
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Distinguishing photodegradation pathways of organic semiconductors on ITO and Ag electrode contacts using IR reflectance–absorbance spectroscopy with multivariate analysis†
Sarah M. Tyler and
Jeanne E. Pemberton*
and
Jeanne E. Pemberton*
Department of Chemistry and Biochemistry, University of Arizona, 1306 East University Boulevard, Tucson, AZ 85721, USA. E-mail: pembertn@arizona.edu
First published on 10th July 2025
Abstract
Although many efforts to study photodegradation of organic semiconductor (OSC) thin films have been reported, few consider the effect of substrate material on degradation pathways. Given the many electrode contacts available, elucidation of the contributions of substrate material to OSC degradation is essential to facilitate the design of more durable devices. Here, we investigate the effect of substrate material on photodegradation of a model organic semiconductor, 4,7-bis(9,9-dimethyl-9H-fluoren-2-yl)benzo[c][1,2,5]thiadiazole (FBTF), an oligomer of the widely used OSC polymer poly(9,9-dioctylfluorene-alt-benzothiadiazole) (F8BT), by monitoring spectral changes in FTIR spectra coupled with multivariate analysis. While the identified degradation products are similar on both substrates, the differences suggest substrate-dependent degradation pathways. Along with the expected polyfluorene ketonic degradation product, strong spectral evidence of anhydride formation implies that a previously unreported interchain coupling mechanism is also present. Additionally, new bands in the 2100–2200 cm−1 region observed in spectra from films on Ag are suggestive of ring opening and rearrangement in the benzothiadiazole unit only on the metallic substrate. Although spectra on different substrates appear visually similar during early-stage degradation, principal component analysis (PCA) and linear discriminant analysis (LDA) reveal differences based on substrate type and degradation extent. This work demonstrates the utility of data-driven, vibrational spectroscopy coupled with multivariate methods for the characterization of OSC degradation and the potential for such approaches as diagnostic tools in operando devices.
Introduction
The use of photovoltaics as a renewable energy source has grown appreciably in recent years, with 2000–2019 seeing yearly growth at a rate of 40%.1 While silicon modules currently comprise the vast majority of the total market share, the use of organic semiconductors (OSCs) in photovoltaic modules has garnered warranted interest due to attractive features such as semi-transparency, flexibility, and low-cost solution processability.1,2 As the use of organic photovoltaics (OPVs) becomes more prevalent, a continuous need exists to better understand the degradation behavior of the active layer materials in these devices. Numerous investigations of OSC photodegradation from solar exposure in OPVs have been reported over the past two decades.3–8 However, no universally-accepted substrate for photodegradation experiments exists, with some studies using multiple substrate types without consideration of possible substrate-dependent effects.7,9–11 In certain cases, this variability is unavoidable due to substrate requirements associated with specific techniques, such as the need for IR-transparent materials for transmission FTIR spectroscopy or metallic contacts for surface enhanced Raman spectroscopy (SERS); however, substrate choice is often at the discretion of the researcher with little consideration of possible effects, leading to somewhat arbitrary choices. For example, for Raman, photoluminescence, or X-ray photoelectron spectroscopy (XPS) studies, which offer flexibility in substrate choice, researchers often choose quartz9,11,12 or indium tin oxide (ITO)-coated glass,7,13 with little discussion or justification for substrate choice. The common usage of ITO occurs even though its surfaces are notoriously unstable when exposed to O2 and/or H2O and rapidly react to form metal-hydroxides which in turn become active sites for further chemical reaction.14–16 In addition, efficiency studies17 and spectroscopic studies18–21 performed on model OPVs suggest that inherent photo-instability at metallic/organic interfaces is a main cause of device degradation. Moreover, evidence has been reported that chemical degradation at the ITO/organic interface upon radiant exposure leads to deterioration of charge transport in devices,22 and a more recent study suggests that the instability of OPVs utilizing non-fullerene acceptors (NFAs) originates from chemical changes at the organic/inorganic interfaces bounding the bulk heterojunction active region.23 Thus, greater attention to possible substrate effects in photodegradation experiments is warranted.In this work, the effect of substrate type on the photodegradation of 4,7-bis(9,9-dimethyl-9H-fluoren-2-yl)benzo[c][1,2,5]thiadiazole (FBTF), an oligomer of poly(9,9-dioctylfluorene-alt-benzothiadiazole) (F8BT), is investigated with IR reflectance–absorbance spectroscopy (IRRAS) coupled with multivariate analysis. F8BT was originally used in high-performance OLEDs,24 but more recently has been used as a NFA in OPVs.25,26 It consists of alternating fluorene (F) acceptor and benzothiadiazole (BT) donor units, and has a high electron affinity of ∼2.8–3.3 eV which helps facilitate favorable charge transfer.27–30 Although F8BT itself is not considered a high-efficiency material for OPVs, the F and BT units are present in various other OSC systems, and the polymer remains a reasonable experimental choice as a model NFA.31,32 FBTF was chosen as a model oligomer for F8BT to enhance spectral clarity and facilitate identification of changes due to degradation.
Substrates selected for this study, Ag and ITO, were chosen based on their use as electrode contacts in OPVs. In conventional OPV devices, ITO is commonly used as an anode material33 while Ag can serve as a cathode material,34 although these roles are reversed in inverted devices.35 In addition, these two materials have been used recently in tandem to create a surface phase-matched, transmission-enhancement top electrode for semi-transparent OPVs.36 Although the active layer material in operational devices is commonly separated from the electrodes by OSC hole or electron selective transport layers, low work function metals and metal oxides can also be utilized as interfacial charge transporting material,35 further justifying the choice of Ag and ITO in this study.
Experimental methods
Materials
FBTF was synthesized via two Suzuki cross-coupling methods. The first method was a more traditional procedure adapted from Zhong37 while the second method utilized a bio-based solvent in a procedure adapted from Edwards,38 including preparation of the melamine–palladium catalyst. Palladium(II) acetate (98%), 4,7-dibromo-2,1,3-benzothiadiazole (98%) and 2-(9,9-dimethyl-9H-fluoren-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (97%) were purchased from AmBeed. Palladium-tetrakis(triphenylphosphine) (Pd(PPh3)4) (≥99.5%) was purchased from VWR. Potassium phosphate, tribasic, (97%) and tetrabutylammonium bromide (TBAB) (98.0+%) were purchased from Fisher Scientific. Ethyl lactate (>98.0%) and melamine monomer (>98.0%) were purchased from TCI. Na2CO3 was purchased from Sigma-Aldrich. Benzene was purchased from EM Science. Toluene was purchased from Macron. Tetrahydrofuran (THF) was purchased from Sigma Aldrich. Ethanol was purchased from Decon Laboratories. Deuterated chloroform (CDCl3) was purchased from Cambridge Isotope Laboratories. Potassium bromide (KBr) was purchased from Mallinckrodt Pharmaceuticals. Methanol, acetonitrile, dichloromethane, and formic acid were purchased from Fisher Chemical. Petroleum ether was obtained from BDH. All chemicals were used as received. H2O (>18 MΩ resistivity, <8 ppb total organic carbon) was provided by a Waters Milli-Q UV Plus purification system (Millipore Corp.).ITO-coated glass microscope slides were obtained from Colorado Concepts. Ag sample stubs were cut from Ag rod obtained from Alfa Aesar (99.9% metals basis).
Synthesis of 4,7-bis(9,9-dimethyl-9H-fluoren-2-yl)benzo[c][1,2,5]thiadiazole (FBTF) – method 1
FBTF was initially synthesized according to the procedure outlined in Scheme 1. 4,7-Dibromo-2,1,3-benzothiadiazole (100 mg, 0.34 mmol), 2-(9,9-dimethyl-9H-fluoren-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (220 mg, 0.68 mmol), Pd(PPh3)4 (8 mg, 0.007 mmol), 2 mL of 2 M aqueous potassium phosphate solution, and TBAB (1.2 mg, 0.004 mmol) were dissolved in 11 mL of toluene. The mixture was heated to 80 °C and mixed with a stir bar for 48 h. The solvent was removed with a rotoevaporator (Buchi) and the crude product was loaded onto a silica gel column. The crude product collected ranged in color from green to yellow and was recrystallized in small aliquots from a THF/ethanol solvent system. | ||
| Scheme 1 Pd-catalyzed Suzuki–Miyaura coupling reaction scheme for synthesis of FBTF via method 1. | ||
1H and 13C NMR spectra and a mass spectrum from the purified FBTF obtained from method 1 are shown in Fig. S1–S3 of the ESI.† 1H NMR and 13C NMR spectra were obtained on a Bruker AVIII 400 spectrometer with CDCl3 as the solvent. Fourier-transform ion cyclotron resonance (FT-ICR) mass spectra were obtained on a Bruker SolariX 2XR FTMS with samples dissolved in dichloromethane and diluted with an acetonitrile/formic acid solution with 2–5 drops of H2O.
Synthesis of FBTF – method 2
The melamine–palladium catalyst for Suzuki–Miyaura cross-coupling was synthesized following a procedure adapted from Edwards.38 Palladium(II) acetate (56 mg, 0.25 mmol) and melamine monomer (126 mg, 1 mmol) were added to a 25 mL volumetric flask. 20 mL of ethyl lactate was added, and the mixture was stirred at 60 °C for ∼20 min until fully dissolved. After dissolution, the stir bar was removed and the solution was diluted to 25 mL with ethyl lactate. The resulting clear yellow liquid was stored in a capped bottle at room temperature.FBTF was synthesized according to the procedure outlined in Scheme 2. To a 25 mL round bottom flask were added 4,7-dibromo-2,1,3-benzothiadiazole (300 mg, 1 mmol), 2-(9,9-dimethyl-9H-fluoren-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (650 mg, 2 mmol), and Na2CO3 (320 mg, 3 mmol) dissolved in 2 mL of Milli-Q H2O (1.5 M). 8 mL of ethyl lactate and 2 mL of the melamine–palladium catalyst solution were added, the temperature of the resulting mixture was raised to 95 °C, and the mixture was stirred overnight. With the presence of solid product, the flask was removed from heat and allowed to cool. After filtration and collection of the solid product, 1 mL of 1.5 M Na2CO3 aqueous solution was added to the reaction mixture and the flask was reheated to 95 °C under stirring. This process was repeated until no additional solid product was formed. Typical crude product yields were between 21–77%. The crude product collected ranged in color from light green to yellow and was recrystallized in small aliquots from a THF/ethanol solvent system as needed for experimentation.
 | ||
| Scheme 2 Pd-catalyzed Suzuki–Miyaura coupling reaction scheme for synthesis of FBTF using ethyl lactate as the solvent. | ||
1H and 13C NMR and mass spectrometry data for the purified synthetic materials from method 2 can be found in Fig. S4–S6 of the ESI.† 1H NMR and 13C NMR spectra were obtained on a Bruker NEO-500 spectrometer with CDCl3 as the solvent. Atmospheric pressure chemical ionization (APCI) mass spectra were acquired on a Bruker amaZon SL with samples dissolved in dichloromethane and diluted with methanol.
Sample preparation
Ag substrates were mechanically and chemically polished using a procedure originally developed by Smolinski39 and later modified in this laboratory.40–42 ITO-coated glass microscope slides were cleaned by sonicating in detergent, H2O, and isopropanol for 5 min each, before drying at 350 °C for 10 min.After substrate cleaning, FBTF was dissolved in benzene to create a 5 mg mL−1 solution. Substrates were placed on a hotplate heated to 80 °C, and two 100 μL aliquots of the FBTF solution were drop cast on the substrate. The solvent from the first aliquot was allowed to evaporate fully before deposition of the second aliquot. These drop cast films were then subsequently photodegraded in 15–90 min intervals using a Ushio UXL-75XE short-arc Xenon lamp housed in a PTI Quanta Master 40 spectrofluorometer. The fluorometer illumination assembly was modified to accommodate a 50 mm dia, 200 mm focal length collimating lens placed in front of the sample surface to provide uniform radiant power across the FBTF film. This arrangement provided the desired accelerated degradation conditions of ∼2 W cm−2 of radiant power to the film (∼20 suns) with a spectral distribution known to closely mimic the solar spectrum.
IRRAS with multivariate analysis
Transmission FTIR spectra were obtained on solid FBTF powder samples suspended in a KBr pellet using a ThermoFisher Scientific Nicolet Nexus 6700 FTIR. IRRAS measurements were made on the same ThermoFisher Scientific Nicolet Nexus 6700 FTIR fitted with a tabletop optical module (TOM) containing a liquid N2-cooled type A HgCdTe detector. IRRA spectra were collected at an angle of 76°. Samples for spectral backgrounds were prepared similarly except with FBTF left out of the benzene aliquots. For these background samples, cleaned Ag and ITO substrates were placed on a hotplate and heated to 80 °C; then, two 100 μL aliquots of benzene were drop cast on the surfaces and evaporated prior to spectral background collection. For each spectrum acquired, the TOM box was first purged with dry air for 20 min prior to collection of 2000 scans at 4 cm−1 resolution.Baseline subtraction of raw spectral data was performed in OriginPro using asymmetric least squares smoothing. Parameters used to calculate the baseline for subtraction include an asymmetric factor of 10−4, threshold value of 0.005, a smoothing parameter of 1.5–4.0, and 25 iterations. Principal component analysis (PCA) was performed either in OriginPro or with a Python program written in-house as adapted from code obtained from Nirpy Research.43 Linear discriminant analysis (LDA) and combined PCA–LDA analyses were performed with similarly adapted Python programs written in-house. One-way analysis of variance (ANOVA) and post hoc Tukey's honest significant difference (HSD) testing on PCA and LDA scores were performed in OriginPro. Code used for PCA, LDA, and PCA–LDA can be found on Github.44
Results & discussion
Qualitative assessment of FBTF degradation using IRRAS
Although FBTF has been synthesized and characterized previously,37,45 the FTIR spectrum of FBTF has not been reported. A transmission FTIR spectrum of FBTF in KBr is shown in Fig. S7a (ESI†). Spectral band assignments are given in Table S1 (ESI†) based on previous assignments46–49 for the related polymeric systems F8BT, polyfluorene, polyfluorenone, and thiadiazole derivatives. IRRA spectra from pristine FBTF thin films on Ag and ITO are shown as the orange and purple traces in Fig. S7b and c (ESI†), respectively. These spectra exhibit bands similar to those in the transmission spectrum of FBTF, although on ITO the spectral envelope around the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) band at 1446 cm−1 is broadened due to the likely presence of δ(M–O–H)50 or other metal-oxide bands51 from the ITO substrate. It is noted that the drop cast films studied here are assumed to be isotropic thin films of FBTF without preferential orientation with respect to the substrate. This assumption is verified by the similarity of the pristine FBTF film IRRA spectra on both substrates to that of the transmission spectrum for FBTF in KBr.
C) band at 1446 cm−1 is broadened due to the likely presence of δ(M–O–H)50 or other metal-oxide bands51 from the ITO substrate. It is noted that the drop cast films studied here are assumed to be isotropic thin films of FBTF without preferential orientation with respect to the substrate. This assumption is verified by the similarity of the pristine FBTF film IRRA spectra on both substrates to that of the transmission spectrum for FBTF in KBr.
FBTF films on Ag and ITO substrates were photodegraded in air under a Xe arc lamp in 15, 30, or 90-min intervals, with spectra obtained after each subsequent exposure interval. In preliminary experiments in which FBTF films were exposed in 90-min or 30-min intervals, one sample on each substrate was analyzed initially to elucidate general patterns of spectral change upon cumulative photodegradation. In the 90-min trials the films were exposed to five cumulative intervals (7.5 h of accumulated exposure) while in the 30-min trials the films were exposed to four cumulative intervals (2 h of accumulated exposure). Once these patterns of spectral change were established, five FBTF samples on Ag and five FBTF samples on ITO were exposed in air to six 15-min intervals of irradiation for a total of 1.5 h of accumulated exposure.
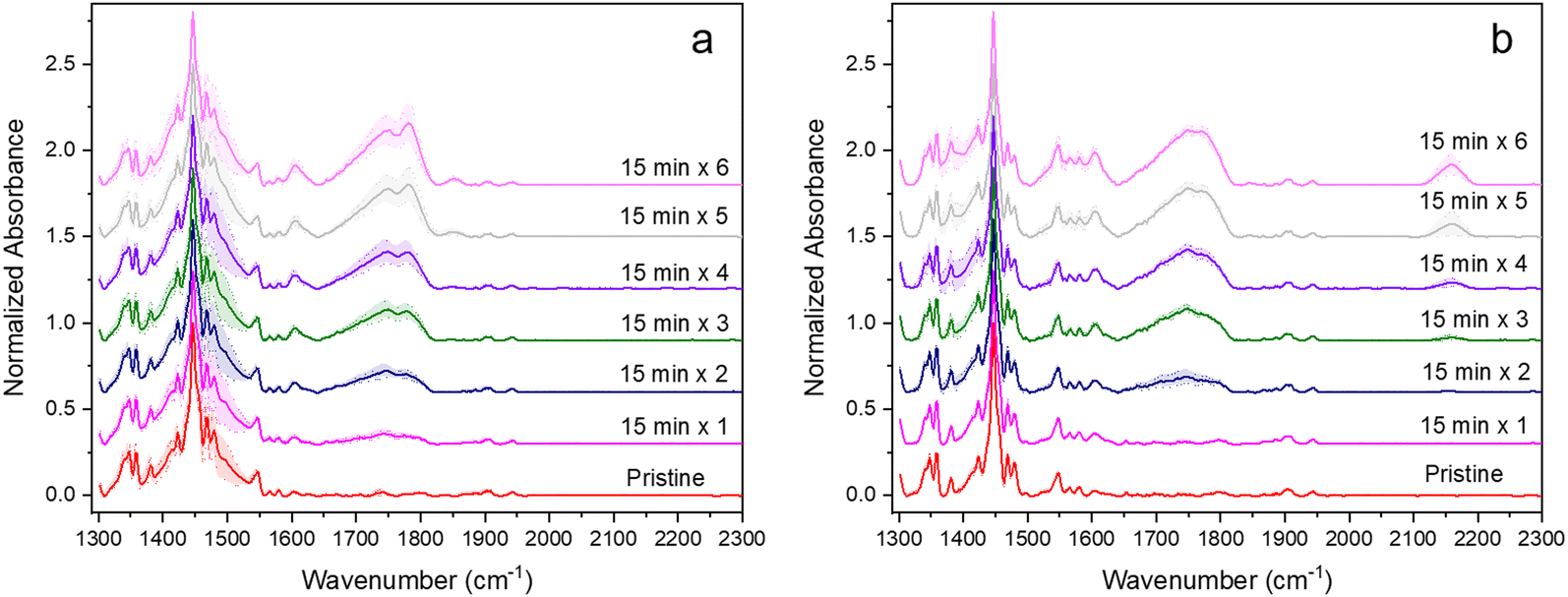
The resulting pristine IRRA spectra on ITO and Ag and those from films after exposure to the full five sequential 90-min periods of irradiation are shown in Fig. 1. To compensate for small differences in absorbance values due to sampling slightly different regions of the FBTF thin films upon repeated insertion and removal of samples, the intensity scale in these spectra is reported in normalized absorbance units with raw absorbance values normalized to most intense band in the pristine spectrum, which is the ν(CC) band at 1446 cm−1. New bands that appear in these spectra of the degraded samples relative to the pristine films are indicated with dashed lines in Fig. 1. These new bands and their tentative assignments are given in Table 1. The full set of normalized spectra for each of five 90-min periods of radiant exposure for ITO and Ag are shown in Fig. S8a, b and S9a, b (ESI†), respectively.
 | ||
| Fig. 1 IRRA spectra of pristine FBTF films (black) and FBTF films after irradiation for five 90-min periods on (a) and (b) ITO (green) and (c) and (d) Ag (red) substrates. Spectra are normalized to the most intense band in each spectrum. Spectra on ITO between ∼1180 and 1320 cm−1 are set to zero normalized absorbance values due to significant changes in the ITO background upon FBTF degradation. New bands due to irradiation designated by vertical dashed lines: black indicate new bands from 9,9-dimethylfluorene subunit, red indicates new bands from benzo[c][1,2,5]thiadiazole subunit. | ||
| ITO | ||||
|---|---|---|---|---|
| 5 × 90 min exposures | 6 × 15 min exposures | Assignment | ||
| Frequency (cm−1) | Relative intensity | Frequency (cm−1) | Relative intensity | |
| a Intensities on ITO set to zero from ∼1180–1320 cm−1 due to significant changes in ITO background upon FBTF film degradation. | ||||
| 1010 | vs | 1050 | w | ν(SO) |
| 1430 | vs | 1440 | m | νs(COO−) |
| 1600 | s | 1606 | w | νring + νas(COO−) |
| 1690 | w | ν(CO)COOH |
||
| 1734 | vs (broad) | 1747 | m | ν(CO)ketone + ν(CO)COOH |
| 1780 | s (sharp) | 1781 | m | ν(CO)anhydride |
| 1850 | m (sharp) | 1853 | w | ν(CO)anhydride |
| 3250 | m | 3250 | w | ν(O–H)COOH |
| Ag | ||||
|---|---|---|---|---|
| 6 × 90 min exposures | 5 × 15 min exposures | Assignment | ||
| Frequency (cm−1) | Relative intensity | Frequency (cm−1) | Relative intensity | |
| 958 | m | δ(C–H)aldehyde | ||
| 1040 | w | ν(SO) |
||
| 1078 | s | νs(SO2) + ν(C–N)NO2 | ||
| 1380 | vs | 1390 | m | νs(NO2) + νas(SO2) + δ(C–H)aldehyde |
| 1550 | vs | 1550 | m | νas(NO2) + νring |
| 1600 | s | 1600 | m | νring |
| 1690 | w | 1690 | w | ν(CO)COOH |
| 1726 | s | 1747 | m | ν(CO)ketone + ν(CO)aldehyde |
| 1768 | m (sharp) | 1768 | m | ν(CO)anhydride |
| 1848 | w (sharp) | ν(CO)anhydride |
||
| 2135 | w | ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) N) |
||
| 2158 | w | 2156 | m | ν(CN) |
| 2850 | w (sharp) | ν(C–H)aldehyde | ||
| 3240 | w | ν(O–H)COOH | ||
The spectral changes observed are substantial on both contact materials with 7.5 h of accumulated exposure indicating significant FBTF photooxidation and degradation. Differences between the two contact materials are also apparent both in the spectra in Fig. 1 and visually from the appearance of the samples. The pristine films on both substrates were initially a bright green color, but after exposure, films on Ag were brown while films on ITO were brownish-yellow. The IRRA spectra of these degraded FBTF films clearly exhibit the growth of new vibrational bands that differ between the two substrates. Detailed analysis of the IRRA spectra provides useful insight into this degradative chemistry. It is considered here separately for the 9,9-dimethylfluorene and benzothiadiazole subunits on each substrate.
The C-9 position of fluorenes is known to be the degradative weak point of fluorene and polyfluorene structures,11,12,52–60 and new optical band behavior has been speculated to be the result of eventual interchain radical coupling after the oxidative degradation of polyfluorene chains.58 The initial step in the chemistry associated with the C-9 position of fluorenes under aerobic radiative stress has been generally agreed to be a radical-based H abstraction mechanism of either a C-9 H defect resulting from inadequate purification of dialkylated fluorene units or a C-α H on a attached alkyl unit at the C-9 position,52–56,58,60 which in this case would be a methyl group. Upon reaction of these radical sites with O2, fluorenone and related ketones with a CO at the C-α position of alkylated fluorenes have been reported on the basis of strong ν(CO) bands in the vicinity of 1630–1750 cm−1.11,52–54 This assertion is further consistent with the concomitant decrease of the ν(CH3) band intensities in the region of 2800–3000 cm−1. In the study of FBTF on ITO reported here, substantial and complex intensity growth with multiple new spectral bands in the ν(CO) region from 1650–1900 cm−1 is observed indicating the appearance of multiple carbonyl-containing species that arise from oxidative degradation of the 9,9-dimethylfluorene subunit. New bands resulting from degradation of the 9,9-dimethylfluorene subunit are indicated by black dashed lines in Fig. 1a, b and listed in Table 1. It is noted that, due to significant changes in the ITO background relative to the ITO reference spectrum upon FBTF film deposition and subsequent degradative reaction, intensities from ∼1180–1320 cm−1 are zeroed out in the spectra in Fig. 1a; therefore, confirmation of assigned species from any bands expected in this region are eliminated from consideration. Despite the absence of unreliable spectral information in this frequency region, these significant changes in the spectral background support the possible involvement of the ITO surface in the degradation process.
Careful analysis of the IRRA spectra reveals previously unidentified reaction products from degradation of the 9,9-dimethylfluorene subunit. A broad band at 1734 cm−1 is assigned to a combination of ketone and carboxylic acid ν(CO) modes. This latter assignment is also consistent with the new broad ν(O–H) band observed at 3250 cm−1. Bands at 1430 and 1600 cm−1 are consistent with the presence of carboxylate moieties as represented by νs(COO−) and νas(COO−) modes, respectively. Significantly, the two sharp bands at 1780 and 1850 cm−1 are strongly suggestive of the presence of anhydride species.
On Ag (Fig. 1c and d), changes similar to those on ITO for degradation of the fluorene subunit are observed, albeit with some notable differences. Bands assigned to ketone (1726 cm−1), carboxylic acid (1690 cm−1) and anhydride (1768 and 1848 cm−1) ν(CO) species are similarly observed for FBTF on Ag. However, bands due to carboxylate modes are not clearly observed on Ag, suggesting that the surface may be involved in their formation on ITO; however, the frequency region in which νs(COO−) and νas(COO−) bands would be observed on Ag is dominated by substantial spectral changes from degradation products associated with the benzothiadiazole subunit that differ from those on ITO, as discussed below. Also distinguishing on Ag in contrast to ITO is the relatively definitive observation of bands attributable to an aldehyde species, with the ν(CO) band at ∼1726 cm−1 and the distinct aldehydic ν(C–H) band at 2850 cm−1. Although the complexity of the ν(CO) region makes assignment of the aldehyde ν(CO) less certain, the clear presence of the aldehyde ν(C–H) band is definitive, as few other modes can possibly be attributed to this sharp but weak band. Overall, the oxidative degradation of FBTF on Ag appears somewhat similar to that observed on ITO, although the products identified in the IRRA spectra are suggestive of a slightly less oxidizing environment for the same radiative conditions. We surmise that these differences can rationalized by the involvement of the ITO surface, which may provide H2O or OH− species that become involved in the FBTF degradation chemistry and/or provide coordinative metal ion sites for carboxylate species. Regardless of these differences, both substrates exhibit the presence of anhydride bands in the degraded film, which can only result from inter-FBTF coupling. Extension of this chemistry to interchain coupling in polymer systems might represent an additional pathway that explains the growth of the so-called green band in the photoluminescence spectrum for degraded polyfluorenes.52,53,55,56,58–60
The proposed degradation chemistry of the 9,9-dimethylfluorene subunit on both substrates is shown in Fig. 2. In both cases, the generally accepted initiation of radiative aerobic degradation is H abstraction by a radical species that provides a point of attack for O2 leading to ketone and/or aldehyde formation.54–56,58–60 Subsequent oxidative chemistry that is contact material-specific can lead to a suite of more highly oxidized species, most significantly culminating in anhydride species that must result from inter-FBTF coupling.
 | ||
| Fig. 2 Proposed pathways for oxidative degradation of 9,9-dimethylfluorene subunit of FBTF on (a) ITO and (b) Ag based on IRRAS results. | ||
Previous work on polyfluorene systems predominantly focuses on ketonic species such as fluorenone as the main degradation products upon photo- or thermal degradation.11,52–58,60 However, the results here convincingly suggest that a much larger range of oxygenated species are formed in the fluorene subunit. Most importantly, the strong spectral evidence of anhydride formation adds another possible interchain coupling degradation mechanism that could contribute towards the characteristic long wavelength emission observed in degraded polyfluorenes. Furthermore, the discrepancies in degradation products observed between FBTF on ITO and Ag highlight the importance of considering the nature of interfacial contacts when investigating degradation mechanisms of OSC materials. Much of the previous work done on the degradation of polyfluorene species involves the use of salt plates for FTIR measurements54–56,60 and quartz substrates for photoluminescence or UV-absorption measurements,52–56,58,60 neither of which are used as interfacial layers in OSC devices. Even when model working devices are photodegraded alongside isolated polyfluorene films, the differences between collected spectra are not covered in great detail.52 Although polyfluorenes such as F8BT are not considered state-of-the-art by today's standards, it is still important to understand their degradation because many new materials still incorporate the core fluorene unit.61–66 Therefore, experimental design of photodegradation studies should consider what substrate material is likely to be in contact with the polyfluorene material to better mimic the realistic degradation products of working devices.
The IRRA spectra in Fig. 1 also support significant oxidative degradation of the benzo[c][1,2,5]thiadiazole subunit in processes that differ substantially on the two contact materials. The new bands that emerge from degradation of this subunit are indicated in Fig. 1 by red dashed lines. The oxidative process on ITO results only in the appearance of a band at 1010 cm−1 attributed to a ν(SO) mode from oxidation of the S in the thiadiazole ring to the sulfoxide. Additional evidence for sulfoxide formation comes from Choi,13 who reported XPS evidence for sulfoxide in experiments on F8BT photodegradation. This product must be quite stable for FBTF on ITO, as even under these severe aerobic photodegradative conditions, no further reaction chemistry of this subunit is evident in the IRRA spectra.
In contrast, multiple new vibrational bands from degradation of the benzo[c][1,2,5]thiadiazole subunit are observed on Ag indicating extensive degradation of both the S and N components of the thiadiazole ring. Specifically, strong bands at 1078, 1120 and 1380 cm−1 are consistent with the ν(SO) of sulfoxide species and the νs(SO2) and νas(SO2) modes of sulfone species, respectively. In addition, the very strong bands at 1380 and 1550 cm−1 are consistent with νs(NO2) and νas(NO2) modes indicating the presence of nitro species. Finally, the band at 2158 cm−1 is attributed to a ν(CN) mode suggestive of a nitrile-containing degradation product.
The proposed degradation chemistry of the benzo[c][1,2,5]thiadiazole subunit on both substrates is shown in Fig. 3. For the S oxidation, this chemistry is similar to that of the related 1,2,5-thiadiazole moiety which exhibits two oxidized forms: 1,2,5-thiadiazole 1-oxide, with a single, strong ν(SO) mode,67 and 1,2,5-thiadiazole 1,1-dioxide,68 giving rise to two characteristic νs(SO2) and νas(SO2) bands in the IR spectrum.48,69,70 This combination of spectral evidence suggests that both sulfoxide and sulfone species are likely degradation products of FBTF. Although no direct evidence for the sulfoxide are observed in the spectra from Ag, the S oxidation to the sulfone must be a stepwise process through the sulfoxide.
 | ||
| Fig. 3 Proposed pathways for oxidative degradation of benzo[c][1,2,5]thiadiazole subunit of FBTF on (a) ITO and (b) Ag based on IRRAS results. | ||
Previous accounts of photodegradation of similar heterocycles provide evidence for mechanisms that result in the formation of –CN species with ν(CN) bands between 2100–2300 cm−1.71 Moreover, early studies of plasmonically enhanced photochemistry of pyridine and pyrazine on Ag noted the emergence of a band at 2200 cm−1, assigned as a ν(CN) mode.72 An additional degradation mechanism of the benzo[c][1,2,5]thiadiazole subunit that could explain the new band in the 2100–2200 cm−1 region comes from previous reports of benzofuran photodegradation. Benzofuran, the oxygen congener of the thiadiazole ring, has been more extensively investigated with evidence for formation of a nitrile oxide intermediate that rearranges into an isocyanate.73,74 Using this mechanism as a model, Scheme 3 outlines an analogous process for the thiadiazole, with final rearrangement following a ring opening mechanism resulting in an isothiocyanate. Organic isothiocyanates have characteristic ν(NSC) bands between 2060–2190 cm−1,75 which matches well the frequency of the new envelope centered at 2157 cm−1 for FBTF on Ag. Other recent work on degradation of a BT-based dye further supports a ring opening mechanism with a desulfurization step that results in two nitrile bonds observed in NMR.76
 | ||
| Scheme 3 Proposed degradation mechanism of BT unit based on previously reported benzofuran mechanism from Heinzelmann.68 The final step is a Curtius or Hofmann-like rearrangement. | ||
Overall, differences observed in the degradation of FBTF on Ag and ITO are directly related to the properties of the two substrates. The preparation procedure used in this work for Ag39–41 is known to result in relatively smooth surfaces with root-mean-squared roughness values on the order of nm.42 However, it is well known that enhanced chemical reactivity can occur on slightly roughened plasmonic substrates such as Ag.77–82 Thus, some plasmonic enhancement of photodegradation may be occurring on Ag here, leading to different products upon extended aerobic photodegradative conditions, especially for the benzo[c][1,2,5]thiadiazole subunit. Conversely, ITO, has distinct active sites on its surface stemming from hydrolysis reactions in ambient conditions that appear to play a role in degradation of the 9,9-dimethylfluorene subunit.
The major spectral changes observed after a single 90-min exposure led to exploration of the photodegradation behavior for shorter exposure intervals of 30 min in an effort to clarify the likely order of subunit degradation. Spectra from pristine films and those exposed sequentially to four 30-min intervals are shown in Fig. S8c and d and S9c and d (ESI†) for ITO and Ag, respectively. Even after shortening the total interval exposure time by 5.5 h, significant spectral changes relative to the pristine films were observed on both substrates; indeed, the changes observed are similar to the changes observed in the 90-min exposures, albeit with lower intensities.
Since changes in the spectra after one 30-min exposure are still significant, experiments with 15-min exposure intervals on five FBTF films on Ag and five films on ITO were undertaken. Pristine spectra and spectra acquired after exposure in six 15 min intervals for an accumulated exposure time of 1.5 h are shown in Fig. 4a and b. New spectral bands observed are tabulated in Table 1. The full set of six spectra with 15 min exposures is shown in Fig. S8e, f and S9e, f (ESI†) for ITO and Ag, respectively. Spectra on both ITO and Ag exhibit significant growth of new bands in the ν(CO) region from 1600–1850 cm−1 due to degradation of the 9,9-dimethylfluorene subunit, but less pronounced changes are observed in the 1300–1600 cm−1 region dominated by changes in the benzo[c][1,2,5]thiadiazole subunit. Indeed, IRRA spectra from both substrates in this latter frequency region are largely unchanged from their pristine condition. One notable difference however is the presence of a broad ν(O–H) band in spectra on Ag during these shorter time intervals that is not observed during the longer 90-min interval. We attribute the absence of this ν(O–H) band at longer exposures to complete decarboxylation of the oxidized fluorene carboxylic acid due to anhydride formation and/or loss of CO2 that is not observed under shorter degradation times. Collectively, these observations are strong evidence that degradation of the 9,9-dimethylfluorene subunit is the initial stage in degradation of FBTF, occurring to a significant extent prior to degradation of the benzo[c][1,2,5]thiadiazole subunit. Interestingly, however, the new band on Ag at 2156 cm−1 attributed to either ν(CN) or ν(NSC) modes appears with higher relative intensity than other bands arising due to the degradation of the thiadiazole unit. Although the exact source of this band is not clear from these data, it may result from an additional contribution from a ν(CC) mode associated with degradation of the fluorene unit.
 | ||
| Fig. 4 IRRA spectra of pristine FBTF films (black) and FBTF films after irradiation for six 15-min periods on (a) and (b) ITO (green) and (c) and (d) Ag (red) substrates. Spectra are normalized to the most intense band in each spectrum. Spectra on ITO between ∼1180 and 1320 cm−1 are set to zero normalized absorbance values due to significant changes in the ITO background upon FBTF degradation. New bands due to irradiation designated by vertical dashed lines: black indicate new bands from 9,9-dimethylfluorene subunit, red indicates new bands from benzo[c][1,2,5]thiadiazole subunit. | ||
Multivariate analysis of FTIR spectral data
The above qualitative analysis of the IRRA spectra provides insight into differences in degradation between the two contact materials. However, a more quantitative, less subjective analysis methodology was sought to further explore the utility of FTIR spectroscopy for its potential use as an operando diagnostic tool to evaluate extent of degradation. Toward this end, two multivariate analysis methods, principal component analysis (PCA) and linear discriminant analysis (LDA), and their combination (PCA–LDA) were explored for analysis of the IRRA spectra from FBTF on Ag and ITO exposed to 15 min radiant illumination intervals. PCA and LDA are tools that reduce the dimensionality of the spectral data to encompass only those variables that contain the most variance of the data as a whole. LDA, a supervised approach, can be further utilized after PCA to predict the identity of an unknown sample from previously assigned user-defined groups that comprise the dataset. IRRA spectra from the 15-min irradiation intervals were selected here for this analysis in lieu of those from 30- or 90-min intervals to assess the ability of PCA and LDA to distinguish spectra early in the degradation process with subtle, and sometimes visually imperceptible, qualitative spectral differences.Averaged normalized IRRA spectra from FBTF films were first subjected to PCA dimensionality reduction to determine if any identifying patterns or trends exist in the dataset. PCA is an unsupervised method that does not consider user-designated group labels; as such, it is not a classification tool. Instead, it allows only identification of new variables, known as principal components (PCs), that maximize differences in spectra from different samples. As such, it serves as an exploratory method for identifying distinguishing characteristics in the spectral data.83 Each PC accounts for a percentage of variance in the data, and for visualization purposes, it is common practice to plot the two PC scores that represent the largest variance. For the work reported here, PCA was applied to spectral data from the series of 15-min radiant exposures in the more restricted frequency region between 1300–1900 cm−1. This range was chosen for its more reliable background correction leading to more robust spectral reproducibility; the shaded regions shown around the spectra in Fig. 5 demonstrate the variance in spectral intensities around the average for both ITO and Ag. The 2100–2300 cm−1 region was intentionally excluded to test the multivariate techniques on IRRA spectral data where differences might be imperceptible to the untrained eye. An even more restrictive region, 1300–1500 cm−1, was also analyzed but did not provide as good a separation of the variables as analysis of the 1300–1900 cm−1 region. The multivariate analysis results for the 1300–1500 cm−1 frequency region are shown in Fig. S10 (ESI†).
 | ||
| Fig. 5 Normalized IRRA spectra of pristine FBTF thin films and those exposed to a series of six 15-min exposures on (a) ITO and (b) Ag. Spectra collected with 2000 scans at 4 cm−1 resolution. Intensities plotted as normalized values with respect to the 1446 cm−1 band; stacked spectra plotted with offset of 0.3 normalized a.u. Shaded regions represent the variance in normalized spectral intensities across the sample set for each substrate. | ||
Within the 1300–1900 cm−1 region, PC1 accounts for 51.1% of the variance while PC2 accounts for 36.3% of the variance, collectively capturing 87.4% of the sample-to-sample variation (Table S2, ESI†). Plots of these first two PC scores for FBTF on ITO and Ag are shown in Fig. 6a. A clear distinction between IRRA spectra from the two substrates is revealed, and a trend in PC scores with degradation extent is noted for both substrates. In general, the PC1 scores are more sensitive to substrate type, with Ag having more negative PC1 scores and ITO having more positive PC1 scores. Conversely, the PC2 scores are more sensitive to degradation extent, with more negative scores corresponding to spectra of less degraded films on both substrates. These sensitivities are better visualized in the box-and-whisker plots of each PC score shown in Fig. S11 (ESI†). For PC1 (Fig. S11a, ESI†), all the median values for the groups on Ag are negative whereas the median values on ITO are positive. For PC2 (Fig. S11b, ESI†), the median values for groups from films exposed to the same accumulated irradiation time are similar on each substrate. Analysis of variance (ANOVA) testing reveals that at the p < 0.05 level, the population means of the PC scores in one or more of the groups are significantly different (Table S3, ESI†). However, post hoc Tukey's honest significant difference (HSD) testing for means comparisons identified several group means that are not significantly different for both PCs (Table S4, ESI†). Comparable analyses for the 1300–1500 cm−1 region are included in Fig. S12 and Tables S5, S6 (ESI†).
 | ||
| Fig. 6 Analysis of IRRA spectral data by (a) PCA, (b) LDA, and (c) PCA–LDA in 1300–1900 cm−1 frequency region for FBTF degradation on ITO and Ag under sequential 15 min exposures to a 75 W Xe lamp. Samples color-coded by substrate type and exposure time. Red circles in PCA results indicate outlier data set for FBTF on one ITO sample; this set retained for subsequent LDA and PCA–LDA analyses. | ||
As expected from visual analysis of the box and whisker plot, the group means in PC1 that are not significantly different are those whose boxes extensively overlap and tend to be of the same substrate type. The outlier samples that are also visualized in Fig. 6a (data points circled in red) are also easily recognized in the box and whisker plot. For PC2, the groups with means that are not significantly different tend to be those with similar levels of degradation extent irrespective of substrate type. This is again well visualized by the box and whisker plot for PC2 which shows similar mean values for groups of similar degradation extent between the two substrate types. Considering the unsupervised nature of PCA, the overlap of means is somewhat expected and exemplifies how PCA is a less desirable approach for predictive analysis on its own. However, the general trends clearly observed in the PC scores provide evidence that, even between samples with little difference in radiant exposure time, quantifiable spectral differences are identified that might not be obvious through visual inspection alone.
Based on the success of PCA, an unsupervised multivariate technique for revealing quantifiable differences in IRRA spectral data based on substate type and extent of photodegradation, we explored whether the supervised multivariate tool of LDA, which calculates linear discriminants (LDs) that maximize differences between user-defined classes, might allow this approach to be used for true sample classification.83 LDA can be used to classify unknown samples after creating a training set of data with labeled classes. Fig. 6b shows the results of LDA applied to the IRRA spectral data for FBTF films on Ag and ITO exposed in 15-min intervals. The linear discriminants LD1 and LD2 (Table S7, ESI†) contain 50.9% and 35.1% of the group-to-group variation, respectively. Plotting LD1 and LD2 produces tight groups for each class, although overlap in LD1 scores, for longer exposure times corresponding to greater degradation, could result in some classification inaccuracy. LD2, however, well separates the samples based on substrate type, with results for Ag exhibiting positive LD2 values and results for ITO giving negative LD2 values. Box plot analysis of the medians of LD1 and LD2 scores in Fig. S13 (ESI†) further support these observations that either substrate type or degradation extent more heavily influences each LD score. For LD1, the less degraded samples on each substrate tend to have more positive scores; for LD2, almost all scores for samples on Ag have negative values while scores for samples on ITO have positive values. The high F values from ANOVA (Table S8, ESI†) indicate that the variance within each group is less than the variance between groups and that the differences in mean values are statistically significant. Tukey's HSD testing reveals fewer groups with means that are not statistically significant when compared against each other (Table S9, ESI†) which implies that the first two LDA scores for the dataset could potentially be utilized for accurate classification of unknown samples. A similar linear discriminant analysis of the 1300–1500 cm−1 region was also undertaken with the results shown on Fig. S14 and Tables S10, S11 (ESI†).
One potential concern in using LDA only for predictive analysis based on this dataset is that the number of spectral variables (i.e. each frequency) is greater than the sample size, which can lead to inaccurate estimated covariance matrices and overall low performance from a predictive standpoint.84 To mitigate this problem of ill-conditioned data, combined PCA–LDA was explored. First, 40 PCs were calculated for each spectrum to reduce the data dimensionality prior to performing LDA on these PCs. In this way, only 40 variables are utilized in the LD analysis instead of the hundreds of variables intrinsic to the raw spectral data. The new LDs (Table S12, ESI†) account for 51.5% and 32.8% of the variance, respectively. As shown in Fig. 6c, plotting LD1 and LD2 still provides good discrimination between substrate type based on LD2, although there is again some overlap in LD1 for the ITO samples for the longer exposure (greater degradation) times. However, the sample size of each unique group in this classification is small, meaning that outliers can have a greater impact on the results and potentially reduce the predictive accuracy. Similar to the box plot analysis of the LDA scores calculated without prior PCA, the median value analysis supports the evidence for discrimination of each LD for either substrate type or degradation extent. These box plots are shown in Fig. S15 (ESI†). The LD1 scores calculated after PCA show much better discrimination between degradation extent, with samples on both substrates irradiated for the same duration having similar score values. The new LD2 scores are also noticeably better segregated, with all Ag samples having negative scores and all ITO samples having positive values. ANOVA testing reveals significant population means at the p < 0.05 level (Table S13, ESI†), and the Tukey HSD results show few means that are not significantly different (Table S14, ESI†). Similar analysis of the IRRA spectral data between 1300–1500 cm−1 showed results almost as good (Fig. S16 and Tables S15, S16, ESI†).
Overall, the good separation between groups suggests that PCA–LDA could be useful for accurately predicting degradation extent of an OSC sample. The fact that the PCA–LDA model successfully separates spectral data from such short exposure time differences (with only 15 min of radiant exposure separating each group) is promising for the possible development of a diagnostic tool for detecting early-stage degradation based on FTIR spectroscopy. Although these results indicate the potential for predictive multivariate analysis in OSC research, future datasets will need to comprise larger sample sizes for each unique class if multivariate analysis is to be utilized as an accurate diagnostic tool of OSC samples and devices.
Conclusions
This work has demonstrated through analysis of IRRA spectral data coupled with multivariate analysis that electrode contact type can affect the nature and extent of photodegradation of FBTF films. FBTF films on both Ag and ITO substrates exhibit spectral changes due to photodegradation upon short sequential exposures to a 75 W Xe lamp intended to model early stage photodegradation. While IRRA spectra of FBTF on both substrates exhibit new strong bands in the ν(CO) region after photo-illumination, differences in identified species are surmised to be from involvement of the reactive ITO surface. However, the observation of bands assigned to anhydrides on both substrates strongly suggests the presence of an inter-FBTF coupling degradation mechanism that when extrapolated to polymer systems might provide an additional explanation for the formation of long wavelength green emission that notoriously plagues degraded polyfluorenes. The growth of new bands between 2100–2300 cm−1 hypothesized to stem from the benzothiadiazole unit observed only in the photodegraded FBTF films on Ag suggest that plasmonic effects on Ag support enhanced photochemistry and lead to a greater diversity of degradation pathways and products beyond those observed for FBTF on ITO.
Multivariate analysis of spectral data can be used to reduce the dimensionality of complex spectra to allow accurate sample classification based on substrate type and degradation extent even in cases when visual differences between spectra are not apparent.
From a device design perspective, these results highlight the importance of careful consideration of the multiple effects of different contact materials on active layer performance in operational devices. However, it should be noted that organic electronic devices are manufactured and encapsulated in inert environments to avoid the instabilities outlined here, although eventual oxygen penetration is inevitable. To better understand the intrinsic instability of OSC material on different contact materials, future work will focus on photodegradation in an inert environment. Moreover, this work supports the potential utility of FTIR with multivariate analysis as a powerful predictive tool for assessing state of active layer material degradation within OSC devices. If a comprehensive spectral dataset of a commercial OSC material taken at industrially relevant photodegradation exposure intervals is collected and utilized as a training set, PCA–LDA could be used as an accurate diagnostic tool to assess the degradation extent for an operable OPV device. Although not commonly used in the spectral analysis of OSCs, this work highlights the potential utility of multivariate analysis as a possible new tool for assessing and diagnosing the status of essential electronic devices.
Conflicts of interest
There are no conflicts to declare.Data availability
The spectral and chemometric analysis data supporting this article have been included as part of the ESI.† The Python code for the chemometric analysis can be found at https://github.com/styler808/OSC-Chemometrics.Acknowledgements
The authors are grateful for support of this research by the National Science Foundation through grant award DMR-2003631. S. M. T. acknowledges financial support through an ARCS Foundation Scholarship. NMR data were collected in the NMR facility of the Department of Chemistry and Biochemistry at the University of Arizona, RRID:SCR_012716. The purchase of the Bruker NEO 500 MHz spectrometer was supported by the National Science Foundation under grant award CHE-1920234 and the University of Arizona. The purchase of the Bruker AVANCE III 400 MHz spectrometer was supported by the National Science Foundation under Grant Number 840336 and the University of Arizona. Mass spectra were acquired in the University of Arizona Analytical & Biological Mass Spectrometry Facility.References
- F. M. van der Staaij, I. M. van Keulen and E. von Hauff, Sol. RRL, 2021, 5, 2100167 CrossRef.
- M. Riede, D. Spoltore and K. Leo, Adv. Energy Mater., 2020, 11, 2002653 CrossRef.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686–714 CrossRef.
- N. Grossiord, J. M. Kroon, R. Andriessen and P. W. M. Blom, Org. Electron., 2012, 13, 432–456 CrossRef CAS.
- A. Distler, P. Kutka, T. Sauermann, H.-J. Egelhaaf, D. M. Guldi, D. Di Nuzzo, S. C. J. Meskers and R. A. J. Janssen, Chem. Mater., 2012, 24, 4397–4405 CrossRef CAS.
- A. Rivaton, A. Tournebize, J. Gaume, P.-O. Bussière, J.-L. Gardette and S. Therias, Polym. Int., 2014, 63, 1335–1345 CrossRef CAS.
- N. Wang, X. Tong, Q. Burlingame, J. Yu and S. R. Forrest, Sol. Energy Mater. Sol. Cells, 2014, 125, 170–175 CrossRef CAS.
- H. Liu, Y. Li, S. Xu, Y. Zhou and Z. A. Li, Adv. Funct. Mater., 2021, 31, 2106735 CrossRef CAS.
- S. Linde and R. Shikler, J. Appl. Phys., 2013, 114, 164506 CrossRef.
- S. Kim, M. A. M. Rashid, T. Ko, K. Ahn, Y. Shin, S. Nah, M. H. Kim, B. Kim, K. Kwak and M. Cho, J. Phys. Chem. C, 2020, 124, 2762–2770 CrossRef CAS.
- H. Ozaki, T. Fukushima, Y. Koshiba, S. Horike and K. Ishida, Jpn. J. Appl. Phys., 2020, 59, 11 CrossRef.
- S. Nabha-Barnea, N. Maman, I. Visoly-Fisher and R. Shikler, Macromolecules, 2016, 49, 6439–6444 CrossRef CAS.
- S. Choi, W. Kim, W. Shin, J. Oh, S. Jin, Y. M. Jung, M.-Y. Ryu and H. Lee, Curr. Appl. Phys., 2020, 20, 1359–1365 CrossRef.
- C. Zhou, J. Li, S. Chen, J. Wu, K. R. Heier and H. Cheng, J. Phys. Chem. C, 2008, 112, 14015–14020 CrossRef CAS.
- M. Brumbach, P. A. Veneman, F. S. Marrikar, T. Schulmeyer, A. Simmonds, W. Xia, P. Lee and N. R. Armstrong, Langmuir, 2007, 23, 11089–11099 CrossRef CAS PubMed.
- N. R. Armstrong, P. A. Veneman, E. L. Ratcliff, D. Placencia and M. Brumbach, Acc. Chem. Res., 2009, 42, 1748–1757 CrossRef CAS PubMed.
- G. Williams, Q. Wang and H. Aziz, Adv. Funct. Mater., 2013, 23, 2239–2247 CrossRef CAS.
- D. L. Matz, E. L. Ratcliff, J. Meyer, A. Kahn and J. E. Pemberton, ACS Appl. Mater. Interfaces, 2013, 5, 6001–6008 CrossRef CAS PubMed.
- L. Sang, D. L. Matz and J. E. Pemberton, J. Phys. Chem. C, 2015, 119, 24290–24298 CrossRef CAS.
- L. Sang and J. E. Pemberton, J. Phys. Chem. C, 2019, 123, 18877–18888 CrossRef CAS.
- L. Sang and J. E. Pemberton, Chem. Mater., 2019, 31, 6908–6917 CrossRef CAS.
- Q. Wang, G. Williams and H. Aziz, Org. Electron., 2012, 13, 2075–2082 CrossRef CAS.
- Y. Li, X. Huang, K. Ding, H. K. M. Sheriff, Jr., L. Ye, H. Liu, C. Z. Li, H. Ade and S. R. Forrest, Nat. Commun., 2021, 12, 5419 CrossRef CAS.
- Y. He, S. Gong, R. Hattori and J. Kanicki, Appl. Phys. Lett., 1999, 74, 2265–2267 CrossRef CAS.
- A. C. Arias, J. D. MacKenzie, R. Stevenson, J. J. M. Halls, M. Inbasekaran, E. P. Woo, D. Richards and R. H. Friend, Macromolecules, 2001, 34, 6005–6013 CrossRef CAS.
- Y. Kim, S. Cook, S. A. Choulis, J. Nelson, J. R. Durrant and D. D. C. Bradley, Chem. Mater., 2004, 16, 4812–4818 CrossRef CAS.
- J.-S. Kim, L. Lu, P. Sreearunothai, A. Seeley, K.-H. Yim, A. Petrozza, C. E. Murphy, D. Beljonne, J. Cornil and R. H. Friend, J. Am. Chem. Soc., 2008, 130, 13120–13131 CrossRef CAS PubMed.
- H. L. Smith, J. T. Dull, E. Longhi, S. Barlow, B. P. Rand, S. R. Marder and A. Kahn, Adv. Funct. Mater., 2020, 30, 2000328 CrossRef CAS.
- J. Zaumseil, C. L. Donley, J. S. Kim, R. H. Friend and H. Sirringhaus, Adv. Mater., 2006, 18, 2708–2712 CrossRef CAS.
- Y. Zhang and P. W. M. Blom, Appl. Phys. Lett., 2011, 98, 43504 CrossRef.
- C. R. McNeill and N. C. Greenham, Adv. Mater., 2009, 21, 3840–3850 CrossRef CAS.
- R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2012, 51, 743–768 CrossRef.
- S. Holliday, Y. Li and C. K. Luscombe, Prog. Polym. Sci., 2017, 70, 34–51 CrossRef CAS.
- N. Wang, J. Yu, Y. Zang, J. Huang and Y. Jiang, Sol. Energy Mater. Sol. Cells, 2010, 94, 263–266 CrossRef CAS.
- R. Steim, F. R. Kogler and C. J. Brabec, J. Mater. Chem., 2010, 20, 2499 RSC.
- B. H. Jiang, H. E. Lee, J. H. Lu, T. H. Tsai, T. S. Shieh, R. J. Jeng and C. P. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 39496–39504 CrossRef CAS.
- W. Zhong, J. Liang, S. Hu, X.-F. Jiang, L. Ying, F. Huang, W. Yang and Y. Cao, Macromolecules, 2016, 49, 5806–5816 CrossRef CAS.
- G. A. Edwards, M. A. Trafford, A. E. Hamilton, A. M. Buxton, M. C. Bardeaux and J. M. Chalker, J. Org. Chem., 2014, 79, 2094–2104 CrossRef CAS PubMed.
- S. Smolinski, P. Zelenay and J. Sobkowski, J. Electroanal. Chem., 1998, 442, 41–47 CrossRef CAS.
- D. J. Tiani and J. E. Pemberton, Langmuir, 2003, 19, 6422–6429 CrossRef CAS.
- D. J. Tiani, H. Yoo, A. Mudalige and J. E. Pemberton, Langmuir, 2008, 24, 134843 CrossRef PubMed.
- M. C. Schalnat and J. E. Pemberton, Langmuir, 2010, 26, 11862–11869 CrossRef CAS PubMed.
- D. Pelliccia, Classification of NIR Spectra by Linear Discriminant Analysis in Python, https://nirpyresearch.com/classification-nir-spectra-linear-discriminant-analysis-python/ Search PubMed.
- S. M. Tyler, OSC-Chemometrics, https://github.com/styler808/OSC-Chemometrics, (accessed 06/09/2024) Search PubMed.
- J. Liu, Z. Li, T. Hu, X. Wei, R. Wang, X. Hu, Y. Liu, Y. Yi, Y. Yamada-Takamura, Y. Wang and P. Wang, Adv. Opt. Mater., 2018, 7, 1801190 CrossRef.
- S. Pagliara, M. S. Vitiello, A. Camposeo, A. Polini, R. Cingolani, G. Scamarcio and D. Pisignano, J. Phys. Chem. C, 2011, 115, 20399–20405 CrossRef CAS.
- W. Zhao, T. Cao and J. M. White, Adv. Funct. Mater., 2004, 14, 783–790 CrossRef CAS.
- P. Pakulski and D. Pinkowicz, Molecules, 2021, 26, 4873 CrossRef CAS PubMed.
- R. Venkatesan, N. Somanathan and N. Rajeswari, Chin. J. Polym. Sci., 2014, 32, 667–674 CrossRef CAS.
- G. M. Silva, E. H. de Faria, E. J. Nassar, K. J. Ciuffi and P. S. Calefi, Quim. Nova, 2012, 35, 473–476 CrossRef CAS.
- I. Boehme, U. Weimar and N. Barsan, Sens. Actuators, B, 2021, 326, 129004 CrossRef CAS.
- V. N. Bliznyuk, S. A. Carter, J. C. Scott, G. Klarner, R. D. Miller and D. C. Miller, Macromolecules, 1999, 32, 361–369 CrossRef CAS.
- J.-I. Lee, G. Klaerner and R. D. Miller, Chem. Mater., 1999, 11, 1083–1088 CrossRef CAS.
- L. Romaner, G. Heimel, H. Wiesenhofer, P. Scandiucci de Freitas, U. Scherf, J.-L. Bredas, E. Zojer and E. J. W. List, Chem. Mater., 2004, 16, 4667–4674 CrossRef CAS.
- L. Liu, S. Qui, B. Wang, W. Zhang, P. Lu, Z. Xie, M. Hanif, Y. Ma and J. Shen, J. Phys. Chem. B, 2005, 109, 23366–23370 CrossRef CAS PubMed.
- R. Grisorio, G. P. Suranna, P. Mastrorilli and C. F. Nobile, Adv. Funct. Mater., 2007, 17, 538–548 CrossRef CAS.
- H. Jiang, W. JunHua and H. Wei, Sci. China, Ser. B: Chem., 2008, 51, 497–520 CrossRef CAS.
- R. Grisorio, G. Allegretta, P. Mastrorilli and G. P. Suranna, Macromolecules, 2011, 44, 7977–7986 CrossRef CAS.
- B. Kobin, F. Bianchi, S. Halm, J. Leistner, S. Blumstengel, F. Henneberger and S. Hecht, Adv. Funct. Mater., 2014, 24, 7717–7727 CrossRef CAS.
- L. Liu, S. Tang, M. Liu, Z. Xie, W. Zhang, P. Lu, M. Hanif and Y. Ma, J. Phys. Chem. B, 2006, 110, 13734–13740 CrossRef CAS.
- L. S. Albuquerque, J. J. R. Arias, B. P. Santos, M. D. F. V. Marques and S. N. Monteiro, J. Mater. Res. Technol., 2020, 9, 7975–7988 CrossRef CAS.
- Y. Che, M. R. Niazi, T. Yu, T. Maris, C.-H. Liu, D. Ma, R. Izquierdo, I. F. Perepichka and D. F. Perepichka, J. Mater. Chem. C, 2023, 11, 8186–8195 RSC.
- X. Ke, L. Meng, X. Wan, Y. Sun, Z. Guo, S. Wu, H. Zhang, C. Z. Li and Y. Chen, Mater. Chem. Front., 2020, 4, 3594–3601 RSC.
- Y. Liu, L. Yin, B. Xie, C.-H. Liu and Y. Li, Mater. Adv., 2022, 3, 6496–6505 RSC.
- Suman, J. Kovvuri and N. Islavath, J. Photochem. Photobiol., A, 2024, 446, 115162 CrossRef CAS.
- Z. Zhou, N. Luo, X. Shao, H. Zhang and Z. Liu, ChemPlusChem, 2023, 88, e202300261 CrossRef CAS PubMed.
- J. S. Amato, S. Karady, R. A. Reamer, S. Bernhard, J. P. Spriung and L. M. Weinstock, J. Am. Chem. Soc., 1981, 104, 1375–1380 CrossRef.
- I. Rozas, J. Phys. Org. Chem., 1992, 5, 74–82 CrossRef CAS.
- V. J. Arán, J. R. Ruiz, E. Dávila, I. Alkorta and M. Stud, Liebigs Ann. Chem., 1988, 1988, 337–341 CrossRef.
- V. J. Arán, P. Goya and C. Ochoa, Adv. Heterocycl. Chem., 1988, 44, 81–197 CrossRef.
- T. S. Cantrell and W. S. Haller, Chem. Commun., 1968, 977–978 RSC.
- G. M. Goncher, C. A. Parsons and C. B. Harris, J. Phys. Chem., 1984, 88, 4200–4209 CrossRef CAS.
- W. Heinzelmann and P. Gilgen, Helv. Chim. Acta, 1976, 59, 2727–2737 CrossRef CAS.
- I. Yavari, S. Esfandiari, A. J. Mostashari and P. W. W. Hunter, J. Org. Chem., 2002, 40, 2880–2883 CrossRef.
- E. Lieber, C. N. R. Rao and J. Ramachandran, Spectrochim. Acta, 1959, 13, 296–299 CrossRef CAS.
- C. C. Warner, A. M. Thooft, S. R. Norris, B. J. Lampkin, S. K. Demirci and B. VanVeller, ChemistrySelect, 2020, 5, 7016–7020 CrossRef CAS.
- A. Nitzan and L. E. Brus, J. Chem. Phys., 1981, 75, 2205–2214 CrossRef CAS.
- C. J. Chen and R. M. Osgood, Phys. Rev. Lett., 1983, 50, 1705–1708 CrossRef CAS.
- R. A. Wolkow and M. Moskovits, J. Chem. Phys., 1987, 87, 5858–5869 CrossRef CAS.
- K. Ueno and H. Misawa, J. Photochem. Photobiol., C, 2013, 15, 31–52 CrossRef CAS.
- C. Zhan, X.-J. Chen, J. Yi, J.-F. Li, D.-Y. Wu and Z.-Q. Tian, Nat. Rev. Chem., 2018, 2, 216–230 CrossRef.
- C. Zhan, X. J. Chen, Y. F. Huang, D. Y. Wu and Z. Q. Tian, Acc. Chem. Res., 2019, 52, 2784–2792 CrossRef CAS PubMed.
- C. L. M. Morais, K. M. G. Lima, M. Singh and F. L. Martin, Nat. Protoc., 2020, 15, 2143–2162 CrossRef CAS.
- X. Yang, K. Elkhalil, A. Kammoun, T. Y. Al-Naffouri and M.-S. Alouini, 2018 IEEE International Symposium on Information Theory (ISIT), Web, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00488h |
| This journal is © The Royal Society of Chemistry 2025 |