
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enargite (Cu3AsS4): a ductile mid-temperature thermoelectric material†
Prakash
Govindaraj
 ab,
Hern
Kim
*b and
Kathirvel
Venugopal
*a
ab,
Hern
Kim
*b and
Kathirvel
Venugopal
*a
aDepartment of Physics and Nanotechnology, SRM Institute of Science and Technology, Kattankulathur, Chengalpattu, Tamil Nadu 603 203, India. E-mail: kathirvv@srmist.edu.in
bDepartment of Energy Science and Technology, Environment Waste Recycle Institute, Myongji University, Yongin, Gyeonggi-do 17058, Republic of Korea. E-mail: hernkim@mju.ac.kr
First published on 10th June 2025
Abstract
Despite their intrinsic large band gap, low carrier concentration, and electrical conductivity, sulfide-based thermoelectric materials have been explored extensively because of their abundance and feasibility. This report uncovers the thermoelectric performance of Cu3AsS4 by combining density functional theory, the modified Debye–Callaway model, and phonon Boltzmann transport equations. The overall assessment of thermal, mechanical, and dynamical stability is confirmed through the ab initio molecular dynamics simulations, elastic constants, and phonon dispersion computations. The ultra-low lattice thermal conductivity of 0.327 and 1.020 W m−1 K−1 at 900 K obtained through various approaches can be attributed to the scattering of phonons induced by bonding heterogeneity and large lattice anharmonicity. Also, to improve the reliability of electronic transport properties, the carrier relaxation time is calculated by including acoustic, optical, and impurity phonon scattering mechanisms. The favourable band features and electron and phonon characteristics collectively facilitate a larger optimum power factor accompanied by the figure-of-merit of 1.07 to 2.31 at 900 K for p-type Cu3AsS4. These results highlight the potential applicability of Cu3AsS4 for mid-temperature thermoelectric applications. Also, this work elaborates the relationship between physical and mechanical characteristics of the crystal structure, which intensifies the understanding from materials to devices.
1. Introduction
The substantial enlargement and advancement of renewable energy sectors are essential to overcome global warming and the energy crisis. The solar and wind power sectors are evolving day-by-day and contributing their maximum to society.1,2 Along these lines, the unique thermoelectric energy conversion technique has the potential to address both issues. Briefly, thermoelectric (TE) devices can convert heat into electricity (generator) and vice versa (refrigerator). The performance of TE devices is determined by the material's dimensionless figure of merit, zT = (σS2/κ)T, where S, σ, κ and T are the Seebeck coefficient, electrical conductivity, thermal conductivity and temperature, respectively.3 That is, a good TE material should have high S and σ with low κ. Conceptually, it is difficult to realize high S and σ simultaneously as they are intrinsically coupled through carrier concentration (n). The complex relationship between σ and κ also resists the tailoring process. Besides these complications, several approaches are developed to enhance the zT of the TE materials.At least zT > 1 or an efficiency above 15% is required to commercialize the TE devices effectively.4 The continuous search in this aspect led to the discovery of several well-performing materials like Bi0.5Sb1.5Te3 (1.47 at 350 K), SnSe (1.4 at 300 K), PbTe (1.5 at 773 K), and GeTe-based compounds (2.14 at 670 K).5–7 But, the high cost of the precursor elements and the need for sophisticated synthesis approaches drastically increase the cost of electricity production using TE devices.8 It is also well-known that ductile materials are crucial to prevent a device's mechanical fractures and failures. However, recent reports disclose the shortage of ductile TE materials.9,10 Hence, over the past two decades, numerous techniques have been developed and employed to enhance the TE properties of earth-abundant sustainable materials, particularly, (i) to tailor power factor (PF = S2σ) – resonant level doping and band, strain and pressure engineering and (ii) to reduce thermal conductivity (κTotal = κe + κL, where κe – electronic thermal conductivity and κL – lattice thermal conductivity) – nano-structuring, introducing complex crystals, and defect and grain boundary engineering.11–18 Research has been extended to various materials, including semiconductors, polymers, and nanostructured materials, with considerable improvements in nanostructuring that helps in reducing thermal conductivity via phonon scattering. Additional interest in low-dimensional materials, like 2D materials and nanowires, which exhibit enhanced thermoelectric properties, has been rising. Further advancements emphasize the significance of doping, interface engineering, and the design of heterostructures to further boost the thermoelectric performance.19–21 In addition to these strategies, it is essential to explore new functional materials for effective implementation of TE technology. The role of high-throughput computations and first principles calculations are unavoidable in this direction.22,23
Recently, the pnictogen containing I3–V–VI4 (I = Cu; V = As, Sb; VI = S, Se) materials have gained widespread attention in various areas, including photovoltaic, photocatalysis and energy storage applications.24–26 Some of these candidates exhibit noticeable thermoelectric performance ascribed to their attractive carrier concentration and low thermal conductivity. Particularly, Sn, Bi doped Cu3SbS3 synthesized by mechanical alloying exhibits a large zT of 0.76 at 623 K. This behaviour is associated with the low κL (∼0.75 W m−1 K−1 at 623 K) ascribed to inherent mass fluctuation. Reports also show the possible enhancement of PF by 150% through hole activation via doping.27 Similarly, the Cu3SbSe4–CuAlSe2 composite yields a peak zT of 1.4 at 723 K with a low κ of ∼0.4 W m−1 K−1. This large zT is ascribed to the increase in carrier concentration boosted by creating cation vacancies.28
Also, one of their family members, Cu3AsS4, is investigated for photovoltaic, optical, and other semiconducting applications.29,30 Nevertheless, the reports on the electron, phonon, and thermoelectric properties of Cu3AsS4 are uncommon. The enargite (chemically, Cu3AsS4) mineral is rarely found in the Longfellow mine, Red Mountain district, San Juan County, Colorado. The enargite crystal exists in a dimorphic state (orthorhombic and tetragonal). The second phase is very rare, while the orthorhombic phase is commonly available in several mines along with the copper ores.31 The orthorhombic phase can also be experimentally synthesized by various physical and chemical methods.32,33
Favourably, the enargite mineral exhibits semiconducting behaviour with holes as the dominating carriers. Allen Pratt analyzed the oxidation states of Cu (1+), As (5+) and S (2−) through X-ray photoemission spectroscopy (XPS).34 Pauporté et al. reported the electrical and optical properties of the natural mineral (NM) and synthetic sample (SS) of Cu3AsS4. The highlights are: (i) the carrier concentrations of NM and SS are 1 × 1017 and 7.8 × 1019 cm−3 at 295 K; (ii) electrical resistances are 6–8 Ω cm−1 (for NM) and 10.5 Ω cm−1 (for SS); and (iii) display semiconducting nature with a band gap (Eg) of 1.43 eV.35 The inherently large n and σ of Cu3AsS4 mineral are expected to result in interesting thermoelectric properties. However, to the best of our knowledge, the thermoelectric properties of Cu3AsS4 remain unexplored. Hence, the present work is motivated and intended to systematically investigate the transport and thermoelectric properties of Cu3AsS4.
To ensure a comprehensive understanding of the thermoelectric properties, multiple state-of-the-art computational approaches are employed to evaluate phonon transport properties, specifically, the modified Debye–Callaway (mDC) model, Cahill–Pohl (CP) model, Slack model and phonon Boltzmann transport equation under single-mode relaxation time approximation (PBTE-RTA) as well as iterative solution (PBTE-Iterative) approaches. This enables the cross-validation of results obtained from different underlying methodologies. Consequently, four different values of zT are presented, each corresponding to a combination of electron and specific phonon transport. Notably, for all combinations, the zT exceeds 1 at 900 K, indicating the potential of Cu3AsS4 for mid-temperature thermoelectric applications. The use of multiple approaches not only facilitates cross-comparison but also enhances the reliability of the predicted thermoelectric performance.
2. Computational methods
2.1. Electronic structure calculation
The electronic band structure and related characteristics are calculated using the Vienna ab initio simulation package (VASP) under the plane augmented wave (PAW) method.36–38 To treat the core and valence electrons, the Perdew–Burke–Ernzerhof (PBE) exchange correlational functional based on generalised gradient approximation (GGA) is used.39 The kinetic energy cutoff of 550 eV along with the converged 7 × 7 × 7 k-mesh is utilized for the entire calculations. The crystal structure is relaxed under the energy and force convergence condition of 1 × 10−8 eV and 1 × 10−7 eV Å−1, respectively. Even though the lattice parameters match well with the experimental values, the band gap is strongly underestimated by the PBE functional. In order to resolve this issue, the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional is adopted for electronic structure calculation.40 Moreover, the Effmass code is used to compute the effective mass of the carriers and the Elastic code to calculate the elastic constant of the present system.41,422.2. Relaxation time and electron transport calculation
The electron transport coefficients are obtained by solving the Boltzmann transport equation (BTE), which is employed in the highly efficient AMSET code.43 It solves the BTE based on momentum relaxation time approximation to calculate scattering rates. This code computes the relaxation time (τ) by considering various band and k-point-dependent scattering approaches. In this work, the τtot is calculated by accounting: (i) acoustic deformation potential (τaco); (ii) polar optical phonon (τpop); and (iii) impurity (τimp) scattering mechanisms. To obtain the aforesaid scattering rates, the band structure, deformation potential, polar phonon frequency dielectric, and elastic constants are computed using VASP. Furthermore, they are averaged by Matthiessen's rule i.e., 1/τtot = 1/τaco + 1/τpop + 1/τimp to obtain τtot. Based on the performed convergence test, the interpolation factor of 21 is used to control the interpolation density (refer to Fig. S1, ESI†).2.3. Phonon properties and lattice thermal conductivity
The phonon dispersion of Cu3AsS4 is obtained using a supercell approach under the finite displacement method (FDM) using the phonopy program.44 To calculate lattice dynamical properties, the 2 × 2 × 2 supercell with 128 atoms is used. Using the finite displacement method, the supercell is displaced (according to its symmetry). The force of each displaced structure is obtained by performing single point calculation using 3 × 3 × 3 k-point mesh. The dynamical matrix is constructed by collecting all the forces. This dynamical matrix is used to calculate the phonon properties such as phonon dispersion spectra, group velocity, Grüneisen parameters and specific heat.The κL is calculated by various approaches like the modified Debye–Callaway (mDC) model, Cahill–Pohl (CP) model, Slack model and phonon Boltzmann transport equation under single-mode relaxation time approximation (PBTE-RTA) as well as iterative solution (PBTE-Iterative). The mDC, PBTE-RTA and PBTE-Iterative approaches are implemented in AICON2, Phono3py and ShengBTE programs, respectively.44–46 In the mDC model, the second order IFCs required for calculation are obtained for three different volumes (including equilibrium, slightly smaller and larger volumes). In addition, the group velocity and Grüneisen parameters are also essential. After computing all the necessary parameters, the AICON2 is triggered to calculate κL and phonon relaxation time. Usually, the AICON2 calculates κL based on the following relation:
| κ = κTA + κTA′ + κLA + κO | (1) |
Here, κTA, κTA′, κLA and κO represent the thermal conductivity contribution of the transverse, longitudinal acoustic and optical phonon modes. The CP and Slack models are applied manually using elastic constants to estimate minimum and temperature-dependent κL, respectively. The detailed description of the relations employed can be found in the ESI,† S1.1. For the PBTE-RTA approach, Phono3py is used, incorporating third-order IFCs and employing the tetrahedron method for the Brillouin zone integration.44 The convergence test and direction dependent lattice thermal conductivities are shown in Fig. S2 (ESI†). Here, the converged q-mesh of 15 × 15 × 15 is used. Subsequently, the iterative solution of full linearized phonon Boltzmann transport equation (PBTE-Iterative) is obtained using ShengBTE. Here, the third order IFCs are computed using the thirdorder.py script. The convergence test of κL performed as a function of the number of neighbours (nn) is shown in Fig. S3(a) (ESI†). Based on this test, the third order IFCs up to the 22nd nearest neighbour (0.56 nm) are considered, which requires single-point calculations on 3056 displaced structures. A converged q-mesh grid of 13 × 13 × 13 and a scaleboard value of 0.5 are used. All convergence tests are performed with a convergence threshold for a ΔκL of 0.05 W m−1 K−1. The results of convergence tests and direction dependent lattice thermal conductivities are given in Fig. S3(a–d) (ESI†). The non-analytical term corrections are included both in PBTE-RTA and PBTE-iterative approaches.
3. Results and discussion
3.1. Crystal structure and bonding nature
The enargite mineral in orthorhombic symmetry with the Pmn21 space group (no. 31) is studied.47 The unit cell fully relaxed using the PBE exchange–correlational functional is displayed in Fig. 1(a), generated with the help of the VESTA program.48 The relaxed unit cell parameters a = 6.485 Å, b = 7.470 Å, and c = 6.191 Å match well with earlier results (refer to Table 1). The unit cell consists of six copper, two arsenic, and eight sulfur atoms. Both the cations (Cu and As) are individually surrounded by the four anions (S) in tetrahedral coordination; they are represented in Fig. 1(b) and (c). | ||
| Fig. 1 (a) Crystal structure along the a-axis, tetrahedral coordination of (b) Cu–S and (c) As–S and (d) 2D electron localization function for the 2 × 2 × 2 supercell projected along the (0 1 0) plane. | ||
In addition, the distance between the cation and the anion is analysed. The bond lengths of Cu–S1, S2, S3, and S4 are 2.312 Å, 2.888 Å, 2.296 Å, and 2.306 Å, whereas the bond lengths of As–S1, S2, S3, and S4 are 2.245 Å, 2.293 Å, 2.272 Å and 2.293 Å. The unequal bond lengths between the cation and the anion are observed in both the tetrahedra. In order to identify the bonding characteristics, the 2D electron localization function (ELF) is computed for the supercell and illustrated in Fig. 1(d). The ELF values 0 (blue), 0.5 (green) and 1.0 (red) indicate no, equal and complete localization of electrons. Moreover, the ELF value between 0.7 and 0.8 is observed in As–S, which manifests the stronger bonding (mostly covalent) characteristics. On the other hand, lower electron population is noted between Cu and S, indicating lower bonding strength (probably ionic bonding). These unequal bond lengths and bonding heterogeneity can trigger the lattice anharmonicity, which resists phonon propagation and thus lowers the lattice thermal conductivity.
To further understand the thermodynamic stability of the studied compound, the formation and cohesive energies are calculated using eqn (2) and (3). Formation energy gives the idea about the possibility of forming the required phase experimentally and the cohesive energy describes the stability of the compound after synthesis.
 | (2) |
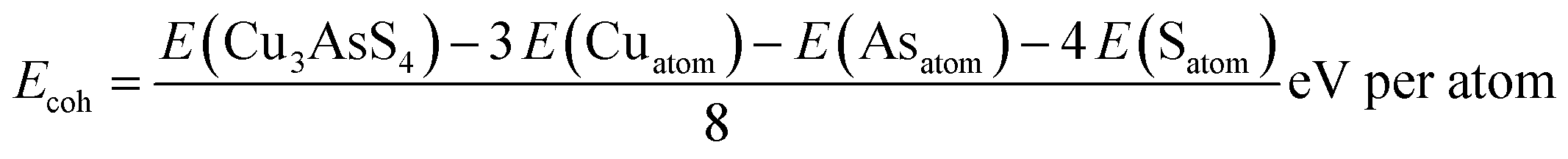 | (3) |
Here, E(Cu3AsS4) represents the total energy of the system and E(Cusolid), E(Assolid), and E(Ssolid) represent the total energy of the respective elements in their bulk form (in terms of eV per atom), while E(Cuatom), E(Asatom), and E(Satom) correspond to the total energy of constituent elements as isolated atoms.49,50 The calculated formation and cohesive energies of Cu3AsS4 are −0.20 eV per atom and −4.08 eV per atom, respectively. The negative sign in both these parameters indicates that Cu3AsS4 can be experimentally synthesized and is thermodynamically stable.
3.2. Thermal stability
To examine the thermal stability of enargite mineral, AIMD simulations are performed using the Nóse–Hoover thermostat for 5 ps. The structures are initially equilibrated for 1 ps at the corresponding temperatures. After neglecting the initial 1 ps, the evaluation of free energy as a function of time (up to 4 ps) is shown in Fig. 2(a). The free energy remains almost the same with negligible fluctuations at 300, 600 and 900 K. The evolved structures after the simulation time of 5 ps are shown for both 300 and 900 K in Fig. 2(b) and (c), which indicates that the structure is stable up to 900 K. At 300 K, the structure retains its original atomic arrangements with small distortion, whereas this distortion is intense at 900 K.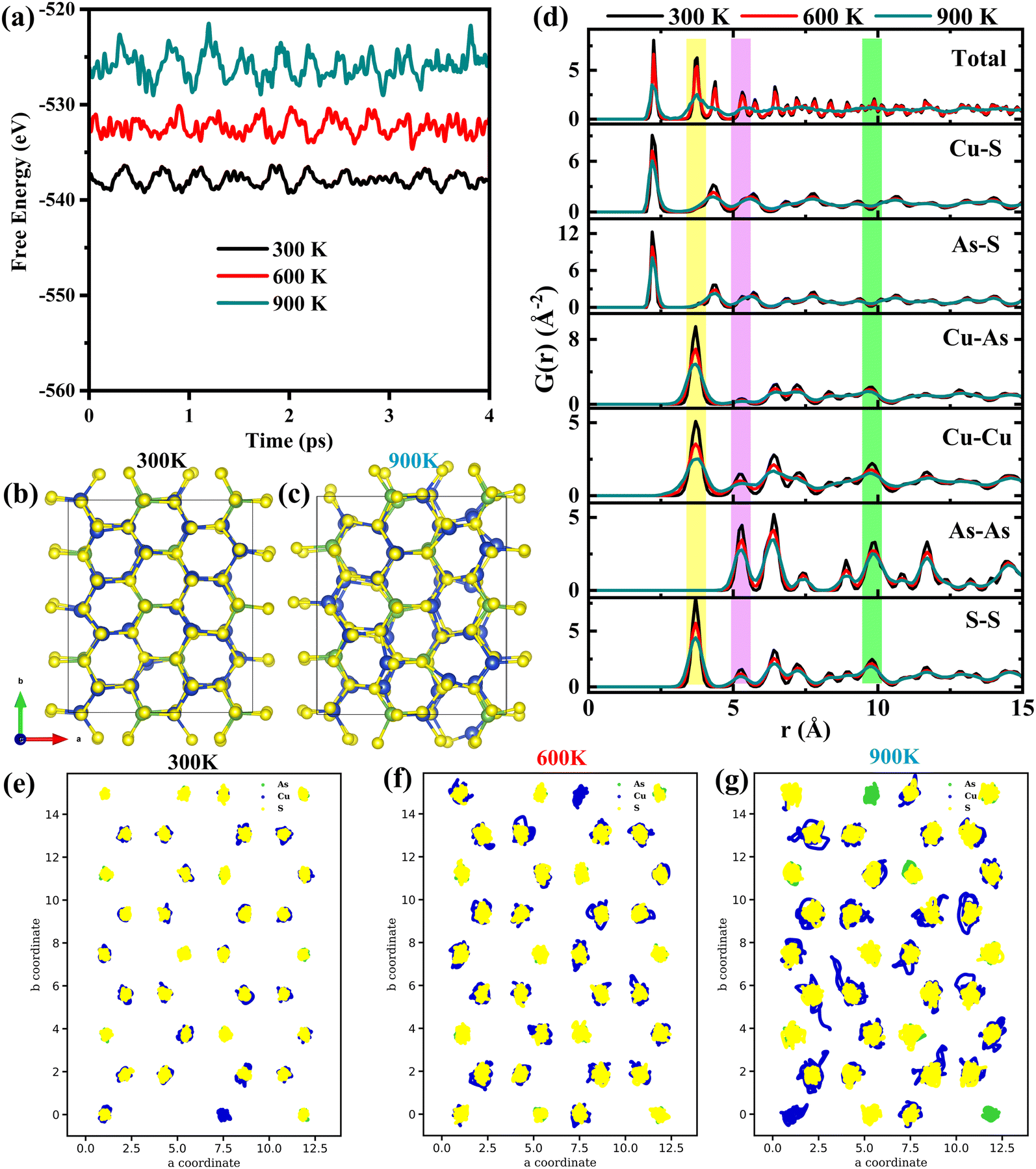 | ||
| Fig. 2 (a) Evolution of free energy at different simulation temperatures. The final structures at (b) 300 K and (c) 900 K. (d) Averaged total and partial pair distribution functions. The atomic trajectory of atoms at (e) 300 K, (f) 600 K and (g) 900 K, respectively. | ||
Fig. 2(d) depicts the calculated radial pair distribution function (PDF) to examine the crystallinity of Cu3AsS4. The peaks are well consistent and they broaden at higher temperature up to 600 K. Even though the observed peaks are present up to 900 K, the peaks above 5 Å radius start disappearing. This behaviour can be attributed to the solid to liquid phase transformation. However, the experimental melting point of Cu3AsS4 is 944 K.51 Hence, it is believed that the melting starts around 900 K. To get further insight into this argument, the partial PDF for each possible pair is calculated and depicted along with the total PDF. The first peak at 2.25 Å arises from the superposition of Cu–S and As–S pairs, whereas the second peak (highlighted in yellow colour) at 3.75 Å can be due to Cu–As, Cu–Cu and S–S pairs. It is noted that the fourth peak (highlighted in purple colour) at 5.3 Å is definite both at 300 K and 600 K, while at higher temperature (900 K) it completely disappears. This peak is contributed by Cu–Cu, As–As and S–S pairs. However, the peak is detectable for all pairs even at 900 K, except for the Cu–Cu pair. This behaviour is also observed near 10 Å (highlighted in green). Hence, it can be expected that the solid to liquid phase transition begins with the diffusion of Cu atoms. These characteristics are quite common in chalcogenides such as AgCrSe2 and Cu3SbSe3.54,55
In order to verify this, the simulated trajectory of atoms in the a–b plane is presented in Fig. 2(e–g). It is noted from Fig. 2(e) that the movement of all atoms in the lattice is similar at 300 K. Upon further heating to 600 K, As and S atoms vibrate at a similar amplitude to the previous one. In contrast, the Cu atoms cover a relatively larger area. This movement of Cu atoms becomes more vibrant at a higher temperature of 900 K. It can also be associated with the weakly bonded (i.e., ionic bonding) Cu–S tetrahedra. In other words, the Cu atoms are weakly bound to the lattice, resulting in large vibrations upon heating. Hence, the Cu atoms move back and forth more vibrantly. This can be the possible reason for the absence of definite peaks above 5 Å in the total PDF. However, there is no bond breakage in Cu–S and As–S tetrahedra, ensuring the crystallinity. This vibration of Cu atoms at higher temperatures results in enhanced tetrahedral distortion and large anharmonicity, which drives the phonon scattering process frequently.
3.3. Elastic constants and mechanical stability
The study of elastic constants plays a vital role in understanding the mechanical stability of the solids. For Cu3AsS4, these constants are calculated by the conventional strain approach. According to orthorhombic symmetry, there exist 9 independent elastic constants whose values are listed in Table 2. The Born's criterion is a well-known condition to ensure the mechanical stability of the crystalline solids. It depends on the crystal symmetry. Eqn (4) represents the Born's stability criteria for orthorhombic structure.56| C11 > 0; C11C22 > C122; C11C22C33 + 2C12C13C23 − C11C122 − C22C132 − C33C122 > 0; C44 > 0; C55 > 0; C66 > 0 | (4) |
| Elastic constants (GPa) | Mechanical properties | ||
|---|---|---|---|
| C 11 | 146.02 | B (GPa) | 81.52 |
| C 12 | 44.35 | G (GPa) | 27.43 |
| C 13 | 27.55 | Y (GPa) | 73.99 |
| C 22 | 34.52 | B/G | 2.97 |
| C 23 | 25.88 | υ | 0.35 |
| C 33 | 173.81 | CP (GPa) | 111.50 |
| C 44 | 34.52 | ||
| C 55 | 36.11 | ||
| C 66 | 49.52 | ||
The enargite mineral satisfies all these six conditions, which ensures its mechanical stability. Using Voigt–Reuss–Hill approximation, bulk modulus (B), Young's modulus (Y) and shear modulus (G) are calculated (refer to Table 2).57–59 The calculated bulk and shear moduli are 81.52 GPa and 27.43 GPa, respectively. The larger B, G and Y values indicate the resistive nature of Cu3AsS4 towards deformation, fracture, and stiffness. The value of Pugh's ratio (B/G) > 1.75, υ > 0.26 and positive Cauchy's pressure (CP) suggest the ductile nature of the system.60 The Pugh's ratio of Cu3AsS4 (2.97) is higher than that of Ag2S (∼2.8) and AgCuS (2.3).61,62 The compared sulphur based thermoelectric materials are well-known for their excellent mechanical stability (ductility). But, Ag2S has the constraint of adjusting the carrier concentration via doping without affecting its ductility, while zT of AgCuS requires significant improvement. In this aspect, Cu3AsS4 is regarded as an advantageous candidate, which intrinsically possesses excellent metallic like ductility and can outperform well in the device aspect.
3.4. Phonon dispersion and dynamical stability
The phonon dispersion spectra along with phonon DOS are displayed in Fig. 3(a) and (b). The phonon dispersion is calculated along high symmetry points i.e., Γ (0, 0, 0) − X (½, 0, 0) − S (½, ½, 0) − Y (0, ½, 0) − Γ (0, 0, 0) − Z (0, 0, ½) − U (½, 0, ½) − R (½, ½, ½) − T (0, ½, ½) − Z (0, 0, ½) − X (½, 0, 0) − U (½, 0, ½) − Y (0, ½, 0) − T (0, ½, ½) − S (½, ½, 0) − R (½, ½, ½). The presence of positive (real) frequency in the phonon spectrum reveals the dynamical stability of Cu3AsS4. Furthermore, 48 branches are observed in the spectra with 3 acoustic and 45 optical modes. Generally, the acoustic mode of vibrations is the primary contributor to the heat transport in many solids.63 Hence, transverse acoustic (TA, TA′) and longitudinal acoustic (LA) modes are highlighted with red, green, and violet colours, respectively. The observed highest acoustic mode frequency is 1.97 THz. And the branches spread up to a maximum of 10.12 THz. Generally, the A–O phonon mode coupling promotes the phonon scattering process and decreases the phonon relaxation time. The acoustic and low-frequency optical modes are primarily contributed by the Cu and As atoms, whereas high-frequency optical modes by the S atoms, as inferred from the phonon density of states (PHDOS) (refer to Fig. 3b). The low frequency A–O coupling in the Cu3AsS4 system is due to the vibrations of Cu and As elements, with nearly equivalent atomic masses. The optical–optical (O–O) frequency gap is noted near 5 THz. The temperature dependent PHDOS at various temperatures are extracted from the AIMD simulations and shown in Fig. 3c–e. Also, the population of phonon modes in this O–O phonon band gap region increases with temperature (refer to Fig. 3c–e, yellow highlighted region). The slope of the optical modes decreases, indicating the flattening of these branches. Furthermore, the phonon branches shift towards the low frequency regime, manifesting the reduction of group velocity and thus decrease of lattice thermal conductivity with temperature is expected.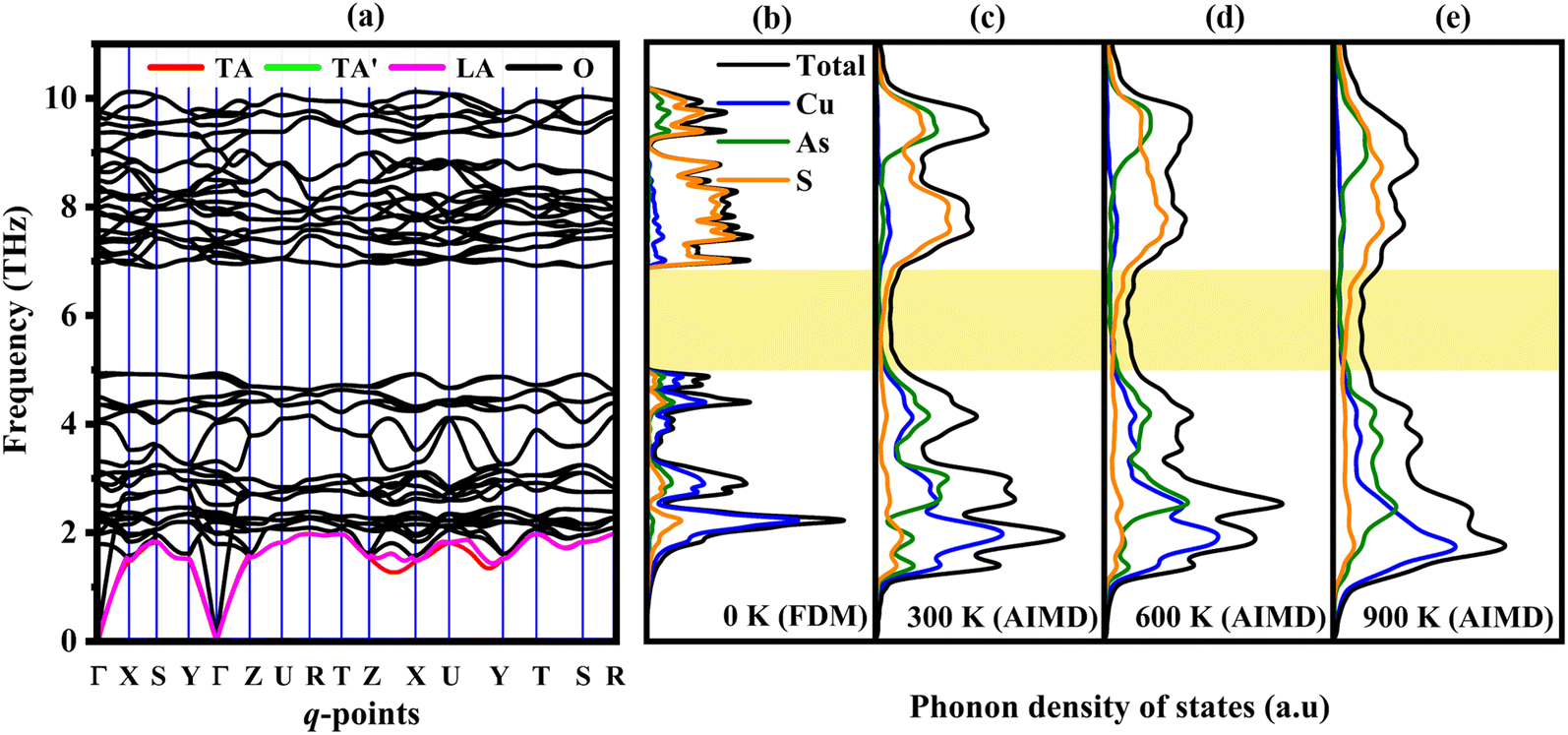 | ||
| Fig. 3 (a) Phonon dispersion spectrum and phonon density of states at (b) 0 K, (c) 300 K, (d) 600 K and (e) 900 K for enargite. | ||
3.5. Phonon transport properties
 .64 The frequency-dependent vg is depicted in Fig. 4(a). It shows that the predominant contribution to the phonon propagation arises from the acoustic mode, particularly LA modes. Thus, vibration of Cu atoms determines the thermal transportation in Cu3AsS4. For better clarity, mode dependent group velocity is illustrated in Fig. 4(b). From this, the average vg of individual acoustic modes can be obtained from the mathematical average of maximum vg at the respective Γ-point. The calculated average vg for TA, TA′ and LA is 2635, 1437 and 1865 m s−1, respectively. Comprehensively, the average vg for acoustic modes is 2186 m s−1, derived from
.64 The frequency-dependent vg is depicted in Fig. 4(a). It shows that the predominant contribution to the phonon propagation arises from the acoustic mode, particularly LA modes. Thus, vibration of Cu atoms determines the thermal transportation in Cu3AsS4. For better clarity, mode dependent group velocity is illustrated in Fig. 4(b). From this, the average vg of individual acoustic modes can be obtained from the mathematical average of maximum vg at the respective Γ-point. The calculated average vg for TA, TA′ and LA is 2635, 1437 and 1865 m s−1, respectively. Comprehensively, the average vg for acoustic modes is 2186 m s−1, derived from 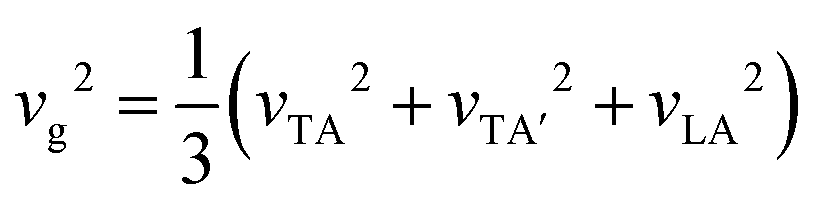 .65 This value for Cu3AsS4 is comparable with those of the benchmark TE materials, as evident from Table 3.
.65 This value for Cu3AsS4 is comparable with those of the benchmark TE materials, as evident from Table 3.
 | ||
| Fig. 4 Group velocity as a function of (a) frequency and (b) q-points. The Grüneisen parameter as a function of (c) frequency, (d) q-points and (e) temperature. (f) Specific heat against temperature. | ||
![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) g), Debye temperature (
g), Debye temperature (![[small theta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c9.gif) D), Grüneisen parameter (
D), Grüneisen parameter (![[small gamma, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c4.gif) ) and lattice thermal conductivity (κL) (at 300 K) of enargite with promising TE materials
) and lattice thermal conductivity (κL) (at 300 K) of enargite with promising TE materials
The Grüneisen parameter, which determines the degree of anharmonicity in the lattice, is calculated and shown in Fig. 4(c). Generally, the positive and negative γ implies the degree of anharmonicity and negative thermal expansion. Since the acoustic modes are important from the TE perspective, the mode dependent γ is calculated with respect to q-points using the relation:  and displayed in Fig. 4(d).64 Furthermore, the average Grüneisen parameter,
and displayed in Fig. 4(d).64 Furthermore, the average Grüneisen parameter,  calculated for TA, TA′ and LA is 1.91, 1.86 and 0.38, respectively. It is further averaged mathematically to obtain material's γ. The value of for the Cu3AsS4 system is 1.39. To understand the effect of temperature on lattice anharmonicity, the temperature dependent γ is calculated by implementing quasi harmonic approximation (QHA) and shown in Fig. 4(e). Interestingly, the γ value rises from 1.50 at 300 K to 1.76 at 900 K (∼18% increase). This rapid increase in γ can be attributed to tetrahedral distortion, as evident from the analysis of AIMD simulation. Also, the obtained values of Cu3AsS4 are compared with other minerals and chalcogenides in Table 3. The observed anharmonicity facilitates the phonon scattering process at high temperatures.
calculated for TA, TA′ and LA is 1.91, 1.86 and 0.38, respectively. It is further averaged mathematically to obtain material's γ. The value of for the Cu3AsS4 system is 1.39. To understand the effect of temperature on lattice anharmonicity, the temperature dependent γ is calculated by implementing quasi harmonic approximation (QHA) and shown in Fig. 4(e). Interestingly, the γ value rises from 1.50 at 300 K to 1.76 at 900 K (∼18% increase). This rapid increase in γ can be attributed to tetrahedral distortion, as evident from the analysis of AIMD simulation. Also, the obtained values of Cu3AsS4 are compared with other minerals and chalcogenides in Table 3. The observed anharmonicity facilitates the phonon scattering process at high temperatures.
Due to the direct relationship between κL and specific heat capacity, understanding the Cv of the present system is also important. Fig. 4(f) displays the computed Cv as a function of T. Initially, Cv abruptly increases with temperature, obeying the T3 law. Then it gets saturated at higher temperatures, reaching the Dulong–Petit limit. The classical limit according to the Dulong–Petit law (= 3NR, N – the number of atoms in the unit cell, R – ideal gas constant) for the enargite mineral is 399.11 J mol−1 K−1. The calculated Cv falls within this limit.
It is equally important to calculate the Debye temperature of the materials from the aspect of thermoelectrics. The θD values for individual acoustic modes are calculated. The values for TA, TA′ and LA are 126, 69 and 90 K, respectively. The global θD of enargite calculated from the averaged vg ( , where N and V are the number of atoms and volume of the unit cell respectively) is 245 K.66 This value is relatively higher than those of most of the reported thermoelectric materials (see Table 3).
, where N and V are the number of atoms and volume of the unit cell respectively) is 245 K.66 This value is relatively higher than those of most of the reported thermoelectric materials (see Table 3).
 | ||
| Fig. 5 (a) Lattice thermal conductivity and (b) phonon relaxation time as a function of temperature for enargite obtained in mDC. | ||
3.6. Electronic structure
Fig. 6 demonstrates the calculated electronic properties of Cu3AsS4. The projected electronic band structure for the Cu3AsS4 system is depicted in Fig. 6(a). The enargite mineral is a direct band gap semiconductor with the VBM and the CBM located at the Γ-point (0, 0, 0). The calculated band gap (Eg) is 1.37 eV, which is in reasonable agreement with experimental reports (refer to Table 4). In addition, the total and projected DOS (Fig. 6b and c) reveal that the VBM is mostly composed of the combination of Cu (d-orbital) and S (p-orbital) atoms, whereas the CBM predominantly arises from the S (p-orbital) atoms. The nearly degenerate bands are observed near the CBM rather than the VBM, named CBM1 and CBM2. The energy difference between the CBM and adjacent degenerate bands (such as CBM1 and CBM2) is 0.030 and 0.086 eV. The observed band convergence effect has a positive impact from the aspect of improving the electrical conductivity. Even with a slight increase in temperature, the carriers in CBM1 and CBM2 will also take part in conduction and thus lead to a significant increase in σ for n-type Cu3AsS4. To look in detail, the Fermi surfaces are drawn using the IFermi package and given in Fig. 6(d) and (e).70 Here, the Fermi surface is plotted 0.1 eV below the VBM and 0.1 eV above the CBM. It is noted that the Fermi surface near the VBM of Cu3AsS4 has only one closed sphere-shaped hole pocket at the centre of the BZ, whereas the CBM has three electron pockets due to the presence of degenerate bands at Γ–Z and Z–X. It is also inferred that the group velocity of the electrons is higher than the holes. This degenerate band and higher carrier group velocity may result in larger electrical conductivity for n-type Cu3AsS4. | ||
| Fig. 6 (a) Orbital-resolved electronic band structure, (b) total and (c) projected density of states. Fermi surface at (d) 0.1 eV below the VBM and (e) 0.1 eV above the CBM. Band decomposed charge density near the (f) VBM and (g) CBM. | ||
Furthermore, the quantification of the effective mass of the carriers (m*) plays a significant role in understanding the transport properties of the material. Here m* is calculated by fitting the band edges. Compared to the CBM, a slightly larger band dispersion is observed at the VBM, resulting in 8% heavier holes 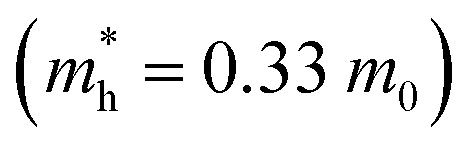 than electrons
than electrons 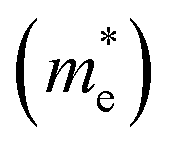 , i.e., 0.31 m0. Here m0 is the rest mass of the electron. Generally, large mass leads to less mobility. According to this observation, electrons possess higher mobility in the Cu3AsS4 lattice. Furthermore, the charge density near the VBM and the CBM is demonstrated to understand the conduction pathways of the carriers (refer to Fig. 6f and g). Even though both band extrema show similar carrier density, the VBM has slightly higher density, leading to larger carrier pathways for holes. The heavier holes may yield a large Seebeck coefficient for p-type Cu3AsS4.
, i.e., 0.31 m0. Here m0 is the rest mass of the electron. Generally, large mass leads to less mobility. According to this observation, electrons possess higher mobility in the Cu3AsS4 lattice. Furthermore, the charge density near the VBM and the CBM is demonstrated to understand the conduction pathways of the carriers (refer to Fig. 6f and g). Even though both band extrema show similar carrier density, the VBM has slightly higher density, leading to larger carrier pathways for holes. The heavier holes may yield a large Seebeck coefficient for p-type Cu3AsS4.
3.7. Electron transport properties
 | ||
| Fig. 7 (a) Carrier relaxation time and (b) electrical conductivity of hole-doped Cu3AsS4 against carrier concentration at different temperatures. | ||
The calculated σ at various temperatures is shown in Fig. 7(b). The σ increases with n following the relation, σ = neμ. For example, at 300 K, the observed σ is 5019 S m−1 for a specific carrier concentration of 1 × 1019 cm−3. And it is increased to 260![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 015 S m−1 at n = 1 × 1021 cm−3 in Cu3AsS4. At the carrier concentration of 1 × 1019 cm−3 and elevated temperature of 900 K, σ is 1397 S m−1 for the hole-doped system. The drastic decrease of τ with temperature leads to relatively higher carrier scattering, which results in the decrease in σ with temperature. The electron-doped system yields larger σ when compared to its counterpart (refer to Fig. S4b, ESI†). This can be ascribed to the larger m* of holes than electrons and to the presence of degenerate bands near the CBM. The lighter electrons exhibit more oscillations and can undergo frequent collisions and scattering at higher temperatures. This characteristic yield decreased σ at 900 K for n-type Cu3AsS4. The details about the magnitude of the calculated parameters are recorded in Table S1 (ESI†).
015 S m−1 at n = 1 × 1021 cm−3 in Cu3AsS4. At the carrier concentration of 1 × 1019 cm−3 and elevated temperature of 900 K, σ is 1397 S m−1 for the hole-doped system. The drastic decrease of τ with temperature leads to relatively higher carrier scattering, which results in the decrease in σ with temperature. The electron-doped system yields larger σ when compared to its counterpart (refer to Fig. S4b, ESI†). This can be ascribed to the larger m* of holes than electrons and to the presence of degenerate bands near the CBM. The lighter electrons exhibit more oscillations and can undergo frequent collisions and scattering at higher temperatures. This characteristic yield decreased σ at 900 K for n-type Cu3AsS4. The details about the magnitude of the calculated parameters are recorded in Table S1 (ESI†).
 | ||
| Fig. 8 (a) Seebeck coefficient and (b) power factor of hole-doped Cu3AsS4 against carrier concentration at different temperatures. | ||
Since S and σ are indirectly related and they are in the numerator of zT relation, it is important to understand the compensation between S and σ. This can be done by calculating PF (S2σ). The PF is one of the deciding parameters in TE materials (i.e.): higher TE performance requires larger PF. Hence, the PF is calculated with respect to n at different temperatures and displayed in Fig. 8(b). The PF increases with carrier concentration up to some extent for both carrier doping and then decreases. Furthermore, the peak PF noted for p-type is 1.49 mW m−1 K−2 at 300 K. This value is higher than that of the n-type counterpart (0.82 mW m−1 K−2), which clearly shows that the major contribution to PF is acquired from S. In other words, even though σ is higher for the electron-doped system, the PF is higher for the hole doped system, indicating the higher reliance of PF on S. The PF further increases with temperature. At 900 K, the peak PF reaches 1.80 mW m−1 K−2 for p-type Cu3AsS4, whereas 0.36 mW m−1 K−2 for n-type Cu3AsS4 (refer to Fig. S5b, ESI†). For better comparison, the peak PF and corresponding carrier concentration are recorded in Table S2 (ESI†).
 | ||
| Fig. 9 (a) Electronic and (b) total thermal conductivity of hole-doped Cu3AsS4 at different temperatures. The light-colored lines indicate the corresponding lattice thermal conductivity at specific temperatures obtained via the PBTE-Iterative approach. | ||
The computed total thermal conductivity (κTotal) for hole-doped Cu3AsS4 is depicted in Fig. 9(b). Since κL is obtained using different approaches, κTotal is also calculated accordingly. For simplicity, only the κTotal obtained using the PBTE-Iterative approach is presented here, while the results based on remaining approaches are provided in the ESI† (see Fig. S7). Interestingly, κTotal is predominately contributed by the lattice part at low carrier concentration, whereas the electronic contribution is larger at higher concentrations. For instance, at 300 K and 1 × 1018 cm−3, the value of κTotal is almost equal to κLi.e., 3.16 W m−1 K−1. It gets increased by nearly ∼61% at higher concentrations (1 × 1021 cm−3, i.e., 1.89 W m−1 K−1). This change is more vibrant at higher temperatures due to the larger κe contributed by higher carrier oscillations. Furthermore, the κTotal for the electron-doped system is given in Fig. S8 (ESI†). The values are also tabulated in Table S3 (ESI†) for better clarity.
3.8. Figure of merit
As observed from the earlier discussion, the lattice thermal conductivity computed by adopting various models has significant difference. Hence, it is important to calculate the zT of Cu3AsS4 by incorporating κL values from different models, in addition to the electron transport properties calculated from AMSET. Fig. 10(a)–(d) displays the calculated zT values as a function of temperature and carrier concentration for hole-doped Cu3AsS4. Regardless of the models, zT increases consistently with temperature. For example, the peak zT of p-Cu3AsS4 (using mDC) at 300 K is 0.26, which increases by 9 times (i.e., 2.31) at 900 K. This behavior can be attributed to the larger carrier scattering and contribution from degenerate bands at higher temperatures. Also, the maximum zT attained at 900 K with κL obtained via Slack, PBTE-RTA and PBTE-Iterative is 2.25, 1.12 and 1.07, respectively. The zT for electron doped Cu3AsS4 is displayed in Fig. S9(a–d) (ESI†). The peak zT values and corresponding carrier concentrations are summarized in Table S4 (ESI†) for better clarity. The larger zT in Cu3AsS4 can be ascribed to: (i) ultralow lattice thermal conductivity: originating from mixed bonding nature (phonon scattering), A–O coupling (reduced phonon relaxation time), low group velocity (limited phonon propagation), larger Grüneisen parameter (enhanced lattice anharmonicity) and (ii) significant power factor: carrier effective masses and nearly degenerate bands (moderate S and σ). | ||
| Fig. 10 The calculated zT using κL computed using (a) mDC, (b) Slack, (c) PBTE-RTA and (d) PBTE-Iterative for hole-doped Cu3AsS4 and (e) zT and κL are compared with the state-of-the-art materials and As-based materials. Alphabets indication: (a) SnSe,74 (b) PbTe,75 (c) Bi2Te3−xSex,76 (d) Sb doped GeTe,77 (e) MgAgSb,78 (f) CeFe4As12,79 (g) β-As2Te3,80 (h) Re3GeAs6,81 (i) CoAsSb,82 (j) Eu5Sn2As683 and (k)–(n) Cu3AsS4. The unit of κL is W m−1 K−1. | ||
The obtained zT and κL are compared with other arsenic-based and benchmark candidates in Fig. 10(e). It is noted from the state-of-the-art that the enargite mineral has the tendency to compete with the Te/Se based benchmark materials. Also, the thermoelectric performance of Cu3AsS4 is higher than that of CuFeS2, Cu12Sb4S13, and Cu2SnS3 – sulfur-based compounds.84–87 In the case of its applicability towards real-time applications, the use of earth-abundant sulphur and arsenic (despite their toxicity) rather than critical elements (like Se/Te) helps in maintaining the low cost of the device. Although As is toxic, the toxicity of the compound should be prioritized at two key stages: (i) during handling at the synthesis stage, (ii) integration into the devices, and (iii) during disposal into the environment. The first and second stages can be managed by carefully reviewing the safety data sheet, adhering to safety protocols, and using appropriate personnel protective equipment. The third stage can be addressed by following proper disposal procedures. Also, Cu3AsS4 exhibits zT greater than 1 at 900 K, which is higher than most of the As-based compounds (blue regime in Fig. 10e). This suggests that the enargite mineral can be an appropriate candidate for mid-temperature TE applications.
Besides the significant thermoelectric performance of Cu3AsS4, adopting a suitable strategy to reduce its lattice thermal conductivity further improves its zT. Generally, vacancy and impurity engineering hold significant promise for tuning the thermoelectric properties of Cu-related materials. Recent studies have increasingly focused on the role of intrinsic and extrinsic defects in enhancing TE performance. For example, in Cu3SbS3, an excess of sulfur content has been shown to increase both hole carrier concentration and mobility.88 Additionally, the elevated sulfur levels enhance phonon scattering, leading to a substantial reduction in the κL. As a result, the thermoelectric zT improves by nearly fourfold (i.e., increased from 0.2 to 0.71) compared to the pristine material. Similarly, in Cu2SnSe4, the introduction of cationic vacancies has been demonstrated to significantly enhance thermoelectric performance, doubling the zT by promoting strong phonon scattering.89 These cationic vacancies effectively reduce κL to reach the amorphous limit at 900 K. For instance, κL is reduced from 1.1 W m−1 K−1 to 0.6 W m−1 K−1. Given these promising results in related materials, similar defect-engineering strategies could be highly effective in optimizing the TE properties of Cu3AsS4. These results highlight an important research avenue and invite further experimental and theoretical research on the use of defects to enhance the thermoelectric efficiency of Cu3AsS4.
4. Conclusions
This study presents a comprehensive computational investigation of the structural, mechanical, thermal, and thermoelectric properties of the enargite mineral. The experimentally existing and unexplored (from the TE perspective) orthorhombic Cu3AsS4 system is chosen. The unit cell comprises two tetrahedra (Cu–S4) and (As–S4), with inequivalent bonding. It led to mixed bonding characters between cations and anions. Mechanical and dynamical stabilities of the present system are confirmed through elastic constants and phonon spectra, respectively. The electronic structure reveals the direct band gap semiconducting nature of enargite. The A–O, O–O coupling at high temperatures, intensive Cu–S tetrahedra distortion, and significant Grüneisen parameter ensure the occurrence of enhanced phonon scattering process and lattice anharmonicity. This favourable structural and phonon properties ended up with an ultralow κL from 0.327 to 1.020 W m−1 K−1 at 900 K. The moderate S and σ yield a noticeable PF. The combined effect of ultralow κL and moderate PF yields the large zT (above 1) at 900 K for hole-doped Cu3AsS4. These findings suggest that enargite can be a suitable system for mid-temperature thermoelectric applications, when compared to other As-based thermoelectric materials. Also, the present work illustrates a clear physical and mechanical picture of Cu3AsS4, which helps to achieve better understanding from the device aspect.Author contributions
Prakash Govindaraj: conceptualization, software, formal analysis, investigation, writing – original draft, visualization, data curation; Hern Kim: supervision, resources, project administration, data curation, funding acquisition, conceptualization; and Kathirvel Venugopal: writing – reviewing and editing, supervision, resources, validation, methodology.Data availability
The computational results that predominantly support our claims were calculated using the Vienna ab initio simulation package (VASP) that can be found at https://www.vasp.at/ with 10.1103/PhysRevB.54.11169 and 10.1103/PhysRevB.59.1758. The version of the code employed for this study is version 5.4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) grants funded by the Ministry of Education (RS-2020-NR049576), Republic of Korea. Also, the authors thank High Performance Computing Center, SRM Institute of Science and Technology for providing the computational facility.References
- W. Akram, X. Li, S. Ahmed, Z. Ouyang and G. Li, RSC Sustainability, 2025, 3, 21–36 Search PubMed.
- Y. Cao, Y. Meng, Z. Zhang, Q. Yang, Y. Li, C. Liu and S. Ba, Renewable Sustainable Energy Rev., 2024, 196, 114351 Search PubMed.
- X.-L. Shi, J. Zou and Z.-G. Chen, Chem. Rev., 2020, 120, 7399–7515 CrossRef CAS PubMed.
- N. V. Morozova, I. V. Korobeinikov and S. V. Ovsyannikov, J. Appl. Phys., 2019, 125, 220901 CrossRef.
- P. Xu, K. Jin, Y. Yu, M. Huang, Z. Yan, X. Li, X. Gao, L. Fu and B. Xu, Adv. Funct. Mater., 2024, 2401240 Search PubMed.
- B. Qin, D. Wang, T. Hong, Y. Wang, D. Liu, Z. Wang, X. Gao, Z.-H. Ge and L.-D. Zhao, Nat. Commun., 2023, 14, 1366 Search PubMed.
- S. Wang, C. Chang, S. Bai, B. Qin, Y. Zhu, S. Zhan, J. Zheng, S. Tang and L.-D. Zhao, Chem. Mater., 2023, 35, 755–763 Search PubMed.
- Z. Zhang, M. Gurtaran and H. Dong, ACS Appl. Energy Mater., 2024, 7, 5629–5646 CrossRef CAS PubMed.
- Q. Yang, S. Yang, P. Qiu, L. Peng, T.-R. Wei, Z. Zhang, X. Shi and L. Chen, Science, 2022, 377, 854–858 CrossRef CAS PubMed.
- Z. Gao, Q. Yang, P. Qiu, T. R. Wei, S. Yang, J. Xiao, L. Chen and X. Shi, Adv. Energy Mater., 2021, 11, 2100883 CrossRef CAS.
- X.-Y. Xin, J. Ma, H.-Q. Liu, Y.-J. Gu, Y.-F. Wang and H.-Z. Cui, J. Mater. Sci. Technol., 2023, 151, 66–72 CrossRef CAS.
- L. Yu, S.-t Wei, L.-j Wang, Z.-p Zhang, Z. Ji, S.-t Luo, J.-x Liang, W.-y Song and S.-Q. Zheng, ACS Appl. Mater. Interfaces, 2023, 15, 53594–53603 CrossRef CAS PubMed.
- R. Gupta, S. Kakkar, B. Dongre, J. Carrete and C. Bera, ACS Appl. Energy Mater., 2023, 6, 3944–3952 CrossRef CAS.
- J. Yue, S. Guo, J. Li, J. Zhao, C. Shen, H. Zhang, Y. Liu and T. Cui, Mater. Today Phys., 2023, 39, 101283 CrossRef CAS.
- N. Tabachkova, M. Shtern, A. Sherchenkov, Y. Shtern, M. Rogachev, V. Panchenko, A. Babich, M. Voronov, M. Tapero and E. Korchagin, Solid State Sci., 2024, 107609 CrossRef CAS.
- T. Ishibe, J. Chikada, T. Terada, Y. Komatsubara, R. Kitaura, S. Yachi, Y. Yamashita, T. Sato, T. Suemasu and Y. Nakamura, Appl. Phys. Lett., 2021, 119, 141603 CrossRef CAS.
- Y. Zheng, T. J. Slade, L. Hu, X. Y. Tan, Y. Luo, Z.-Z. Luo, J. Xu, Q. Yan and M. G. Kanatzidis, Chem. Soc. Rev., 2021, 50, 9022–9054 Search PubMed.
- S. Li, Z. Huang, R. Wang, C. Wang, W. Zhao, N. Yang, F. Liu, J. Luo, Y. Xiao and F. Pan, J. Mater. Chem. A, 2021, 9, 11442–11449 RSC.
- C. Artini, G. Pennelli, P. Graziosi, Z. Li, N. Neophytou, C. Melis, L. Colombo, E. Isotta, K. Lohani and P. Scardi, Nanotechnology, 2023, 34, 292001 CrossRef CAS.
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 Search PubMed.
- J. Mao, Z. Liu, J. Zhou, H. Zhu, Q. Zhang, G. Chen and Z. Ren, Adv. Phys., 2018, 67, 69–147 Search PubMed.
- B. Andriyevsky, I. Barchiy, I. Studenyak, A. Kashuba and M. Piasecki, Sci. Rep., 2021, 11, 19065 CrossRef CAS PubMed.
- X. Jia, Y. Deng, X. Bao, H. Yao, S. Li, Z. Li, C. Chen, X. Wang, J. Mao and F. Cao, npj Comput. Mater., 2022, 8, 34 CrossRef CAS.
- U. Chalapathi, B. Poornaprakash and S.-H. Park, Ceram. Int., 2017, 43, 5229–5235 CrossRef CAS.
- G. García, J. J. Fernández, P. Palacios and P. Wahnón, J. Electron. Mater., 2019, 48, 6777–6785 CrossRef.
- C. Behera, R. Samal, C. S. Rout, R. S. Dhaka, G. Sahoo and S. L. Samal, Inorg. Chem., 2019, 58, 15291–15302 CrossRef CAS PubMed.
- M. Shen, S. Lu, Z. Zhang, H. Liu, W. Shen, C. Fang, Q. Wang, L. Chen, Y. Zhang and X. Jia, ACS Appl. Mater. Interfaces, 2020, 12, 8271–8279 CrossRef CAS PubMed.
- Y. Huang, B. Zhang, J. Li, Z. Zhou, S. Zheng, N. Li, G. Wang, D. Zhang, D. Zhang and G. Han, Adv. Mater., 2022, 34, 2109952 CrossRef CAS PubMed.
- J. Andler, N. Mathur, F. Zhao and C. Handwerker, Assessing the Potential Environmental Impact of Cu3AsS4 PV Systems, 2019 IEEE 46th Photovoltaic Specialists Conference (PVSC), Chicago, IL, USA, 2019, pp. 1669–1674 DOI:10.1109/PVSC40753.2019.8981146.
- T. Shi, W.-J. Yin, M. Al-Jassim and Y. Yan, Appl. Phys. Lett., 2013, 103, 152105 Search PubMed.
- P. W. Pohwat, Rocks Miner., 2012, 87, 342–351 CrossRef.
- A. Müller and R. Blachnik, Thermochim. Acta, 2002, 387, 153–171 CrossRef.
- M. Fantauzzi, D. Atzei, B. Elsener, P. Lattanzi and A. Rossi, Surf. Interface Anal., 2006, 38, 922–930 CrossRef CAS.
- A. Pratt, Surf. Interface Anal., 2004, 36, 654–657 CrossRef CAS.
- T. Pauporté and D. Lincot, Adv. Mater. Opt. Electron., 1995, 5, 289–298 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- L. D. Whalley, J. Open Source Softw., 2018, 3, 797 CrossRef.
- R. Golesorkhtabar, P. Pavone, J. Spitaler, P. Puschnig and C. Draxl, Comput. Phys. Commun., 2013, 184, 1861–1873 CrossRef CAS.
- A. M. Ganose, J. Park, A. Faghaninia, R. Woods-Robinson, K. A. Persson and A. Jain, Nat. Commun., 2021, 12, 2222 CrossRef CAS PubMed.
- A. Togo, J. Phys. Soc. Jpn., 2023, 92, 012001 CrossRef.
- T. Fan and A. R. Oganov, Comput. Phys. Commun., 2021, 266, 108027 CrossRef CAS.
- W. Li, J. Carrete, N. A. Katcho and N. Mingo, Comput. Phys. Commun., 2014, 185, 1747–1758 Search PubMed.
- J. Henao, G. D. de Delgado, J. Delgado, F. Castrillo and O. Odreman, Mater. Res. Bull., 1994, 29, 1121–1127 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. Zhang, Y. Wang, C. Hua, S. Yang, Y. Liu, J. Luo, T. Liu, J. Nai and X. Tao, Phys. Rev. B, 2022, 106, 235401 CrossRef CAS.
- L. Cheng, M. Li, L. Zhang and H. Wang, Phys. Rev. B, 2024, 110, 155202 CrossRef CAS.
- R. R. Seal, R. A. Robie, B. S. Hemingway and H. T. Evans Jr, J. Chem. Thermodyn., 1996, 28, 405–412 CrossRef CAS.
- L. Pauling and S. Weinbaum, Z. Kristallogr. - Cryst. Mater., 1934, 88, 48–53 CrossRef CAS.
- P. Velásquez, D. Leinen, J. Pascual, J. Ramos-Barrado, R. Cordova, H. Gómez and R. Schrebler, J. Electroanal. Chem., 2000, 494, 87–95 Search PubMed.
- C. Wang and Y. Chen, npj Comput. Mater., 2020, 6, 26 CrossRef CAS.
- C. Wang, Y. Wu, Y. Pei and Y. Chen, npj Comput. Mater., 2020, 6, 155 CrossRef CAS.
- F. Mouhat and F.-X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- R. Hill, Proc. Phys. Soc., Sect. A, 1952, 65, 349 CrossRef.
- W. Voigt, BG Teubner, Lehrbuch der Kristallphysik (Textbook of crystal physics), Leipzig und Berlin, 1928 Search PubMed.
- A. Reuss, J. Appl. Math. Mech., 1929, 9, 49–58 CAS.
- M. Woodcox, J. Young and M. Smeu, Phys. Rev. B: Condens. Matter Mater. Phys., 2019, 100, 104105 CrossRef CAS.
- Y. Deng, M. Wei, Y. Lei, J. Lu, P. Peng, Y. Zhang and Z. Zheng, CrystEngComm, 2025, 27, 1055–1077 RSC.
- M. Zhu, X.-L. Shi, H. Wu, Q. Liu and Z.-G. Chen, Chem. Eng. J., 2023, 473, 145236 CrossRef CAS.
- W. Kim, J. Mater. Chem. C, 2015, 3, 10336–10348 RSC.
- X.-L. Zhu, C.-H. Hou, P. Zhang, P.-F. Liu, G. Xie and B.-T. Wang, J. Phys. Chem. C, 2019, 124, 1812–1819 CrossRef.
- S. Mukhopadhyay, L. Lindsay and D. J. Singh, Sci. Rep., 2016, 6, 37076 CrossRef CAS PubMed.
- R. A. Robie and J. L. Edwards, J. Appl. Phys., 1966, 37, 2659–2663 CrossRef CAS.
- A. Suzumura, M. Watanabe, N. Nagasako and R. Asahi, J. Electron. Mater., 2014, 43, 2356–2361 CrossRef CAS.
- R. Knura, T. Parashchuk, A. Yoshiasa and K. T. Wojciechowski, Dalton Trans., 2021, 50, 4323–4334 RSC.
- L.-D. Zhao, J. He, D. Berardan, Y. Lin, J.-F. Li, C.-W. Nan and N. Dragoe, Energy Environ. Sci., 2014, 7, 2900–2924 RSC.
- A. M. Ganose, A. Searle, A. Jain and S. M. Griffin, J. Open Source Softw., 2021, 6, 3089 CrossRef.
- S. K. Wallace, K. L. Svane, W. P. Huhn, T. Zhu, D. B. Mitzi, V. Blum and A. Walsh, Sustainable Energy Fuels, 2017, 1, 1339–1350 RSC.
- B. Durant and B. A. Parkinson, Photovoltaic response of naturally occurring semiconducting sulfide minerals, 2016 IEEE 43rd Photovoltaic Specialists Conference (PVSC), Portland, OR, USA, 2016, pp. 2774–2779 DOI:10.1109/PVSC.2016.7750157.
- N. Wang, M. Li, H. Xiao, X. Zu and L. Qiao, Phys. Rev. Appl., 2020, 13, 024038 CrossRef CAS.
- L.-D. Zhao, S.-H. Lo, Y. Zhang, H. Sun, G. Tan, C. Uher, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, Nature, 2014, 508, 373–377 CrossRef CAS PubMed.
- T. Fu, X. Yue, H. Wu, C. Fu, T. Zhu, X. Liu, L. Hu, P. Ying, J. He and X. Zhao, J. Materiomics, 2016, 2, 141–149 CrossRef.
- G. Zhang, B. Kirk, L. A. Jauregui, H. Yang, X. Xu, Y. P. Chen and Y. Wu, Nano Lett., 2012, 12, 56–60 CrossRef CAS PubMed.
- R. K. Vankayala, T. W. Lan, P. Parajuli, F. Liu, R. Rao, S. H. Yu, T. L. Hung, C. H. Lee, S. I. Yano and C. R. Hsing, Adv. Sci., 2020, 7, 2002494 Search PubMed.
- H. Zhao, J. Sui, Z. Tang, Y. Lan, Q. Jie, D. Kraemer, K. McEnaney, A. Guloy, G. Chen and Z. Ren, Nano Energy, 2014, 7, 97–103 CrossRef CAS.
- A. Watcharapasorn, R. Feigelson, T. Caillat, A. Borshchevsky, G. Snyder and J.-P. Fleurial, J. Appl. Phys., 2002, 91, 1344–1348 CrossRef CAS.
- J.-B. Vaney, J. Carreaud, G. Delaizir, C. Morin, J. Monnier, E. Alleno, A. Piarristeguy, A. Pradel, A. P. Goncalves and E. B. Lopes, J. Electron. Mater., 2016, 45, 1786–1791 Search PubMed.
- N. Soheilnia, H. Xu, H. Zhang, T. M. Tritt, I. Swainson and H. Kleinke, Chem. Mater., 2007, 19, 4063–4068 CrossRef CAS.
- X. Tan, K. P. Devlin, X. Deng, C.-J. Kang, M. Croft, C. E. Frank, C. Pak, S. Lapidus, S. M. Kauzlarich and V. Taufour, Chem. Mater., 2018, 30, 4207–4215 CrossRef CAS.
- J. Wang, S.-Q. Xia and X.-T. Tao, Inorg. Chem., 2012, 51, 5771–5778 CrossRef CAS PubMed.
- A. V. Powell, J. Appl. Phys., 2019, 126, 100901 Search PubMed.
- T. Bernard, M. A. Malagutti, K. Lohani, M. D’Incau, N. Ataollahi and P. Scardi, J. Mater. Sci., 2024, 59, 15491–15503 CrossRef CAS.
- B. S. Shah, J. B. Raval, D. Kumar, S. H. Chaki and M. Deshpande, J. Alloys Compd., 2023, 938, 168566 CrossRef CAS.
- M. A. Malagutti, K. Lohani, I. Caño Prades, A. Navarro-Güell, T. Bernard, A. Chiappini, E. Saucedo, N. Ataollahi and P. Scardi, ACS Appl. Electron. Mater., 2024, 7, 21–32 CrossRef.
- L. Li, J. Wan, C. Lin, Y. Wu, Z. Cheng, Y. Lin, S. Yang and M. Luo, Small, 2025, 2503137 Search PubMed.
- W. Li, S. Lin, X. Zhang, Z. Chen, X. Xu and Y. Pei, Chem. Mater., 2016, 28, 6227–6232 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The convergence tests to calculate electron transport properties; the detailed description of Cahill–Pohl and Slack models; the convergence test and lattice thermal conductivity calculated through phono3py and ShengBTE; the relaxation time, electrical conductivity, Seebeck coefficient, thermoelectric power factor, electronic and total thermal conductivity, and figure-of-merit for the electron doped system; and the transport and thermoelectric properties at specific carrier concentration and temperature. See DOI: https://doi.org/10.1039/d5tc00351b |
| This journal is © The Royal Society of Chemistry 2025 |