
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc oxide-decorated MIL-53(Al)-derived porous carbon for supercapacitor devices†
Arpad Mihai
Rostas‡
 a,
Ahmet
Gungor‡
bd,
Angela M.
Kasza
a,
Feray Bakan
Misirlioglu
c,
Alexandru
Turza
a,
Lucian
Barbu-Tudoran
a,
Emre
Erdem
*bd and
Maria
Mihet
*a
a,
Ahmet
Gungor‡
bd,
Angela M.
Kasza
a,
Feray Bakan
Misirlioglu
c,
Alexandru
Turza
a,
Lucian
Barbu-Tudoran
a,
Emre
Erdem
*bd and
Maria
Mihet
*a
aNational Institute for Research and Development of Isotopic and Molecular Technologies-INCDTIM, Donat Street, 67-103, 400293 Cluj-Napoca, Romania. E-mail: maria.mihet@itim-cj.ro
bFaculty of Engineering and Natural Sciences, Sabancı University, 34956 Istanbul, Turkey. E-mail: emre.erdem@sabanciuniv.edu
cSabancı University, Nanotechnology Research and Application Center (SUNUM), 34956 Istanbul, Turkey
dCenter of Excellence for Functional Surfaces and Interfaces for Nano-Diagnostics (EFSUN), Sabancı University, Tuzla 34956, Istanbul, Turkey
First published on 15th April 2025
Abstract
In this study, we present a facile and direct approach for the synthesis of ordered mesoporous metal–organic framework (MOF)-derived carbon materials, uniformly adorned with zinc oxide (ZnO), to serve as electrode materials for supercapacitor applications. The method involves the impregnation of zinc nitrate into both the as-synthesized (as) and activated low-temperature (lt) forms of the MIL-53(Al) metal–organic framework, which are subsequently employed as precursors to fabricate ZnO-decorated carbon structures (ZnO@C) through simultaneous decomposition under thermal treatment in an Ar atmosphere. The resultant ZnO@C(as) and ZnO@C(lt) materials exhibit a channel-like carbon morphology with uniformly distributed ZnO and residual alumina nanoparticles and a bimodal porous structure with pores approximately 8.5 and 15 nm in size. Additionally, a greater concentration of carbon-related defect centers was identified in ZnO@C(as) relative to ZnO@C(lt), as evidenced by Raman, and electron paramagnetic resonance spectroscopy. When utilized as electrode materials in both symmetric and asymmetric supercapacitor devices, the ZnO@C materials demonstrated exceptional performance, achieving energy and power densities of up to 30.5 W h kg−1 and 388 kW kg−1, respectively, and exhibiting coulombic efficiencies exceeding 95% in all instances.
1. Introduction
The increasing use of renewable energy and the growing demand for electricity for daily electronic devices have accelerated research on electrochemical energy storage.1,2 The objective is not only to store energy but also to efficiently transmit the stored energy; therefore, the demand for energy storage devices with high power density is increasing.3,4 Among the different devices developed so far, the low power density and limited charge/discharge cycles of batteries, as well as the low energy density of conventional capacitors, have limited their use in advanced technologies, giving rise to the development of supercapacitors (SCs), which bridge the gap between the former two.1,5–8Depending on the charge storage mechanism, SCs are classified into electrochemical double-layer capacitors (EDLCs), pseudocapacitors (PSCs), and hybrid supercapacitors (HSCs).9,10 In the case of EDLCs, the charge is stored electrostatically at the interface between a solid electrode and an electrolyte, with no electrons passing between the two.7,10,11 High capacitance is achieved by the use of carbon-based electrode materials, such as active carbon (AC), graphene oxide (GO), reduced graphene oxide (rGO), or carbon nanotubes (CNTs), with high surface area and adequate porosity (pore size distribution and pore volume).1,7,10,12,13 For pseudocapacitors, on the other hand, the charge storage mechanism is based on the faradaic reaction between electroactive species, resulting in higher energy density.7,14 Pseudocapacitors use transition metal oxides (MnO2, ZnO, NiO, Co3O4), transition metal hydroxides, or conducting polymers (polyaniline, polypyrrole)4,14–17 as electrode materials, and reach at least one order of magnitude higher capacitance than EDLCs.1,7 By combining the properties of EDLCs and pseudocapacitors, hybrid supercapacitors are obtained, which use both the non-faradaic and faradaic storage mechanisms to achieve high energy and power density simultaneously.10,18–20
Regardless of the SC type, the used electrode material is essential in achieving high electrochemical performance. Therefore, the SC research focuses on developing efficient electrode materials to ensure high energy and power density, exceptional gravimetric capacitance, and long cycle life.10,21
Metal–organic frameworks (MOFs) have emerged as promising candidates due to their exceptional properties such as high surface area, ordered porosity, structural diversity, multiple redox activity, as well as active sites for charge storage.7,22 First introduced by Yaghi23 in 1995, MOFs are highly crystalline materials obtained by coordination bonds between diverse metal nodes and organic linkers, leading to structures with well-defined porosity.
Despite their exceptional textural properties, which make them attractive for SCs, the direct use of MOFs as electrode materials is limited due to their usually low electrical conductivity, as well as their low chemical and structural stability during the charge–discharge process.10,11 Thus, Diaz et al.24 first reported the use of pristine MOFs for SC devices, that is Co8-MOF-5, MOF-5 with Zn partially substituted by Co, while Sheberla et al.11 used (Ni3(HITP)2), a MOF with high electrical conductivity, as the sole electrode material in an EDLC, resulting in a MOF-based device with an areal capacitance that outperforms most carbon-based materials.
Besides their direct use, MOFs can be used as precursors and templates to obtain derived nanostructured materials by different thermal treatments. Thus, depending on the chemical atmosphere during the heat treatment, nanoporous carbon materials (inert atmosphere) or nanoporous metal oxides (air) can be obtained, with inherited characteristics from the parent MOFs and, more importantly, improved electrical conductivity and electrochemical performance, besides enhanced stability.1,10,25 Moreover, MOF-derived carbons stand out among the carbonaceous materials due to the generally simple synthesis procedure and their ordered porous architecture, which ensures large surface areas and porosity.10,26,27
Due to the large variety of known MOFs as a consequence of the diversity of employed organic ligands and metals used as secondary building units, there are numerous reports on the use of MOF-derived carbon materials for supercapacitors. Thus, Liu et al. were the first to report the use of MOF-5 with furfuryl alcohol filled pores to produce nanoporous carbon for EDLCs,28,29 showing specific capacitance of 204 F g−1 at 5 mV s−1.28 Further on, MOFs such as ZIF-8,30 UiO-66,31 H-KUST-1,32 M-MOF-74,33 Al-BTC,34 MIL-53(Al),21,35,36 Al-PCP32 subjected to various thermal treatments resulted in MOF-derived carbons with various textural and electrochemical properties.
In the present study, MIL-53(Al)-derived carbon decorated with ZnO was used as electrode material for symmetric and asymmetric SC devices. MIL-53 metal–organic-frameworks are flexible three-dimensional materials possessing a unique porous structure given by the 1D-diamond-shaped channels formed as a result of the interconnection of corner-sharing MO4(OH)2 octahedra (M = Cr3+ Al3+, Fe3+) with benzene-dicarboxylate units.37 Among these MIL-53 metal–organic-frameworks, the aluminum-based representative has received more attention due to the affordability of the Al precursors. However, previous works on MIL-53(Al)-derived carbons as electrode materials reported complex synthesis procedures in dimethyl formamide (DMF) and HCl to obtain the MOF(Al) precursor, while the alumina-doped nanoporous carbon obtained after thermal treatment in an inert atmosphere was subjected to alumina removal by HCl treatment at elevated temperature.35,36 The novelty of this work is given by the following ideas: (1) the MOF precursor, MIL-53(Al), was obtained by an optimized hydrothermal synthesis procedure previously reported by our group (190 °C, 12 h, under autogenous pressure) using only water as solvent;38 (2) both the as-synthesized (as) and the activated low-temperature (lt) forms of MIL-53(Al) were used as supports for the impregnation with zinc nitrate, followed by the simultaneous decomposition under inert atmosphere of Zn(NO3)2 to ZnO and of MIL-53(Al) to Al2O3-decorated C structure; (3) direct use of MIL-53(Al)-derived carbon structures with the remaining Al2O3 from the pristine MOF after the thermal treatment, avoiding thus any HCl treatment to remove alumina. Several advantages arise from this approach. Not only is the synthesis procedure of MIL-53(Al) green (only water as solvent) and more economical due to the shorter synthesis time and the lower reaction temperature employed, but the starting materials are cheap and affordable. Also, direct use of the as-synthesized form of MIL-53(Al) (with unreacted benzene-dicarboxylic acid trapped in the pores of the MOF) as a precursor prevents the tedious activation procedure to obtain the activated low-temperature form. Moreover, obtaining ZnO and MIL-53(Al)-derived carbon simultaneously during the thermal treatment brings an additional economic advantage.
2. Experimental
All chemicals were used as purchased without further purification, and their source and purity are indicated in every case.The starting MOF, MIL-53(Al), was synthesized by an optimized synthesis procedure as detailed in our previous work.38 Briefly, a reaction mixture consisting of aluminum nitrate nonahydrate Al(NO3)3·9H2O (Carl Roth, >98%), 1,4-benzenedicarboxylic acid (BDC) (Merck, >98%), and water in the molar ratio Al![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BDC:H2O = 2:1:160 was hydrothermally treated at 190 °C for 12 h, under autogenous pressure. The as-synthesized white product (MIL-53(Al)(as)) was filtered and washed thoroughly with deionized water and dried at room temperature overnight. The activated form of the MOF, namely MIL-53(Al)(lt), was obtained by heating under reflux in dimethylformamide (DMF, Chempur, >99%) for 24 h, followed by a heat treatment in air at 300 °C (1 °C min−1) for 24 h, to remove the unreacted BDC trapped in the pores of the as-synthesized MOF. The MOF-derived ZnO@C samples were obtained in two steps. First, both the as-synthesized (as) and activated (lt) forms of MIL-53(Al) were impregnated with an aqueous solution of zinc nitrate (Zn(NO3)2·4H2O, Merck, >98%) calculated in such a way as to give a final ZnO loading of 40 wt% on the MOF-derived C, and then dried overnight, at room temperature. In the second step, the white-impregnated MIL-53(Al) samples were thermally treated in a tubular furnace under Ar flow (Linde Gas, Romania, 99.999%) using the following thermal sequence: (a) heating with 0.5 °C min−1 from room temperature to 600 °C, (b) 5 h at 600 °C, and (c) cooling down to room temperature under the same Ar flow. This final temperature was chosen to ensure the decomposition of the MIL-53(Al) forms to the corresponding C structures decorated with the remaining Al2O3 (as indicated by the TGA profiles presented in ESI,† Fig. S1). Alongside the formation of the MOF-derived C during the heat treatment in Ar, Zn(NO3)2 is also decomposed to ZnO. As a reference material, ZnO was also obtained by decomposing Zn(NO3)2 under the same heat treatment (600 °C for 5 h, 0.5 °C min−1, under an Ar flow). It should be emphasized here that although ZnO typically undergoes sublimation at temperatures above 600 °C, the sublimation rate of ZnO under an inert atmosphere is very low,39 as also confirmed by mass balance performed for each prepared ZnO-based sample. A schematic of the synthesis steps is illustrated in Scheme 1.
:BDC:H2O = 2:1:160 was hydrothermally treated at 190 °C for 12 h, under autogenous pressure. The as-synthesized white product (MIL-53(Al)(as)) was filtered and washed thoroughly with deionized water and dried at room temperature overnight. The activated form of the MOF, namely MIL-53(Al)(lt), was obtained by heating under reflux in dimethylformamide (DMF, Chempur, >99%) for 24 h, followed by a heat treatment in air at 300 °C (1 °C min−1) for 24 h, to remove the unreacted BDC trapped in the pores of the as-synthesized MOF. The MOF-derived ZnO@C samples were obtained in two steps. First, both the as-synthesized (as) and activated (lt) forms of MIL-53(Al) were impregnated with an aqueous solution of zinc nitrate (Zn(NO3)2·4H2O, Merck, >98%) calculated in such a way as to give a final ZnO loading of 40 wt% on the MOF-derived C, and then dried overnight, at room temperature. In the second step, the white-impregnated MIL-53(Al) samples were thermally treated in a tubular furnace under Ar flow (Linde Gas, Romania, 99.999%) using the following thermal sequence: (a) heating with 0.5 °C min−1 from room temperature to 600 °C, (b) 5 h at 600 °C, and (c) cooling down to room temperature under the same Ar flow. This final temperature was chosen to ensure the decomposition of the MIL-53(Al) forms to the corresponding C structures decorated with the remaining Al2O3 (as indicated by the TGA profiles presented in ESI,† Fig. S1). Alongside the formation of the MOF-derived C during the heat treatment in Ar, Zn(NO3)2 is also decomposed to ZnO. As a reference material, ZnO was also obtained by decomposing Zn(NO3)2 under the same heat treatment (600 °C for 5 h, 0.5 °C min−1, under an Ar flow). It should be emphasized here that although ZnO typically undergoes sublimation at temperatures above 600 °C, the sublimation rate of ZnO under an inert atmosphere is very low,39 as also confirmed by mass balance performed for each prepared ZnO-based sample. A schematic of the synthesis steps is illustrated in Scheme 1.
 | ||
| Scheme 1 Synthesis route for the ZnO@C samples derived from MIL-53(Al) metal–organic framework. | ||
Powder X-ray diffraction measurements were carried out on a Bruker D8 advance diffractometer equipped with a Ge monochromator used to obtain CuKα radiation, LYNXEYE detector, and the X-ray tube operating at 40 kV and 40 mA. The patterns were recorded with a scan rate of 0.01° s−1 in the 10–85° 2θ range.
Nitrogen sorption isotherms were measured on a Sorptomatic 1990 apparatus (Thermo Electron). The specific surface area was calculated in the relative pressure range of 0.01–0.25 p/p0 (BET method), and the total pore volume and pore size distribution were determined by the Dollimore–Heal method using the desorption branch. Pretreatment of the samples consisted of degassing under vacuum at 200 °C for 4 h.
Scanning and transmission electron microscopy (SEM-TEM) was carried out on a Hitachi HD-2700 STEM microscope (Hitachi, Japan) operating at 200 kV. Before STEM analysis, each sample was finely grounded and dispersed in ethanol by sonication (3 min).
Raman spectroscopy was employed to gain insights into the lattice dynamics of ZnO nanostructures at the molecular scale. The measurement utilized a Renishaw Raman InVia System with a 532 nm green laser.
Fourier transform infrared spectroscopy (FTIR) was used to analyze the chemistry and functional groups in the synthesized MOF-derived carbon materials. Analyses were carried out using a Bruker Tensor II spectrophotometer (Germany).
Electron parametric resonance (EPR) spectroscopy was performed on a continuous-wave X-band spectrometer, a Bruker ELEXSYS E500. The X-band measurements were performed at 9.87 GHz frequency with an X-SHQ 4119HS-W1 Bruker microwave cavity.
The electrochemical properties of the synthesized materials were characterized using a Biologic (VMP300) electrochemical workstation. In the two-electrode system, ZnO and ZnO@C were employed as electrode materials. A 6 M KOH aqueous solution was selected as the electrolyte due to its high ionic conductivity, compatibility with carbon-based electrode materials, and ability to provide a stable electrochemical environment, ensuring high coulombic efficiency and long-term cycling stability.40,41 Whatman glass fiber was used as a separator. Graphite was used as the secondary electrode in asymmetric designs. The electrode material was deposited onto a surface area of 0.63 mg cm−2. A circular glass fiber separator with an area of 0.786 cm2 was placed between the two electrodes, and 40 μL of 6 M KOH electrolyte was added to ensure proper ionic conductivity. The electrodes were then assembled with stainless steel current collectors, which were tightly compressed to form the supercapacitor device. Also, the electrode area and the integration of the ZnO@C MOF-derived samples are illustrated in the scheme provided below (Table 1). The mass loading of the ZnO@C MOF-derived samples was integrated onto the electrode surface at 0.63 mg cm−2, based on our group's previous studies and supported by literature data.42–44 This value was maintained consistently across all electrochemical measurements. The electrochemical characteristics of the prepared SC designs were investigated through potentiostatic electrochemical impedance spectroscopy (PEIS), cyclic voltammetry (CV), and galvanostatic cycling with potential limitation (GCPL) techniques. CV analyses were conducted at different scan rates ranging from 5 mV s−1 to 200 mV s−1, and the applied potential spanned from 0 V to 1 V. PEIS analyses were performed with an AC perturbation of 10 mV across a frequency range of 10 MHz to 1 MHz. GCPL analysis was carried out over a range of current densities from 0.1 A g−1 to 2.4 A g−1, maintaining the same voltage range.
| SC design | Electrode 1 | Electrode 2 | Type |
|---|---|---|---|
| ZnO-sym | ZnO | ZnO | Symmetric |
| ZnO@C(as)-sym | ZnO@C(as) | ZnO@C(as) | Symmetric |
| ZnO@C(lt)-sym | ZnO@C(lt) | ZnO@C(lt) | Symmetric |
| ZnO-asym | ZnO | Graphite | Asymmetric |
| ZnO@C(as)-asym | ZnO@C(as) | Graphite | Asymmetric |
| ZnO@C(lt)-asym | ZnO@C(lt) | Graphite | Asymmetric |

|
|||
3. Results and discussions
3.1. Characterization of materials
It is known that the thermal treatment of MOFs in an inert atmosphere (Ar, N2, etc.) leads to the formation of the corresponding metal or metal oxide carbon-based nanomaterials, depending on the reduction potential of the metal present in the initial MOF.45,46 Since Al has a reduction potential of −1.66 V, lower than −0.27 V, it is expected that the carbonization of MIL-53(Al) will produce aluminum oxide decorated carbon-based materials.45On the other hand, since both the as-synthesized (as) and activated low-temperature (lt) forms of MIL-53(Al) were first impregnated with Zn(NO3)2 before heat treatment in Ar, the derived materials thereof will contain C, Al2O3, and ZnO. However, powder X-ray diffraction analysis of the MIL-53(Al)-derived ZnO-impregnated samples highlights the mineral form of ZnO, with no evidence of the corresponding reflections for C or Al2O3 (Fig. 1).
 | ||
| Fig. 1 XRD patterns of the ZnO@C MIL-53(Al)-derived samples. | ||
The crystal structure of ZnO belongs to the hexagonal crystal system, P63mc space group with the lattice parameters a = 3.249 Å and c = 5.207 Å.47 Due to the lower degree of crystallinity in both MIL-53(Al)-derived ZnO samples, the (004), (202), and (014) diffraction lines are not visible in the diffraction patterns of ZnO@C(as) and ZnO@C(lt). The average size of crystallites was evaluated based on the Scherrer formula,48 and the obtained values are listed in Table 2. The crystallite sizes in ZnO@C(as) and ZnO@C(lt) are comparable, only slightly larger in the former. On the other hand, for the reference ZnO sample, it can be noticed that the diffraction peaks are considerably narrower, which is related to significantly larger crystallite sizes compared to the ZnO-decorated MIL-53(Al)-derived carbon samples. For ZnO@C(lt), two diffraction peaks at 2θ = 14.8 and 17.3° are also observed, which can be assigned to traces of MIL-53(Al) left in the sample. The absence of characteristic diffraction lines for C and Al2O3 is attributed to the formation of small and well-dispersed amorphous phases. Indeed, XRD patterns of the un-impregnated (as) or (lt) MIL-53(Al) carbonized samples presented in Fig. S2 in ESI† reveal the rather amorphous character of the MIL-53(Al)-derived carbons, with the low and broad diffraction lines centered at 22 and 43° corresponding to the (002) and (101) planes of amorphous graphitic carbon.49
| Sample | S BET (m2 g−1) | V t (cm3 g−1) | D pore (nm) | ZnO content (wt%) | d XRD (nm) | I D/IG |
|---|---|---|---|---|---|---|
| MIL-53(Al)(lt) | 1216 | 0.57 | 1.2 | — | — | — |
| ZnO@C(as) | 222 | 0.96 | 10/13.5 | 40 | 25.1 | 0.98 |
| ZnO@C(lt) | 168 | 0.55 | 8.5/15 | 40 | 20.8 | 0.80 |
| ZnO | 0 | 0 | 0 | 100 | 71.0 | — |
The morphology and microstructure of the ZnO@C MIL-53(Al)-derived samples were examined by SEM, which revealed the ordered porous structure of these carbon materials. The carbon samples obtained by deriving the un-impregnated MIL-53(Al) forms reveal a channel-like morphology (Fig. S3 in ESI†), which is preserved to a large extent in the case of the ZnO-decorated MIL-53(Al)-derived samples (Fig. 2, and Fig. S4 in ESI†). Additionally, TEM images confirm the channel-like structure of the formed C as a consequence of the carbonization of the initial MIL-53(Al) with 1D-rhombic-shaped channels.50 EDS elemental mapping performed during TEM analysis highlights the uniform distribution of C, Al, and O, confirming the formation of the Al2O3-doped C structure (Fig. 2). ZnO obtained during the simultaneous thermal decomposition of the Zn(NO3)2 impregnated MIL-53(Al) samples is uniformly distributed within the MIL-53(Al)-derived C-structure, as demonstrated by the corresponding Zn mapping.
 | ||
| Fig. 2 Scanning and transmission electron microscopy images, with EDS elemental mapping of the ZnO@C samples. | ||
The materials' specific surface area, pore volume, and pore size distribution were evaluated from the N2 sorption isotherms. The starting MIL-53(Al) is a microporous material, as indicated by the type I isotherm given in Fig. 3(a). Indeed, the corresponding pore size distribution is very narrow, and the average pore size is around 1.2 nm (Fig. 3(b) and Table 2). The ZnO@C materials obtained by carbonization of the Zn(NO3)2 impregnated MIL-53(Al) (as) and (lt) samples present a type IV isotherm with a slit-type hysteresis loop, characteristic of mesoporous materials with polydisperse pore size distribution.51 Both ZnO@C samples present two types of mesopores ranging from 8 to 17 nm. This bimodal pore structure is more clearly evidenced in the case of ZnO@C(lt), with pores of 8.5 and 15 nm, while ZnO@C(as) shows pores around 10 and 13.5 nm, respectively. This narrow porosity is also confirmed by the small pores evidenced by the high-magnification SEM images (Fig. S4 in ESI†). In terms of specific surface area, carbonization of the Zn(NO3)2 impregnated MIL-53(Al) samples (the (as) and (lt) forms) leads to a decrease of at least 6 times, as compared to the pristine MIL-53(Al) (see Table 2). However, ZnO@C(as) shows a larger surface area as compared to ZnO@C(lt) (222 m2 g−1, and 168 m2 g−1, respectively). Interestingly, the total pore volume is significantly larger for ZnO@C(as) as compared to the initial MOF scaffold, while ZnO@C(lt) shows a slightly lower, but comparable value (see Table 2). These surface area and pore size distribution results are consistent with previous reports on MOF-derived carbon materials with remanent metal oxides from the initial MOF scaffolds.52,53
 | ||
| Fig. 3 (a) N2 sorption isotherms, and (b) pore size distribution for the ZnO@C samples. | ||
Further structural characterization was achieved by Raman spectroscopy, with spectra illustrated in Fig. 4. The hexagonal wurtzite (P63mc) ZnO structure exhibits 12 phonons, with three being acoustic. A prominent peak at 437 cm−1 corresponds to the E2high mode, serving as a distinctive Raman fingerprint for the wurtzite ZnO. Although the E2low mode was not detected, a vibration around 331 cm−1 is typically associated with the second-order Raman process, assigned to the E2high − E2low difference mode.54–56 In the wurtzite structure, each cation is encompassed by anions arranged in a tetrahedron, leading to polar symmetry along the c-axis and resulting in A1 mode oscillation. Due to polarity, the A1 and E1 modes are further split into transverse-optical (TO) and longitudinal-optical (LO) phonons. The E1(TO) and A1(TO) modes exhibit weak peaks at approximately 408 and 380 cm−1, respectively. TO modes depend on the incident and scattered light being perpendicular to the c-axis, while LO phonon modes appear when the incident and scattered light are parallel to the c-axis.54 The A1(LO) mode was identified at 583 cm−1. A second-order Raman process occurs when the material structure contains surface and/or aggregate defects, resulting in the emergence of combination bands that influence the total frequencies of the optical branches. The two-phonon difference modes were observed within the frequency range of the one-phonon branches. In contrast, frequencies above ∼600 cm−1 are primarily influenced by two-phonon sum modes and higher-order multi-phonon processes, as illustrated in Fig. 4. The broad band spanning from 1020 to 1200 cm−1 is probably associated with a multiphoton process, while the wide peak approximately at 1150 cm−1 might be indicative of 2LO (two-phonon longitudinal-optical) scattering. Regarding the occurrence of carbon in the structure, peaks at approximately 1340 and 1598 cm−1 indicate the D and G bands, respectively.55,56 The manifestation of spectral bands encompassing the wavenumber range of 1000–1800 cm−1 aligns with the existence of sp2 hybridized C–C bonds and the presence of aromatic six-membered sp2 carbon clusters, thus corroborating the carbonaceous phase.55 The ratio between the intensities of the D and G bands is used as a measure of carbon defects in carbonaceous materials; therefore, the higher the ID/IG ratio, the larger the degree of disorder.57,58 In this regard, given the ID/IG ratios listed in Table 2, ZnO@C(as) presents slightly more carbon defects than ZnO@C(lt). On the other hand, since the carbon bands dominate the spectrum, the ZnO peaks were not observed in the ZnO@C samples, as evidenced in Fig. 4.
 | ||
| Fig. 4 First-order and second-order Raman spectra of the ZnO@C samples (λ = 532 nm). | ||
FTIR spectra presented in Fig. S5 (ESI†) indicate the prevalence of MIL-53(Al)-derived carbon related functional groups, with no clear evidence of Zn-related bands, in good agreement with the Raman spectra previously discussed. Thus, the broad band around 3450 cm−1 is ascribed to the stretching vibration of the surface O–H groups, the band at 1630 cm−1 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration,59 while the band at 1385 cm−1 can be ascribed to the aromatic CC.14 It may be observed that both ZnO decorated C materials exhibit an additional band around 1510 cm−1 attributed to CC, as well as a broad and weak band around 1035 cm−1 assigned to C–O.59 The vibrations due to atmospheric CO2 can be observed in all spectra around 2360 cm−1.
C stretching vibration,59 while the band at 1385 cm−1 can be ascribed to the aromatic CC.14 It may be observed that both ZnO decorated C materials exhibit an additional band around 1510 cm−1 attributed to CC, as well as a broad and weak band around 1035 cm−1 assigned to C–O.59 The vibrations due to atmospheric CO2 can be observed in all spectra around 2360 cm−1.
The X-band EPR spectrum of the pure ZnO material is presented in the inset of Fig. 5, showing two distinct signals attributed to two different defect centers such as Zn vacancies (VZn) and oxygen vacancies (VO), with g = 1.96 and g = 2.002, respectively.60 To explain the presence of these two signals, a core–shell model was introduced by Erdem.61 The model suggests that the EPR signal at g = 1.96 originates from bulk defects (core), representing the negatively charged Zn vacancies (V−/2Zn) while the resonance signal near g = 2.002 arises from surface defects (shell), and such defects have been attributed to the positively charged oxygen vacancies (V+/2+O).
 | ||
| Fig. 5 X-band EPR spectra of the ZnO@C MOF-derived samples. The inset presents the X-band EPR spectrum of pure ZnO. | ||
The EPR spectra of the ZnO containing carbon structures, ZnO@C(as) and ZnO@C(lt), are depicted in Fig. 5. The above described ZnO-related EPR signals are barely distinguishable, superimposed to a sharp, very intense EPR signal originating from the carbon-related defect centers of the MOF-derived materials. The EPR signal has a g-value of 2.0064, characteristic of such defect centers,62 while the intensity of the signals is slightly different, ZnO@C(as) having a higher EPR signal, which indicates a higher concentration of defect centers in the material. These findings are in good agreement with the Raman results. The presence of slightly more carbon defects in the ZnO@C(as) may be attributed to the trapped benzene-dicarboxylic acid (BDC) in the pores of the as-synthesized MIL-53(Al), providing in this way an additional source of carbon in the final composite material.
3.2. Electrochemical characterization results
Various techniques were employed to evaluate the performance of the ZnO@C electrodes in comparison to ZnO, in both symmetric and asymmetric SC designs.Potentiostatic electrochemical impedance spectroscopy (PEIS), including the circuit model, is an important technique that facilitates modeling molecular interactions and real transport mechanisms of electrical components, allowing thus a deeper comprehension of events occurring at the electrode and electrolyte interface and/or within the electrolyte itself.63Fig. 6 depicts the Nyquist curves obtained from PEIS analysis. Furthermore, each Nyquist curve was fitted using the Z-fit software, and the calculated parameter values are provided in Table S1 of ESI.†
 | ||
| Fig. 6 Experimental PEIS and fit results of the designed SC devices: (a)–(c) symmetric, and (d)–(f) asymmetric designs. | ||
Regarding the value of R1, both ZnO-sym and ZnO-asym SC designs show larger values than their C-based counterparts, implying a decrease in electrical resistance at the electrolyte/electrode interface when MOF-derived C-based electrodes are used. This behavior might be attributed to the large surface area of ZnO@C composite materials obtained by carbonization of MIL-53(Al), as evidenced by the BET analysis (Table 2). The interfacial resistance in a SC is related to the transmission resistance between the electrode material and the electrolyte. Therefore, electrodes with a high surface area often facilitate enhanced interfacial communication, potentially reducing the resistance between the electrode and electrolyte, thereby contributing to an overall performance enhancement of the SC. In addition, when the R2 values are examined, an increase in the resistance value of the ZnO@C-based electrodes is observed compared to the ZnO ones (ZnO-sym and ZnO-asym), indicating that the EDLC capacitance resistance of the SC increases when ZnO@C-based electrodes are used. Essentially, this is to be expected because, after 10000 cycles, the electrodes in the SC can deteriorate due to repeated charge/discharge cycles or exposure to harsh operating conditions. This degradation could increase the resistance at the electrode–electrolyte interface, resulting in a higher R2 value, which is compatible with the capacitance retention value calculated as a result of the GCPL analysis. Compared to the ZnO designs (ZnO-sym and ZnO-asym), the capacitance retention values of the MOF-derived carbon-based electrodes decreased, albeit partially, after 10000 cycles.
When examining the Nyquist plots of the designed SC configurations, no semicircles are observed in the high-frequency region. Therefore, this absence suggests low charge transfer resistance at the electrode and electrode/electrolyte interface, indicating a high ion diffusion rate. Furthermore, the Nyquist plots of the symmetric designs of MOF-derived carbon ZnO electrodes (ZnO@C(as) and ZnO@C(lt)) exhibit an almost linear form in the low-frequency region, meaning a low equivalent series resistance (ESR) in these designs.64 Upon reviewing Table S1 (ESI†) it is evident that, compared to asymmetric designs, the symmetric designs possess lower ESR, that is, R2 values. Moreover, considering the −Zim values, it can be inferred that ZnO@MOF-derived carbon electrodes attain higher impedance values in both symmetric and asymmetric designs, indicating an increased presence of reactive components within the electrode.65 The increased surface reactance due to the larger BET surface area and the highly defective carbon structure, evidenced by EPR and Raman analysis, significantly contribute to this outcome.
Cyclic voltammetry (CV) is frequently employed to investigate the electrochemical properties of adsorbed materials on electrodes and explore molecular species' reduction and oxidation processes.66 In Fig. 7, the CV curves acquired at various scan rates for the SCs prepared using ZnO and ZnO@MOF-derived carbon electrodes are presented, exhibiting nearly rectangular shapes, which indicates that the utilized electrodes manifest an EDLC behavior. This implies that the charge storage occurs by forming electric double layers (EDLs) at the electrode/electrolyte interface.67
 | ||
| Fig. 7 CV curves of the symmetric and asymmetric SC devices obtained at different scanning rates. | ||
The appearance of a fusiform shape in the CV curve obtained at the highest scan rate of 200 mV s−1 can be attributed to the limitations in ion transport and adsorption processes within the EDL under such ultrafast scanning speeds. At high scan rates, the time available for ions to diffuse and adsorb at the electrode–electrolyte interface during the CV cycle becomes limited. As a result, the charge storage and release rate at the electrode surface is accelerated, leading to a higher current response in the CV curve.68 When the applied voltage reaches approximately 0.9 V, the voltage at which the employed KOH electrolyte undergoes decomposition, deviations from the rectangular shape occur in the CV curves.69 Furthermore, in the case of ZnO@MOF-derived carbon, the current values slightly decrease at higher scan rates. This reduction suggests a faster depletion of reactants during the electrochemical reaction. The simultaneous decrease in the electrode/electrolyte interface resistance and the equivalent series resistance for the ZnO@C designs might contribute to the accelerated consumption of reactants.70
The GCPL graphs of the fabricated symmetric and asymmetric SCs are given in Fig. 8. The GCPL data were determined using 0.1 A g−1 after 10000 cycles. Upon examining the specific capacity data concerning the applied potential, a specific capacity increase is observed for the symmetric and asymmetric designs when using ZnO@MOF-derived porous carbon as an electrode. The charge and discharge capacities for all SC designs are summarized and provided in Table 3. This table presents the specific capacity of the SC designs under the specified operating conditions, providing a comprehensive overview of their charge storage and release capabilities performance. The large surface area and the augmentation of pore diameter and volume have enhanced the specific charge storage capacity per unit mass. This is attributed to the fact that a higher surface area facilitates the adsorption of more ions and enhances the ion diffusion rate. Furthermore, the utilization of graphite as a second electrode in the asymmetric designs also contributes to this enhancement due to its carbon-based nature, high surface area, and conductivity. Additionally, upon examining Fig. 10, it is evident that the designed SC configurations do not exhibit a distinct voltage plateau in the GCPL curves. This absence signifies the dominance of non-faradaic capacitive behavior.71
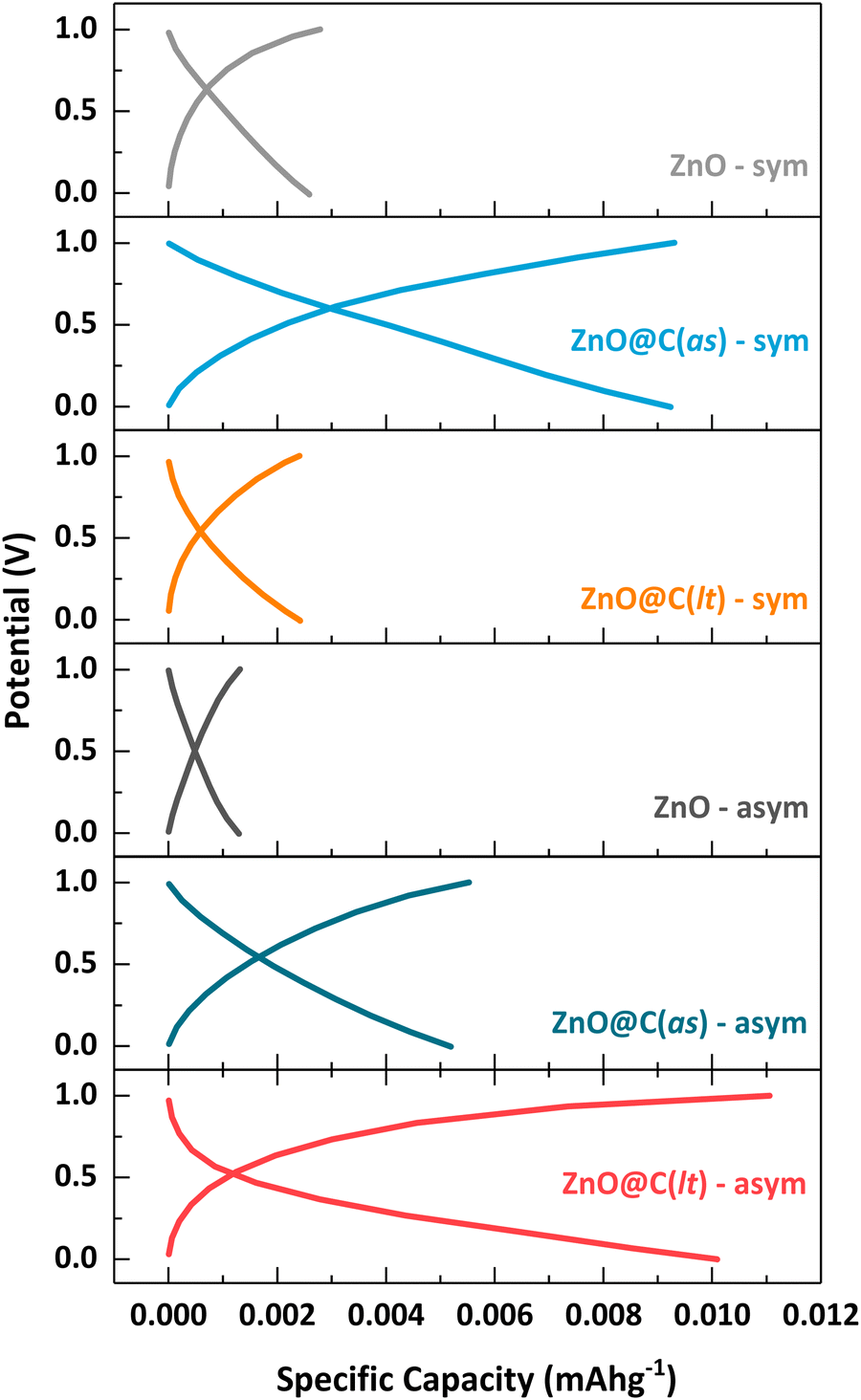 | ||
| Fig. 8 GCD profiles of the symmetric and asymmetric SCs at a current density of 0.1 A g−1. | ||
| SC design | Capacity (μA h g−1) | C e (%) | C p (F g−1) | E D (W h kg−1) | P D (kW kg−1) | |
|---|---|---|---|---|---|---|
| Charge | Discharge | |||||
| ZnO-sym | 2.8 | 2.6 | 95.2 | 64.6 | 9.0 | 264 |
| ZnO@C(as)-sym | 9.3 | 9.2 | 98.7 | 146.6 | 20.4 | 219 |
| ZnO@C(lt)-sym | 2.4 | 2.4 | 99.9 | 88.5 | 12.3 | 388 |
| ZnO-asym | 1.3 | 1.3 | 98.8 | 30.2 | 4.2 | 86 |
| ZnO@C(as)-asym | 5.5 | 5.2 | 95.0 | 105.1 | 14.6 | 261 |
| ZnO@C(lt)-asym | 1.1 | 10 | 97.5 | 219.3 | 30.5 | 186 |
Fig. 9(a) and (b) illustrate the variation in Coulombic efficiency and capacitance retention values for ZnO and ZnO@MOF-derived carbon-based electrodes over 10000 cycles. Coulombic efficiency is a crucial parameter for assessing the performance of electrochemical energy storage systems and enhancing efficiency in energy storage/indicator applications.72 It may be observed that the coulombic efficiencies of the prepared SCs range between 95.0% and 99.9% even after 10000 cycles. This indicates the successful energy conversion and electron transfer in the anticipated chemical reactions occurring at the electrolyte/electrode or electrolyte interfaces. As a consequence of larger surface area and porosity, the ZnO@C(as)-based SC design demonstrates the ability to store more energy per unit mass facilitating enhanced electron interactions and electrochemical reactions. Furthermore, the presence of paramagnetic centers in these materials has been confirmed through EPR spectroscopy, revealing a significant concentration of C-related defect centers. These defect centers also significantly impact the material's electrical properties, improving the supercapacitive Coulombic efficiency. Capacitance retention refers to the ability of a capacitor or SC to maintain its capacitance over time or after undergoing certain operating conditions. It measures how well the device retains its charge storage capacity relative to its initial or maximum capacitance value. As depicted in Fig. 9(b), the capacitance retention values of all SCs are nearly above 100%. Maintaining high cyclic stability in all SCs even after 10000 cycles is desirable for energy storage systems. This is because chemical reactions occurring throughout the entire charge/discharge process and potential disturbances can adversely affect the cyclic stability of electrodes.
 | ||
| Fig. 9 (a) Coulombic efficiency, and (b) capacitance retention of the investigated SC designs. | ||
The time-dependent potential graphs of the prepared SCs at 0.1 A g−1 are presented in Fig. 10(a) and 10(b) for the symmetric and asymmetric SC devices, respectively. According to Fig. 10, it is evident that both SC designs experience approximately a threefold increase in charge/discharge time when MOF-derived carbon-based electrodes are employed. The total charge/discharge time for ZnO-sym is 0.25 s, extending to 0.35 s in the asymmetric design. In the case of ZnO@MOF-derived carbon-based designs, these times are prolonged to approximately 0.66 s in the symmetric design and up to 1.22 s in the asymmetric designs. The utilization of high surface area porous graphite is believed to contribute to this increase in the asymmetric design. On the other hand, the rise in the symmetric design can be attributed to the reduction in equivalent series resistance, which leads to enhanced and faster ion transfer, consequently resulting in an elongated charge/discharge time.
 | ||
| Fig. 10 Potential vs. time graphs of the designed SCs at the 1st cycle (current density of 0.1 A g−1): (a) symmetric designs, and (b) asymmetric designs. | ||
As a result of the conducted PEIS, CV, and GCPL analyses, some crucial electrochemical parameters have been provided in Table 3. Accordingly, specific capacitance, energy, and power density values have significantly changed in the case of ZnO@MOF-derived carbon-based designs. The specific capacitance of the ZnO-sym design is approximately 65 F g−1, whereas it reaches around 147 F g−1 in the ZnO@C(as)-sym design. Similarly, while the specific capacitance of ZnO-asym is measured at 30.2 F g−1, the ZnO@C(lt)-asym design shows an approximate value of 220 F g−1. The energy and power density values also show a similar increasing trend. It is a well-established fact that the relationship between surface area, defect structure, and specific capacitance strongly affects the storage capacity of electrodes. As per the results of the surface area measurements (see Table 2), ZnO@C(as) has a surface area of 222 m2 g−1, which is larger than for ZnO@C(lt), having a surface area of 168 m2 g−1. A higher surface area electrode provides more electrochemical interactions and more active material. Additionally, the bimodal pore structure of the materials should be recalled, with ZnO@C(as) exhibiting pore sizes around 10 nm and 13.5 nm, and ZnO@C(lt) showing pore sizes around 8.5 nm and 15 nm (Table 2). This bimodal distribution enhances the electrochemical performance of the materials by combining the advantages of smaller pores, which increase the surface area for charge storage, and larger pores, which improve ion diffusion and electrolyte accessibility. Hence, the specific capacitance of the SC prepared using ZnO@C(as) as electrode material in the symmetric design is higher than for the other designs. However, upon analyzing Table 3, it becomes apparent that utilizing the ZnO@C(lt) sample in the asymmetric design results in a much higher specific capacitance. Despite having a higher surface area, the specific capacitance of ZnO@C(as) decreases when using graphite as a secondary electrode due to possible diffusion limitations. This is because an increase in surface area can only reach a limited capacity at a certain point; the electrode's surface may become completely covered with active material, and the higher surface area does not enhance the specific capacitance. This may result in a decline in specific capacitance. Given these constraints, the ZnO@C(lt) asymmetric construct, which exhibits a reduced surface area, is projected to have a superior specific capacitance. The electrochemical test results showed that the MOF-derived porous carbon-based electrodes increase the ion transfer rate and the surface area of the electrodes, ensuring a more active region. Furthermore, the reduction in equivalent series resistance and diffusion resistance has led to an increase in both energy and power density.
The ZnO@C materials synthesized in this study exhibit remarkable energy and power densities, positioning them as competitive candidates among MOF(Al)-derived carbon materials used in supercapacitors (see Table 4). For instance, the symmetric supercapacitor device based on ZnO@C(as) achieved an energy density of 20.4 W h kg−1 and a power density of 219 kW kg−1, while the asymmetric device based on ZnO@C(lt) reached an energy density of 30.5 W h kg−1 and a power density of 186 kW kg−1. These values not only align with but in some cases surpass those reported for MOF-derived carbon materials and transition metal oxide-based supercapacitors, which typically achieve energy densities of 10–25 W h kg−1 and power densities of 100–200 kW kg−1.
| Electrode/SC system | Starting MOF | Electrolyte | C P (F g−1) | I (A g−1) | Efficiency (%) | E D (W h kg−1) | P D (kW kg−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| SPC (spindle-like porous carbon)/(2 electrodes, sym) | Al-BTEC (1,2,4,5-benzenetetracarboxylic acid) | 6 M KOH | 173 | 1.0 | 95 | 15.3 | 0.336 | 52 |
| CPC (coin shaped porous carbon)/(2 electrodes, sym) | Al-BDC (1,4-benzenedicarboxylic acid) | KOH/PVA | 117.2 | 0.2 | 88.5 | 36.5 | 0.165 | 21 |
| CPMD/(3 electrodes) | Al-BDC (1,4-benzenedicarboxylic acid) | 6 M KOH | 391 | 0.5 | — | — | — | 36 |
| CPMD–Zn/(2 electrodes, asym) | Al-BDC (1,4-benzenedicarboxylic acid) | 3 M ZnSO4 | 335 | 0.1 | 92.2 | 150.3 | 0.090 | 36 |
| AMUPC (3 electrodes) | Al-BDC (1,4-benzenedicarboxylic acid) | 6 M KOH | 355 | 0.5 | 88.2 | 20.1 | 225 | 35 |
| DHTAC-2/(3 electrodes) | Al-DHTA (2,5-dihydroxi-1,4-benzenedicarboxylic acid) | 6 M KOH | 298.8 | 1.0 | — | — | — | 53 |
| ZnO@C(as) (2 electrodes, sym) | Al-BDC (1,4-benzenedicarboxylic acid) | 6 M KOH | 146.6 | 0.1 | 98.7 | 20.4 | 219 | This work |
| ZnO@C(lt) (2 electrodes, asym) | Al-BDC (1,4-benzenedicarboxylic acid) | 6 M KOH | 219.3 | 0.1 | 97.5 | 30.5 | 186 | This work |
As highlighted in Table 4, the ZnO@C(lt)-asym device demonstrates a significantly higher energy density (30.5 W h kg−1) compared to spindle-like porous carbon (SPC), which achieves 15.3 W h kg−1.52 Similarly, the ZnO@C(as)-sym device delivers a power density of 219 kW kg−1, placing it among the highest values reported for MOF(Al)-derived carbon materials. This superior performance can be attributed to the synergistic effects of the channel-like carbon morphology, uniform distribution of ZnO nanoparticles, and the defect-rich carbon structure, which collectively enhance ion transport, charge storage, and electrical conductivity.
4. Conclusions
Supercapacitor (SC) devices incorporating ZnO-decorated carbon derived from MIL-53(Al) have been engineered, utilizing MIL-53(Al) metal–organic frameworks as a precursor for the synthesis of carbon-based electrode materials. In summary, both the as-synthesized and activated low-temperature variants of MIL-53(Al) underwent impregnation with zinc nitrate, followed by a concurrent decomposition via thermal treatment in an inert atmosphere, transforming Zn(NO3)2 into ZnO and MIL-53(Al) into an Al2O3-decorated carbon structure. Comprehensive analyses of the ZnO@C materials revealed a well-defined channel-like configuration in the produced carbon with surface areas reaching approximately 220 m2 g−1. Electron paramagnetic resonance (EPR) spectroscopy identified the presence of paramagnetic centers in these materials, demonstrating a high concentration of carbon-related defect centers, which significantly influence the electrical properties of the materials. The electrical characteristics of these materials indicate enhanced energy and power densities, achieving up to 20.4 W h kg−1 and 219 kW kg−1 in the most efficient symmetric SC device, and 30.5 W h kg−1 and 186 kW kg−1 in the most efficient asymmetric device utilizing a graphite second electrode. The efficiency of all SC devices exceeded 95%.Author contributions
Arpad Mihai Rostas: Conceptualization, Methodology, Investigation, Validation, Formal Analysis, Data Curation, Visualization, Writing – Original Draft, Writing – Review & Editing. Ahmet Gungor: Methodology, Investigation, Validation, Formal Analysis, Visualization, Writing – Original Draft, Writing – Review & Editing. Angela M. Kasza: Methodology, Investigation, Validation, Formal Analysis. Feray Bakan Misirlioglu: Investigation, Formal Analysis, Writing – Original Draft. Alexandru Turza: Investigation, Formal Analysis, Data Curation, Writing – Original Draft. Lucian Barbu–Tudoran: Investigation. Emre Erdem: Conceptualization, Methodology, Validation, Formal Analysis, Resources, Writing – Review & Editing, Supervision. Maria Mihet: Conceptualization, Methodology, Investigation, Validation, Formal Analysis, Resources, Data Curation, Visualization, Writing – Original Draft, Writing – Review & Editing, Supervision.Data availability
Original data are available upon request and can be obtained by contacting the corresponding author.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was partially funded through the “Nucleu” Program within the National Research Development and Innovation Plan 2022–2027, Romania, carried out with the support of MEC, project no. 27N/03.01.2023, component project code PN 23 24 01 01.References
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, Metal-Organic Framework-Derived Nanoporous Metal Oxides toward Supercapacitor Applications: Progress and Prospects, ACS Nano, 2017, 11, 5293–5308, DOI:10.1021/acsnano.7b02796.
- H. Jiang, Q. Mu, H. Kimura, R. Liu, W. Yang, L. Liu, W. Du and C. Hou, Advanced ferroferric oxide-based composites for lithium-ion battery: Recent developments and future perspectives, Prog. Nat. Sci. Mater. Int., 2023, 33, 743–753, DOI:10.1016/j.pnsc.2023.12.017.
- I. Dincer, Renewable energy and sustainable development: a crucial review, Renewable Sustainable Energy Rev., 2000, 4, 157–175 CrossRef.
- K. O. Otun, M. S. Xaba, S. Zong, X. Liu, D. Hildebrandt, S. M. El-Bahy, M. T. Alotaibi and Z. M. El-Bahy, ZIF-8-derived ZnO/C decorated hydroxyl-functionalized multi-walled carbon nanotubes as a new composite electrode for supercapacitor application, Colloid Interface Sci. Commun., 2022, 47, 100589, DOI:10.1016/j.colcom.2022.100589.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Achieving high energy density and high power density with pseudocapacitive materials, Nat. Rev. Mater., 2020, 5, 5–19, DOI:10.1038/s41578-019-0142-z.
- B. Li, J. Zheng, H. Zhang, L. Jin, D. Yang, H. Lv, C. Shen, A. Shellikeri, Y. Zheng and R. Gong, et al., Electrode materials, electrolytes, and challenges in nonaqueous lithium-ion capacitors, Adv. Mater., 2018, 30, 1705670 CrossRef PubMed.
- P. E. Lokhande, S. Kulkarni, S. Chakrabarti, H. M. Pathan, M. Sindhu, D. Kumar, J. Singh, A. Kumar, Y. Kumar Mishra, D. C. Toncu, M. Syväjärvi, A. Sharma and A. Tiwari, The progress and roadmap of metal–organic frameworks for high-performance supercapacitors, Coord. Chem. Rev., 2022, 473, 214771, DOI:10.1016/j.ccr.2022.214771.
- M. Chen, H. Kimura, X. Yang, H. Huang, C. Hou, X. Xie, F. Meng, X. Sun, P. Ren, W. Du and X. Yang, Carbon reduction assist structured preparation stable three-dimensional V6O13 for durable aqueous zinc-ion batteries, J. Power Sources, 2023, 580, 233332, DOI:10.1016/j.jpowsour.2023.233332.
- J. Libich, J. Máca, J. Vondrák, O. Čech and M. Sedlaříková, Supercapacitors: Properties and applications, J. Energy Storage, 2018, 17, 224–227, DOI:10.1016/j.est.2018.03.012.
- W. Zhao, Y. Zeng, Y. Zhao and X. Wu, Recent advances in metal-organic framework-based electrode materials for supercapacitors: A review, J. Energy Storage, 2023, 62, 106934, DOI:10.1016/j.est.2023.106934.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dinca, Conductive MOF electrodes for stable supercapacitors with high areal capacitance, Nat. Mater., 2017, 16, 220–224, DOI:10.1038/nmat4766.
- W. Yang, D. Peng, H. Kimura, X. Zhang, X. Sun, R. A. Pashameah, E. Alzahrani, B. Wang, Z. Guo, W. Du and C. Hou, Honeycomb-like nitrogen-doped porous carbon decorated with Co3O4 nanoparticles for superior electrochemical performance pseudo-capacitive lithium storage and supercapacitors, Adv. Compos. Hybrid Mater., 2022, 5, 3146–3157, DOI:10.1007/s42114-022-00556-6.
- Y. Wang, Y. Zhang, H. Kimura, J. Wang, C. Hou, X. Xie, X. Sun, Y. Zhang, W. Du and X. Yang, Nitrogen-doped expanded graphite derived from spent graphite induce uniform growth of lithium peroxide for high-performance Li-oxygen battery, J. Cleaner Prod., 2023, 414, 137703, DOI:10.1016/j.jclepro.2023.137703.
- B. Hosseinzadeh, B. Nagar, R. Benages-Vilau, P. Gomez-Romero and S. H. Kazemi, MOF-derived conformal cobalt oxide/C composite material as high-performance electrode in hybrid supercapacitors, Electrochim. Acta, 2021, 389, 138657, DOI:10.1016/j.electacta.2021.138657.
- Y. Han and L. Dai, Conducting polymers for flexible supercapacitors, Macromol. Chem. Phys., 2019, 220, 1800355 CrossRef.
- K. D. Kumar, Y. A. Kumar, T. Ramachandran, A. A. Al-Kahtani and M. C. Kang, Cactus-Like Ni-Co/CoMn2O4 composites on Ni foam: Unveiling the potential for advanced electrochemical materials for pseudocapacitors, Mater. Sci. Eng. B., 2023, 296, 116715, DOI:10.1016/j.mseb.2023.116715.
- K. D. Kumar, T. Ramachandran, Y. A. Kumar, A. A. A. Mohammed and M. C. Kang, Hierarchically fabricated nano flakes-rod-like CoMoO–S supported Ni-foam for high-performance supercapacitor electrode material, J. Phys. Chem. Solids., 2024, 185, 111735, DOI:10.1016/j.jpcs.2023.111735.
- D. P. Chatterjee and A. K. Nandi, A review on the recent advances in hybrid supercapacitors, J. Mater. Chem. A, 2021, 9, 15880–15918, 10.1039/d1ta02505h.
- P. A. Shinde, Q. Abbas, N. R. Chodankar, K. Ariga, M. A. Abdelkareem and A. G. Olabi, Strengths, weaknesses, opportunities, and threats (SWOT) analysis of supercapacitors: A review, J. Energy Chem., 2023, 79, 611–638, DOI:10.1016/j.jechem.2022.12.030.
- W. Du, X. Wang, J. Zhan, X. Sun, L. Kang, F. Jiang, X. Zhang, Q. Shao, M. Dong, H. Liu, V. Murugadoss and Z. Guo, Biological cell template synthesis of nitrogen-doped porous hollow carbon spheres/MnO2 composites for high-performance asymmetric supercapacitors, Electrochim. Acta, 2019, 296, 907–915, DOI:10.1016/j.electacta.2018.11.074.
- W. Jiang, J. Pan, J. Wang, J. Cai, X. Gang, X. Liu and Y. Sun, A coin like porous carbon derived from Al-MOF with enhanced hierarchical structure for fast charging and super long cycle energy storage, Carbon, 2019, 154, 428–438, DOI:10.1016/j.carbon.2019.08.035.
- Y. Z. Chen, R. Zhang, L. Jiao and H. L. Jiang, Metal–organic framework-derived porous materials for catalysis, Coord. Chem. Rev., 2018, 362, 1–23, DOI:10.1016/j.ccr.2018.02.008.
- O. M. Yaghi and H. Li, Hydrothermal Synthesis of a Metal-Organic Framework Containing Large Rectangular Channels, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- R. Díaz, M. G. Orcajo, J. A. Botas, G. Calleja and J. Palma, Co8-MOF-5 as electrode for supercapacitors, Mater. Lett., 2012, 68, 126–128, DOI:10.1016/j.matlet.2011.10.046.
- T. Mageto, F. M. de Souza, J. Kaur, A. Kumar and R. K. Gupta, Chemistry and potential candidature of metal-organic frameworks for electrochemical energy storage devices, Fuel Process. Technol., 2023, 242, 107659, DOI:10.1016/j.fuproc.2023.107659.
- D. Muthu, R. K. Dharman, S. E. Muthu and T. H. Oh, Recent developments in metal-organic framework-derived transition metal oxide@carbon nanostructure and carbon nanostructure for supercapacitor applications, J. Energy Storage, 2025, 119, 116365, DOI:10.1016/j.est.2025.116365.
- J. Song, L. Chai, M. Zhao, Y. Sun, X. Li and J. Pan, Research progress on metal organic framework derived porous carbon though interfacial engineering and synergistic effect, J. Power Sources, 2024, 604, 234471, DOI:10.1016/j.jpowsour.2024.234471.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, Metal-organic framework as a template for porous carbon synthesis, J. Am. Chem. Soc., 2008, 130, 5390–5391, DOI:10.1021/ja7106146.
- B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Metal-organic framework (MOF) as a template for syntheses of nanoporous carbons as electrode materials for supercapacitor, Carbon, 2010, 48, 456–463, DOI:10.1016/j.carbon.2009.09.061.
- H. Yi, H. Wang, Y. Jing, T. Peng and X. Wang, Asymmetric supercapacitors based on carbon nanotubes@NiO ultrathin nanosheets core-shell composites and MOF-derived porous carbon polyhedrons with super-long cycle life, J. Power Sources, 2015, 285, 281–290, DOI:10.1016/j.jpowsour.2015.03.106.
- W. Gao, D. Chen, H. Quan, R. Zou, W. Wang, X. Luo and L. Guo, Fabrication of Hierarchical Porous Metal-Organic Framework Electrode for Aqueous Asymmetric Supercapacitor, ACS Sustain, Chem. Eng., 2017, 5, 4144–4153, DOI:10.1021/acssuschemeng.7b00112.
- X. Yan, X. Li, Z. Yan and S. Komarneni, Porous carbons prepared by direct carbonization of MOFs for supercapacitors, Appl. Surf. Sci., 2014, 308, 306–310, DOI:10.1016/j.apsusc.2014.04.160.
- K. M. Choi, H. M. Jeong, J. H. Park, Y. B. Zhang, J. K. Kang and O. M. Yaghi, Supercapacitors of nanocrystalline metal-organic frameworks, ACS Nano, 2014, 8, 7451–7457, DOI:10.1021/nn5027092.
- Y. Sun, S. Guo, W. Li, J. Pan, C. Fernandez, R. A. Senthil and X. Sun, A green and template-free synthesis process of superior carbon material with ellipsoidal structure as enhanced material for supercapacitors, J. Power Sources, 2018, 405, 80–88, DOI:10.1016/j.jpowsour.2018.10.034.
- X. Xu, J. Li, A. Dymerska, J. J. Koh, J. Min, S. Liu, J. Azadmanjiri and E. Mijowska, MIL-53(Al) assisted in upcycling plastic bottle waste into nitrogen-doped hierarchical porous carbon for high-performance supercapacitors, Chemosphere, 2023, 340, 139865, DOI:10.1016/j.chemosphere.2023.139865.
- J. Li, S. Zhang, Y. Hua, Y. Lin, X. Wen, E. Mijowska, T. Tang, X. Chen and R. S. Ruoff, Facile synthesis of accordion-like porous carbon from waste PET bottles-based MIL-53(Al) and its application for high-performance Zn-ion capacitor, Green Energy Environ., 2024, 9, 1138–1150, DOI:10.1016/j.gee.2023.01.002.
- P. L. Llewellyn, S. Bourrelly, C. Serre, Y. Filinchuk and G. Férey, How Hydration Drastically Improves Adsorption Selectivity for CO2 over CH4 in the Flexible Chromium Terephthalate MIL-53, Angew. Chem., 2006, 118, 7915–7918, DOI:10.1002/ange.200602278.
- O. Grad, A. M. Kasza, A. Turza, M. Dan, L. Barbu-Tudoran, M. D. Lazar and M. Mihet, Facile and efficient synthesis of ordered mesoporous MIL-53 (Al)-derived Ni catalysts with improved activity in CO2 methanation, J. Environ. Chem. Eng., 2023, 11, 109456, DOI:10.1016/j.jece.2023.109456.
- D. F. Anthrop and A. W. Searcy, Sublimation and thermodynamic properties of zinc oxide, J. Phys. Chem., 1964, 68, 2335–2342, DOI:10.1021/j100790a052.
- S. Yu, Y. Li and N. Pan, KOH activated carbon/graphene nanosheets composites as high performance electrode materials in supercapacitors, RSC Adv., 2014, 4, 48758–48764, 10.1039/c4ra06710j.
- Y. C. Wang, W. B. Li, L. Zhao and B. Q. Xu, MOF-derived binary mixed metal/metal oxide@carbon nanoporous materials and their novel supercapacitive performances, Phys. Chem. Chem. Phys., 2016, 18, 17941–17948, 10.1039/c6cp02374f.
- Y. Anil Kumar, S. Singh, D. K. Kulurumotlakatla and H. J. Kim, A MoNiO4 flower-like electrode material for enhanced electrochemical properties: Via a facile chemical bath deposition method for supercapacitor applications, New J. Chem., 2019, 44, 522–529, 10.1039/c9nj05529k.
- Y. Anil Kumar, K. Dasha Kumar and H. J. Kim, A novel electrode for supercapacitors: Efficient PVP-assisted synthesis of Ni3S2 nanostructures grown on Ni foam for energy storage, Dalton Trans., 2020, 49, 4050–4059, 10.1039/d0dt00191k.
- Y. Anil Kumar, K. Dasha Kumar and H. J. Kim, Facile preparation of a highly efficient NiZn2O4-NiO nanoflower composite grown on Ni foam as an advanced battery-Type electrode material for high-performance electrochemical supercapacitors, Dalton Trans., 2020, 49, 3622–3629, 10.1039/d0dt00268b.
- D. Liu, W. Gu, L. Zhou, L. Wang, J. Zhang and Y. Liu, Recent advances in MOF-derived carbon-based nanomaterials for environmental applications in adsorption and catalytic degradation, Chem. Eng. J., 2022, 427, 131503, DOI:10.1016/j.cej.2021.131503.
- X. Liu, L. Zhang and J. Wang, Design strategies for MOF-derived porous functional materials: Preserving surfaces and nurturing pores, J. Mater., 2021, 7, 440–459, DOI:10.1016/j.jmat.2020.10.008.
- Y. N. Xu and W. Y. Ching, Electronic, optical, and structural properties of some wurtzite crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4335–4351, DOI:10.1103/PhysRevB.48.4335.
- Y. Waseda, E. Matsubara and K. Shinoda, X-ray diffraction crystallography: introduction, examples and solved problems, Springer Science & Business Media, 2011 Search PubMed.
- R. Wang, P. Wang, X. Yan, J. Lang, C. Peng and Q. Xue, Promising Porous Carbon Derived from Celtuce Leaves with Outstanding Supercapacitance and CO 2 Capture Performance, Appl. Mater. Interfaces, 2012, 4, 5800–5806 CrossRef CAS PubMed.
- S. Bureekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, D. Tanaka, N. Yanai and S. Kitagawa, One-dimensional imidazole aggregate in aluminium porous coordination polymers with high proton conductivity, Nat. Mater., 2009, 8, 831–836, DOI:10.1038/nmat2526.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87, 1051–1069, DOI:10.1515/pac-2014-1117.
- X. Liu, X. Wang, R. Miao, Y. Sun, Y. Chen, Y. Tang, P. Wan and J. Pan, Al-MOF-derived spindle-like hierarchical porous activated carbon for advanced supercapacitors, Dalton Trans., 2022, 51, 2538–2546, 10.1039/d1dt03395f.
- J. Zhao, N. Liu, Y. Sun, Q. Xu and J. Pan, Nitrogen-modified spherical porous carbon derived from aluminum-based metal-organic frameworks as activation-free materials for supercapacitors, J. Energy Storage, 2023, 73, 109070, DOI:10.1016/j.est.2023.109070.
- R. Cuscó, E. Alarcón-Lladó, J. Ibáñez, L. Artús, J. Jiménez, B. Wang and M. J. Callahan, Temperature dependence of Raman scattering in ZnO, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 1–11, DOI:10.1103/PhysRevB.75.165202.
- C. T. Altaf, T. O. Colak, E. Erdem, U. Unal, F. B. Misirlioglu, G. G. Condorelli, N. D. Sankir and M. Sankir, Disulfonated polyarylene ether sulfone membrane for graphitic carbon nitride/zinc oxide based photo-supercapacitors, Electrochim. Acta, 2023, 456, 142415, DOI:10.1016/j.electacta.2023.142415.
- C. T. Altaf, T. O. Colak, A. M. Rostas, M. Mihet, M. D. Lazar, I. Iatsunskyi, E. Coy, I. D. Yildirim, F. B. Misirlioglu, E. Erdem, M. Sankir and N. D. Sankir, GO/ZnO-based all-solid-state photo-supercapacitors: Effect of GO:ZnO ratio on composite properties and device performance, J. Energy Storage, 2023, 68, 107694, DOI:10.1016/j.est.2023.107694.
- Y. Wang, J. Xu, X. Lin, B. Wang, Z. Zhang, Y. Xu and Y. Suo, Facile synthesis of MOF-5-derived porous carbon with adjustable pore size for CO2 capture, J. Solid State Chem., 2023, 322, 123984, DOI:10.1016/j.jssc.2023.123984.
- E. I. Biru and H. Iovu, in Chapter 9, Graphene Nanocomposites Studied by Raman Spectroscopy, ed. G. M. Do Nascimento, Raman Spectrosc., IntechOpen, 2018, pp. 179–201 Search PubMed.
- A. Fiszka Borzyszkowska, A. Sulowska, P. Czaja, A. Bielicka-Giełdoń, I. Zekker and A. Zielińska-Jurek, ZnO-decorated green-synthesized multi-doped carbon dots from Chlorella pyrenoidosa for sustainable photocatalytic carbamazepine degradation, RSC Adv., 2023, 13, 25529–25551, 10.1039/d3ra04188c.
- A. U. Ammar, I. D. Yildirim, M. H. Aleinawi, M. Buldu-Akturk, N. S. Turhan, S. Nadupalli, A. M. Rostas and E. Erdem, Multifrequency EPR spectroscopy study of Mn, Fe, and Cu doped nanocrystalline ZnO, Mater. Res. Bull., 2023, 160, 112117, DOI:10.1016/j.materresbull.2022.112117.
- E. Erdem, Defect induced p-type conductivity in zinc oxide at high temperature: Electron paramagnetic resonance spectroscopy, Nanoscale, 2017, 9, 10983–10986, 10.1039/c7nr03988c.
- B. Wang, A. J. Fielding and R. A. W. Dryfe, Electron Paramagnetic Resonance as a Structural Tool to Study Graphene Oxide: Potential Dependence of the EPR Response, J. Phys. Chem. C., 2019, 123, 22556–22563, DOI:10.1021/acs.jpcc.9b04292.
- A. C. Lazanas and M. I. Prodromidis, Electrochemical Impedance Spectroscopy–A Tutorial, ACS Meas. Sci. Au., 2023, 3, 162–193, DOI:10.1021/acsmeasuresciau.2c00070.
- M. Buldu-Akturk, Ö. Balcl-Çaǧlran and E. Erdem, EPR investigation of point defects in HfB2 and their roles in supercapacitor device performances, Appl. Phys. Lett., 2022, 120, 153901, DOI:10.1063/5.0089931.
- W. Choi, H.-C. Shin, J. M. Kim, J.-Y. Choi and W.-S. Yoo, Modeling and Applications of Electrochemical Impedance Spectroscopy (EIS) for Lithium-ion Batteries, J. Electrochem. Sci. Technol., 2020, 11, 1–13, DOI:10.33961/jecst.2019.00528.
- D. H. Evans, K. M. O’Connell, R. A. Petersen and M. J. Kelly, Cyclic voltammetry, J. Chem. Educ., 1983, 60, 290–293, DOI:10.1007/978-3-642-02915-8_4.
- A. Gopalakrishnan, T. D. Raju and S. Badhulika, Green synthesis of nitrogen, sulfur-co-doped worm-like hierarchical porous carbon derived from ginger for outstanding supercapacitor performance, Carbon, 2020, 168, 209–219, DOI:10.1016/j.carbon.2020.07.017.
- N. Liu, X. Liu and J. Pan, A new rapid synthesis of hexagonal prism Zn-MOF as a precursor at room temperature for energy storage through pre-ionization strategy, J. Colloid Interface Sci., 2022, 606, 1364–1373, DOI:10.1016/j.jcis.2021.08.105.
- K. Fic, M. Meller and E. Frackowiak, Interfacial Redox Phenomena for Enhanced Aqueous Supercapacitors, J. Electrochem. Soc., 2015, 162, A5140–A5147, DOI:10.1149/2.0251505jes.
- A. U. Ammar, M. Stefan, S. G. Macavei, S. Tripon, O. Pana, C. Leostean, I. D. Vlaicu, A. M. Rostas and E. Erdem, Characterization of defect structures in nanoscaled W-doped TiO2 tested as supercapacitor electrode materials, J. Mater. Sci. Mater. Electron., 2023, 34, 1–18, DOI:10.1007/s10854-022-09540-8.
- J. Liu, J. Wang, C. Xu, H. Jiang, C. Li, L. Zhang, J. Lin and Z. X. Shen, Advanced Energy Storage Devices: Basic Principles, Analytical Methods, and Rational Materials Design, Adv. Sci., 2018, 5, 1700322, DOI:10.1002/advs.201700322.
- S. Güz, M. Buldu-Akturk, H. Göçmez and E. Erdem, All-in-One Electric Double Layer Supercapacitors Based on CH3NH3PbI3 Perovskite Electrodes, ACS Omega, 2022, 7, 47306–47316, DOI:10.1021/acsomega.2c06664.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00966a |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |