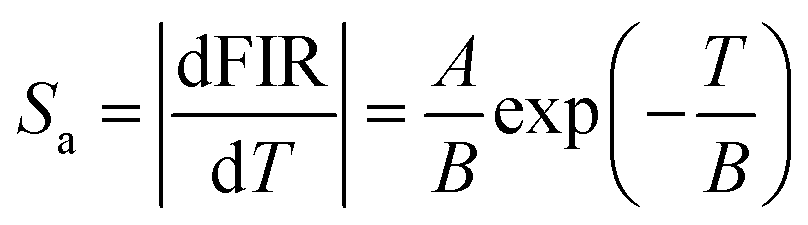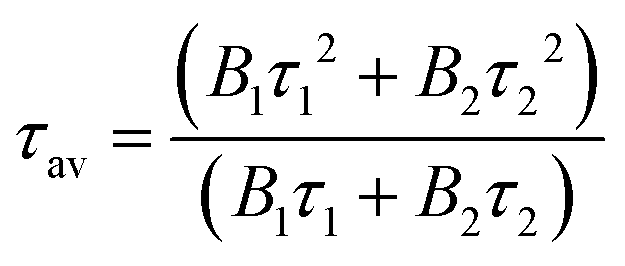DOI:
10.1039/D5TC00156K
(Paper)
J. Mater. Chem. C, 2025,
13, 8032-8042
Crystal growth and temperature dependence of luminescence characteristics of Pr3+ and Tb3+ doped solid-solution sesquioxide single crystals
Received
14th January 2025
, Accepted 3rd March 2025
First published on 5th March 2025
Abstract
In recent decades, optical thermometry has gained significant attention due to its promising properties, such as non-contact measurement, temperature mapping and immunity to electromagnetic interference. It overcomes the limitations of conventional temperature measurement methods and offers additional benefits. However, the widespread adoption of optical thermometry requires an expanded operating temperature range and improved sensitivity. Therefore, in this study, we focused on solid-solution sesquioxides, which are expected not only to enable the growth of single crystals with a cubic structure but also to allow for a wider selection of luminescence centers. We evaluated the applicability of (Lu, Y, Sc)2O3 single crystals doped with Pr3+ and Tb3+, which exhibit different temperature dependent behaviors, for optical thermometry. The optical temperature sensing properties evaluated using the fluorescence lifetime method revealed a maximal relative sensitivity of 1.53% K−1 in the temperature range of 78–790 K for Pr3+, Tb3+:(Lu, Y, Sc)2O3. These results suggest that (Lu, Y, Sc)2O3 co-doped with Pr3+ and Tb3+ is a promising candidate for optical thermometry.
1. Introduction
Sesquioxides such as Sc2O3, Lu2O3 and La2O3 have drawn considerable attention due to their promising properties, high thermal and chemical stability, high thermal conductivity, relatively wide band gap, high density, wide range of optical transparency and low phonon energy.1–3 Therefore, sesquioxides are attractive optical host materials for applications such as substrates, lasers, scintillators and optical thermometry. These optical materials require single crystals with low scattering and absorption inside the crystal; however, sesquioxides have a high melting point (above 2400 °C) and undergo phase transitions, making it difficult to obtain high quality and large size single crystals. For crystal growth above the melting point of 2400 °C, the iridium crucible (softening point temperature: 2200 °C), which is generally used for crystal growth from the melts, cannot be used. Other candidates for crucible materials are rhenium (softening point temperature: 2800 °C) and tungsten (softening point temperature: 3000 °C); however, rhenium is extremely expensive and tungsten is reactive with raw materials. Recently, crystal growth using a tungsten crucible has been investigated, and it has been reported that La2Hf2O7 (melting point: ∼2418 °C) single crystals were successfully grown.4 Concerning the structural phase transitions, most sesquioxides undergo polymorphic transitions and have five phases as a function of temperature and ionic radius.5 Crystals that undergo a phase transition upon cooling have low structural quality due to severe cracking caused by lattice expansion and contraction.6 Here, the structural phase transition of sesquioxides as a function of temperature and ionic radius was reported, according to which it might be possible to grow sesquioxides in the cubic phase by adjusting the average ionic radius of rare-earth ions below the Lu3+ ionic radius.5,6 Furthermore, expanding the choice of luminescence centers by mixing multiple sesquioxides will be considered for optical thermometry applications.
In recent years, interest in optical thermometry has grown rapidly due to its wide range of possibilities.7,8 Optical thermometry enables accurate, fast and non-invasive measurement in scientific research, industry, production and biomedicine. It has the potential to be used in harsh environments exposed to high temperature, high pressure and strong electromagnetic radiation, e.g., high-voltage power plants, volcanic fire detection, oil refineries and corrosive circumstances unlike any of the conventional thermometry.9 Focusing on the utilization of sesquioxides as host materials, investigations of optical thermometers doped with Yb3+/Er3+, Ho3+/Yb3+ as luminescence centers have been reported.10,11 This study focused on Pr3+ and Tb3+ as luminescence centers with different characteristics: Pr3+ has a red emission wavelength, whereas Tb3+ has a green emission wavelength. Moreover, Pr3+ is characterized by its resistance to thermal quenching, while Tb3+ has a temperature dependence at high temperatures.12 In this study, we adjust the average ionic radius using solid solutions of several rare-earth ions to grow novel sesquioxide solid solution single crystals and characterize their optical thermometry.
2. Materials and methods
(PrxTbyLu0.250Y0.250–x–ySc0.500)2O3 (Pr3+, Tb3+:(Lu, Y, Sc)2O3, (x, y) = (0.005, 0.000), (0.000, 0.005), and (0.005, 0.005)) crystals were grown by the micro-pulling-down (μ-PD) method.13 The starting raw materials, Y2O3 (99.999%), Lu2O3 (99.995%), Sc2O3 (99.9%), Pr2O3 (99.9%) and Tb4O7 (99.99%) powders were mixed and filled into a W crucible with ϕ4 mm die with ϕ0.8 mm capillary. The crystals were grown using an μ-PD furnace with a radio-frequency induction heating system and a vacuum-tight chamber under the following growth conditions: a pulling down rate of 0.05 mm min−1, deoxygenated ZrO2 insulators, W rod as the seed, and Ar + 2% H2 gas flow. The liquid–solid interface was observed using a charge-coupled device camera during the crystal growth. After crystal growth, the crystals were annealed at 1200 °C for 12 hours under an air atmosphere to suppress oxygen defects. A 1 mm thick plate was cut from the grown crystal and mirror polished for optical characterization.
The crystalline system and space group of the grown crystals were identified by the powder X-ray diffraction (XRD) analysis with the Cu-Kα (λ = 0.15418 nm) target (D8 DISCOVER, Bruker). All samples were measured in the 2θ range of 10° to 80° with a step range of 0.01° to 0.02°. The chemical composition was determined by electron probe microanalysis (EPMA, S-3400N, Hitachi). Transmittance spectra were measured using a UV/vis spectrophotometer (V-550, JASCO) in the wavelength range of 190–800 nm. The photoluminescence (PL) emission and excitation spectra were measured using a spectrometer (FLS 1000, Edinburgh Instruments) with a Xe lamp. The temperature dependence of PL spectra and PL decay time was determined using the spectrometer combined with a cryostat (VPF-100-H, Lake Shore Cryotronics) and using a microsecond flashlamp (μF2, Edinburgh Instruments). During the measurement, the temperature stabilization time was set to 600 s and temperature tolerance to ±1 K. The temperature dependence evaluation was performed in the temperature range of 78–790 K.
3. Results and discussion
3.1 Structural properties
Transparent crystals were obtained in all compositions by the μ-PD method (see Fig. 1). After air annealing at 1200 °C, the color of all samples changed from black to orange. Such change from black to transparent is attributed to oxygen defects, which have been observed in the growth of single crystals using a W crucible.4,14 It seems to be caused by the result from oxygen defects caused by growing under a reducing atmosphere. Meanwhile, the color change to orange is supposed to be due to the tetravalent ions in the luminescence center (see Section 3.2). To confirm the crystalline system and space group, powder XRD was performed (Fig. 2). All diffraction peaks were consistent with the reference (CCDC 1992594,15 Nd3+:YScO316), and the crystalline system and space group were identified to be cubic and Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , respectively. These crystal structures belong to the bixbyite-type cubic structure and have two symmetrically different cation sites.16Table 1 shows the EPMA results of the grown samples. The actual composition of the host materials Lu2O3, Y2O3, and Sc2O3 approximately matches the nominal composition; on the other hand, the actual composition of the luminescence center is lower than the nominal composition. Pr3+ and Tb3+ used as luminescence centers are considered to be substituted into the Y3+ site with the closest ionic radius. However, these ionic radii of six coordination are Pr3+ (0.1013 nm), Tb3+ (0.0923 nm), and Y3+ sites (0.0892 nm),17 and these differences in the ionic radii of Y3+ and the luminescence centers caused the difference between the nominal and actual compositions.
, respectively. These crystal structures belong to the bixbyite-type cubic structure and have two symmetrically different cation sites.16Table 1 shows the EPMA results of the grown samples. The actual composition of the host materials Lu2O3, Y2O3, and Sc2O3 approximately matches the nominal composition; on the other hand, the actual composition of the luminescence center is lower than the nominal composition. Pr3+ and Tb3+ used as luminescence centers are considered to be substituted into the Y3+ site with the closest ionic radius. However, these ionic radii of six coordination are Pr3+ (0.1013 nm), Tb3+ (0.0923 nm), and Y3+ sites (0.0892 nm),17 and these differences in the ionic radii of Y3+ and the luminescence centers caused the difference between the nominal and actual compositions.
 |
| | Fig. 1 Photographs of the grown (a) Pr3+:(Lu, Y, Sc)2O3, (b) Tb3+:(Lu, Y, Sc)2O3 and (c) Pr3+, Tb3+:(Lu, Y, Sc)2O3 crystals (left panel: before annealing, right panel: after annealing). | |
 |
| | Fig. 2 XRD patterns of the Pr3+, Tb3+:(Lu, Y, Sc)2O3 specimens. | |
Table 1 Result of composition analysis by the EPMA [at%]. (C0: nominal concentration, C: actual concentration)
|
|
|
Pr3+:(Lu, Y, Sc)2O3 |
Pr3+, Tb3+:(Lu, Y, Sc)2O3 |
Tb3+:(Lu, Y, Sc)2O3 |
| Lu |
C
0
|
25.00 |
25.00 |
25.00 |
|
C
|
24.83 |
26.10 |
23.93 |
|
C/C0 |
0.99 |
1.04 |
0.96 |
| Y |
C
0
|
24.50 |
24.00 |
24.5 |
|
C
|
24.34 |
22.75 |
25.54 |
|
C/C0 |
0.99 |
0.95 |
1.04 |
| Sc |
C
0
|
50.00 |
50.01 |
50.00 |
|
C
|
50.53 |
50.60 |
49.98 |
|
C/C0 |
1.01 |
1.01 |
1.00 |
| Pr |
C
0
|
0.50 |
0.50 |
— |
|
C
|
0.29 |
0.19 |
— |
|
C/C0 |
0.58 |
0.34 |
— |
| Tb |
C
0
|
— |
0.49 |
0.50 |
|
C
|
— |
0.37 |
0.55 |
|
C/C0 |
— |
0.75 |
1.10 |
3.2 Optical properties
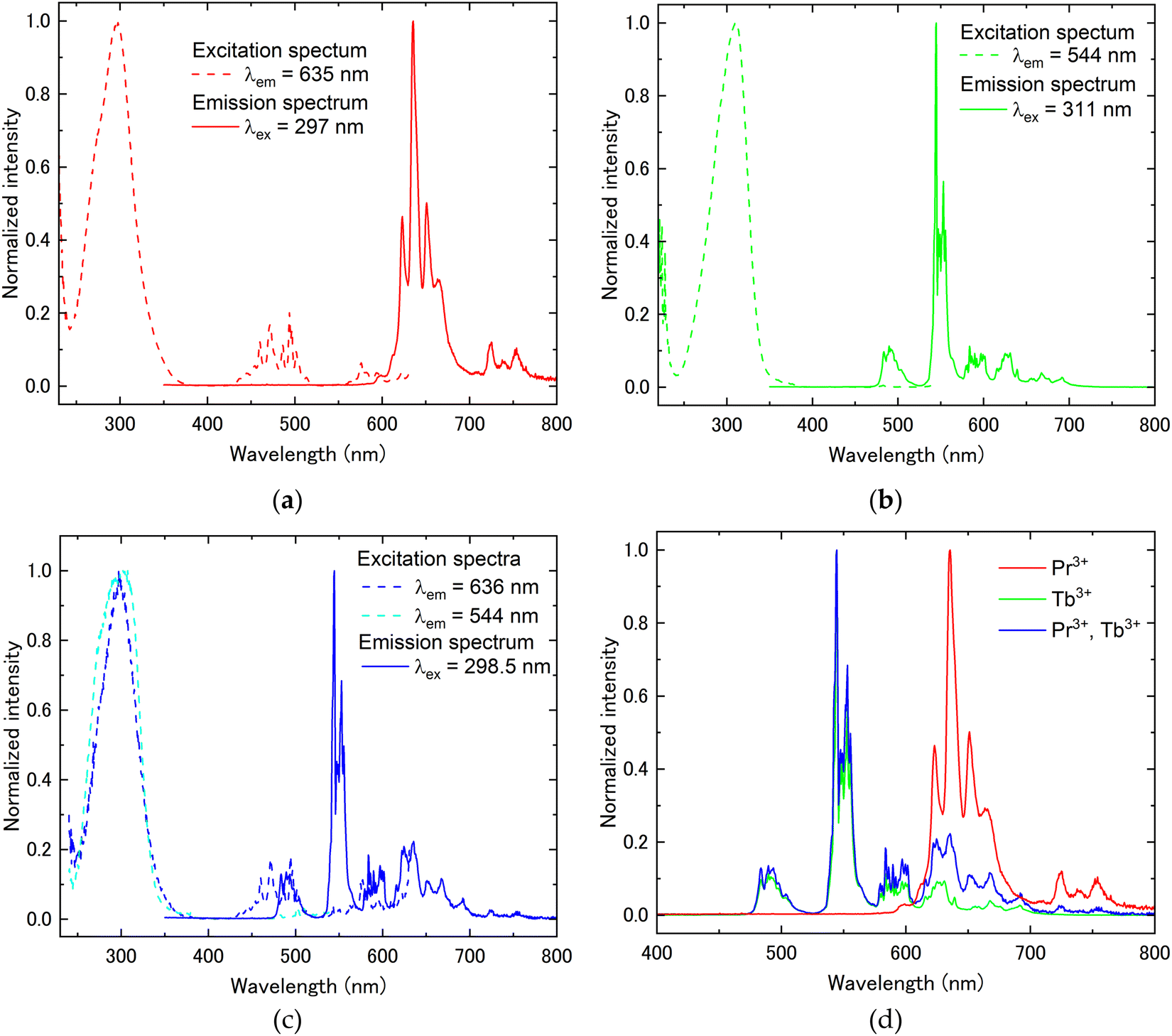
Fig. 3 shows the transmittance spectra in the wavelength range of 200–800 nm. All samples showed high transparency of around 80% in the wavelength region where there is no absorption. In the Pr3+-doped and Pr3+, Tb3+ co-doped samples, sharp absorption peaks originating from the Pr3+ 4f–4f transition were observed in the wavelength region of 450–600 nm. Additionally, the broad absorption peak due to interconfigurational 4f–5d transitions of Pr3+ or Tb3+ was observed in the wavelength range of below 400 nm. The broad band of absorption spectra at around 400–600 nm was attributed to the charge transfer (CT) between O2− and Tb4+, as well as between O2− and Pr4+.18,19 It has been reported that annealing at high temperatures induces CT states, resulting in the orange coloration of the sample. The orange coloration observed in this study is consistent with this phenomenon. Based on the transmittance measurement results, PL excitation and emission spectra were subsequently measured. Fig. 4(a) presents the PL excitation and emission spectra of the Pr3+:(Lu, Y, Sc)2O3 sample. In the excitation spectrum, the broad excitation peak attributed to the Pr3+ 4f–5d transition and the sharp excitation peaks attributed to Pr3+ 3H4–3P0,1,2 transitions were observed at around 240–380 nm and 430–530 nm, respectively. In the emission spectrum excited by the Pr3+ 4f–5d transition (302 nm), some sharp peaks from 590 to 780 nm relative to Pr3+ 1D2–3H4 and 1D2–3H5 transitions were observed. The emission from the Pr3+ 5d–4f transition was not observed due to the narrow bandgap of sesquioxide. Other than the emission from the Pr3+ 5d–4f transition, the emission from the Pr3+ 3Pj level was also not observed. The Pr3+ 4f15d1 level is localized within the conduction band, resulting in the formation of the self-trapped exciton-like state, [Pr4+ + eCB], where eCB is an electron in the conduction band. As a result, the emission from 3Pj[=0,1,2] is quenched as the excited electrons bypass the 3Pj level by the exciton-like state and preferentially cause a non-radiative transition to the 1D2.20 The excitation and emission spectra of Tb3+:(Lu, Y, Sc)2O3 are depicted in Fig. 4(b). In the excitation spectrum, at the emission peak wavelength of 544 nm, only the broad excitation peak attributed to the Tb3+ 4f–5d transition was observed at around 240–375 nm because the intensities of Tb3+ 4f–4f transitions were quite low. In the emission spectrum excited at 311 nm (Tb3+ 4f–5d transition), several sharp peaks were observed from 460 nm to 710 nm, which were ascribed to the Tb3+ 4f–4f transitions (5D4–7F6 (470–525 nm), 5D4–7F5 (530–575 nm), 5D4–7F4 (575–610 nm), and 5D4–7F3 (610–645 nm)). Fig. 4(c) displays the excitation and emission spectra of Pr3+, Tb3+:(Lu, Y, Sc)2O3. The excitation spectra were measured for two different emissions: the Pr3+ 1D2–3H4 transition (636 nm) and the Tb3+ 5D4–7F5 transition (544 nm). Both excitation peaks were observed at approximately the same wavelength (300 nm). In the emission spectrum excited at 298.5 nm, sharp peaks attributed to the Tb3+ and Pr3+ 4f–4f transitions were observed between 470 and 765 nm. For a better understanding of the Pr3+ and Tb3+ luminescence, the emission spectra for the enlarged wavelength range from 400 to 800 nm are shown in Fig. 4(d). From Fig. 4(d), the sharp emission peaks at 470 to 605 nm are considered to be the emission from the Tb3+ 5D4–7Fj transitions. In contrast, there are several sharp emission peaks at wavelengths from 605 to 765 nm associated with the transitions from the Tb3+ 5D4 level and Pr3+ 1D2 level. As mentioned above, (Lu, Y, Sc)2O3 has two crystallographically unique rare earth (RE) sites, the C2 symmetry site (Wyckoff-24d) and the C3i symmetry site (Wyckoff-8b).21 In order to evaluate the luminescence from each RE site, the emission spectra were evaluated using different excitation wavelengths. Fig. 5(a) shows the PL excitation spectra at emission wavelengths of 612.5 and 635 nm (dashed lines) and the PL emission spectra at the excitation wavelength of 297 and 319 nm (solid lines) for Pr3+:(Lu, Y, Sc)2O3. The excitation spectrum at λem = 612.5 nm shows double broad peaks at 285 nm and 320 nm, whereas at λem = 635 nm, it shows a single broad peak at 296 nm. It was caused by the luminescence from the Pr3+ occupying different RE sites. To gain a more detailed understanding of luminescence from different RE sites, the PL decay time was evaluated. The PL decay curve measured at λex = 297 nm and λem = 635 nm (D1) or λex = 319 nm and λem = 612.5 nm (D2) are shown in Fig. 5(b). The decay curves were fitted by the exponential function and the decay times were calculated (Table 2). Both decay curves exhibit decay time components of approximately 40–50 μs and 140–150 μs, with only D2 showing an additional slower component of 466 μs. The emission peak wavelengths at 635 nm and 612.5 nm corresponded to the Pr3+ 1D2–3H4 transitions, which are the electric dipole transitions. Here, it has been reported that electric dipole transitions are suppressed at the C3i site with the inversion symmetry in the bixbyite-type structure, resulting in low transition probabilities and long decay times.22 Therefore, a comparison of the PL decay times revealed that the double broad excitation peaks around 319 nm and the broad excitation peak at 297 nm are due to the Pr3+ 4f–5d transition occupying the C3i symmetry site and the C2 symmetry site, respectively.22 The fact that the decay curve contains more than two components might be explained by the difference in the adjacent coordination spheres of Pr3+, as also suggested in ref. 16. Furthermore, the decay time at an excitation wavelength of 297 nm (Pr3+ 4f–5d transition) was slower than that at the excitation wavelength of 494 nm (Pr3+ 4f–4f transition). This seems to be caused by the complex energy transition processes of the electrons excited in the Pr3+ 5d level, as mentioned earlier. The excitation and emission spectra of Tb3+:(Lu, Y, Sc)2O3 at different excitation and emission peak wavelengths are depicted in Fig. 6(a). The PL decay curves measured at the same excitation and emission wavelength pairs as the PL spectra are shown in Fig. 6(b). The excitation spectra showed that the excitation peak wavelength changes depending on the emission wavelength. The PL decay times were evaluated from the decay curves in Fig. 6(b). It was found that both decay curves have two decay components of approx. 1000 μs and 4000 μs (Table 2). Focusing on the ratio of the two components, the decay curve with (λex, λem) = (288 nm, 542.5 nm) has a higher ratio of the fast component (88.5%), while (λex, λem) = (311 nm, 544 nm) has a higher ratio of the slow component (76.0%). Considering the difference in the symmetry of the RE sites as in Pr3+:(Lu, Y, Sc)2O3, we conclude that the emission peak at 542.5 nm and 544 nm are dominated by luminescence from Tb3+ in the C2 and the C3i sites, respectively.23 The two components originating from the two RE sites were obtained in both decay curves, which might be due to the overlap of the excitation and emission wavelengths. Since the Pr3+, Tb3+ co-doped sample showed a complex overlap of Pr3+ and Tb3+ luminescence, we focused on the Tb3+ luminescence for the effect of the different RE sites
occupied by the luminescence centers. According to Fig. 7(a), similar changes were observed for the Pr3+, Tb3+ co-doped sample as in Tb3+:(Lu, Y, Sc)2O3. The decay curves also exhibit two components (Table 2), consistent with the Tb3+:(Lu, Y, Sc)2O3, see Fig. 7(b). In addition, the decay curves and decay times of Pr3+, Tb3+:(Lu, Y, Sc)2O3 associated with the Pr3+ 1D2–3H4 transition, measured at λex = 297 nm and λem = 651 nm, are shown in Fig. 7(b) and Table 2, respectively. A slow component of about 1 ms, which was not observed for the same transition in Pr3+:(Lu, Y, Sc)2O3, was obtained in the Pr3+, Tb3+ co-doped sample. This is attributed to energy transfer from Tb3+ 5D4 to Pr3+ 3P0.24,25
 |
| | Fig. 3 Transmittance spectra of Pr3+ and/or Tb3+:(Lu, Y, Sc)2O3. | |
 |
| | Fig. 4 PL excitation and emission spectra of (a) Pr3+:(Lu, Y, Sc)2O3, (b) Tb3+:(Lu, Y, Sc)2O3, (c) Pr3+, Tb3+:(Lu, Y, Sc)2O3 and (d) emission of all samples overlapping at 400 to 800 nm. | |
 |
| | Fig. 5 (a) PL excitation and emission spectra and (b) PL decay curves of Pr3+:(Lu, Y, Sc)2O3. | |
Table 2 PL decay time under various excitation and emission wavelengths
|
|
Excitation [nm] |
Emission [nm] |
PL decay time (ratio [%]) [μs] |
|
τ
1
|
τ
2
|
τ
3
|
ave. |
| Pr3+:(Lu, Y, Sc)2O3 |
297 |
635 |
52.8 (70.0) |
140.5 (30.0) |
|
79.1 |
| 319 |
612.5 |
40.1 (17.6) |
151.8 (31.9) |
466.7 (50.5) |
291.0 |
| 494 |
635 |
31.4 (46.1) |
74.4 (53.9) |
|
54.6 |
| Tb3+:(Lu, Y, Sc)2O3 |
311 |
544 |
1180 (24.0) |
4423 (76.0) |
|
3644 |
| 288 |
542.5 |
1050 (88.5) |
3834 (11.5) |
|
1371 |
| Pr3+, Tb3+:(Lu, Y, Sc)2O3 |
298.5 |
544 |
932.3 (41.4) |
4142.4 (58.6) |
|
2814.9 |
| 281 |
542.5 |
912.3 (91.8) |
2619.4 (8.2) |
|
1052.8 |
| 297 |
651 |
23.6 (28.6) |
75.6 (51.9) |
1137 (19.5) |
267.6 |
| 494 |
651 |
30.3 (52.6) |
69.3 (47.4) |
|
48.7 |
 |
| | Fig. 6 (a) PL excitation and emission spectra and (b) PL decay curves of Tb3+:(Lu, Y, Sc)2O3. | |
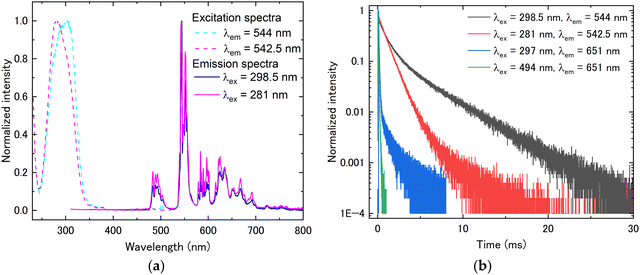 |
| | Fig. 7 (a) PL excitation and emission spectra and (b) PL decay curves of Pr3+, Tb3+:(Lu, Y, Sc)2O3. | |
3.3 Temperature characterization
The potential for application as an optical thermometer was assessed using the fluorescence intensity ratio (FIR) method and the fluorescence lifetime (FL) method based on the temperature dependence of the PL decay time. Initially, the FIR method was used for the evaluation. As shown in Fig. 4, the Pr3+:(Lu, Y, Sc)2O3 and Tb3+:(Lu, Y, Sc)2O3 exhibited luminescence from the Pr3+ 1D2 excited level and Tb3+ 5D4 excited level, respectively. Thus, only the Pr3+, Tb3+ co-doped sample was investigated by the FIR method. Fig. 8(a) illustrates the temperature dependence of the PL emission spectra for the co-doped sample excited at 298.5 nm, measured over the temperature range of 78 to 790 K. The emission peaks originating from the Tb3+ 5D4–7F5 (527–574 nm) transition significantly decreased with increasing temperature beyond 360 K. On the other hand, the emission peaks attributed to the Pr3+ 1D2–3H4 transition (645–660 nm) gradually decreased with increasing temperature beyond 450 K. Using the integral areas of these two emissions, the FIR was calculated using eqn (1):26,27| |  | (1) |
where A, B and C represent constants and T represents absolute temperature [K]. The FIR fitting parameters and fitting curve are depicted in Fig. 8(b), where FIR (IPr3+/ITb3+) values monotonously increased with increasing temperature in the temperature range of 450–660 K. The absolute sensitivity (Sa) and the relative sensitivity (Sr) are important factors to evaluate the optical temperature sensing properties. The Sa and Sr were described using the following equations, respectively:26| |  | (2) |
| |  | (3) |
The maximal Sr value of Pr3+, Tb3+:(Lu, Y, Sc)2O3 was 0.72% K−1 at 660 K in the temperature range of 450–660 K (Fig. 8(c)). Some reported optical thermometry materials and their associated parameters are listed in Table 3. A comparison with the data in Table 3 indicates that the Sr values of the Pr3+, Tb3+:(Lu, Y, Sc)2O3 are relatively higher than those of other optical thermometry materials.
 |
| | Fig. 8 (a) Temperature dependence of the PL spectra (λex = 298.5 nm) in the temperature range of 78–790 K, (b) the FIR versus temperature and fitted curve and (c) the absolute and relative sensitivity as a function of temperature at 450–660 K of Pr3+, Tb3+:(Lu, Y, Sc)2O3. | |
Table 3 Optical temperature sensing properties of Pr3+ and/or Tb3+:(Lu, Y, Sc)2O3 samples and conventional materials
| Host material |
Luminescence center |
Transitions |
MAX Sr [% K−1] |
ΔT [K] |
Method |
Ref. |
| La2O3 |
Tm3+, Yb3+ |
1G4–3H6 |
0.16 |
298–333 |
FL |
28
|
| Y2O3 |
Yb3+, Er3+ |
2H11/2–4F2/9 |
0.50 |
298–338 |
FL |
10
|
| Y2O3 |
Er3+ |
2H11/2–4I15/2 |
1.1 |
RT–1473 |
FL |
29
|
| Gd2ZnTiO6 |
Pr3+ |
1D2–3H4 |
1.48 |
433–593 |
FL |
30
|
| Y2O3 |
Tm3+, Yb3+ |
1G4(a)–3H6 |
0.35 |
303–753 |
FIR |
31
|
|
1G4(b)–3H6 |
| NaYF4 |
Yb3+, Er3+ |
Er3+ 520, 540 nm |
0.46 |
283–338 |
FIR |
32
|
| LaB3O6 |
Bi3+, Eu3+ |
Bi3+ 395–405 nm |
0.579 |
298–548 |
FIR |
33
|
| Eu3+ 695–705 nm |
| YNbO4 |
Pr3+, Tb3+ |
5D4–7F5 |
1.01 |
298–538 |
FIR |
34
|
|
1D2–3H4 |
| Y2O3 |
Ho3+, Yb3+ |
5F4/5S2–5I8 |
1.03 |
298–473 |
FIR |
11
|
|
5F5–5I8 |
| (Lu, Y, Sc)2O3 |
Tb3+ |
5D4–7F5 |
1.71 |
380–790 |
FL |
This work |
| Pr3+, Tb3+ |
5D4–7F5 |
1.53 |
390–790 |
FL |
|
1D2–3H4 |
1.53 |
78–790 |
FL |
|
5D4–7F5 |
0.72 |
450–660 |
FIR |
|
1D2–3H4 |
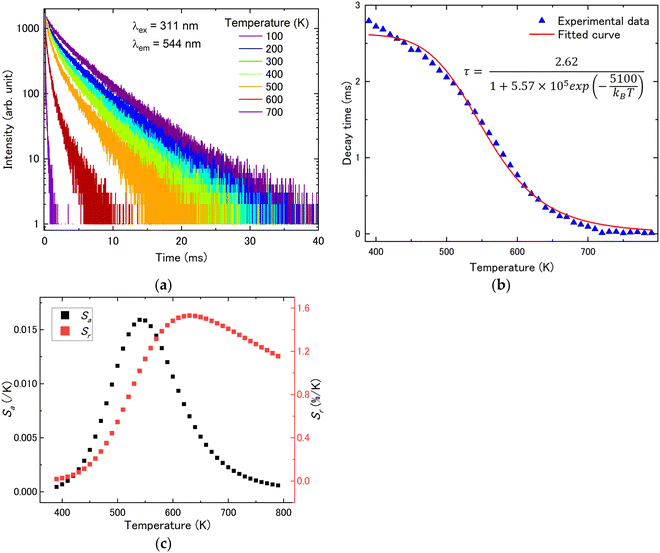
Subsequently, the FL method was used for evaluation. The temperature dependence of the PL decay times for Pr3+:(Lu, Y, Sc)2O3 (λex = 297 nm and λem = 635 nm), Tb3+:(Lu, Y, Sc)2O3 (λex = 311 nm and λem = 544 nm) and Pr3+, Tb3+:(Lu, Y, Sc)2O3 (λex = 311 nm and λem = 544 nm, λex = 297 nm and λem = 651 nm) from 78 K to 790 K is shown in Fig. 9(a), 10(a), 11(a) and 12(a). The average decay times were calculated using the following equation and are displayed in Fig. 9(b), 10(b), 11(b) and 12(b):35
| |  | (4) |
where
B1 and
B2 are constants and
τ1 and
τ2 are decay times. In Pr
3+:(Lu, Y, Sc)
2O
3, the PL decay time was accelerated due to the thermal quenching from around 700 K, however, it was not fully quenched even at 790 K. Therefore, Pr
3+:(Lu, Y, Sc)
2O
3 is expected to have applications at even higher temperatures. For Tb
3+:(Lu, Y, Sc)
2O
3 and Pr
3+, Tb
3+:(Lu, Y, Sc)
2O
3, a similar acceleration of the PL decay time due to the thermal quenching was observed from around 500 K. The relationship between the temperature and decay time is expressed by the following equation:
36–38| |  | (5) |
where
D,
τ,
τ0, Δ
E and
kB represent the pre-exponential constant, decay time, decay time at 0 K, the activation energy for the thermal quenching and the Boltzmann constant, respectively. The fitting parameters and fitting curve are indicated in
Fig. 10(b),
Fig. 11(b) and
Fig. 12(b). Tb
3+:(Lu, Y, Sc)
2O
3, Pr
3+, Tb
3+:(Lu, Y, Sc)
2O
3 (
λex = 311 nm and
λem = 544 nm) and Pr
3+, Tb
3+:(Lu, Y, Sc)
2O
3 (
λex = 297 nm and
λem = 651 nm) were well fitted in the temperature range of 380–790 K, 390–790 K and 78–790 K, respectively. Notably, here is the thermal quenching process of Pr
3+, Tb
3+ co-doped (Lu, Y, Sc)
2O
3, focusing on the Pr
3+ 1D
2–
3H
4 transition. In the Pr
3+:(Lu, Y, Sc)
2O
3, thermal quenching did not occur completely even at 790 K. In contrast, in the Pr
3+, Tb
3+ co-doped (Lu, Y, Sc)
2O
3, thermal quenching was observed starting from around 500 K. This temperature is consistent with the onset of thermal quenching for the Tb
3+ 5D
4–
7F
5 transition in Tb
3+:(Lu, Y, Sc)
2O
3. Here, focusing on the thermal quenching mechanism, thermal quenching can occur not only through the thermal ionization of electrons excited to the excitation level of the luminescence center into the conduction band but also
via nonradiative relaxation through an intermediate level (
e.g. intervalence charge transfer (IVCT),
34,39,40 Ce
4+ + e
CB41). In the thermal quenching process of IVCT, which is formed by electron transfer from Pr
3+ and Tb
3+ to metal ions with d
0 configurations, electrons in the excited states relax to the ground state through the IVCT state.
34,42 Regarding the thermal quenching of Ce
3+, it can occur that excited electrons relax to the ground state
via the Ce
4+ + e
CB state.
41 Thus, in this study, considering that self-trapped exciton-like states are formed when 5d levels of Pr
3+ and Tb
3+ are located within the conduction band, thermal quenching through these self-trapped exciton-like levels might have occurred, leading to the difference in the quenching temperature between Pr
3+:(Lu, Y, Sc)
2O
3 and Tb
3+:(Lu, Y, Sc)
2O
3. Moreover, in the Pr
3+, Tb
3+ co-doped (Lu, Y, Sc)
2O
3, the slow decay component associated with energy transfer from Tb
3+ 5D
4 to Pr
3+ 3P
0, as described in Section 3.2, may have contributed to thermal quenching when focusing on the luminescence of the Pr
3+ 1D
2–
3H
4 transition. Based on the fitted results, we calculated
Sa and
Sr using the following equations, respectively:
36,37| |  | (6) |
| |  | (7) |
For the Tb
3+:(Lu, Y, Sc)
2O
3, Pr
3+, Tb
3+:(Lu, Y, Sc)
2O
3 (
λex = 311 nm and
λem = 544 nm) and Pr
3+, Tb
3+:(Lu, Y, Sc)
2O
3 (
λex = 297 nm and
λem = 651 nm), the maximal
Sr reached 1.71% K
−1 at 610 K, 1.53% K
−1 at 630 K and 1.53% K
−1 at 630 K, respectively (
Fig. 10(c),
Fig. 11(c) and
Fig. 12(c)). It is worth noting that the high
Sr values of >0.5% K
−1 are achieved at temperatures of 490–790 K for Tb
3+:(Lu, Y, Sc)
2O
3 and 500–790 K for both Pr
3+, Tb
3+:(Lu, Y, Sc)
2O
3. The optical temperature sensing properties of the Pr
3+ and/or Tb
3+:(Lu, Y, Sc)
2O
3 and the conventional materials are summarized in
Table 3. The Pr
3+ and/or Tb
3+:(Lu, Y, Sc)
2O
3 were found to be promising materials for optical thermometer applications due to their relatively high
Sr values over a wide temperature range.
 |
| | Fig. 9 Temperature dependence of (a) PL decay curves in the temperature range of 78–790 K (λex = 297 nm and λem = 635 nm) and (b) average decay times of Pr3+:(Lu, Y, Sc)2O3. | |
 |
| | Fig. 10 (a) Temperature dependence of PL decay curves in the temperature range of 78–790 K (λex = 311 nm and λem = 544 nm), (b) average decay times and (c) the absolute and relative sensitivity as a function of temperature at 380–790 K of Tb3+:(Lu, Y, Sc)2O3. | |
 |
| | Fig. 11 (a) Temperature dependence of PL decay curves in the temperature range of 78–790 K (λex = 311 nm and λem = 544 nm), (b) average decay times and (c) the absolute and relative sensitivity as a function of temperature at 390–790 K of Pr3+, Tb3+:(Lu, Y, Sc)2O3. | |
 |
| | Fig. 12 (a) Temperature dependence of PL decay curves in the temperature range of 78–790 K (λex = 297 nm and λem = 651 nm), (b) average decay times and (c) the absolute and relative sensitivity as a function of temperature at 78–790 K of Pr3+, Tb3+:(Lu, Y, Sc)2O3. | |
4. Conclusions
We focused on (Lu, Y, Sc)2O3 solid-solution sesquioxides in order to grow single crystals without phase transition. (PrxTbyLu0.250Y0.250–x–ySc0.500)2O3 single crystals have been grown using the micro-pulling-down method with a W crucible. From the powder XRD results, the crystalline system and space group were identified to be cubic and Ia, respectively. Moreover, (Lu, Y, Sc)2O3 has two symmetrically different RE sites (C2 symmetry and C3i symmetry), and the PL excitation and emission spectra showed luminescence originating from the luminescence centers (Pr3+ or Tb3+) occupying each RE site. From the results of the optical temperature sensing properties by the FIR method, the maximal Sr of 0.72% K−1 was observed at 660 K for Pr3+, Tb3+:(Lu, Y, Sc)2O3. In addition, evaluation by the FL method revealed the maximal Sr value of 1.71% K−1 at 610 K for Tb3+:(Lu, Y, Sc)2O3 and 1.53% K−1 at 630 K for the Pr3+, Tb3+:(Lu, Y, Sc)2O3. In particular, when focusing on the Pr3+ transition, the Pr3+, Tb3+:(Lu, Y, Sc)2O3 exhibited a significantly broadened temperature range from 78 K to 790 K based on the energy transfer from Tb3+ to Pr3+. It was found that Pr3+ and/or Tb3+ doped (Lu, Y, Sc)2O3 exhibit great optical thermometry performance and relatively high relative sensitivity.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the Japan Society for the Promotion of Science (JSPS) KAKENHI, the Grant-in-Aid for Scientific Research [K22K144690] is gratefully acknowledged. Moreover, this work is supported by (i) Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) from the Japan Science and Technology Agency (JST) Japan Grant Number JPMJTR232C and (ii) GIMRT Program of the Institute for Materials Research, Tohoku University and Cooperative Research and Development Center for Advanced Materials (Proposal No. 202312-CRKEQ-0410).
References
- G. Feng, Y. Wu, H. Lu, R. Zhang, S. Wang and S. Wu, J. Mater. Chem. C, 2023, 11, 2863, 10.1039/d2tc05277f.
- D. Yin, J. Wang, Y. Wang, P. Liu, J. Ma, X. Xu, D. Shen, Z. Dong, L. B. Kong and D. Tang, J. Eur. Ceram. Soc., 2020, 40, 444, DOI:10.1016/j.jeurceramsoc.2019.09.051.
- A. Kruk, Materials, 2020, 13, 4928, DOI:10.3390/ma13214928.
- T. Suda, Y. Yokota, T. Horiai, A. Yamaji, M. Yoshino, T. Hanada, H. Sato, S. Toyoda, Y. Ohashi, S. Kurosawa, K. Kamada and A. Yoshikawa, J. Cryst. Growth, 2022, 583, 126547, DOI:10.1016/j.jcrysgro.2022.126547.
- G. Y. Adachi and N. Imanaka, Chem. Rev., 1998, 98, 1479 CrossRef CAS PubMed.
- M. Pianassola, K. L. Anderson, J. Safin, C. Agca, J. W. McMurray, B. C. Chakoumakos, J. C. Neuefeind, C. L. Melcher and M. Zhuravleva, J. Adv. Ceram., 2022, 11, 1479, DOI:10.1007/s40145-022-0625-z.
- C. D. S. Brites, P. P. Lima, N. J. O. Silva, A. Millán, V. S. Amaral, F. Palacio and L. D. Carlos, New J. Chem., 2011, 35, 1177, 10.1039/c0nj01010c.
- M. D. Dramićanin, J. Appl. Phys., 2020, 128, 040902, DOI:10.1063/5.0014825.
- X. Wang, Q. Liu, Y. Bu, C. S. Liu, T. Liu and X. Yan, RSC Adv., 2015, 5, 86219, 10.1039/c5ra16986k.
- L. F. D. Santos, J. A. O. Galindo, K. D. O. Lima, A. R. Pessoa, A. M. Amaral, L. D. S. Menezes and R. R. Gonçalves, J. Lumin., 2023, 262, 119946, DOI:10.1016/j.jlumin.2023.119946.
- N. An, L. Ye, R. Bao, L. Yue and L. G. Wang, J. Lumin., 2019, 215, 116657, DOI:10.1016/j.jlumin.2019.116657.
- Z. Zhang, Q. Meng, L. Bai and W. Sun, J. Lumin., 2022, 251, 119229, DOI:10.1016/j.jlumin.2022.119229.
- A. Yoshikawa, M. Nikl, G. Boulon and T. Fukuda, Opt. Mater., 2007, 30, 6, DOI:10.1016/j.optmat.2006.10.030.
- T. Suda, Y. Yokota, T. Horiai, A. Yamaji, M. Yoshino, T. Hanada, H. Sato, S. Toyoda, Y. Ohashi, S. Kurosawa, K. Kamada and A. Yoshikawa, J. Cryst. Growth, 2021, 575, 126357, DOI:10.1016/j.jcrysgro.2021.126357.
-
O. Alimov, E. Dobretsova, D. Guryev, V. Kashin, G. Kiriukhina, S. Kutovoi, S. Rusanov, S. Simonov, V. Tsvetkov, V. Vlasov, V. Voronov and O. Yakubovich, CSD 1992594, 2020, Experimental Crystal Structure Determination DOI:10.25505/fiz.icsd.cc24wg7r.
- O. Alimov, E. Dobretsova, D. Guryev, V. Kashin, G. Kiriukhina, S. Kutovoi, S. Rusanov, S. Simonov, V. Tsvetkov, V. Vlasov, V. Voronov and O. Yakubovich, Cryst. Growth Des., 2020, 20, 4593, DOI:10.1021/acs.cgd.0c00389.
- R. D. Shannon and C. T. Prewitt, Acta Cryst., 1969, B25, 925 CrossRef.
- G. López-Pacheco, I. Padilla-Rosales, A. Miguel-Eugenio, E. Barrera-Calva, R. Rosas and F. González, J. Lumin., 2022, 242, 118596, DOI:10.1016/j.jlumin.2021.118596.
- R. Oka, T. Nouchi and T. Masui, Colorants, 2022, 1, 347, DOI:10.3390/colorants1030020.
- A. M. Srivastava, C. Renero-Lecuna, D. Santamaría-Pérez, F. Rodríguez and R. Valiente, J. Lumin., 2014, 146, 27, DOI:10.1016/j.jlumin.2013.09.028.
- C. R. Stanek, K. J. McClellan, B. P. Uberuaga, K. E. Sickafus, M. R. Levy and R. W. Grimes, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 134101, DOI:10.1103/PhysRevB.75.134101.
- G. C. Aumuller, W. Kostler, B. C. Grabmaier and R. Frey, J. Phys. Chem. Solids, 1994, 55, 767 CrossRef.
- D. den Engelsen, P. G. Harris, T. G. Ireland, G. Fern and J. Silver, ECS J. Solid State Sci. Technol., 2015, 4, R105, DOI:10.1149/2.0251507jss.
- L. X. Lovisa, Y. L. R. L. Fernandes, L. M. P. Garcia, B. S. Barros, E. Longo, C. A. Paskocimas, M. R. D. Bomio and F. V. Motta, Opt. Mater., 2019, 96, 109332, DOI:10.1016/j.optmat.2019.109332.
- Z. Mu, Y. Hu, L. Chen, X. Wang and G. Ju, ECS J. Solid State Sci. Technol., 2012, 1, R153–R157, DOI:10.1149/2.010206jss.
- C. Jin, J. Zhang, W. Lu and Y. Fei, J. Lumin., 2019, 214, 116581, DOI:10.1016/j.jlumin.2019.116581.
- Q. Wang, M. Liao, Q. Lin, M. Xiong, Z. Mu and F. Wu, J. Alloys Compd., 2021, 850, 156744, DOI:10.1016/j.jallcom.2020.156744.
- A. Siaï, P. Haro-González, K. Horchani-Naifer and M. Férid, Sens. Actuators, B, 2016, 234, 541–548, DOI:10.1016/j.snb.2016.05.019.
- J. I. Eldridge, J. Lumin., 2019, 214, 116535, DOI:10.1016/j.jlumin.2019.116535.
- Y. Gao, Y. Cheng, T. Hu, Z. Ji, H. Lin, J. Xu and Y. Wang, J. Mater. Chem. C, 2018, 6, 11178, 10.1039/c8tc03851a.
- D. Li, Y. Wang, X. Zhang, K. Yang, L. Liu and Y. Song, Opt. Commun., 2012, 285, 1925, DOI:10.1016/j.optcom.2011.12.075.
- F. Xu, Z. Ba, Y. Zheng, Y. Wang, M. Hu, X. Xu, J. Wang and Z. Zhang, J. Mater. Sci., 2018, 53, 15107–15117, DOI:10.1007/s10853-018-2702-9.
- X. Wu, L. Lou, H. Feng, G. Lv, Q. Wang, D. Zhu, C. Jiang and Z. Mu, Optik, 2021, 243, 167459, DOI:10.1016/j.ijleo.2021.167459.
- S. Yuan, S. Zhao, L. Lou, D. Zhu, Z. Mu and F. Wu, Powder Technol., 2022, 395, 83–92, DOI:10.1016/j.powtec.2021.09.053.
- S. Liu, J. Du, Z. Song, C. Ma and Q. Liu, Light: Sci. Appl., 2023, 12, 181, DOI:10.1038/s41377-023-01219-x.
- H. Luo, X. Li, X. Wang and M. Peng, Chem. Eng. J., 2020, 384, 123272, DOI:10.1016/j.cej.2019.123272.
- H. Zhang, Y. Liang, H. Yang, S. Liu, H. Li, Y. Gong, Y. Chen and G. Li, Inorg. Chem., 2020, 59, 14337, DOI:10.1021/acs.inorgchem.0c02118.
- Y. Zhuo, F. Wu, Y. Niu, Y. Wang, Q. zhang, Y. Teng, H. Dong and Z. Mu, Laser Photonics Rev., 2024, 18, 2400105, DOI:10.1002/lpor.202400105.
- P. Boutinaud, R. Mahiou, E. Cavalli and M. Bettinelli, Chem. Phys. Lett., 2006, 418, 185–188, DOI:10.1016/j.cplett.2005.10.120.
- P. Boutinaud, P. Putaj, R. Mahiou, E. Cavalli and A. Speghini, Spectrosc. Lett., 2007, 40, 209, DOI:10.1080/00387010701247019.
- P. Dorenbos, J. Lumin., 2018, 197, 62–65, DOI:10.1016/j.jlumin.2018.01.013.
- P. Boutinaud, E. Cavalli and M. Bettinelli, J. Phys.: Condens. Matter, 2007, 19, 386230, DOI:10.1088/0953-8984/19/38/386230.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Takahiko
Horiai
*bc,
Yuui
Yokota
*ab,
Takahiko
Horiai
*bc,
Yuui
Yokota