
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiquid crystal-refilled polymer network templates formed by photo-polymerisation in an orientationally ordered liquid crystal†
Chung-Hao
Chen
and
Ingo
Dierking
 *
*
Department of Physics and Astronomy, University of Manchester, Oxford Road, Manchester, M139PL, UK. E-mail: Ingo.Dierking@manchester.ac.uk
First published on 10th March 2025
Abstract
Polymer networks, formed via photo-polymerisation in a thermotropic liquid crystal, will largely follow the self-organised order of the phase in which they form. Using the wash out – refill technique, where a network is formed, the liquid crystal is washed out and the network refilled with a different liquid crystal material produces devices which are largely dominated by the interaction between the polymer network and the refilled liquid crystal. We have used this technique to determine the lower pitch boundary of a chiral nematic which will allow enhancing the selective reflective intensity of light beyond the theoretical limit to approximately P ≈ 1 μm. We further demonstrate that a helical polymer network is not sufficient to induce a twist grain boundary TGB-like structure in an achiral SmA phase and provide an explanation in terms of elasticity. A third aspect of the investigations is the drastic enhancement of alignment and orientation of a refilled lyotropic nematic phase to achieve excellent dark states over large areas. Using a helical network, it is shown that helicity can be transferred to refilled lyotropic phases when a polymer network was formed in a helical thermotropic phase.
1. Introduction
Liquid crystal-polymer composites1 have attracted increasing attention over the last two decades, ever since polymer dispersed liquid crystals (PDLCs)2,3 have reached maturity as privacy windows4 and polymer stabilised liquid crystals (PSLCs)5 show promise as smart glass6,7 and in scattering displays.8,9 The concept of PSLCs is straight forward:10 the rod-like molecules of the liquid crystal are mixed with a small amount, typically less than 5% by weight, of a photo-curable, bifunctional monomer which is also of rod-shape and is often mesogenic by itself. A small amount of a photo-initiator is added, often in quantities of about 1–2% of the monomer concentration. The monomer molecules orient with their long molecular axis parallel to the director field n(r) of the liquid crystal host, and photo-polymerisation via UV illumination is initiated. Prior to polymerisation, the liquid crystal director field n(r) can be pre-determined via the phase structure and boundary conditions. During photo-polymerisation, the polymer network phase separates from the liquid crystal such that a bi-continuous, phase-separated structure evolves where a continuous polymer network is embedded in a continuous liquid crystal. The resulting polymer network that is formed is thus a template of the liquid crystal in which it forms. This can, for example, be the uniform orientation of a nematic,11 the director field around topological defects12 or the helical structure of a cholesteric phase,13 which has been shown by scanning electron microscopy (SEM). In the latter case of a cholesteric helical template, the polymer network can be left-handed or right-handed, depending on the helicity of the cholesteric phase. This exhibits the quite remarkable phenomenon of selective reflection of a narrow (∼50 nm) wavelength band around a centre wavelength λ0 = 〈n〉 P, which is related to the helical pitch P and the average refractive index 〈n〉. The width of the band, Δλ, is generally related to the birefringence Δn by Δλ = Δn P. Selective reflection can thus be observed in the UV, VIS, and IR, depending on the pitch, and device application. For lasers, this photonic band gap (PBG) is often sought to be in the visible range of the spectrum, while for heat reflectors, telecommunication or scattering devices, the IR regime is often useful.Much attention has been paid in the past to achieve a broadband reflection of light from a cholesteric liquid crystal structure.14–16 All of the different approaches to widen the reflection band essentially depend on the formation of a helical polymer network with a pitch gradient through the cell.17,18 The pitch variation is then due to monomer diffusion during polymerisation caused by a UV-intensity gradient. This can be achieved by single-sided irradiation at varying UV intensities but possibly better controlled by the addition of a small amount of a UV absorption dye.19–21 Another method employed to obtain pitch gradients is polymerisation during the change of temperature, exploiting the temperature dependence of the pitch P(T).22–25 This may also be performed with chiral monomers.25,26 A third commonly used method is the application of an electric field.27–29 A lesser employed methodology is the use of reactive surface coating layers.30 Broadband reflection bandgaps are useful in applications, such as widely tuneable cholesteric lasers28 or Bragg–Berry mirrors.31
Another aspect of cholesteric selective reflectivity which has attracted much attention is the intensity of reflected light. In an ideal structure, reflectivity is limited to 50% of the incoming intensity of linearly polarised light. The latter can be thought of as a superposition of left- and right-handed circular polarised, LCP and RCP, light. For a cholesteric helix of a particular handedness, one of these components is transmitted without loss, while the opposite component is selectively reflected. For applications, it is of course desirable to have as much light reflected as possible, ideally 100%. Again, several methodologies are available to achieve an increased reflection, which have been demonstrated. Obviously, the easiest way to obtain higher reflectivity is to use a two-cell double layer, one filled with a left- and the other with a right-handed cholesteric.32 This does work for all wavelengths, visible and IR, and has been shown to provide a reflectivity of 75% (which could surely be increased for samples with less defects etc.), yet it is not a single cell device and does not incorporate polymer networks.
An innovative approach which provided close to 100% reflectivity was proposed by Mitov et al.33,34 The single cell device exploited the properties of a twist inversion compound, a cholesteric liquid crystal which changed its pitch handedness as a function of temperature.35 The polymer network was formed at one handedness and the device exhibited a 50% reflectivity. When the temperature was changed the CLC adopted opposite handedness to that of the helical polymer network, thus achieving close to 100% reflectivity at a wavelength of λ0 = 3.5–4 μm. This approach was further elaborated by the addition of a UV dye, causing a polymer rich and a polymer-deprived region within the cell.36 This gave rise to two reflection bands with opposite handedness. On tuning the temperature to match the pitches of both bands, a 90% reflectivity was obtained. It has further been shown for different materials that the twist inversion approach may be supercooled by quenching most likely into a glass at room temperature, again maintaining a reflectivity of 80% at a reflection wavelength of λ0 = 2.7 μm.37 Using a liquid crystal of certain handedness and a photo-monomer of opposite handedness, in visible wavelength regions, reflectivities of up to only 55% have been achieved.38 Finally, another obvious approach is the formation of a polymer network in a liquid crystal of one handedness, washing out the LC and refilling the structure with a liquid crystal of opposite handedness.39 This gave reflectivities of about 85% at a wavelength of λ0 = 1.8 μm.
We note that all efforts to increase the reflectivity of a single device beyond the reflectivity limit were successful at pitch values in the micrometre regime. One of the aims of this investigation is to determine the lower limit of applicability of this approach. Another lies in the question if helicity can be transferred onto orthogonal smectic phases to produce artificial TGB-like structures. In a third part, we will demonstrate that polymer networks formed in a thermotropic nematic liquid crystal can aid the macroscopic alignment of lyotropic phases. For these studies, we will use the wash out/refill – technique,39 where a polymer network is formed in a particular liquid crystal which will then be washed out and replaced by a different LC because this has recently been employed in a number of application devices such as diffraction gratings,40 optical thermal sensors,41 lasing at different colours42 or colour memory devices.43
2. Experimental method
2.1. Materials
The thermotropic nematic liquid crystal chosen as a host material was 4-cyano-4′-pentylbiphenyl (5CB), which displays a phase sequence of Cr 22.5 N 35 I on heating (all temperatures in °C). Since this material is achiral, investigations involving helical superstructures needed 5CB to be doped with a chiral dopant at varying concentrations with either (S-(+)-2-octyl 4-(4-hexyloxybenzoyloxy) benzoate, abbreviated as S811, or (11)-9-(4-propylcyclohexyl)-9-10-dihydro-8H-dinaphtho[2,1-f:1′,2′-h]1,5dioxonine, abbreviated as R/S5011. As a thermotropic smectic phase, 4-cyano-4′-octylbiphenyl (8CB) with a phase sequence of Cr 21.5 SmA 33 N 41 I on heating was employed. Polymer networks were obtained from the bifunctional, UV-curable mesogen 2-methyl-1,4-phenylene bis(4-(3-(acryloyloxy) propoxy)benzoate), (RM257), in combination with a small amount of benzoin methyl ether, (BME), as a photo-initiator. Investigations with lyotropic liquid crystals were carried out with the chromonic dye disodium,6-oxo-5-[(4-sulfonatophenyl) hydrazinylidene] naphthalene-2-sulfonate, (sunset yellow, SSY), in water at concentrations in excess of the biphasic region (weight% 25 SSY/75 H2O). The liquid crystals 5CB and 8CB were purchased from Fluorochem, UK. The chiral dopant S811 was purchased from Xian Hua, China, and R/S5011 from Daken, China. The monomer RM257 was obtained from Synthon, Germany, and the photo-initiator BME was obtained from Aldrich. Sunset Yellow was purchased from Instant Sunshine. All materials are well-known standard compounds, were used as received and their chemical structures are shown in Fig. 1. | ||
| Fig. 1 Chemical structures of the thermotropic liquid crystals 5CB and 8CB, the lyotropic liquid crystal Sunset Yellow, chiral dopants, S811, R5011 and S5011, the bifunctional photo-reactive monomer RM257 and the photo-initiator BME employed in the investigations. | ||
2.2. Sample preparation
The nematic liquid crystal 5CB was mixed with less than 5% by weight of RM257 and a tiny amount of photo-initiator BME (2 wt% of monomer content) in the presence of isopropanol or dichloromethane. Monomer concentrations were chosen as to avoid phase separation above the solubility limit. For chiral template networks also, a respective amount of chiral dopant was added and the weight percentage depending on the pitch was adjusted. The mixture was sonicated for 15 minutes and kept at a temperature of T = 40 °C on a hot plate (IKA C-MAG HP 10) until the solvent had completely evaporated.For the construction of sandwich cells, glass substrates were cleaned with 2-propanol in a sonication bath for 15 minutes and the glasses were then baked in an oven at 100 °C for 10 minutes to evaporate all solvents. Furthermore, the substrates were placed in a plasma chamber for 10 minutes to remove organic residues. Planar boundary conditions for the liquid crystal were achieved by spin-coating the substrates with an alignment layer of a polyvinyl alcohol (PVA) and water solution (0.5 mg mL−1) and further baking at 100 °C to evaporate the water. The PVA layer was then unidirectionally rubbed with a velvet cloth. Finally, the substrates were assembled antiparallel with a film spacer determining the sandwich cell thickness between 23 and 85 μm (Fig. S1, ESI†).
The LC-monomer mixture was filled into the cell by capillary action under darkroom conditions. The samples were polymerised by UV irradiation (NeoLab UV lamp, 0.06 mW cm−2, for 1.5 h). After polymerisation, the sample was immersed into cyclohexane on a magnetic stirrer (Gallenkamp) for 48 h to remove the liquid crystal from the phase separated polymer to leave a polymer network template of the liquid crystal order which can then be refilled (Fig. S2, ESI†). To verify whether 5CB has been completely removed from the polymer network, FTIR analysis (Bruker Alpha II) was performed on 5CB/S5011, RM257 and the polymer network after washing out the liquid crystal. Fig. 2 shows the spectra of 5CB/S5011 and the polymer network after washing out the liquid crystal from the PSLC. The intensity of the polymer network spectrum has been multiplied by 30 because the concentration of RM257 in the PSLC was 3 wt%. A strong absorption near 2200 cm−1 (black) is distinguished due to the CN triple bonds in 5CB. In the polymer network spectrum, no such absorption peak is observed anywhere near that wavenumber, indicating that the liquid crystal has been completely removed from the polymer network. The polymer network exhibits an absorption peak between 2800 and 3000 cm−1. This very weak peak is also found in the monomer RM257 and is rather related to the latter than the liquid crystal mixture (see also Fig. S3, ESI†). The full FTRI spectra of the investigation are provided in Fig. S3 of the ESI.†
 | ||
| Fig. 2 FTIR spectra of the 5CB/S5011 mixture (black) and the polymer network after washing out the liquid crystal from the PSLC (red). | ||
Sample morphologies and textures were observed with a polarised optical microscope (Leica, DMLP) equipped with a hot stage (Mettler Toledo, FP90) and a temperature controller for accuracies of relative temperatures to 0.1 K. Images between crossed polarisers were recorded using a digital camera (IDS uEye camera, 2048 × 1088 pixel resolution) attached to the microscope. The optical measurements use the same microscope-based setup with an in-house built photodiode and a voltage metre (Agilent, 34401A) to detect and record the light intensity during sample rotation between crossed polarisers. The handedness of the helical structures can be determined by using the POM with monochromatic light while rotating the top polariser in clockwise and counterclockwise directions to determine in which direction the transmission becomes darker and brighter. This is equivalent to a qualitative circular dichroism (CD) measurement.
Reflection and transition properties were measured by using an optical microscope (Leica, DM2500P) equipped with a UV-VIS spectrometer (Ocean Optics, QE65000) connected via an optical fibre to collect the intensity in the range of visible light. Data for larger ranges of wavelengths were measured by UV-VIS-NIR spectrometry (Cary 5000) collecting the reflection spectra for wavelengths scanned from 3000 to 600 nm at a rate of 10 nm s−1. The handedness of chiral liquid crystals was determined by circular dichroism measurements (Jasco J810) for wavelengths scanned from 550 to 300 nm at a rate of 1 nm s−1.
3. Experimental results and discussion
3.1. Refilled thermotropic liquid crystal in a polymer network
Cholesteric liquid crystals (CLCs) exhibit unique optical properties, such as selective reflection with the reflectance limit at 50%. Mitov et al.33 constructed a device where a chiral polymer network of a certain handedness is formed at a specific temperature. The chiral nematic host exhibits an inversion of its helicity at a different temperature. This implied a right-handed helical polymer network embedded in a left-handed helical liquid crystal below the inversion temperature and a structure exceeding the theoretical reflectance limit at wavelengths of several micrometres.In this study, we investigated the limitations with respect to the reflection wavelength of the polymer network, aiming to determine the minimum pitch at which the polymer network could still significantly enhance the reflection (absorption) above the theoretical limit of 50%. In our experiment, the LC blends consisted of the standard nematic 5CB, 3% photoreactive monomer RM257 and various concentrations of chiral dopant S5011 (0.7%, 1.0%, 1.2%, and 1.5%) to induce helicity, resulting in selective reflection of λ0 ≈ 1700–800 nm for increasing chiral dopant concentration (with 〈n〉 = 1.5, corresponding to the pitch of approximately P ≈ 1150–550 nm). The LC blends were introduced into planar cells, polymerised by UV illumination, the LC washed out by subsequent immersion in cyclohexane, and then refilled with 5CB/R5011 mixtures of the same pitch but opposite handedness.
The original composite (Fig. 3, red) was polymerised in the presence of the left-handed dopant S5011 and thus represents a system of left-handed polymers in a left-handed chiral nematic liquid crystal. The refilled system (Fig. 3, black) is composed of the right-handed dopant R5011 and represents a left-handed polymer in a right-handed chiral nematic LC of the same pitch. Dopant concentrations of the original system (S5011) and the refilled system (R5011) are equal within limits of error, which may occur during the weighing and mixing process; the detailed concentration data are shown in Table S1 of the ESI.† These deviations in chiral dopant concentration explain the shift of the selective reflection bands with respect to each other (red and black curves).
 | ||
| Fig. 3 Vis-near IR reflection spectrum of polymer stabilised liquid crystal 5CB/3% RM257. (a) Dopant concentration 1.5%, P = 550 nm, (b) 1.2% dopant, P = 670 nm, (c) 1.0% dopant, P = 730 nm and (d) 0.7% dopant, P = 1150 nm. | ||
The reflection spectrum was investigated by Vis-NIR spectrometry between 600 and 3000 nm. Fig. 3 depicts the reflection spectra of the originally polymerised 5CB/S5011 cells (red curves) compared to the refilled 5CB/R5011 cells (black curves) for varying selective reflections between 800 and 1700 nm (pitch 550–1150 nm) observed for decreasing dopant concentrations from (a) 1.5%, (b) 1.2%, (c) 1.0% and (d) 0.7%, respectively. The original cells exhibited maximum reflection intensities reaching approximately 50% as expected. The reflection spectra of the refilled 5CB/R5011 cells are slightly larger for small wavelengths in the optical regime and markedly larger in the IR. From the original selective reflection of the equally handed polymer network and liquid crystal (left–left) and the one observed by the system with the opposite handed polymer network and liquid crystal (left–right), one can infer the relative percentage increase of the selective reflection intensity as a function of pitch. This is found to be approximately 10% for the short pitch samples at high dopant concentrations (Fig. 3(a) and (b)), as compared to about 33% for the longer pitch, low dopant concentration samples (Fig. 3(c) and (d)). These results are summarised in Fig. 4.
 | ||
| Fig. 4 Relative percentage increase of the reflection intensity as a function of pitch. | ||
These measurements illustrate that there is a regime where the polymer network cannot follow the tight winding of the cholesteric helix for strongly doped nematics with a short pitch and thus has little influence on the selective reflection properties for composites of opposite polymer and liquid crystal helicity, i.e. the helical order of the LC is in fact not transferred onto the polymer network. Only at larger helical pitches, one can observe an increase in selective reflection beyond the theoretical limit, when the helical structure is transferred onto the polymer network during the photo-polymerisation process, and the composite of opposite polymer and liquid crystal handedness influences the selective reflection properties. It is likely that this is the reason why all the (successful) previous experiments on shifting the reflection limit beyond theory were in fact carried out at wavelengths of several micrometres, not hundreds of nanometres, although desirable for applications. Further evidence for such a behaviour can also be found in the polymer network formation of other structures in liquid crystals. For example, polymer networks are expelled from highly twisted cholesteric defects such as oily streaks.44 Also, in polymer stabilised Blue Phases, the polymer networks are not formed in the highly twisted double twist cylinders, but in the defect regions between them.45 Furthermore, polymer networks are not formed in the highly twisted regions of twist grain boundary (TGB) phases, but in the untwisted smectic A regions.46 And as a last example, in topological defects of nematics, the polymer networks are expelled from the highly deformed regions of the singularities.12 From the systems under investigation here, we can infer that the lower limit of helical polymer formation is of the order of 650–750 nm, so roughly of the order of one micrometre. This is a reasonable order of magnitude because the individual polymer network strands formed during polymerisation exhibit a width of the order of 100 nm.
In Fig. 4, it can be seen that for small pitch, the polymer network only slightly influences the reflection intensity, indicating that the network cannot follow the strong twist of the liquid crystal and that the helical transfer is at best incomplete. This observation also explains why the reflectance of polymer stabilised cholesteric liquid crystals can only reach to 55% in the visible region.38 For a larger pitch, the reflection intensity is clearly improved for the refilled composite, indicating that both left- and right-handed components of circularly polarised light are reflected. This implies the helical transfer of the chiral nematic order onto the polymer network during polymerisation. The crossover between both regimes is located at about P ≈ 700 nm (λ0 ≈ 1100 nm), which is in accordance with the size of the network strands being approximately one order of magnitude smaller (Fig. 5).
 | ||
| Fig. 5 (a) Schematic illustration of a cholesteric liquid crystal and a Bouligand cut of the polymer network. (b) SEM image of a helical polymer network at low magnification, revealing an arc structure due to the helical superstructure (reproduced by permission from ref. 5). (c) SEM image of the polymer network templating a cholesteric liquid crystal (reproduced by permission from ref. 5). (d) Width distribution of the polymer network strands, with an average polymer stand width of approximately 110 nm. The size of the network voids is about 800 nm on average. | ||
Fig. 5(a) illustrates a cholesteric liquid crystal and chiral polymer network in a planar-aligned glass cell. The Bouligand cut reveals the arc structure of the obliquely cut helix, where the width of each arc represents half of the CLC pitch, as shown in Fig. 5(b). Fig. 5(c) shows the SEM image of a polymer network templating the cholesteric liquid crystal with the LC removed for network preparation for electron microscopy. Cross-linking is clearly visible, indicating that the original samples are bi-continuous, with a continuous polymer network penetrating the continuous liquid crystal phase. A comparison of Fig. 5(b) and (c) illustrates that the polymer network density is uniformly similar at large as well as smaller scales. The average width of the polymer strands is approximately 110 nm as determined from the distribution depicted in Fig. 5(d). Over 50% of the strands have widths ranging from 80 to 120 nm, which implies that the polymer network cannot follow the cholesteric helix when the pitch of the liquid crystal is below 700 μm. This is due to the fact that a π-twist would need to be templated by only three polymer strands. Additionally, the voids of the polymer network were measured on the surface of the sample to avoid shielding by higher polymer layers. The average size of the voids is about 800 nm, voids being slightly deformed to an ellipsoidal shape, Nevertheless, this free volume in the polymer network is sufficient for the refilled liquid crystal to form a helical or at least twisted structure. It should be pointed out that for uniform UV illumination, the structural uniformity of the formed polymer networks with respect to strand width and pore size is indeed given, as has been shown in ref. 47 and 48. It is noted that the structures somewhat change for different polymerisation temperatures and also UV dose and intensity variations, yet these changes are not significant enough to invalidate the above argumentation.
3.2. Smectic A phase in a helical polymer network
Given the fact that we can transfer helicity from a chiral polymer network onto an achiral nematic phase, it provokes the questions if it is possible to also induce helicity in an orthogonal fluid smectic A phase. The reason why this is a fundamentally interesting question is due to the existence of a spontaneous, discontinuous helicity of the frustrated twist grain boundary (TGB) phase.49 In some chiral materials, this phase, which is the liquid crystal analogue to the Abrikosov flux lattice phase of a type II superconductor in a magnetic field,50 is observed in a generally small temperature interval between the cholesteric and the smectic A* phase. The question is, can one induce a TGB-like structure in an achiral smectic A phase through the chirality field of a twisted polymer network?A sample was prepared of 2 wt% RM257 with a tiny amount of photo-initiator BME, doped in nematic 5CB with S5011 to produce a range of pitch values, namely (i) P = 5 μm, (ii) P = 10 μm, and (iii) P = 30 μm in cells of thickness 30 μm. This corresponds to twists of (i) 6 × 2π, (ii) 3 × 2π, and (iii) 1 × 2π, respectively, through the cell gap. These twists were chosen deliberately because for chiral nematics, one would expect case (i) to behave optically like a cholesteric phase, i.e. with a constant transmission intensity when the sample is rotated around the helix axis by an angle α between crossed polarisers. Case (ii) would behave like a slightly deformed helix, i.e. with slight intensity variations, and case (iii) like a strongly deformed helix with a clear variation of intensity, yet less than a standard well oriented nematic.
The mixture was then polymerised, the liquid crystal was washed out and the cells were refilled with 8CB, which exhibits a SmA phase at room temperature, and a phase sequence on cooling of: Iso 41 N 33 SmA 20 Cryst. (temperatures in °C). As a control sample, the transmitted light intensity of a non-stabilised, well oriented 8CB SmA phase was also measured upon rotation between crossed polarisers, exhibiting the expected sin2(α) behaviour.
Fig. 6(a) depicts the obtained relative transmission intensity curves for the 30 μm thick cell with a polymer network of pitch P = 5 μm. The black squares represent the data obtained for the SmA phase without any polymer present, showing the expected transmission variations from black to bright with a periodicity of 90°. Cooling the isotropic phase to the nematic at T = 40 °C in the presence of the helical polymer network indicates a transfer of helical order onto the liquid crystal because the related transmission (red circles) hardly varies when rotating the sample between crossed polarisers. Cooling further into the SmA phase at T = 32 °C (blue triangles) appears to provide an indication for a partially twisted SmA phase, i.e. a distorted TGB-like structure, because the transmission does not become zero upon sample rotation between crossed polarisers, albeit not staying constant either. Nevertheless, this interpretation turns out to be a fallacy when viewing the corresponding texture (Fig. 6(b)). This clearly exhibits a SmA phase, indeed a partial fan-shaped appearance with the optic axis pointing in different directions in different fans. This accounts for the fact that the transmission never reaches values close to zero. We thus do not find evidence for a network-induced TGB-like helix in SmA.
 | ||
| Fig. 6 (a) Relative transmission intensity by rotation between crossed polarisers of the SmA phase without the polymer network (black squares), the nematic phase in the presence of a chiral polymer network of pitch P = 5 μm, indicating a transfer of helicity (red circles), and the SmA phase in the presence of a helical network (blue triangles). (b) The respective texture of the SmA phase clearly indicates the absence of a TGB-like helical structure for the SmA phase in the helical network, but it rather exhibits a fan-shaped structure with different directions of the smectic layer normal, and thus an optic axis. | ||
In a very similar experiment, we varied the pitch of the helical structure in which the polymer network is formed via photo-polymerisation from P = 5 μm, P = 10 μm and P = 30 μm, again at the 2 wt% monomer concentration and the d = 30 μm cell gap. This approach should vary the exerted elastic twist energy of the polymer network at constant interaction energy between the liquid crystal and polymer because only the direction of polymer strands is varied, not the polymer density. The resulting relative transmission intensity behaviour is shown in Fig. 7(a).
 | ||
| Fig. 7 (a) Relative transmitted intensity as a function of the rotation angle of the sample between crossed polarisers for the SmA phase in a helical polymer of pitch P = 5 μm (red circles), P = 10 μm (blue up triangles), and P = 30 μm (green down triangles). Black squares represent the SmA control sample without any polymer network. (b) Corresponding textures of the samples, which all exhibit a largely non-oriented director distribution. | ||
Again, as a control experiment, the SmA phase of 8CB without a polymer exhibits a transmission variation between black and bright with a 90° periodicity between crossed polarisers, as expected (black squares). As soon as a helical polymer is introduced, the relative transmitted intensity varies much less, and no dark state is observed (Fig. 7(a)), as can be seen for the P = 5 μm (red circles), the P = 10 μm (blue up triangles), and the P = 30 μm networks (green down triangles). Yet again, this result is not due to an induced helical SmA, or TGB-like state but is rather due to the formation of a SmA fan-like texture with numerous domains where the optic axis points in different directions (Fig. 7(b)).
The results might be understood in terms of the interaction energy between the polymer network and the liquid crystal, compared to the smectic layer elasticity. Similar to a normal liquid, the compressibility of a liquid crystal phase is very small of the order of less than 1%. In this case, the elastic free energy density of a SmA phase is given by:
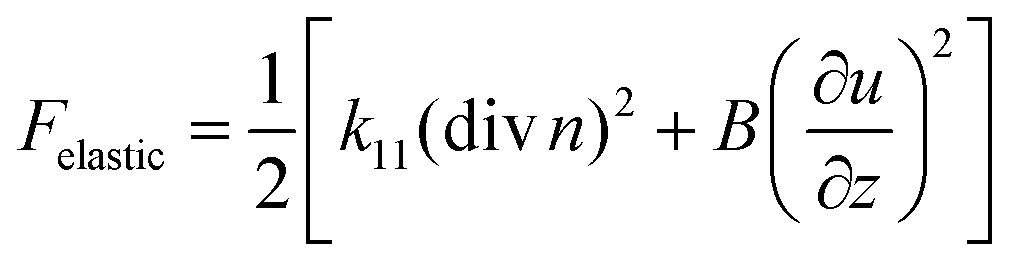
3.3. Lyotropic liquid crystal refilled polymer networks
The polymer network stabilisation of liquid crystal structures and refilling of networks formed in liquid crystals is generally used in the field of thermotropic LC phases. The former involves templating, thus resulting in a transfer of liquid crystal order onto the dispersed polymer network, while the latter is connected to the elastic influence of a structured polymer network onto a liquid crystal phase. A quite different class of liquid crystals is lyotropic liquid crystals (LLCs), where amphiphilic molecules or shape anisotropic particles are dissolved in an appropriate isotropic solvent to form a liquid crystalline phase once a specific concentration is reached. Only few reports discuss the polymer stabilisation of lyotropic phases. One of these describes the production of uniform nanostructures using polymerizable surfactants.54 Another is related to non-aqueous bicontinuous cubic phases being photo-polymerized.55 Non-amphiphilic, colloidal lyotropic systems are discussed for nanotubes in a solution with surfactants, monomers and crosslinkers to form oriented films of nanotubes via photo-polymerization.56 Similarly, also for lyotropic graphene oxide, a polymerisation step was introduced to prepare polyacrylonitrile-grafted GO.57 Systems like the ones discussed above could find application in soft actuators, opto-electronics or nano-photonics, and are thus of importance to be studied further.Lyotropic chromonic liquid crystals (LCLCs) are one type of lyotropic liquid crystal, employing dye molecules in aqueous solution.58 We are here using a different approach: the formation of polymer network structures in thermotropic liquid crystals, namely in the achiral nematic and the chiral, helical nematic phase, combined with subsequent removal of the LC and refilling with the lyotropic phase. Since lyotropic liquid crystals are often investigated in thick, and therefore poorly oriented, samples, the primary aim of this part of our studies is the achievement of oriented lyotropic nematic phases and the transfer of helicity from a network onto the lyotropic nematic.
At first, we compare the sunset yellow, SSY, lyotropic nematic liquid crystal at a wt% ratio of SSY/water (25/75) in a planer cell of gap 85 μm without any polymer (Fig. 8(a)) and with a 5% nematic polymer network (Fig. 8(b)).
 | ||
| Fig. 8 (a) Lyotropic nematic texture of SSY/water at (25/75) wt% in a 85 μm thick cell without a polymer network. (b) Lyotropic nematic under the same conditions, refilled into a 5 wt% stabilising polymer network with a nematic structure. (c) and (d) Quantification of the observed behaviour of (a) and (b). | ||
The orienting effect of the polymer network with a nematic structure onto the nematic lyotropic phase can already clearly be seen by visually inspecting the texture (Fig. 8(b)) in comparison to that of the non-stabilised LLC (Fig. 8(a)). The texture is not only much smoother in appearance, but the nematic director does indeed point into the direction of the polariser, which leads to a very good dark state of the oriented lyotropic. This can also be quantified by plotting the grayscale value of a cut through the texture as a function of position. Variations of the transmitted intensity are approximately 10× larger for the nematic LLC which relies solely on boundary conditions for orientation (Fig. 8(c)), while the polymer network stabilised material is much better oriented with grayscale variation being an order of magnitude smaller (Fig. 8(d)). We thus clearly observe the transfer of nematic orientational order of the polymer network onto the refilled continuous lyotropic nematic phase.
As a second demonstration of the enhancement of the optic properties of the lyotropic nematic SSY/water phase, we now refilled the cell at a 5% polymer network with nematic order in a 30 μm thick cell (Fig. 9).
 | ||
| Fig. 9 Nematic LLC SSY/water refilled into a 30 μm thick cell with a 5% polymer network of nematic orientational order. (a) Nematic director oriented parallel to the polariser, α = 0°, (dark state) and (b) at a position α = 45° (maximum transmission). We note that in both cases, a very good orientational alignment of the nematic LCLC due to the polymer network is observed. | ||
Fig. 9(a) shows the nematic lyotropic phase aligned with its director parallel to one of the polariser directions, which leads to extinction, i.e. the dark state, between crossed polarisers. A very good alignment of the refilled lyotropic liquid crystal is observed, due to the transfer of nematic order from the polymer network. Rotating the cell by 45° leads to the orientation of maximum light transmission, i.e. the bright state, which can equally be seen to be very well oriented, producing a decent contrast ratio for a lyotropic phase (Fig. 9(b)).
In a comparison between refilled samples with nematic polymer networks at different concentrations, 2% and 5% by weight, one can observe very good alignment and a good dark state for both cases, as seen in Fig. 10(a) and (b), respectively. A quantitative analysis via grayscale cuts reveals that the larger polymer content leads to a slightly better alignment and orientation of the nematic director of the LLC. The minimum transmitted intensity of the dark state is lower for the 5% polymer sample (Fig. 10(d)) than for the 2% sample (Fig. 10(c)), while also the grayscale variations throughout the texture are lower. This implies that not only the refilled lyotropic liquid crystal is aligned by the order transfer from the polymer network, but also the alignment improves with the increasing polymer content.
 | ||
| Fig. 10 Textures of the dark state of the refilled nematic LLC in an 85 μm thick cell with a polymer network formed under nematic order at a monomer concentration of (a) 2 wt% and (b) 5 wt%. Both configurations display a visually very good dark state. A more quantitative analysis via a grayscale cut through the texture (c) for a 2% polymer network and (d) for a 5% polymer network, exhibits that the increase in polymer concentration leads to a better orientation of the nematic LLC, as expected. | ||
It is now of further interest, whether a twist of the polymer network, formed through photo-polymerisation of the monomers in the chiral nematic phase of thermotropic 5CB also can be transferred to a refilled lyotropic nematic system of SSY/water at a (25/75) ratio by weight. To this end, the chiral dopant S811 was added to the 5CB/2% RM257 mixture to produce a pitch of P = 10 μm in an 85 μm thick cell at a 2% monomer concentration. The sample was photo-polymerised and then washed and refilled with the lyotropic nematic.
Due to the rotation of the sample between crossed polarisers, Fig. 11 illustrates that the polymer network structure as well as the refilled LLC in the polymer have adopted a helical superstructure. This is transferred from the helical polymer network onto the nematic lyotropic phase, thus inducing a chiral nematic lyotropic phase in the refilled system. The handedness of the refilled LLC has been confirmed by circular dichroism measurements. Both 5CB/S5011 and refilled SSY in a helical polymer network showed left-handed signals, as shown in Fig. 11(c). The full CD investigation is shown in S4 of the ESI.† However, the pitch of the refilled lyotropic is difficult to measure, as the reflection band is in the far infrared spectrum. In addition, the Grandjean-Cano method cannot be used to determine the pitch because it would deform the polymer network. The pitch of the polymer network was adjusted to 10 μm, which suggests that the refilled lyotropic pitch is also approximately 10 μm. The 85 μm cell gap provides space for about 8 full 2π-twists. By observing the transmission light intensity while rotating the sample between crossed polarisers, the intensity remained constant, as shown by the red regions in Fig. 11(a and b). This implies that the refilled lyotropic liquid crystal exhibits a proper, non-deformed helical nematic phase.
 | ||
| Fig. 11 (a) Partially refilled cell leaving a part of the polymer network (yellow) unfilled with the LLC and another part refilled (red). (b) Rotating the sample between crossed polarisers does not produce any transmitted intensity variation, which evidences a helical superstructure. (c) Circular dichroism measurements of the cholesteric PSLC system (black) and the refilled lyotropic nematic SSY system (red) show the transfer of helicity of the polymer network onto the lyotropic nematic. In both cases, a left-handed superstructure is observed. | ||
4. Conclusions
We have investigated several different systems consisting of a polymer network formed in a helical as well as untwisted thermotropic liquid crystal via photo-polymerisation, washing to remove the LC and refilling the network with a different thermotropic or lyotropic material. Refilling a helical network of a specific handedness with a thermotropic chiral nematic LC of opposite handedness increases the reflected light intensity beyond the theoretically predicted 50%. This behaviour is dependent on the pitch of the original liquid crystal in which the network was formed. Below a pitch of approximately P ≈ 1 μm, the polymer strands cannot form a helical superstructure, due to the spatial dimension of the strands themselves. Only pitches larger than this limiting value allow an increased selective reflection intensity.Investigations of networks of several pitch values, refilled with an achiral thermotropic liquid crystal, have shown that the helicity of the network is transferred onto the refilled LC in the nematic phase, while a twist grain boundary-like structure at the transition to the SmA phase cannot be observed in any case. This is due to the elasticity of the smectic layers, where even a small smectic layer compression requires an energy that is several orders of magnitude larger than the interaction energy between the polymer network and the liquid crystal.
A polymer network of nematic order, obtained by photo-polymerisation in a nematic thermotropic liquid crystal can be refilled with a nematic lyotropic LC to drastically benefit the alignment and orientation of the polymer stabilised LLC as compared to its un-stabilised counterpart. Very good dark states can thus be obtained for lyotropic nematics, leading to good contrast ratios. Stabilisation is improved for increasing polymer network concentrations. When refilling a helical polymer network, this helicity is transferred onto the nematic LLC to produce an induced chiral nematic lyotropic state.
Data availability
Most of the data used in the discussion are available in the paper. Raw data can be made accessible upon reasonable request to the authors.Conflicts of interest
There are no conflicts to declare.References
- Polymer-modified liquid crystals, ed. I. Dierking, Royal Society of Chemistry, Cambridge, 2019 Search PubMed.
- D. Coates, Polymer-dispersed liquid crystals, J. Mater. Chem., 1995, 5(12), 2063–2072 CAS.
- L. Bouteiller and P. L. Barny, Polymer-dispersed liquid crystals: Preparation, operation and application, Liq. Cryst., 1996, 21(2), 157–174 CrossRef CAS.
- W. Shen and G. Li, Recent progress in liquid crystal-based smart windows: materials, structures, and design, Laser Photonics Rev., 2023, 17(1), 2200207 CrossRef CAS.
- I. Dierking, Polymer network–stabilized liquid crystals, Adv. Mater., 2000, 12(3), 167–181 CAS.
- X. Meng, S. Lin, S. Chen, X. Shen, D. Guo and J. Guo, Recent Advances in Smart Windows Based on Photo-Responsive Liquid Crystals Featuring Phase Transition, ChemPlusChem, 2024, e202300700 CrossRef CAS PubMed.
- R. Yamaguchi and T. Takasu, Hybrid aligned nematic liquid crystal smart glass with asymmetrical daylight controls, J. Soc. Inf. Disp., 2015, 23(8), 365–370 CrossRef CAS.
- K. M. Lee, Z. M. Marsh, E. P. Crenshaw, U. N. Tohgha, C. P. Ambulo, S. M. Wolf, K. J. Carothers, H. N. Limburg, M. E. McConney and N. P. Godman, Recent advances in electro-optic response of polymer-stabilized cholesteric liquid crystals, Materials, 2023, 16(6), 2248 CrossRef CAS PubMed.
- Y. Ye, L. Guo and T. Zhong, A review of developments in polymer stabilized liquid crystals, Polymers, 2023, 15(13), 2962 CrossRef CAS PubMed.
- R. Hikmet, Electrically induced light scattering from anisotropic gels, J. Appl. Phys., 1990, 68(9), 4406–4412 CrossRef CAS.
- Y. Fung, D.-K. Yang, S. Ying, L.-C. Chien, S. Zumer and J. Doane, Polymer networks formed in liquid crystals, Liq. Cryst., 1995, 19(6), 797–801 CrossRef CAS.
- I. Dierking and P. Archer, Imaging liquid crystal defects, RSC Adv., 2013, 3(48), 26433–26437 RSC.
- I. Dierking, L. Kosbar, A. Afzali-Ardakani, A. Lowe and G. Held, Two-stage switching behavior of polymer stabilized cholesteric textures, J. Appl. Phys., 1997, 81(7), 3007–3014 CrossRef CAS.
- M. Mitov, Cholesteric liquid crystals with a broad light reflection band, Adv. Mater., 2012, 24(47), 6260–6276 CrossRef CAS PubMed.
- L.-Y. Zhang, Y.-Z. Gao, P. Song, X.-J. Wu, X. Yuan, B.-F. He, X.-W. Chen, W. Hu, R.-W. Guo, H.-J. Ding and J.-M. Xiao, Research progress of cholesteric liquid crystals with broadband reflection characteristics in application of intelligent optical modulation materials, Chin. Phys. B, 2016, 25(9), 096101 CrossRef.
- H. Zhou, H. Wang, W. He, Z. Yang, H. Cao, D. Wang and Y. Li, Research progress of cholesteric liquid crystals with broadband reflection, Molecules, 2022, 27(14), 4427 CrossRef CAS PubMed.
- D. Broer, J. Lub and G. Mol, Wide-band reflective polarizers from cholesteric polymer networks with a pitch gradient, Nature, 1995, 378(6556), 467–469 CrossRef CAS.
- D. J. Broer, G. N. Mol, J. A. V. Haaren and J. Lub, Photo-induced diffusion in polymerizing chiral-nematic media, Adv. Mater., 1999, 11(7), 573–578 CrossRef CAS.
- D. Katsis, D. Kim, H. Chen, L. Rothberg, S. Chen and T. Tsutsui, Circularly polarized photoluminescence from gradient-pitch chiral-nematic films, Chem. Mater., 2001, 13(2), 643–647 CrossRef CAS.
- F. Li, L. Wang, W. Sun, H. Liu, X. Liu, Y. Liu and H. Yang, Dye induced great enhancement of broadband reflection from polymer stabilized cholesteric liquid crystals, Polym. Adv. Technol., 2012, 23(2), 143–148 CrossRef CAS.
- W. Shi, X. Zhang, R. Han, H. Li, H. Cao, Y. Chen, D. Wang, Z. Yang and W. He, Preparation of cholesteric polymer networks with broadband reflection memory effect, Liq. Cryst., 2022, 49(2), 153–161 CrossRef CAS.
- A. Lavernhe, M. Mitov, C. Binet and C. Bourgerette, How to broaden the light reflection band in cholesteric liquid crystals? A new approach based on polymorphism, Liq. Cryst., 2001, 28(5), 803–807 CrossRef CAS.
- M. Mitov, E. Nouvet and N. Dessaud, Polymer-stabilized cholesteric liquid crystals as switchable photonic broad bandgaps, Eur. Phys. J. E:Soft Matter Biol. Phys., 2004, 15, 413–419 CrossRef CAS PubMed.
- R. Guo, K. Li, H. Cao, X. Wu, G. Wang, Z. Cheng, F. Wang, H. Zhang and H. Yang, Chiral polymer networks with a broad reflection band achieved with varying temperature, Polymer, 2010, 51(25), 5990–5996 CrossRef CAS.
- J. Guo, J. Sun, L. Zhang, K. Li, H. Cao, H. Yang and S. Zhu, Broadband reflection in polymer stabilized cholesteric liquid crystal cells with chiral monomers derived from cholesterol, Polym. Adv. Technol., 2008, 19(11), 1504–1512 CrossRef CAS.
- D. J. Dyer, U. P. Schröder, K.-P. Chan and R. J. Twieg, Polymer-stabilized reflective cholesteric displays: effects of chiral polymer networks on reflectance properties, Chem. Mater., 1997, 9(7), 1665–1669 CrossRef CAS.
- H. Lin, Y. Zhao, Z. He, H. Gao, J. Gao, D. Wang and Y. Luan, Low-voltage electrochromic behaviour of polymer-stabilised liquid crystal with considerable broadband reflection, Liq. Cryst., 2024, 1–20 Search PubMed.
- H. Lu, C. Wei, Q. Zhang, M. Xu, Y. Ding, G. Zhang, J. Zhu, K. Xie, X. Zhang, Z. Hu and L. Qiu, Wide tunable laser based on electrically regulated bandwidth broadening in polymer-stabilized cholesteric liquid crystal, Photonics Res., 2019, 7(2), 137–143 CrossRef CAS.
- M. Rumi, V. P. Tondiglia, L. V. Natarajan, T. J. White and T. J. Bunning, Non-Uniform Helix Unwinding of Cholesteric Liquid Crystals in Cells with Interdigitated Electrodes, ChemPhysChem, 2014, 15(7), 1311–1322 CAS.
- J. Kim, H. Kim, S. Kim, S. Choi, W. Jang, J. Kim and J.-H. Lee, Broadening the reflection bandwidth of polymer-stabilized cholesteric liquid crystal via a reactive surface coating layer, Appl. Opt., 2017, 56(20), 5731–5735 CrossRef CAS PubMed.
- M. Rafayelyan, G. Agez and E. Brasselet, Ultrabroadband gradient-pitch Bragg-Berry mirrors, Phys. Rev. A, 2017, 96(4), 043862 CrossRef.
- J. S. Gwag, D. S. Kim, G. Bae, B. S. Bae, S. Han and C. G. Jhun, High reflectance of cholesteric liquid crystal reflector by double-layer structure, Mol. Cryst. Liq. Cryst., 2014, 601(1), 64–70 CrossRef CAS.
- M. Mitov and N. Dessaud, Going beyond the reflectance limit of cholesteric liquid crystals, Nat. Mater., 2006, 5(5), 361–364 CrossRef CAS PubMed.
- M. Mitov and N. Dessaud, Cholesteric liquid crystalline materials reflecting more than 50% of unpolarized incident light intensity, Liq. Cryst., 2007, 34(2), 183–193 CrossRef CAS.
- I. Dierking, F. Gieβelmann, P. Zugenmaier, W. Kuczynskit, S. T. Lagerwall and B. Stebler, Investigations of the structure of a cholesteric phase with a temperature induced helix inversion and of the succeeding Sc* phase in thin liquid crystal cells, Liq. Cryst., 1993, 13(1), 45–55 CrossRef CAS.
- S. Relaix and M. Mitov, Polymer-stabilised cholesteric liquid crystals with a double helical handedness: influence of an ultraviolet light absorber on the characteristics of the circularly polarised reflection band, Liq. Cryst., 2008, 35(8), 1037–1042 CrossRef CAS.
- G. Agez and M. Mitov, Cholesteric liquid crystalline materials with a dual circularly polarized light reflection band fixed at room temperature, J. Phys. Chem. B, 2011, 115(20), 6421–6426 CAS.
- T. Zhang, Y. Cong, B. Zhang and W. Zhao, Multistable polymer stabilised cholesteric liquid crystal: exceeding reflection limit in visible region, Liq. Cryst., 2014, 41(12), 1778–1782 CAS.
- J. Guo, F. Liu, F. Chen, J. Wei and H. Yang, Realisation of cholesteric liquid-crystalline materials reflecting both right-and left-circularly polarised light using the wash-out/refill technique, Liq. Cryst., 2010, 37(2), 171–178 CAS.
- A. Bobrovsky, V. Shibaev, B. Ostrovskii, M. Cigl, V. Hamplová and A. Bubnov, Photo-and electro-controllable 2D diffraction gratings prepared using electrohydrodynamic instability in a nematic polymerizable mixture, J. Mater. Chem. C, 2023, 11(33), 11379–11391 RSC.
- Y. Li, Y. Liu and D. Luo, Optical thermal sensor based on cholesteric film refilled with mixture of toluene and ethanol, Opt. Express, 2017, 25(21), 26349–26355 CrossRef CAS PubMed.
- X. Zhan, H. Fan, Y. Li, Y. Liu and D. Luo, Low threshold polymerised cholesteric liquid crystal film lasers with red, green and blue colour, Liq. Cryst., 2019, 46(6), 970–976 CAS.
- D. Zhang, W. Shi, H. Cao, Y. Chen, L. Zhao, P. Gan, Z. Yang, D. Wang and W. He, Reflective band memory effect of cholesteric polymer networks based on washout/refilling method, Macromol. Chem. Phys., 2020, 221(22), 1900572 CrossRef CAS.
- I. Dierking, unpublished results.
- K. Higashiguchi, K. Yasui and H. Kikuchi, Direct Observation of Polymer-Stabilized Blue Phase I Structure with Confocal Laser Scanning Microscope, J. Am. Chem. Soc., 2008, 130, 6326–6327 CrossRef CAS PubMed.
- P. Archer and I. Dierking, Polymer stabilisation of twisted smectic liquid crystal defect states, Soft Matter, 2009, 5, 835–841 RSC.
- I. Dierking, L. L. Kosbar, A. C. Lowe and G. A. Held, Polymer network structure and electro-optic performance of polymer stabilized cholesteric textures I. The influence of curing temperature, Liq. Cryst., 1998, 24(3), 387–395 CrossRef CAS.
- I. Dierking, L. L. Kosbar, A. C. Lowe and G. A. Held, Polymer network structure and electro-optic performance of polymer stabilized cholesteric textures II. The effect of UV curing conditions, Liq. Cryst., 1998, 24(3), 397–406 CrossRef CAS.
- J. W. Goodby, M. A. Waugh, S. M. Stein, E. Chin, R. Pindak and J. S. Patel, “Characterization of a new helical smectic liquid crystal”, Nature, 1989, 337, 449–452 CrossRef CAS.
- S. R. Renn and T. C. Lubensky, Abrikosov dislocation lattice in a model of the cholesteric to smectic-A transition, Phys. Rev. A:At., Mol., Opt. Phys., 1988, 38, 2132–2147 CrossRef PubMed.
- S. Shibahara, J. Yamamoto, Y. Takanishi, K. Ishikawa and H. Takezoe, Layer compression modulus of chiral smectic liquid crystals showing V-shaped switching, Jpn. J. Appl. Phys., 2001, 40, 5026–5029 CrossRef CAS.
- P. Archer and I. Dierking, Elastic coupling in polymer stabilised ferroelectric liquid crystals, J. Phys. D: Appl. Phys., 2008, 41, 155422 CrossRef.
- P. Archer, I. Dierking and M. Osipov, Landau model for polymer stabilised ferroelectric liquid crystals: Experiment and theory, Phys. Rev. E:Stat., Nonlinear, Soft Matter Phys., 2008, 78, 051703 CrossRef PubMed.
- M. E. Tousley, X. Feng, M. Elimelech and C. O. Osuji, “Aligned Nanostructured Polymers by Magnetic-Field-Directed Self-Assembly of a Polymerizable Lyotropic Mesophase”, ACS Appl. Mater. Interfaces, 2014, 6, 19710–19719 CrossRef CAS PubMed.
- B. M. Carter, B. R. Wiesenauer, E. S. Hatakeyama, J. L. Barton, R. D. Noble and D. L. Gin, Glycerol-Based Bicontinuous Cubic Lyotropic Liquid Crystal Monomer System for the Fabrication of Thin-Film Membranes with Uniform Nanopores, Chem. Mater., 2012, 24, 4005–4007 CrossRef CAS.
- M. S. Mauter, M. Elimelech and C. O. Osuji, Nanocomposites of Vertically Aligned Single-Walled Carbon Nanotubes by Magnetic Alignment and Polymerization of a Lyotropic Precursor, ACS Nano, 2010, 4, 6651–6658 CrossRef CAS PubMed.
- Z. Liu, Z. Xu, X. Hu and C. Gao, Lyotropic Liquid Crystal of Polyacrylonitrile-Grafted Graphene Oxide and Its Assembled Continuous Strong Nacre Mimetic Fibers, Macromolecules, 2013, 46, 6931–6941 CrossRef CAS.
- Y. A. Nastishin, et al., Optical characterization of the nematic lyotropic chromonic liquid crystals: Light absorption, birefringence, and scalar order parameter, Phys. Rev. E:Stat., Nonlinear, Soft Matter Phys., 2005, 72(4), 041711 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00641d |
| This journal is © The Royal Society of Chemistry 2025 |