities of the dimer species will be expected to lower the EQE if they are present in OLEDs. This difference in EL spectra suggests that FTPA-Thio is more effective in breaking up dimers in the dopant/host film. The shoulder/peak at ∼600 nm can possibly be assigned to exciplex formation and this inefficient energy transfer from host to dopant may also have impacted on device efficiency for FTPA-OMe and FTPA-TPA.
The change in EL spectra means that for devices containing FTPA-OMe and FTPA-TPA, the emission is more yellow compared to devices using FTPA-Thio host. Thus, the host material influences the emission colour as well as the efficiency of the devices.
Conclusions
This paper describes the synthesis and characterisation of N-HTs for application as p-type host materials for OLEDs. The films exhibited mobilities in the range of 10−5–10−4 cm2 V−1 s−1, which is appropriate for p-type hosts. When used in the emissive layer with a 4CzIPN dopant, OLEDs containing FTPA-Thio host material gave an external quantum efficiency of 6.9%, while the other materials showed reduced performance. This was attributed to better distribution of the dopant in the FTPA-Thio layer, thereby avoiding dimer formation. We have demonstrated that N-HT materials can perform effectively as p-type host materials. As such, these materials may warrant further study with different emissive materials, device structures and/or deposition by thermal evaporation to further improve device performance.Author contributions
SW and GC designed, synthesised and analysed the materials. DW performed analysis. CW refined the X-ray data. DV, JVG, JC, PJS and SW fabricated the devices and carried out the analysis. SW wrote the initial manuscript, and all authors contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the Supplementary Information. Crystallographic data for FTPA-OMe, FTPA-Thio, FTPA-NMe2 and FTPA-TPA have been deposited with the CCDC. These can be found at CCDC 2130319–2130322.Acknowledgements
SW and JC thank the EPSRC for funding (EP/M506539, EP/M508056/1, EP/T022477/1). GC thanks the Leverhulme Trust for the award of a Research Fellowship. This work is also supported by the Research Council of Lithuania (project COLED, No S-MIP-24-2). We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for the mass spectrometry. National crystallography service for the crystal structures.Notes and references
- P. Blanchard, C. Malacrida, C. Cabanetos, J. Roncali and S. Ludwigs, Polym. Int., 2019, 68, 589–606 CrossRef CAS.
- P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348–1373 RSC.
- A. Farokhi, H. Shahroosvand, F. Zisti, M. Pilkington and M. K. Nazeeruddin, J. Mater. Chem. A, 2023, 11, 25136–25215 RSC.
- A. Farokhi, H. Shahroosvand, G. D. Monache, M. Pilkington and M. K. Nazeeruddin, Chem. Soc. Rev., 2022, 51, 5974–6064 RSC.
- M. U. Munshi, G. Berden, J. Martens and J. Oomens, Phys. Chem. Chem. Phys., 2017, 19, 19881–19889 RSC.
- A. N. Sobolev, V. K. Belsky, I. P. Romm, N. Yu. Chernikova and E. N. Guryanova, Acta Crystallogr. C, 1985, 41, 967–971 CrossRef.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef.
- N. Hammer, T. A. Schaub, U. Meinhardt and M. Kivala, Chem. Rec., 2015, 15, 1119–1131 CrossRef.
- D. Hellwinkel and M. Melan, Chem. Ber., 1971, 104, 1001–1016 CrossRef.
- D. Hellwinkel and M. Melan, Chem. Ber., 1974, 107, 616–626 CrossRef.
- N. Karjule and J. Nithyanandhan, J. Mater. Chem. A, 2016, 4, 18910–18921 RSC.
- H. Choi, S. Park, S. Paek, P. Ekanayake, M. K. Nazeeruddin and J. Ko, J. Mater. Chem. A, 2014, 2, 19136–19140 RSC.
- Z. Fang, V. Chellappan, R. D. Webster, L. Ke, T. Zhang, B. Liu and Y.-H. Lai, J. Mater. Chem., 2012, 22, 15397–15404 RSC.
- X.-K. Chen, Y. Tsuchiya, Y. Ishikawa, C. Zhong, C. Adachi and J.-L. Brédas, Adv. Mater., 2017, 29, 1702767 CrossRef PubMed.
- Y. Kervella, J. M. Andrés Castán, Y. A. Avalos-Quiroz, A. Khodr, Q. Eynaud, T. Koganezawa, N. Yoshimoto, O. Margeat, A. Rivaton, A. J. Riquelme, V. M. Mwalukuku, J. Pécaut, B. Grévin, C. Videlot-Ackermann, J. Ackermann, R. Demadrille and C. Aumaître, J. Mater. Chem. C, 2023, 11, 8161–8169 RSC.
- J. Petkus and K. Shubin, Chem. Heterocycl. Compd., 2020, 56, 512–514 CrossRef.
- Z. Jiang, Y. Chen, C. Yang, Y. Cao, Y. Tao, J. Qin and D. Ma, Org. Lett., 2009, 11, 1503–1506 CrossRef.
- Z. Jiang, T. Ye, C. Yang, D. Yang, M. Zhu, C. Zhong, J. Qin and D. Ma, Chem. Mater., 2011, 23, 771–777 CrossRef.
- C. Liu, Y. Li, Y. Zhang, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. – Eur. J., 2012, 18, 6928–6934 CrossRef CAS PubMed.
- M. Zhang, G. Wang, D. Zhao, C. Huang, H. Cao and M. Chen, Chem. Sci., 2017, 8, 7807–7814 RSC.
- F. Wu, L. T. L. Lee, J. Liu, S. Zhao, T. Chen, M. Wang, C. Zhong and L. Zhu, Synth. Met., 2015, 205, 70–77 CrossRef CAS.
- D.-X. Zhao, L.-Y. Bian, Y.-X. Luo, M.-D. Zhang, H. Cao and M.-D. Chen, Dyes Pigm., 2017, 140, 278–285 CrossRef CAS.
- S. S. Reddy, H.-Y. Park, H. Kwon, J. Shin, C.-S. Kim, M. Song and S.-H. Jin, Chem. – Eur. J., 2018, 24, 6426–6431 CrossRef CAS PubMed.
- T. A. Schaub, T. Mekelburg, P. O. Dral, M. Miehlich, F. Hampel, K. Meyer and M. Kivala, Chem. – Eur. J., 2020, 26, 3264–3269 CrossRef CAS PubMed.
- M. Krug, M. Wagner, T. A. Schaub, W.-S. Zhang, C. M. Schüßlbauer, J. D. R. Ascherl, P. W. Münich, R. R. Schröder, F. Gröhn, P. O. Dral, M. Barbatti, D. M. Guldi and M. Kivala, Angew. Chem., Int. Ed., 2020, 59, 16233–16240 CrossRef CAS.
- J. S. Ward, N. A. Kukhta, P. L. dos Santos, D. G. Congrave, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Chem. Mater., 2019, 31, 6684–6695 CrossRef CAS PubMed.
- S. F. Gauthier Jean and M. J. Synthesis, Stuttg, 1987, 4, 383–385 Search PubMed.
- J.-H. Pan, H.-L. Chiu, L. Chen and B.-C. Wang, Comput. Mater. Sci., 2006, 38, 105–112 CrossRef CAS.
- P. Cias, C. Slugovc and G. Gescheidt, J. Phys. Chem. A, 2011, 115, 14519–14525 CrossRef CAS PubMed.
- D. Poplavskyy and J. Nelson, J. Appl. Phys., 2003, 93, 341–346 CrossRef CAS.
- Y. Suzuki, Q. Zhang and C. Adachi, J. Mater. Chem. C, 2015, 3, 1700–1706 RSC.
- R. Komatsu, H. Sasabe, S. Inomata, Y.-J. Pu and J. Kido, Synth. Met., 2015, 202, 165–168 CrossRef CAS.
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov, C. Pflumm, V. Bhalla, M. R. Bryce and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 11109–11117 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Device methodology, NMR, MS, thermal analysis and crystallographic data. CCDC 2130319–2130322. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc01004g |
| ‡ The CIFs of FTPA-OMe, FTPA-Thio, FTPA-NMe2 and FTPA-TPA were deposited with the CCDC. CCDC 2130319–2130322. |
| This journal is © The Royal Society of Chemistry 2025 |