
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple medium-bandgap quinoidal A–D–A non-fullerene acceptor for ternary organic solar cells†
Shyam Shankar
S.
a,
María
Privado
b,
Pilar
de la Cruz
 *b,
Fernando
Langa
*b and
Ganesh D.
Sharma
*a
*b,
Fernando
Langa
*b and
Ganesh D.
Sharma
*a
aDepartment of Physics and Electronic Communication, The LNM Institute of Information Technology, Jamdoli, Jaipur (Rajasthan) 302031, India. E-mail: gdsharma@lnmiit.ac.in; gdsharma273@gmail.com
bInstituto de Nanociencia, Nanotecnología y Materiales Moleculares (INAMOL), Universidad de Castilla-La Mancha, Campus de la Fábrica de Armas, 45071 Toledo, Spain. E-mail: Fernando.Langa@uclm.es; Pilar.Cruz@uclm
First published on 22nd April 2025
Abstract
The composition of the bulk heterojunction active layers in organic solar cells is crucial to their photovoltaic performance. Highly efficient organic solar cells have been constructed by the so-called ternary approach due to its process simplicity and diverse donor and acceptor materials. In the study described here, the simple medium-bandgap closed-shell quinoidal A–D–A non-fullerene small molecule acceptor QDT1, i.e., 4,4-dihexyl-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CPDT), was used as a guest acceptor in the PBDB-T:Y6 host binary active layer to fabricate ternary organic solar cells. QDT1 has an absorption profile that is complementary to those of the host active materials (PBDB-T and Y6). This matching of profiles is beneficial for light-harvesting and ultimately enhances the photocurrent in the OSC devices. Ternary organic solar cells fabricated in ambient conditions with the optimized PBDB-T![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :QDT1:Y6 (1.0:0.2:1.0) active layer gave an overall power conversion efficiency of 13.31%, which is better than that of the PBDB-T:Y6 counterpart (11.17%). The higher PCE in the ternary system is mainly attributed to the higher short circuit photocurrent and fill factor along with a decreased energy loss. The increases in photocurrent and fill factor can both be attributed to faster exciton dissociation, energy transfer from QDT1 to Y6, more rapid charge extraction, extended charge carrier lifetime and lower charge recombination. The organic solar cells based on a PBDB-T:QDT1 active layer provided a PCE greater than 23% under indoor illumination (white LED).
:QDT1:Y6 (1.0:0.2:1.0) active layer gave an overall power conversion efficiency of 13.31%, which is better than that of the PBDB-T:Y6 counterpart (11.17%). The higher PCE in the ternary system is mainly attributed to the higher short circuit photocurrent and fill factor along with a decreased energy loss. The increases in photocurrent and fill factor can both be attributed to faster exciton dissociation, energy transfer from QDT1 to Y6, more rapid charge extraction, extended charge carrier lifetime and lower charge recombination. The organic solar cells based on a PBDB-T:QDT1 active layer provided a PCE greater than 23% under indoor illumination (white LED).
Introduction
Organic solar cells (OSCs) have several advantages over silicon-based solar cells and these include flexibility, a lightweight structure, and the possibility of cost-effective applications in wearable electronic devices, indoor internet of things and building integrated photovoltaics.1–6 In recent years the emergence of non-fullerene acceptors (NFAs)7–11 has led to significant advances in OSCs and the power conversion efficiency (PCE) has reached 20%.12–17 The most effective OSCs generally consist of bulk heterojunction (BHJ) blend layers that consist of electron donors (D) and acceptors (A). In these layers the hierarchical blend structures tend to incorporate interpenetrated D/A nanoscale networks to enhance the exciton dissociation into free charge carriers and their subsequent transport towards electrodes.18,19 The majority of OSCs are based on NFAs but the synthesis of these materials involves complex and lengthy procedures, which result in substantial expenses that create major obstacles for industrialization of OSCs.In recent years non-fused-ring NFAs have emerged as competitive low-cost materials for OSCs due to their simple synthesis routes.20–23 OSCs based on non-fused NFAs have given a PCE value of 17% for a binary BHJ active layer24–26 and 18% for a ternary active layer,27 which is very close to the values obtained for fused-ring NFAs.
Despite the remarkable properties of quinoidal semiconductors and the fact that they are already used in organic transistors,28,29 their application as electron acceptors in organic solar cells (OSCs) remains limited.30,31 The incorporation of quinoidal structures, however, provides an alternative approach to developing high-performance electron acceptors owing to several exclusive advantages: (i) the rigid and flat configuration of the quinoidal unit facilitates compact π–π stacking and efficient intermolecular charge transport and (ii) quinoidal units typically have excellent π-electron delocalization and a strong intramolecular charge-transfer effect, resulting in reduced optical bandgaps and elevated absorption coefficients.
Thiophene and its derivatives play a crucial role in the semiconductor electronic revolution, namely organic electronics, and thiophene-containing polymers and oligomers are prominent in this field in applications that include thin-film transistors, OLEDs and organic photovoltaics.32 4,4-Dihexyl-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CPDT) is a versatile component of opto-electronic materials due to the completely planar nature of the fused-dithiophene, the excellent π-electron donor ability and the possibility of incorporating alkyl chains to afford solubility.33,34
Tetracyanoquinodimethane oligothiophenes have been widely used in the field of organic conjugated molecules35,36 and some of us prepared a quinoidal derivative of bis-CPDT-vinylene that had novel electronic properties and potential applications in organic electronics.37 We recently reported a series of bis(semiquinone) compounds with CPDT bridges of different lengths (n-QDT) that were encapsulated with benzoquinone to give systems that evolve from closed-shell (n = 1) to full diradical (n = 3).38
The majority of efficient OSC active layers still have the BHJ structure proposed by Heeger and co-workers.18 The absorption spectra of most polymer donors are in the short wavelength region while the absorption spectra of most NFAs are in the long wavelength and near-infrared regions and, as a consequence, the absorption spectrum of the binary BHJ active layer makes it difficult to cover the entire solar spectrum. This limitation hinders the effective absorption of solar light, which in turn limits any improvement in photocurrent. In an effort to overcome this challenge, a ternary approach has been developed that involves BHJ active layers with either two donors and one acceptor or one donor and two acceptors. This approach has led to significant enhancements in the PCE of OSCs.39,40 The third component (donor or acceptor) of effective ternary OSCs must have a complementary absorption profile, cascade energy level alignment, and strong compatibility with the existing binary host blend.
We report here the use in OSCs of the shortest member of the aforementioned series, namely QDT1,38 which has a stable quinoidal closed-shell form. QDT1 was employed as a medium-bandgap non-fused small molecule acceptor (NFSMA) for ternary organic solar cells along with PBDB-T as a donor and Y6 as an acceptor. QDT1 showed excellent performance as the third component. The OSC based on PBDB-T:QDT1:Y6 gave a PCE value of 13.31%, which exceeds that of binary counterparts (11.17 and 7.52% for PBDB-T:Y6 and PBDB-T:QDT1, respectively). PL and TRPL spectroscopy revealed that a highly efficient FRET occurred between QDT1 and Y6, which led to more efficient exciton diffusion and subsequent exciton dissociation. TPV and TPC measurements revealed that the introduction of QDT1 into the host PBDB-T:Y6 material led to faster charge extraction and longer charge carrier lifetime, which in turn led to the suppression of both bimolecular and trap-assisted recombination. This resulted in improvements in the JSC, FF and overall PCE. Moreover, the OSC based on PBDB-T:QDT1 gave a PCE greater than 23% under white light LED illumination.
Results and discussion
Electronic and theoretical studies
The skeletal structures of polymer PBDB-T, QDT1, and Y6 are illustrated in Fig. 1a. QDT1 was prepared as reported previously.38 The spectrum of a dilute solution of QDT1 in CH2Cl2 shows an absorption maximum at 656 nm and an absorption edge at around 780 nm (Fig. 1b). In thin film, a broader absorption band with a maximum of 676 nm and a shoulder at 780 nm is observed. This bathochromic shift indicates a highly ordered and compact J-aggregate arrangement of molecules in the solid state, which enhances the excitonic interactions. The absorption edge in the thin film is observed at 850 nm, and this corresponds to an optical bandgap of 1.45 eV. The thin film UV-Vis absorption spectra of PBDB-T, QDT1, and Y6 cast from chloroform solution are shown in Fig. 1b. Both PBDB-T and Y6 show strong absorption in the regions of 350–700 nm and 600–900 nm. QDT1 displays maximum absorption at 676 nm, whilst its predominant absorption ranges from 500–850 nm. Both spectra combine to give a complementary absorption spectrum that should enable substantial photon harvesting with the host PBDB-T and Y6. This system is expected to have a strong capability for photon harvesting and potentially lead to a higher JSC value in the ternary OSCs. | ||
| Fig. 1 (a) Chemical structures of PBDB-T, QDT1, and Y6. (b) UV-Vis-NIR spectra of QDT1 in solution, QDT1 in thin film, PBDB-T in thin film, and Y6 in thin film. (c) Schematic energy level diagrams of materials used in photovoltaic devices. | ||
The energy levels of PBDB-T, Y6, and QDT1, as estimated by electrochemistry, are depicted in Fig. 1c. The results infer that the LUMO (−4.00 eV) and HOMO (−5.40 eV) energy levels of QDT1 are located between those of PBDB-T (−3.53/−5.21 eV) and Y6 (−4.08/−5.68 eV), thus establishing a cascading energy arrangement. This arrangement could help to facilitate efficient charge transfer at the D/A interfaces, modulate the energy losses and promote charge separation.41,42 Furthermore, the relatively upshifted LUMO energy level of QDT1 (−4.00 eV), when compared to that of Y6 (−4.08 eV), indicates possible advantages in achieving greater VOC values for ternary OSCs.
The most stable geometry of QDT1 (Fig. S1a, ESI†) was calculated using a DFT (B3LYP) method with a 6-31G* basis set. These calculations indicate the high planarity of the quinoidal system (QDT1). The molecular dipole moments of Y6 and QDT1 were also calculated (Fig. S1b, ESI†) and it was found that QDT1 has a significantly higher dipole moment (4.859 D) than Y6 (0.523 D). The main difference in these dipole moments lies along the y-axis. In the case of Y6, there are polar groups on both sides (up and down) of the x-axis and this arrangement results in compensation of the dipole moments on both sides. In QDT1, however, all the polar groups are aligned along the ‘north’ axis. The intermolecular packing interactions43,44 are affected by large differences in the dipole moments between QDT1 and Y6. The electrostatic potential (ESP) distributions of Y6 and QDT1 were computed (Fig. S2, ESI†) and the results show that the highest energy value is in the CPDT moiety and the lowest in the quinone terminal units.
Photovoltaic performance
As depicted in Fig. 1c, the LUMO offset between PBDB-T and either QDT1 or Y6 is sufficiently above the threshold force required for the dissociated excitons from the PBDB-T to break it into free charge carriers. This situation facilitates the subsequent transfer of electrons from the donor to the acceptor in both PBDB-T:Y6 and PBDB-T:QDT1 BHJ active layers. There is adequate HOMO offset between PBDB-T and Y6 to ensure that there is effective hole transfer from Y6 to PBDB-T which is followed after the dissociation of excitons generated in Y6. PBDB-T and QDT1 assure a HOMO offset of about 0.19 eV between them. To gain information about exciton dissociation after their generation, and transfer of holes from HOMO of QDT1 to HOMO of PBDB-T, the photoluminescence (PL) spectra of pristine QDT1 film and its blend with PBDB-T were compared and shown in Fig. S3 (ESI†). We can see PL quenching of QDT1 when blended with PBDB-T. This only suggests effective exciton dissociation and subsequent transfer of holes from QDT1 to PBDB-T. The processes occur though the HOMO offset is less. Such exciton dissociation and charge transfer are often reported for OSCs in which NFSMA, and polymer or small molecule donors are employed.45–47The OSCs were fabricated with the conventional architecture of binary and ternary OSCs comprising ITO-coated glass as TCO, PEDOT:PSS as HTL, PFN-Br as ETL and Ag (100 nm) as anode (which was deposited by thermal evaporation technique). All the processes were done in ambient conditions. PBDB-T and Y6 or QDT1 were used as the polymer donor and acceptor, respectively. All devices were prepared by spin-coating solutions of the components with a PBDB-T:Y6 weight ratio of 1:1.2. A concentration of 14 mg mL−1 was maintained. The detailed procedure for device fabrication is provided in the ESI.† The ternary OSCs were prepared by adding different amounts of QDT1. 1-Chloronaphthalene (CN) at 0.25 vol% was included as a solvent additive. The resulting thin films were allowed for thermal annealing at 110 °C for about 5 minutes. The obtained devices were characterized under 1 sun conditions (AM 1.5G spectra).
The current–voltage (J–V) characteristics of the optimized binary (PBDB-T:QDT1 or PBDB-T:Y6) and ternary (PBDB-T:QDT1:Y6) devices are depicted in Fig. 2a and their photovoltaic parameters are tabulated in Table 1. The binary OSCs comprising PBDB-T:Y6 and PBDB-T:QDT1 yield efficiencies of 11.17% and 7.52%, respectively. The dipole moment of QDT1 is higher than that of Y6, and, as a consequence, the exciton binding energy of the QDT1 system is lower. In this case, a lower driving force is required for the dissociation of excitons, and hence the higher value of VOC for the OSCs based on QDT1. Moreover, the inclusion of 20% wt of QDT1 in PBDB-T:Y6 blends gave PBDB-T:QDT1:Y6 (1:0.2:1.0) ternary OSCs with a remarkable PCE value of 13.31%. This high value is due to the simultaneous increase in VOC (0.836 V), JSC (22.84 mA cm−2) and fill factor (0.697) when compared to PBDB-T:Y6 OSCs. The QDT1 film exhibited a complementary absorption with both PBDB-T and Y6, thus enhancing the light-harvesting capability and increasing the JSC in the ternary OSC. The VOC value for the ternary device is higher than the binary Y6 counterpart but still less than PBDB-T:QDT1. The open voltage of the BHJ-based OSC is correlated to the difference in energy levels between the LUMO of the acceptor and the HOMO of the donor. It can, therefore, be envisaged that the higher VOC for the ternary system, when compared to the Y6 binary counterpart, may be due to the fact that the LUMO of QDT1 is upshifted in relation to that of Y6.
 | ||
| Fig. 2 (a) J–V characteristics under 1 sun condition (AM1.5G, 100 mW cm−2), (b) EQE spectra of the optimized binary (PBDB-T:QDT1 and PBDB-T:Y6) and ternary PBDB-T:QDT1:Y6 OSCs. | ||
| Active layer | J SC (mA cm−2) | V OC (V) | FF | PCE |
|---|---|---|---|---|
| a J SC value obtained from the integration of EQE spectra. b Average value of 8 identical devices. | ||||
| PBDB-T:QDT1 | 14.36 (14.09)a | 0.854 | 0.613 | 7.52 (7.34)b |
| PBDBT-T:Y6 | 20.78 (20.57)a | 0.812 | 0.662 | 11.17 (10.97)b |
| PBDB-T:QDT1:Y6 | 22.84 (22.65)a | 0.836 | 0.697 | 13.31 (13.06)b |
The external quantum efficiency spectra (EQE) of all the devices are displayed in Fig. 2b. The spectra of PBDB-T:QDT1 and PBDB-T:Y6 are very similar to the UV-Vis-NIR spectra of their corresponding blends in the thin film. The EQE values of the ternary OSCs are higher than those of the PBDB-T:Y6 counterpart – particularly in the wavelength region 600–740 nm, where the absorption coefficient of QDT1 is high. This indicates that the incorporation of QDT1 into PBDB-T:Y6 mixtures increases the light-harvesting ability and thus leads to a higher JSC value for the ternary OSC. The JSC values of the OSCs are given further validation by the EQE spectra. The JSC values attained by the area of the EQE spectra closely match the JSC measured from the J–V curves (Table 1).
The energy level difference of LUMO of the acceptor material and HOMO of the donor material gives out the VOC of the bulk-hetero junction devices. Due to the same, the VOC of the ternary active layer (0.836 V) lies between the two binary counterparts. Cyclic voltammetry measurements on optimized Y6:QDT1 blend film were done to calculate HOMO–LUMO levels and further their VOC dependence. The HOMO and LUMO energy levels of the optimized acceptor blend are −5.48 and −4.04 eV, respectively. These values lie between those of Y6 and QDT1, which results in an increase in the charge transfer (CT) state (ECT) with dark current density being in a suppressed state for ternary blends.48,49 Moreover, the VOC values of the ternary OSCs increase as the concentration of QDT1 increases (Table S1, ESI†), a trend that is consistent with the formation of an alloy between the two acceptors.50 Thus, the incorporation of QDT1 as a third component can increase the open circuit voltage (VOC).51,52
Exciton generation and charge recombination dynamics
The exciton dissociation (Pdiss) probability was investigated by examining plots of the photocurrent density (Jph) variation with effective voltage (Veff) (Fig. 3). The estimation of both the Jph and Veff values is described in the ESI.†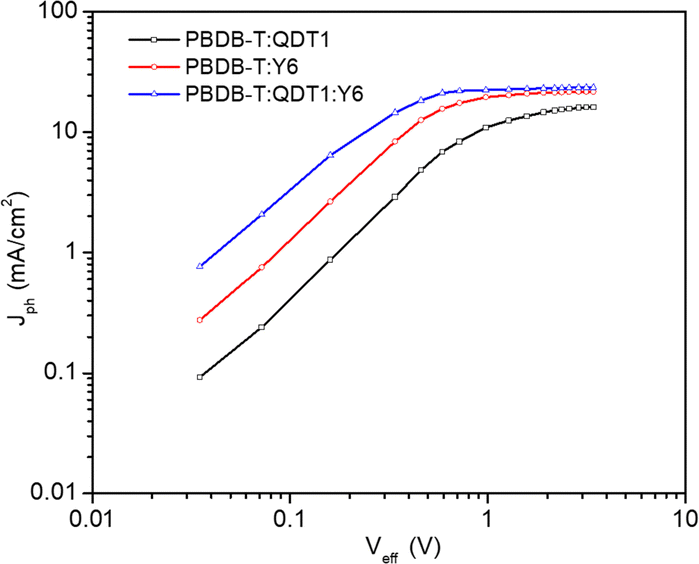 | ||
| Fig. 3 Plot of photocurrent density (Jph) with effective voltage (Veff) for binary (PBDB-T:QDT1 and PBDB-T:Y6) and ternary (PBDB-T:QDT1:Y6) OSCs. | ||
The Jph value increases as Veff increases and the photocurrent then becomes almost saturated when Veff > 1.8 V. The saturation current density (Jsat) in the ternary OSC was found to be higher than those in the binary counterparts and this finding indicates enhanced light-harvesting ability and a more efficient exciton generation rate. The value of Pdiss under short circuit condition was estimated using the expression Pdiss = Jph/Jsat. The Pdiss value for the ternary OSC (0.982) is higher than that for binary counterparts (0.946 and 0.963 for PBDB-T:QDT1 and PBDB-T:Y6, respectively) and this suggests that the ternary OSC exhibited faster dissociation of excitons into free charges and subsequent collection of them by the electrodes, which in turn contributes to the enhancement of JSC and FF.53
The carrier dynamics of devices, including their transport, and recombination processes were evaluated by examining the JSC and VOC correlation with varying illumination power (Pin) in both binary and ternary OSCs. The slopes of VOCversus Pin plots (Fig. 4a) were fitted using the expression VOC = (nkT/q)lnPin and the values were 1.45, 1.32 and 1.23kT/q for PBDB-T:QDT1, PBDB-T:Y6 and PBDB-T:QDT1:Y6 OSCs, respectively. Compared to binary OSCs, the lower slope obtained for the ternary counterpart suggests that trap-assisted recombination was lower in the ternary OSC, which leads to improved charge transport and enhanced JSC and FF.54 The JSC–Pin curves (Fig. 4b) were fitted using the relationship JSC ∝ (Pin)α, where α is used to determine the degree of bimolecular recombination. The α values calculated for ternary structure is 0.974, while binary counterparts PBDB-T:QDT1 (0.912), PBDB-T:Y6 (0.959), has a slightly lower values. The higher α value obtained for the ternary device, when compared to the binary counterparts, indicates relatively weak bimolecular recombination in the ternary OSC.55,56
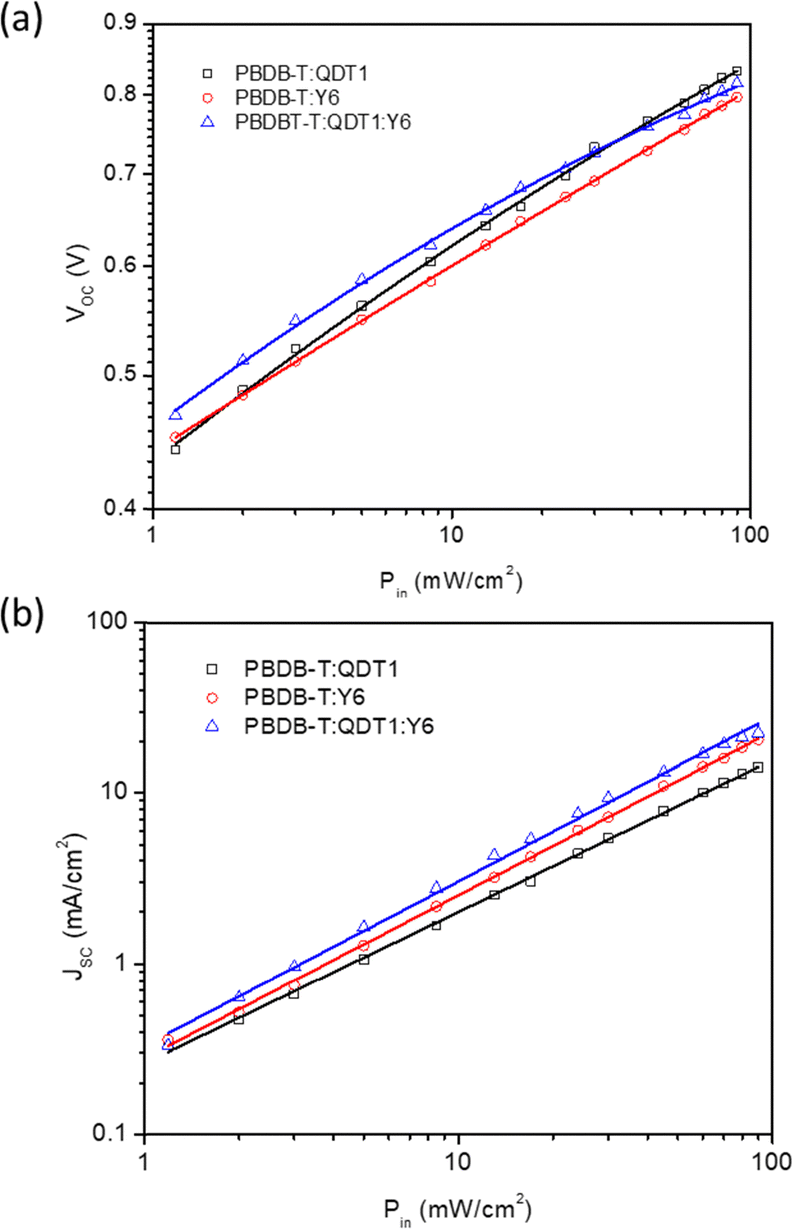 | ||
| Fig. 4 Dependence of (a) VOC and (b) JSC with Pin for optimized binary (PBDB-T:QDT1 and PBDB-T:Y6) and ternary (PBDB-T:QDT1:Y6) OSCs. | ||
The transient photovoltage (TPV) was employed for assessing the carrier recombination kinetics in these devices and the results are depicted in Fig. 5a. The PBDB-T:QDT1:Y6 ternary device exhibited a longer carrier lifetime (7.75 μs) than the binary counterparts (5.38 μs and 3.76 μs for PBDB-T:Y6 and PBDB-T:QDT1, respectively). The longer carrier lifetime attributes to a longer transport distance for photogenerated charge carriers, and this is only possible due to less recombination, which in turn enhances both JSC and FF.57 Moreover, transient photocurrent (TPC) measurements (Fig. 5b) demonstrate that the PBDB-T:QDT1:Y6 device yielded the shortest photocurrent decay time of 0.784 μs when compared to PBDB-T:QDT1 (1.26 μs) and PBDB-T:Y6 (0.965 μs). This difference signifies that the ternary devices have enhanced charge extraction efficiency. The high carrier extraction eased to reduce carrier recombination inside the active layer and this ensures timely extraction and transport of a greater number of photogenerated carriers to the electrodes, thereby improving both JSC and FF.58
 | ||
| Fig. 5 (a) TPV and (b) TPC curves for the binary (PBDB-T:QDT1 and PBDB-T:Y6) and ternary (PBDB-T:QDT1:Y6) OSCs. | ||
The charge transport kinetics in the active layers were further examined by carrying out space charge limited current (SCLC) measurements using hole and electron-only devices (Fig. S4a and b, ESI†). The addition of QDT1 led to an increase in both hole mobility (μh) and electron mobility (μe) for the PBDB-T:QDT1:Y6 device when compared with the PBDB-T:Y6 devices. The μh and μe values increased from 4.56 × 10−4 to 5.67 × 10−4 cm2 V−1 s−1 and from 3.42 × 10−4 to 4.78 × 10−4 cm2 V−1 s−1, respectively. The enhanced charge carrier mobilities and more balanced μh/μe = 1.19 in the PBDB-T:QDT1:Y6 ternary film, which suppresses the charge recombination, is consistent with the enhanced FF for the ternary OSC. These findings clearly show that QDT1, as a guest acceptor, can simultaneously aid in exciton dissociation and carrier generation, thus significantly enhancing the JSC and FF of the OSC.
TRPL spectroscopy is a technique that is used to obtain information about the exciton diffusion and exciton dissociation in the devices and selected spectra are shown in Fig. 6. The decay time represents the exciton dissociation at the D/A interface and exciton diffusion towards the interfaces.59 Compared with binary blends (0.886 ns and 0.781 ns for QDT1 and Y6-based blends, respectively), the shorter decay time (0.506 ns) measured for the ternary blend indicates faster exciton diffusion and transfer behaviour in ternary OSCs. These properties result in increased JSC and PCE values.
 | ||
| Fig. 6 (a) TRPL plots for PBDB-T:QDT1, PBDB-T:Y6 and PBDB-T:QDT1:Y6 and (b) QDT1 and QDT1:Y6. | ||
Energy loss analysis
Energy loss (Eloss) analysis of binary and ternary devices was performed in an effort to understand the VOC enhancement in the ternary OSCs. Eloss is expressed as Eloss = Eg − VOC, where Eg is the optical energy bandgap of the blend film, which can be estimated from the peak observed 1st derivative of EQE of the OSC (Fig. S5, ESI†). The Eg values for the blends were estimated to be 1.58, 1.38 and 1.38 eV for PBDB-T:QDT1, PBDB-T:Y6 and PBDB-T:QDT1:Y6, respectively. The detailed Eloss values are shown in Table 2. In general, the Eloss of a solar cell can be divided into three parts: ΔE1, ΔE2 and ΔE3. ΔE1 is denoted as Eg − qVSQOC, where VSQOC represents the theoretical upper limit for the VOC, as derived from Shockley–Queisser theory. This limit originates from the absorption above the bandgap60 and its value is almost the same for all of the devices. ΔE2 can be determined by the expression ΔE2 = q(VSQOC − VradOC) and it represents the loss of radiation recombination below the bandgap, which is determined by the degree of energy disorder and recombination.61 The ΔE2 value obtained for the ternary device is significantly lower than that for the binary device – a finding that suggests minimal energy disorder in the ternary device. ΔE2 can also be quantified by a parameter called the Urbach energy (EU), where a low EU value signifies low energy disorder. The FTPS-normalized EQE (Fig. S5, ESI†) with Urbach's rule62,63 indicates that the EU of the ternary device is lower (26.65 meV) than that of the binary devices (31.43 meV and 28.25 meV for QDT1 and Y6, respectively). The lower EU value is mainly attributed to the enhanced molecular aggregation, reduced reorganization energy and energy disorder in the ternary blend film.64 ΔE3 represents the non-radiative loss term, which originates from the recombination of charge carriers in the device. The ΔE3 value for the ternary system is 0.219 eV, which is lower than the values for the binary counterparts. The lower ΔE3 value for ternary OSC is related to suppression of the charge carrier recombination.| Device | E g (eV) | ΔE1 (eV) | ΔE2 (eV) | ΔE3 (eV) | Total Eloss (eV) |
|---|---|---|---|---|---|
| PBDB-T:QDT1 | 1.58 | 0.255 | 0.0945 | 0.377 | 0.726 |
| PBDB-T:Y6 | 1.38 | 0.249 | 0.0876 | 0.231 | 0.568 |
| PBDB-T:QDT1:Y6 | 1.38 | 0.249 | 0.076 | 0.219 | 0.544 |
Energy and charge transfer mechanisms in the ternary device
It can be seen from Fig. 7a that the steady state PL of QDT1 has significant overlap with the absorption spectrum of Y6. This observation indicates that Förster resonance energy transfer (FRET) occurs from the medium-bandgap acceptor QDT1 to the low-bandgap acceptor Y6 – a phenomenon that is observed in many ternary OSCs. In an effort to obtain more information about the energy transfer, the steady state PL spectra of QDT1, Y6 and a QDT1:Y6 blend were investigated. It can be seen from Fig. 7b that the emission peaks of QDT1 and Y6 are located at 816 nm and 920 nm, respectively. The incorporation of QDT1 with Y6 led to almost complete quenching of the PL emission intensity of QDT1 along with a simultaneous increase in the PL intensity of Y6. This behavior indicates an efficient energy transfer from QDT1 to Y6. An alternative approach to confirm the energy transfer between QDT1 and Y6 involves creating solar cells with QDT1:Y6 (0.2:1.0) and pristine QDT1 or Y6 as the active layers without a donor, respectively. The JSC value of the QDT1:Y6-based OSC is 0.68 mA cm−2, which is lower than the values for both pristine QDT1 (0.89 mA cm−2) and Y6 (1.05 mA cm−2). These values imply that charge transfer between QDT1 and Y6 is negligible.
 | ||
| Fig. 7 (a) Absorption spectra and PL spectra of Y6 and QDT1, respectively, (b) PL spectra of pristine QDT1, Y6 and QDT1:Y6, and (c) energy and charge transfer in the ternary device. | ||
In accordance with FRET theory, the lifetimes of energy donors in an excited state decrease with increasing acceptor concentration since FRET is an additional non-radiative decay channel for a donor in the system.65 TRPL spectroscopy was used to investigate the energy transfer from QDT1 to Y6 (excitation wavelength of 690 nm and probing 830 nm emission). The average lifetime of pristine QDT1 was 1.55 ns, which is markedly decreased to 0.66 ns for the QDT1:Y6 (0.2:1.0) film. The energy transfer efficiency (η) from the energy donor (QDT1) to the energy acceptor (Y6) can be estimated using the following expression: η = 1 − (τDA/τD), where τD and τDA denote the fluorescence lifetime of the donor with and without the acceptor, respectively. QDT1 and Y6 act as energy donor and energy acceptor, respectively. On using the above expression, the energy transfer efficiency was calculated to be 57.3% and this is consistent with the overlap of the absorption spectrum of Y6 and PL spectrum of the QDT1. The energy transfer and charge transfer mechanisms are illustrated in Fig. 7c. It can be concluded that there is a balance between the exciton dissociation process and the energy transfer process in the optimized ternary OSC.66
The surface morphologies of PBDB-T:Y6 and PBDB-T:QDT1:Y6 blends were investigated by atomic force microscopy (AFM) (Fig. S6, ESI†) in an attempt to elucidate why the ternary OSC gave a high PCE value. Both blends gave smooth films that had low surface roughness. The ternary PBDB-T:QDT1:Y6 film showed slightly higher roughness (2.34 nm) compared to the binary PBDB-T:Y6 counterpart (2.09 nm) and this is mainly due to the improved molecular aggregation. Higher surface roughness is beneficial for both high JSC and FF values in OSCs.67
The surface morphologies of PBDB-T:Y6 and PBDB-T:QDT1:Y6 blends were investigated by atomic force microscopy (AFM) (Fig. S6, ESI†) in an attempt to elucidate why the ternary OSC gave a high PCE value. Both blends gave smooth films that had low surface roughness. The ternary PBDB-T:QDT1:Y6 film showed slightly higher roughness (2.34 nm) compared to the binary PBDB-T:Y6 counterpart (2.09 nm) and this is mainly due to the improved molecular aggregation. Higher surface roughness is beneficial for both high JSC and FF values in OSCs.67
A great deal of research has been carried out in recent years on OSCs for indoor applications68–70 and the PCE has been boosted up to 30%.71,72 Both PBDB-T and QDT1 showed strong absorptions up to 800 nm and this is very similar to the emission spectrum of white LEDs. As a consequence, the J–V characteristics were measured under different absorption spectra. The binary and ternary OSCs were also tested under indoor light illumination (white LED, 1000 lux) using standard white (SW) and cool white (CW). The detail of the J–V measurements is summarized in ESI.† We have measured the intensity of indoor light by luxmeter and it is 300.3 and 317.5 μW cm−2 for SW and CW LEDs, respectively.73 The J–V characteristics are shown in Fig. 8 and the corresponding photovoltaic parameters are compiled in Table 3. The PCE values of OSCs based on indoor light illumination exceeded 23% under SW LED, thus demonstrating the potential applications of medium-bandgap NFAs along with wide-bandgap polymer donors for indoor OSCs.
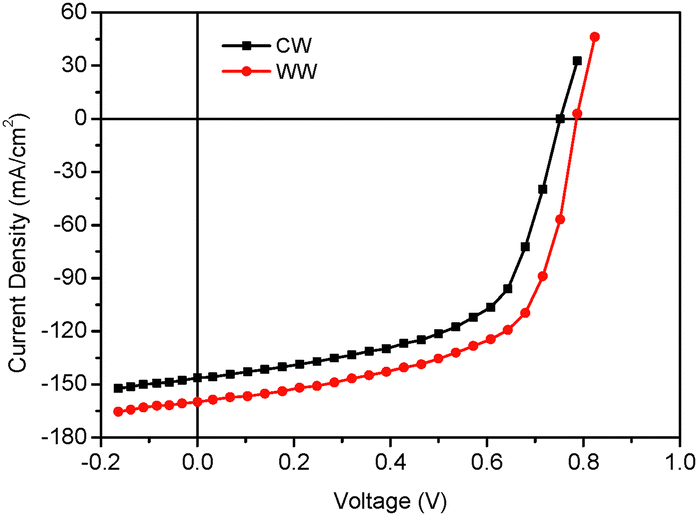 | ||
| Fig. 8 J–V characteristics of the OSC based on PBDB-T:QDT1 under illumination with stanard white (SW) LED (1000 lux). | ||
| LED | J SC (μA cm−2) | V OC (V) | FF | PCE (%) |
|---|---|---|---|---|
| CW | 146.34 | 0.752 | 0.587 | 21.51 |
| SW | 159.45 | 0.786 | 0.602 | 23.76 |
Conclusions
A quinoidal A–D–A non-fullerene small-molecule acceptor QDT1 was utilized as a guest acceptor in the host binary blend PBDB-T:Y6 to fabricate ternary OSCs. It is worth highlighting that addition of only a small amount of QDT1 can significantly improve the PCE of the ternary OSC from 11.17% to 13.31%. This improvement mainly results from the enhanced JSC and FF. The mechanism was investigated by PL and TRPL studies, which showed that QDT1 acts as an additional light absorber to harvest more photons and deliver photon energy to Y6 through FRET. In this process extra excitons were generated and then dissociated into free charge carriers, which resulted in an improvement in the JSC in the ternary OSCs. Moreover, the OSCs based on PBDB-T:QDT1 gave PCE values above 24% under indoor light illumination, thus highlighting the potential of the BHJ active layer consisting of PBDB-T and QDT1 for applications in indoor OSCs.Author contributions
M. P. has synthesized simple medium-bandgap quinoidal A–D–A non-fullerene acceptor QDT1. S. S. S. has been involved in the photovoltaic studies. P. C., F. L. and G. D. S. have directed the experiments and the interpretation of the data.Data availability
The data supporting the findings of this study are available in the ESI† of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the MCIN/AEI of Spain (PID2022-141687OB-I00, RED2022-134939-T and TED2021-131255B-C42), the Junta de Comunidades de Castilla-La Mancha, and European Social Funds (SBPLY/21/180501/000142) are acknowledged.References
- H. Yao and J. Hou, Angew. Chem., Int. Ed., 2022, 61, e202209021 CrossRef CAS PubMed.
- J. Yi, G. Zhang, H. Yu and H. Yan, Nat. Rev. Mater., 2024, 9, 46–62 CrossRef CAS.
- X. Liu, S. Xu, B. Tang and X. Song, Chem. Eng. J., 2024, 497, 154944 CrossRef CAS.
- Y. Li, X. Huang, H. K. M. Sheriff and S. R. Forrest, Nat. Rev. Mater., 2023, 8, 186–201 CrossRef.
- B. Deng, Y. Li, Z. Lu, K. Zheng, T. Xu, S. Wang, X. Luo, B. Grandidier, J. Zhang and F. Zhu, Nat. Commun., 2025, 16, 597 CrossRef CAS PubMed.
- C. Zhu, H. Zheng, L. Zhao and X. Zhu, Sci. China Mater., 2025 DOI:10.1007/s40843-024-3210-y.
- C. Zhang, R. Yu, Q. Lv, S. Li, H. Yuan, B. Huang and Z. Tan, ChemSusChem, 2025, 18, e202401138 CrossRef CAS PubMed.
- Q. Wu, S. Ding, A. Sun and Y. Xia, Mater. Today Chem., 2024, 41, 102290 CrossRef.
- T. Yu and D. Ma, Sol. RRL, 2024, 8, 2300751 CrossRef CAS.
- J. Wang, Y. Xie, K. Chen, H. Wu, J. M. Hodgkiss and X. Zhan, Nat. Rev. Phys., 2024, 6, 365–381 CrossRef CAS.
- D. Luo, W. Jang, D. D. Babu, M. S. Kim, D. H. Wang and A. K. K. Kyaw, J. Mater. Chem. A, 2022, 10, 3255–3295 RSC.
- Y. Jiang, S. Sun, R. Xu, F. Liu, X. Miao, G. Ran, K. Liu, Y. Yi, W. Zhang and X. Zhu, Nat. Energy, 2024, 9, 975–986 CrossRef CAS.
- C. Guo, Y. Sun, L. Wang, C. Liu, C. Chen, J. Cheng, W. Xia, Z. Gan, J. Zhou, Z. Chen, J. Zhou, D. Liu, J. Guo, W. Li and T. Wang, Energy Environ. Sci., 2024, 17, 2492–2499 RSC.
- Y. Sun, L. Wang, C. Guo, J. Xiao, C. Liu, C. Chen, W. Xia, Z. Gan, J. Cheng, J. Zhou, Z. Chen, J. Zhou, D. Liu, T. Wang and W. Li, J. Am. Chem. Soc., 2024, 146, 12011–12019 CrossRef CAS PubMed.
- S. Wang, S. Wang, J. Wang, N. Yu, J. Qiao, X. Xie, C. Li, M. S. Abbasi, R. Ding, X. Zhang, Y. Han, G. Lu, J. Zhang, X. Hao, Z. Tang, Y. Cai and H. Huang, Adv. Energy Mater., 2025, 2405205 CrossRef.
- F. Furlan and N. Gasparini, Nat. Mater., 2025, 24(3), 336–337 CrossRef CAS.
- H. Chen, Y. Huang, R. Zhang, H. Mou, J. Ding, J. Zhou, Z. Wang, H. Li, W. Chen, J. Zhu, Q. Cheng, H. Gu, X. Wu, T. Zhang, Y. Wang, H. Zhu, Z. Xie, F. Gao, Y. Li and Y. Li, Nat. Mater., 2025, 24, 444–453 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- H. Huang, Q. Guo, S. Feng, C. Zhang, Z. Bi, W. Xue, J. Yang, J. Song, C. Li, X. Xu, Z. Tang, W. Ma and Z. Bo, Nat. Commun., 2019, 10, 3038 CrossRef.
- P. Jiang, Y. Liu, J. Song and Z. Bo, Acc. Chem. Res., 2024, 57, 3419–3432 CrossRef CAS PubMed.
- X. Zhang, X. Gu and H. Huang, Acc. Chem. Res., 2024, 57, 981–991 CrossRef CAS.
- R. Yu, S. Li, H. Yuan, Z. Yang, S. Jin and Z. Tan, J. Phys. Chem. Lett., 2024, 15, 2781–2803 CrossRef CAS PubMed.
- D.-L. Ma, Q.-Q. Zhang and C.-Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202214931 CrossRef CAS.
- D. Li, H. Zhang, X. Cui, Y.-N. Chen, N. Wei, G. Ran, H. Lu, S. Chen, W. Zhang, C. Li, Y. Liu, Y. Liu and Z. Bo, Adv. Mater., 2024, 36, 2310362 CrossRef CAS.
- Y. Shao, R. Sun, W. Wang, X. Yang, C. Sun, Y. Li and J. Min, Sci. China: Chem., 2023, 66, 1101–1110 CrossRef CAS.
- H. Tan, W. Fan, M. Zhu, J. Zhu, X. Wang, M. Xiao, R. Yang, W. Zhu and J. Yu, Small, 2023, 19, 2304368 CrossRef CAS PubMed.
- S. Vegiraju, G.-Y. He, C. Kim, P. Priyanka, Y.-J. Chiu, C.-W. Liu, C.-Y. Huang, J.-S. Ni, Y.-W. Wu, Z. Chen, G.-H. Lee, S.-H. Tung, C.-L. Liu, M.-C. Chen and A. Facchetti, Adv. Funct. Mater., 2017, 27, 1606761 CrossRef.
- T. Du, R. Gao, Y. Deng, C. Wang, Q. Zhou and Y. Geng, Angew. Chem., Int. Ed., 2020, 59, 221–225 CrossRef CAS PubMed.
- H. Feng, B. Yin, L. Pan, X. Liu, S. Kim, Y. Zhao, X. Huang, C. Yang and C. Duan, Chem. Commun., 2023, 59, 9529–9532 RSC.
- Z. Zhou, S. Xu and X. Zhu, Bull. Chem. Soc. Jpn., 2021, 94, 929–936 CrossRef CAS.
- I. F. Perepichka and D. F. Perepichka, Handbook of Thiophene-Based Materials: Applications in Organic Electronics and Photonics, 2 Volume Set, John Wiley & Sons, 2009 Search PubMed.
- X. Gu, X. Zhang and H. Huang, J. Mater. Chem. A, 2024, 12, 17973–17991 RSC.
- B.-H. Jiang, Y.-S. Chen, Y.-C. You, Y.-W. Su, C.-Y. Chang, H.-S. Shih, Z.-E. Shi, C.-P. Chen and K.-T. Wong, J. Mater. Chem. C, 2024, 12, 12004–12014 RSC.
- Z. Zeng, S. Lee, J. L. Zafra, M. Ishida, X. Zhu, Z. Sun, Y. Ni, R. D. Webster, R.-W. Li, J. T. López Navarrete, C. Chi, J. Ding, J. Casado, D. Kim and J. Wu, Angew. Chem., Int. Ed., 2013, 52, 8561–8565 CrossRef CAS.
- Z. Zeng, S. Lee, J. L. Zafra, M. Ishida, N. Bao, R. D. Webster, J. T. López Navarrete, J. Ding, J. Casado, D. Kim and J. Wu, Chem. Sci., 2014, 5, 3072–3080 RSC.
- P. Mayorga Burrezo, R. Domínguez, J. L. Zafra, T. M. Pappenfus, P. de la Cruz, L. Welte, D. E. Janzen, J. T. López Navarrete, F. Langa and J. Casado, Chem. Sci., 2017, 8, 8106–8114 RSC.
- M. Privado, S. Moles Quintero, M. Barrejón, P. de La Cruz, J. B. PM, D. Casanova, F. Langa and J. Casado, Angew. Chem., Int. Ed., 2025, 64, e202413988 CrossRef CAS PubMed.
- N. Y. Doumon, L. Yang and F. Rosei, Nano Energy, 2022, 94, 106915 CrossRef CAS.
- J. Liu, X. Liu, J. Xin, Y. Zhang, L. Wen, Q. Liang and Z. Miao, Small, 2024, 20, 2308863 CAS.
- C. Sun, J.-W. Lee, C. Lee, D. Lee, S. Cho, S.-K. Kwon, B. J. Kim and Y.-H. Kim, Joule, 2023, 7, 416–430 CAS.
- L. Xie, A. Lan, Q. Gu, S. Yang, W. Song, J. Ge, R. Zhou, Z. Chen, J. Zhang, X. Zhang, D. Yang, B. Tang, T. Wu and Z. Ge, ACS Energy Lett., 2023, 8, 361–371 CrossRef CAS.
- Z. Zhang, J. Wu, J. Lin, R. Zhang, J. Lv, L. Yu, X. Guo and M. Zhang, J. Mater. Chem. A, 2023, 11, 15553–15560 CAS.
- J. Lin, Q. Guo, Q. Liu, J. Lv, H. Liang, Y. Wang, L. Zhu, F. Liu, X. Guo and M. Zhang, Chin. J. Chem., 2021, 39, 2685–2691 CrossRef CAS.
- L. Wang, C. Zhang, Z. Su, Y. Wang, W. Su, X. Man, Z. Ma, W. Zhang, C. Li, C. Yang and Z. Bo, J. Mater. Chem. C, 2023, 11, 6971–6980 RSC.
- S. Li, L. Zhan, C. Sun, H. Zhu, G. Zhou, W. Yang, M. Shi, C.-Z. Li, J. Hou, Y. Li and H. Chen, J. Am. Chem. Soc., 2019, 141, 3073–3082 CrossRef CAS PubMed.
- S. Chen, Y. Wang, L. Zhang, J. Zhao, Y. Chen, D. Zhu, H. Yao, G. Zhang, W. Ma, R. H. Friend, P. C. Y. Chow, F. Gao and H. Yan, Adv. Mater., 2018, 30, 1804215 CrossRef.
- X. Ma, H. Bin, B. T. van Gorkom, T. P. A. van der Pol, M. J. Dyson, C. H. L. Weijtens, M. Fattori, S. C. J. Meskers, A. J. J. M. van Breemen, D. Tordera, R. A. J. Janssen and G. H. Gelinck, Adv. Mater., 2023, 35, 2209598 CrossRef CAS PubMed.
- G. Simone, M. J. Dyson, S. C. J. Meskers, R. A. J. Janssen and G. H. Gelinck, Adv. Funct. Mater., 2020, 30, 1904205 CrossRef CAS.
- D. Yun, S. Xuyao, S.-Y. Lee, V. V. Sharma, H. Li, S.-J. Park, Y.-H. Kim and G.-H. Kim, ACS Appl. Energy Mater., 2024, 7, 1243–1249 CrossRef CAS.
- G. Zhang, Q. Wu, Y. Duan, W. Liu, M. Zou, H. Zhou, J. Cao, R. Li, X. Xu, L. Yu and Q. Peng, Chem. Eng. J., 2023, 476, 146538 CrossRef CAS.
- J. Wang, L. Wang, R. Sun, L. Ye, B. Zhao, J. Min and S. Tan, Chem. Eng. J., 2024, 492, 152364 CrossRef CAS.
- Z. Liu and N. Wang, J. Mater. Chem. A, 2020, 8, 3211–3221 RSC.
- J. Lv, Q. Yang, W. Deng, H. Chen, M. Kumar, F. Zhao, S. Lu, H. Hu and Z. Kan, Chem. Eng. J., 2023, 465, 142822 CrossRef CAS.
- X. Chen, D. Wang, Z. Wang, Y. Li, H. Zhu, X. Lu, W. Chen, H. Qiu and Q. Zhang, Chem. Eng. J., 2021, 424, 130397 CrossRef CAS.
- M. Zhang, B. Chang, R. Zhang, S. Li, X. Liu, L. Zeng, Q. Chen, L. Wang, L. Yang, H. Wang, J. Liu, F. Gao and Z.-G. Zhang, Adv. Mater., 2024, 36, 2308606 CrossRef CAS PubMed.
- Z. Zhao, J. Zhao, S. Chung, K. Cho, W. Xu and Z. Kan, ACS Mater. Lett., 2023, 5, 1718–1726 CrossRef CAS.
- X. Sun, J. Lv, F. Wang, C. Zhang, L. Zhu, G. Zhang, T. Xu, Z. Luo, H. Lin, X. Ouyang, C. Yang, C. Yang, G. Li and H. Hu, Adv. Energy Mater., 2024, 14, 2302731 CrossRef CAS.
- K.-N. Zhang, Z.-N. Jiang, T. Wang, J.-W. Qiao, L. Feng, C.-C. Qin, H. Yin, S.-K. So and X.-T. Hao, Nano Energy, 2021, 79, 105513 CrossRef CAS.
- X.-K. Chen, D. Qian, Y. Wang, T. Kirchartz, W. Tress, H. Yao, J. Yuan, M. Hülsbeck, M. Zhang, Y. Zou, Y. Sun, Y. Li, J. Hou, O. Inganäs, V. Coropceanu, J.-L. Bredas and F. Gao, Nat. Energy, 2021, 6, 799–806 CrossRef CAS.
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- U. Rau, B. Blank, T. C. M. Müller and T. Kirchartz, Phys. Rev. Appl., 2017, 7, 44016 CrossRef.
- Z. Zhang, Y. Li, G. Cai, Y. Zhang, X. Lu and Y. Lin, J. Am. Chem. Soc., 2020, 142, 18741–18745 CrossRef CAS PubMed.
- J. Li, C. Zhang, X. Sun, H. Wang, H. Hu, K. Wang and M. Xiao, Nano Energy, 2024, 125, 109542 CrossRef CAS.
- J. Lakowicz, Principles of Fluorescence Spectroscopy, 2006, vol. 1 Search PubMed.
- L. Duan, Y. Zhang, R. Deng, H. Yi and A. Uddin, ACS Appl. Energy Mater., 2020, 3, 5792–5803 CrossRef CAS.
- M. Jiang, H.-F. Zhi, B. Zhang, C. Yang, A. Mahmood, M. Zhang, H. Y. Woo, F. Zhang, J.-L. Wang and Q. An, ACS Energy Lett., 2023, 8, 1058–1067 CrossRef CAS.
- S. Biswas, Y. Lee, H. Choi, H. W. Lee and H. Kim, RSC Adv., 2023, 13, 32000–32022 RSC.
- R. Suthar, H. Dahiya, S. Karak and G. D. Sharma, J. Mater. Chem. C, 2023, 11, 12486–12510 RSC.
- X. Liu, S. Xu, B. Tang and X. Song, Chem. Eng. J., 2024, 497, 154944 CrossRef CAS.
- W. Wang, Y. Cui, Y. Yu, J. Wang, C. Wang, H. Hou, Q. Kang, H. Wang, S. Chen, S. Zhang, H. Xia and J. Hou, Nano Energy, 2024, 128, 109893 CrossRef CAS.
- C. Lee, J.-H. Lee, H. H. Lee, M. Nam and D.-H. Ko, Adv. Energy Mater., 2022, 12, 2200275 CrossRef CAS.
- A. Venkateswararao, J. K. W. Ho, S. K. So, S.-W. Liu and K.-T. Wong, Mater. Sci. Eng., R, 2020, 139, 100517 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00724k |
| This journal is © The Royal Society of Chemistry 2025 |