
Reversible photomechanical actuators with liquid crystal polymer graphene oxide nanocomposites†
Guan-Ting
Li‡
a,
Chia-Hsien
Hsu
 ab,
Bhupendra Pratap
Singh‡
c and
Shug-June
Hwang
*ac
ab,
Bhupendra Pratap
Singh‡
c and
Shug-June
Hwang
*ac
aInternational Master Program of Translational Medicine, National United University, Miaoli, Taiwan. E-mail: june@nuu.edu.tw
bInstitute of Biomedical Engineering and Nanomedicine, National Health Research Institutes, Miaoli, Taiwan
cDepartment of Electro-Optical Engineering, National United University, Miaoli, Taiwan
First published on 26th November 2024
Abstract
This study investigates the quantitative photomechanical actuation properties of a bifunctional acrylate liquid crystal (LC242) doped with azo dye and graphene oxide (GO) nanocomposites under a 40 mW cm−2 green laser. A photo-responsive liquid crystal polymer (LCP) was developed via UV curing, utilizing methyl red (MR) for its photoisomerism and GO for its photothermal properties. This investigation focuses on optimizing photo-actuating performance by tuning MR and GO doping concentrations as well as photopolymerization conditions. Experimental results show that with a doping concentration of 20% MR, the LCP achieves a photo-induced bending angle of approximately 35° and a response of 2 seconds and a recovery time of 1 second. Additionally, GO incorporation significantly reduces the critical laser power required for deformation; at a 10% concentration, the critical optical power is lowered to 25 mW cm−2. UV curing for 65 minutes (with a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 front-to-back exposure ratio) optimizes strain characteristics, enhancing both bending deformation and recovery stability in LCP samples. The tailored photomechanical response allows for efficient and controllable actuation, enabling applications such as PDMS film switching in micropump systems. This LCP film demonstrates improved actuation strain and repeatability, promising advancements for smart material systems and biomedical devices. These results underscore the potential of doping strategies and photopolymerization control to enhance LCP photo-actuation capabilities for light-driven innovations.
:1 front-to-back exposure ratio) optimizes strain characteristics, enhancing both bending deformation and recovery stability in LCP samples. The tailored photomechanical response allows for efficient and controllable actuation, enabling applications such as PDMS film switching in micropump systems. This LCP film demonstrates improved actuation strain and repeatability, promising advancements for smart material systems and biomedical devices. These results underscore the potential of doping strategies and photopolymerization control to enhance LCP photo-actuation capabilities for light-driven innovations.
1. Introduction
Stimuli-responsive polymer actuators, which may change shape or move in response to external stimuli, have generated significant interest in the development of artificial muscles,1,2 soft robotics,3–5 scaffolds for tissue engineering,6 adaptive devices,7 biomedical systems,8,9 and micro-manipulation tools.10 Photoresponsive polymer actuators stand out as particularly appealing due to their ability to respond to light, enabling remote and localized activation of the polymers without necessitating alterations to their surrounding environment.11,12 In the realm of actuator applications, the requisite for crosslinking of liquid crystal polymers (LCPs) into either liquid crystal elastomers (LCEs) or liquid crystal networks (LCNs) is paramount. Typically, LCEs have lightly crosslinking, side-chain or main-chain mesogenic units, and a flexible chain backbone.13,14 These polymers (LCEs) often have a low glass transition temperature (Tg) below room temperature (RT) and behave similarly to classic rubbers. Conversely, LCNs are moderate to densely crosslinked structures, commonly synthesized through the polymerization of oriented liquid crystal (LC) monomers, also known as reactive mesogens. LCNs generally have a relatively high Tg than LCEs (generally above RT). LCEs and LCNs have emerged as highly promising material systems for polymer actuators, owing to their capacity to undergo significant reversible changes in shape during the transition from the ordered LC phase to the disordered isotropic phase.Typical photo-responsive polymer actuators primarily depend on the reversible trans–cis photoisomerization of azobenzene (azo) derivatives, which is caused by both UV and visible light.15 LCPs incorporating azo mesogens represent the most extensively investigated polymer actuator systems.16,17 In their trans configuration, azo molecules exhibit a rod-like shape and are compatible with the ordered LC phase. Conversely, in the cis configuration, azo molecules adopt a bent shape, leading to the destabilization of the LC phase.18 Azobenzene-functionalized polymeric materials exhibit shape-adaptive effects when subjected to light. Lee et al.19 conducted a thorough spectroscopic examination to differentiate the photomechanical response of glassy, azobenzene-functionalized liquid crystal polymer networks (azo-LCN) under UV and blue-green irradiation. UV light induces photoisomerization of azobenzene from trans to cis, causing a contractile strain on the absorbing surface in azo-LCNs. Liu et al.20 developed a hydrophobic LCP membrane that responds to humidity and UV light, demonstrating rapid reversible actuation and complicated movements. This facilitated the development of a contact-free electronic device with dual-mode actuation for a variety of applications. Photo-responsive polymers with photomechanical conversion characteristics have emerged as potential smart materials, recognized for their light-based benefits that enabled remote, immediate, localized, and exact control.21–25 In the last century, the initial demonstration of light-induced deformation in photo-responsive polymers involved an azobenzene-containing polymer shrinking by approximately 1% when exposed to light.26,27 The initial demonstration of photoinduced deformation in a crosslinked LCP resulted in a 20% spontaneous contraction, sparking fresh rivalry in the field.28 Researchers have worked to improve the photo-deformability of LCPs for contraction/expansion, as well as develop 3D motion for more complex functions like bending,16,29,30 twisting,31 oscillating,32–34 and soft actuators.35–37 Essentially, any species with a strong photothermal effect, characterized by elevated light absorption and a heightened quantum yield of photothermal conversion, holds potential for integration with LCPs in the fabrication of light-driven polymer actuators.
Azobenzenes, which are commonly employed as photosensitive dyes in LCPs, can induce fully photothermal-driven actuation when incorporated into the network as side-chain molecules.38 This is particularly evident with donor–acceptor substituted azobenzenes, which are favored for their short cis isomer half-life time. Additionally, photothermal dyes exhibit another effect called network photo-softening, leading to a decrease in the storage modulus of the material upon exposure to light. This decrease in modulus is notably more significant than the thermal softening observed in typical glassy polymers. The phenomenon of photo-softening can thus contribute to enhanced actuation capabilities. Photothermal chromophores enable rapid actuation that is promptly reversed upon cessation of excitation light. However, these chromophores restrict the actuation of LCPs in dry environments. This limitation arises because in underwater actuation the dissipation of heat to the surrounding medium significantly hampers motion. Recent studies have revealed the feasibility of underwater actuation in a highly pliable LCP. However, achieving sufficient heating necessitates the utilization of exceedingly high laser intensities.39,40 Photomechanical effects in LCPs entail including photo-switches such as azobenzenes, which cause macroscopic motion upon light absorption. Azobenzenes undergo a shift from stable trans to bending cis states, resulting in network stress and molecular order breakdown, contraction along the molecular direction, and expansion perpendicular to it. Irradiation with polarized blue/green light may also cause photo-reorientation, however with lower stresses than UV-induced trans–cis isomerization.19
To achieve proficient photothermal actuation, LCPs can be incorporated with various photothermal agents, typically as nanofillers. These agents encompass carbon nanotubes,41–43 graphene oxides,44–46 metal nanoparticles,47,48 organic or organometallic dyes,49–51 and conjugated polymers.52 These photothermal agents possess the capability to absorb visible and/or near-infrared (NIR) light, subsequently converting optical energy into thermal energy. This heat is then dissipated into the polymer matrix, thereby initiating the LC to isotropic phase transition. The prevailing control mechanism for micropump systems traditionally relies on electronic power. Polydimethylsiloxane (PDMS), serving as the driving film, functions primarily as the interface between the electronic control device and the microfluid.53–55 In the realm of micro-pump design, PDMS films are routinely employed as substrates for diaphragms. The application of external forces, such as electric, thermal, vapor, or liquid pressure, to the diaphragm induces uneven pressure distribution within the micropump cavity. This uneven distribution directs the directional flow of liquid in the cavity, thereby establishing a functional micropump system. Nevertheless, micropump systems controlled by electronic means exhibit certain drawbacks, notably larger volumes, and limited mobility, posing challenges for operators conducting microfluidic experiments.
To assess the photoactivation capability of the prepared LCP in this report, the LCP was deployed to actuate the PDMS membrane in the micropump system. The photomechanical action of the LCP was employed to compress and deform the PDMS film, inducing controlled stretching and contraction. The experimental findings clarified that the LCP could provide remote switchable characteristics upon the PDMS film in the micropump system, thereby prevailing the limitations associated with traditional electronic control. The observed light-actuated mechanical behaviors of the PDMS film clearly substantiated the photomechanical capacity of the LCP.
2. Materials and methods
2.1. LCP synthesis
The LCP film was synthesized through the formulation of a liquid crystal mixture, comprising reactive mesogen LC242 (BASF Co. Ltd; Δε = −2, Δn = 0.14, TCN = 79 °C and TNI = 118 °C), methyl red with an azoxy structure (Alfa Aesar Co. Ltd), and the photoinitiator 2,2-dimethoxy-1,2-diphenyl-ethanone (IRG651, CIBA Co. Ltd), in a weight ratio of (99 − x):x:1, where x = 0, 5, 10, 15, 20, and 25, respectively. Molecular structures and schematic representations of LC242, methyl red (MR) dye, and I651 are illustrated in Fig. 1(a).
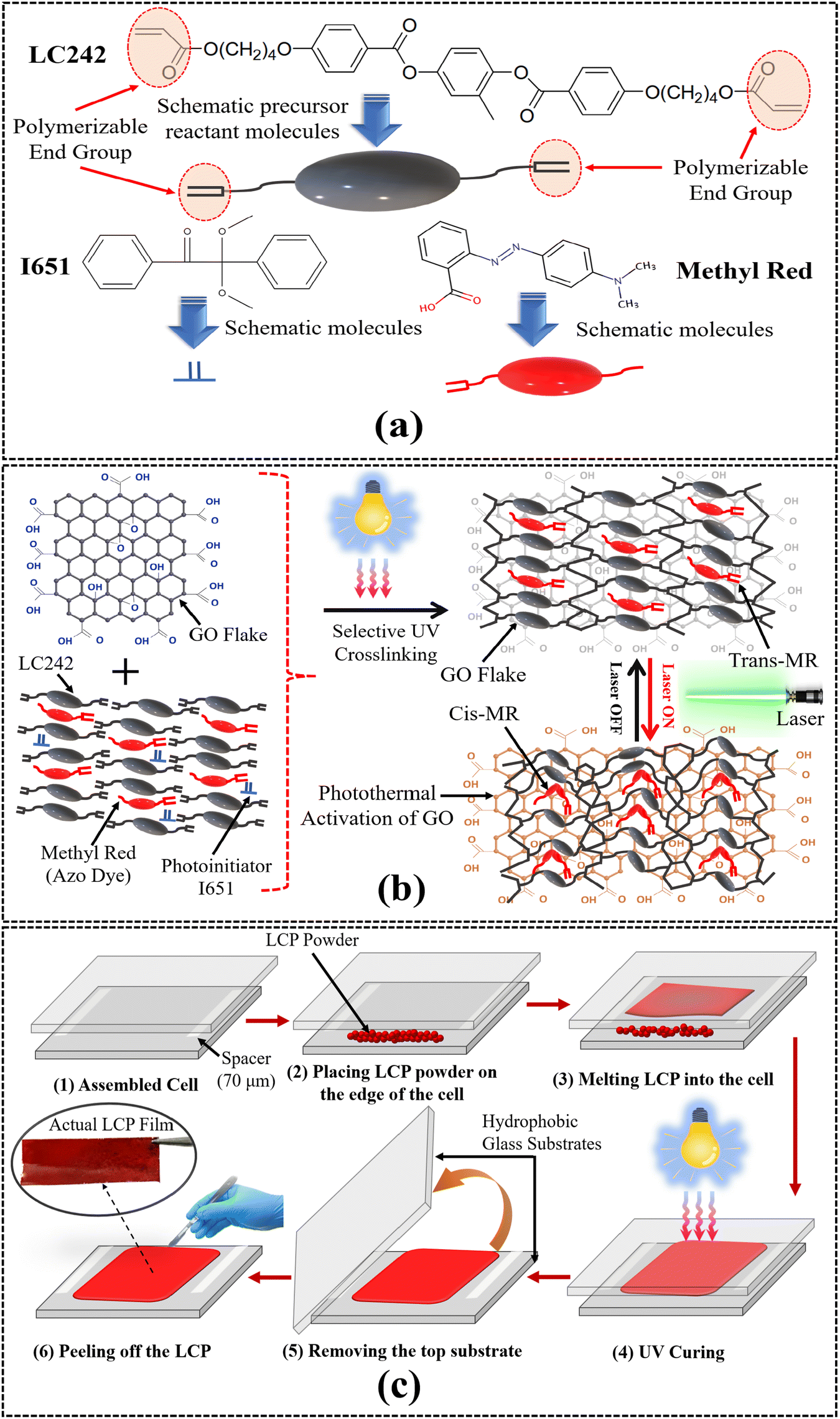 | ||
| Fig. 1 (a) The molecular structures along with schematic representations of the bifunctional acrylate main chain monomer (LC242), I651 (photoinitiator), and methyl red (azo dye), (b) schematic depiction of selective UV irradiation of graphene oxide (photothermal agent) infused LCP synthesis, and (c) schematic illustration of the fabrication process of the LCP with actual LCP film. | ||
To prepare the mixture, all LC blends were dissolved in a methyl ethyl ketone (MEK) solution (4:6) and thoroughly amalgamated with a graphene oxide solution concentrated to 4 mg mL−1 (Product No. P8GO-30; Pin Shuo Tan Corp., Taiwan) in a weight ratio of (100 − y):y, where y = 0, 5, and 10, respectively. Homogeneous mixing was achieved by subjecting the samples to ultrasonic mixing for 30 minutes. Subsequently, the solvent in the LC mixture was evaporated to yield the LC mixture powder.
Furthermore, two optically flat glass substrates (size: 2.5 cm × 1.5 cm), previously cleaned and treated with superhydrophobic nano-coating, were assembled into a 70 μm thick cell (see Determination of the optimal thickness for the LCP section in ESI†). The LC mixture powder was placed at the edge of the glass substrate and heated to 120 °C to induce the transition of the LC mixture into the isotropic phase. During this stage, the LC mixture was introduced into the cell via capillary force, and shear force was applied to the substrate to effectively align the LCs in the shearing direction. To prepare an LCP, the polymerization of the LC mixture was achieved by employing ultraviolet (UV) light (sourced from ORIEL Co. Ltd) with a wavelength of 365 nm and an intensity of 40 mW cm−2. The acrylate groups in LC242, characterized by double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), are prone to radical polymerization. The photoinitiator I651, activated by UV light, initiates the generation of free radicals that act as initiators for polymerization reactions. When LC242, with its acrylate groups, is combined with I651 and exposed to UV light, the photoinitiator absorbs UV energy, leading to the generation of free radicals. These radicals then trigger the polymerization of the acrylate groups in LC242, resulting in the formation of a polymer network. The photopolymerization of LC242 results in the formation of a three-dimensional network structure due to the presence of two reactive sites in the diacrylate LC242 monomer during the polymerization process. The potential addition of methyl red azo dye to the formulation is anticipated to act as a photoisomerization agent, switching azobenzene from trans to cis configuration and thereby inducing a contractile strain on the surface without significantly hindering the photopolymerization process. Fig. 1(b) represents a schematic depiction of selective UV irradiation, network formation, and LCP synthesis.
C), are prone to radical polymerization. The photoinitiator I651, activated by UV light, initiates the generation of free radicals that act as initiators for polymerization reactions. When LC242, with its acrylate groups, is combined with I651 and exposed to UV light, the photoinitiator absorbs UV energy, leading to the generation of free radicals. These radicals then trigger the polymerization of the acrylate groups in LC242, resulting in the formation of a polymer network. The photopolymerization of LC242 results in the formation of a three-dimensional network structure due to the presence of two reactive sites in the diacrylate LC242 monomer during the polymerization process. The potential addition of methyl red azo dye to the formulation is anticipated to act as a photoisomerization agent, switching azobenzene from trans to cis configuration and thereby inducing a contractile strain on the surface without significantly hindering the photopolymerization process. Fig. 1(b) represents a schematic depiction of selective UV irradiation, network formation, and LCP synthesis.
As the doping concentrations of methyl red and graphene oxide, along with the UV exposure time, influenced the performance of the LCP film, experiments were conducted based on these parameters, and the results were discussed. For an in-depth examination of the photomechanical properties of the LCP film under varying curing conditions, adjustments were made to the total UV exposure time on both sides of the substrate. After the complete curing of the LC film, the LCP film was detached from the substrate. Fig. 1(c) illustrates the detailed step-by-step fabrication process of the LCP film.
2.2. Characterization of LCP
To characterize phase transition temperatures within LC242 and blends, such as the nematic to isotropic phase transition temperature (TNI) and the crystal to nematic phase transition temperature (TCN), differential scanning calorimetry (DSC) analysis was carried out using a NETZSCH DSC-200-F3-Maia calorimeter (temperature range −170 °C to 600 °C with an accuracy of 0.1 K; NETZSCH, Germany). Furthermore, the glass transition temperature (Tg) of the final polymerized LCP samples was ascertained by DSC. Nonetheless, it is vital to admit that, below the thermal degradation threshold, finding the TNI of the LCP was extremely challenging. The unpolymerized monomer blends were heated and cooled at a rate of 10 °C min−1 throughout a temperature range of −40 °C to 140 °C. This approach was developed to avoid the Tg and thermal polymerization of the monomer blend, with a 30-minute isothermal period inserted to promote crystallization. In contrast, the polymerized LCP samples were subjected to analogous heating and cooling approaches but with a wider temperature range of up to 250 °C.56,57Fig. 2(a) depicts a schematic illustration of the experimental configuration employed for assessing the photomechanical properties of the LCP film. The LCP film underwent activation through a green laser (Soon-Link Co. Ltd, Taiwan) emitting at a wavelength of 532 nm, inducing bending deformation. Subsequently, the photomechanical properties of the LCP film were observed and quantified. An optical power meter was utilized to measure the laser power output traversing the sample. Concurrently, a charge-coupled device (CCD) was deployed to capture the light deformation image resulting from laser irradiation on the film. The light-induced deformation bending angle of the LCP was determined based on the laser light point irradiated on the LCP as the recording point. The bending angle of the LCP induced by light in the vertical direction was examined to derive the light-induced driving characteristics, as illustrated in Fig. 2(b). The study focused on examining how the photo-responsive characteristics of the LCP film are influenced by incident laser power and fabrication parameters. Furthermore, for the practical utilization of the prepared LCP in the optomechanical application of microstructures, we have applied the principles of cantilever beams and moments of inertia in material mechanics.58,59 A polydimethylsiloxane (PDMS) membrane has been employed in the microstructure design, as delineated in Fig. 2(c). The primary objective of this study is to actualize the micropump effect within the designed PDMS microstructure when activated by the LCP film under laser light irradiation. In this investigation, specific design enhancements have been implemented to augment the downward deformation of the PDMS membrane. A thinner “bridge” has been integrated at both ends of the film, connecting to the beams. Additionally, a “button” positioned on the top of the membrane facilitates the effective downward displacement of the PDMS membrane upon contact with the LCP, as elucidated in Fig. 2(d). Fig. 2(e) depicts the experimental configuration employed to assess the light-induced actuation capabilities of the prepared LCP film. This film was strategically positioned atop the micropump system, predicated on a PDMS membrane. Activation of the LC polymer was ensured through the application of a green laser, inducing bending and subsequent downward compression of the PDMS film. To examine the responsiveness of the PDMS film to the photo-actuation of the LCP, a mirror was deployed to facilitate a side-view observation of both the LCP and the PDMS film. Concurrently, a CCD recorded the dynamic alterations in the downward deformation of the PDMS film under light-induced mechanical pressure. The recorded image of the LCP and the measurement of the deformation angle were executed.
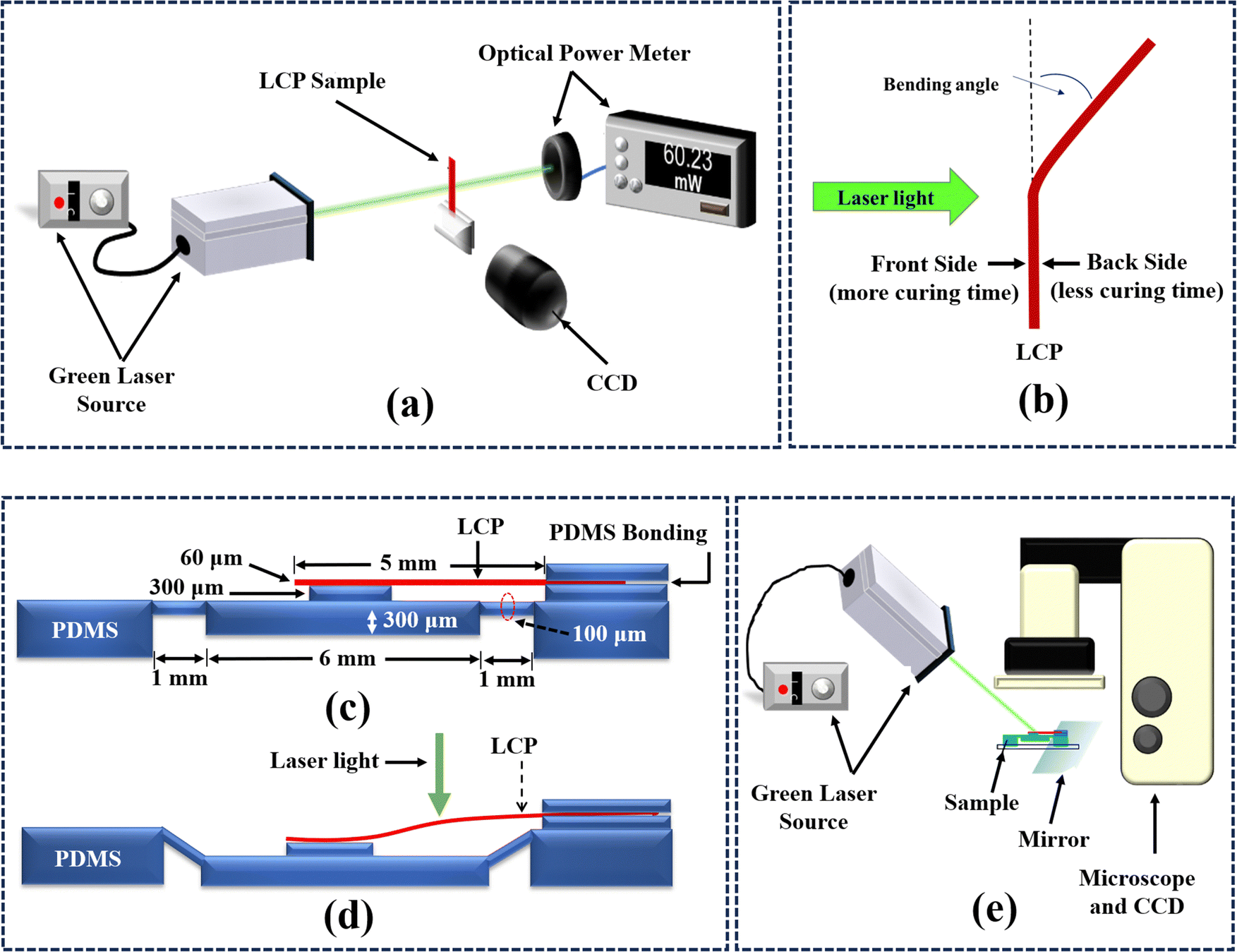 | ||
| Fig. 2 (a) Illustration of the experimental setup devised for measuring the photo-responsive characteristics of the LCP film, (b) light-induced bending angle; defined as the angle between the non-bent state and the bent state utilized for evaluating the deformation in the LCP photo-responsive characteristics, (c) schematic diagram of PDMS microstructure (d) structure deformation of PDMS membrane driven by LCP and (e) the experimental arrangement employed for quantifying the photo-actuated deformation of the PDMS film driven by LCP. | ||
3. Results and discussion
The differential scanning calorimetry (DSC) technique was used to determine the phase transition temperatures of the pristine unpolymerized monomer LC242, from the crystal to nematic phase transition temperature (TCN = 79.4 °C) and from the nematic to isotropic phase transition temperature (TNI = 118.2 °C). Fig. 3 depicts thermal data from the DSC analysis. Notably, the nematic phase exhibits monotropic behavior in both the pristine LC242 monomer and monomer compositions containing up to 25% methyl red (MR; a trans–cis isomerization agent) and 10% graphene oxide (GO; a photothermal agent) (see Table 1). Polymer properties, including mechanical qualities, energy absorption capacity, and thermal coefficients, vary with temperature, especially near the glass transition.60 Designing LCP materials for applications requires careful consideration of their glass transition temperature (TG). DSC was utilized to examine the impact of dopant concentration (MR and GO) on the TG post-polymerization phase of the LCP. The corresponding values can be found in Table 1.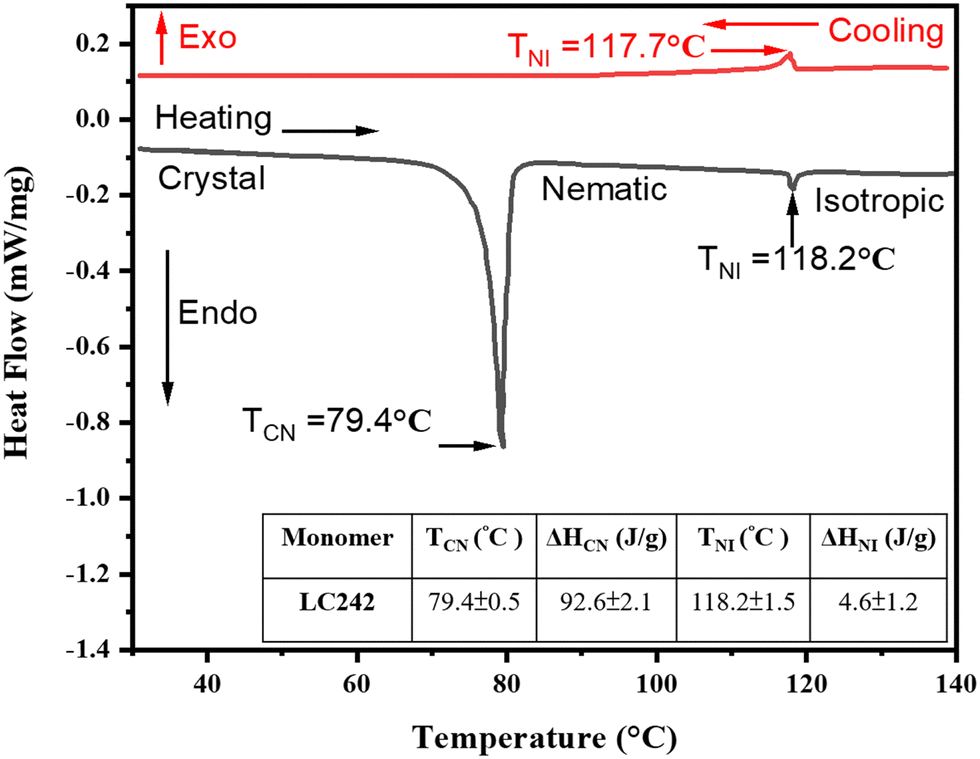 | ||
| Fig. 3 Differential scanning calorimetry (DSC) thermograph of bifunctional acrylate main chain monomer (LC242) during heating and cooling cycle at the scan rate of 10 °C min−1. | ||
| Samples (LCPs with fix 1% I651) | T CN (°C) | T NI (°C) | T g (°C) | T Max (°C) |
|---|---|---|---|---|
| Pure LC242 | 79.4 ± 0.5 | 118.2 ± 1.5 | N/A | N/A |
| LC242 (94%) + MR (5%) | 77.0 ± 0.8 | 112 ± 1.5 | N/A | 120 ± 1.2 |
| LC242 (89%) + MR (10%) | 74 ± 1.1 | 108 ± 1.2 | 95 ± 1.6 | 135 ± 1.4 |
| LC242 (84%) + MR (15%) | 69 ± 1.3 | 105 ± 1.5 | 92 ± 1.5 | 152 ± 2.2 |
| LC242 (79%) + MR (20%) | 67 ± 1.4 | 102 ± 1.7 | 88 ± 1.3 | 163 ± 1.6 |
| LC242 (74%) + MR (25%) | 70 ± 1.2 | 98 ± 1.6 | 85 ± 1.6 | 172 ± 1.8 |
| LC242 (74%) + MR (20%) + GO (5%) | 75 ± 1.6 | 97 ± 1.8 | 62 ± 1.8 | 182 ± 1.7 |
| LC242 (69%) + MR (20%) + GO (10%) | 81 ± 1.9 | 99 ± 2.1 | 48 ± 2.1 | 196 ± 1.8 |
Table 1 depicts alterations in transition temperatures from the crystal to nematic phase (TCN), nematic to isotropic phase (TNI), glass transition (Tg), and maximum attainable temperature (TMax). Concurrently, Fig. 3 exhibits the relationship between heat flow and temperature, as determined via differential scanning calorimetry (DSC) analysis of LC242. The accompanying inset summarizes the resultant phase transition temperatures and enthalpies. It is noteworthy that the characteristics of polymers, encompassing mechanical resilience, energy absorption capacity, and thermal response, undergo significant modifications with temperature variations, particularly during the glass transition phase.60
The Tg of an LCP stands as a pivotal determinant in the formulation of LCP materials for diverse applications. This investigation revealed that Tg is subject to the influence of the crosslinker concentration (LC242) within the composite system. Specifically, diminishing the LC242 crosslinker concentration from 89 to 69 wt% in LCP induced a nearly linear decline in TG from 95 to 48 °C. However, concentrations surpassing 89% of LC242 crosslinker led to either an indiscernible Tg or values falling beyond the measurement range. The decrease in Tg found in LCP samples is directly related to the decreased density of crosslinkers inside the samples. This lowered density allows for greater relaxation of segmental chains, which contributes to a lower glass transition temperature. This finding is consistent with previous research exploring the effect of crosslinker density on Tg in LCPs.56,61,62 The photothermal effect of azo derivatives (organic photothermal agent), renowned for their dye characteristics, plays a pivotal role in LCP actuators incorporating azo mesogens. This effect significantly influences the TMax under 120 mW and 532 nm wavelength laser irradiation, achieved through light absorption and subsequent conversion into heat. Upon exposure to light of suitable wavelength, dictated by dye absorption properties, and at high intensity (100–400 mW cm−2), films comprising liquid-crystalline polymers containing azobenzene moieties can manifest a temperature elevation of up to 80 K.63,64 In the current investigation, an increase in the concentration of MR dye (azo) within the LC monomer (LC242) from 5 wt% to 25 wt% results in an elevation of the TMax from 120 °C to 172 °C. Furthermore, the introduction of an inorganic photothermal agent (GO) from 5 to 10 wt% yields a subsequent increase in TMax from 172 °C to 196 °C. Notably, owing to its exceptionally high molar extinction coefficient and quantum yield of photothermal conversion,44–46 GO can provide the LCP actuator with swift photothermal response, even when present in relatively low concentrations.
To systematically investigate the influence of varying concentrations of MR dopant on the light-induced deformation performance of the LCP, distinct LCP films were prepared with MR dopant concentrations of 0%, 5%, 10% 15%, 20%, and 25%, respectively. The LCP sample underwent UV light irradiation for 40 minutes on the front side and 30 minutes on the backside. Notably, the LCP films, containing MR dopant concentrations (ranging from 5 to 25%), exhibited no deformation or bending when subjected to laser irradiation with intensities up to 20 mW. However, upon further increasing the laser power, deformation in the LCP became apparent (see Fig. 4). This observation could potentially be attributed to inadequate photoactivation energy. Although small concentrations of MR had very high laser thresholds, they yielded relatively low deformation angles. For instance, LCP films containing 5% MR demonstrated a laser threshold of approximately 90 mW, with a maximum deformation of merely 4° (at 120 mW), as depicted in Fig. 4. Notably, LCP films with 25% MR exhibited a relatively lower laser threshold (∼30 mW) and achieved a favorable deformation angle (43° at 120 mW). However, the repeatability and reversibility became challenging when the curvature angle exceeded 15°.
 | ||
| Fig. 4 Deformation angles of LCP under laser power irradiation, varied with different concentrations of methyl red (MR) dye. | ||
Although the LCP with a 20% MR concentration exhibited significant bending, its laser threshold was too high (∼50 mW), which could be attributed to insufficient photoactivation energy from MR alone. Consequently, a suitable concentration of an inorganic photothermal agent (i.e., GO) was introduced to enhance the attainable temperature and the speed of temperature rise of the prepared actuators under light irradiation. Observations revealed that elevating the concentration of the photothermal agent led to higher maximum temperatures and quicker rates of temperature increase under light irradiation. Likewise, maintaining a constant concentration of the photothermal agent while increasing the light power resulted in higher and faster temperature increments.
Fig. 5(a) illustrates the bending angles of the three LCPs in response to different laser powers. Notably, the LCP doped with a 15% MR + 10% GO concentration exhibits a translucent red appearance, requiring a minimum threshold laser power of 15 mW for initiating and demonstrating a light-induced bending angle of 18° at an incident laser power of 120 mW. The LCP infused with a 20% MR + 10% GO concentration exhibits a translucent deep red hue, initiates deformation at a laser power of 10 mW, and achieves a bending angle exceeding 30° under laser irradiation with a power of 120 mW. In contrast to the LCP containing 15% MR + 10% GO, the 20% MR + 10% GO-doped LCP displays a greater bending angle and exhibits commendable reversible recovery performance, nearly returning to its original position within a very short interval (∼2 s) after deactivation of the laser light source. At an MR concentration of 25% + 10% GO, the appearance of the LCP is nearly opaque red-black. Due to a lower content of host-reactive LC compared to the preceding concentrations, its structure is softer. Although it exhibits a lower threshold laser power of 5 mW and a larger deformation angle at 120 mW, the reversible effect is compromised, and recovery to the initial position becomes challenging when the curvature angle exceeds 20°. Consequently, a 25% MR + 10% GO dopant concentration is estimated not suitable for LCP actuator fabrication. The experimental findings indicate an increasing deformation angle of the LCP with rising MR concentrations with fixed GO. Considering factors such as structural rigidity, photo-induced deformation angle, and reversibility, an MR concentration of 20% + 10% GO was selected for further experimentation and characterization of the photo response in the LCP film. Notably, the LCP film bends away from the light irradiation, since the front side of LCP underwent a more extensive UV curing compared to the back side. Consequently, the front of the LCP solidified more, while the back retained a relatively softer state. This differential curing process resulted in the LCP film bending away from the direction of light irradiation, illustrating the influence of the varied curing approach. Video S1 (ESI†) illustrates the photo-actuation behavior of the LCP under laser irradiation.
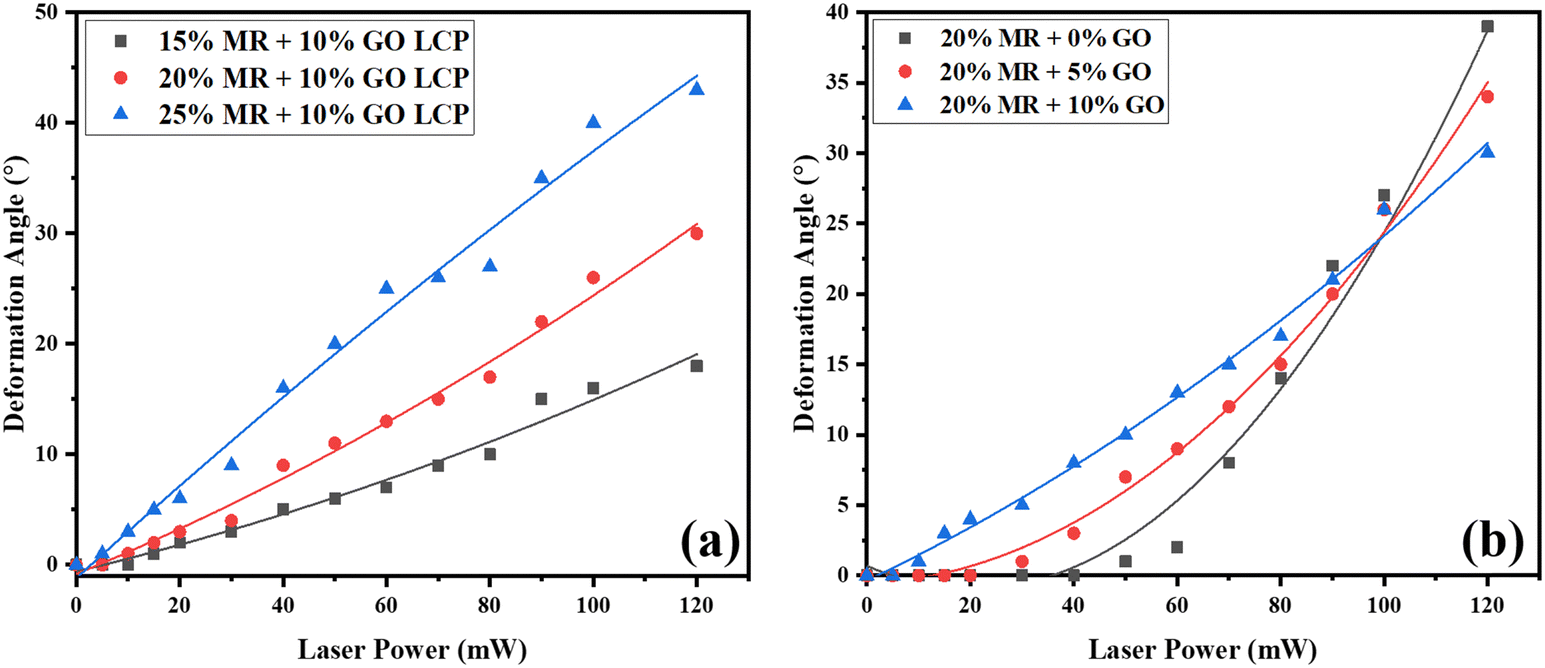 | ||
| Fig. 5 (a) Deformation angles of LCP under laser power irradiation, varied with different concentrations of methyl red with fixed 10% GO doping, (b) the variation in deformation angle resulting from laser power irradiation of LCP doped with varying concentrations of GO solution while maintaining a constant 20% MR concentration. | ||
Furthermore, Fig. 5(b) demonstrates the influence of varying concentrations of GO on the alteration of the deformation angle of LCP induced by laser power irradiation, while maintaining a consistent 20% MR concentration throughout the experiments. Remarkably, as the inorganic photothermal agent is increased from 0 to 10%, the laser threshold for LCP deformation decreases significantly from 50 mW to 10 mW. Moreover, these LCPs exhibit outstanding reversible recovery performance even at greater deformation angles. Particularly noteworthy is the LCP doped with a 20% MR and 10% GO concentration, which yields the most favorable outcome with a minimum threshold laser power of 10 mW for initiation and demonstrates a maximum light-induced deformation angle of 30° at an incident laser power of 120 mW.
The time required for the photopolymerization of the LC film was found to be significantly linked to the thickness of the LCP and the concentration of the host-reactive LC. Since UV light exposure was confined to one side of the LC film, incomplete photopolymerization on the opposite side caused adherence to the glass substrate, complicating the peeling process. To ensure comprehensive polymerization of the LC polymer film, UV light exposure was applied to both sides of the LC film coated on the glass substrate. The concentrations of methyl red and GO solution doped in the LCP sample were maintained at 20% and 10%, respectively. Fig. 6(a) depicts the deformation angle of the LCP film concerning different total UV curing times under various laser power irradiations. The total curing time represented the cumulative UV irradiation time on both the front and back sides of the LC film, maintaining a curing time ratio of 1:1. For instance, 100 minutes signified 50 minutes of exposure on the front side and 50 minutes on the backside. The experimental findings revealed that the LCP obtained with a total curing time of 65 minutes exhibited maximum deformation, followed by curing times of 70 and 50 minutes. Conversely, curing times of 25 and 100 minutes demonstrated the least favorable deformation characteristics. This discrepancy was attributed to the insufficient solidification of the film at a curing time of 25 minutes, making the LCP less responsive to laser light. On the other hand, a curing time of 100 minutes might have led to overexposure, resulting in the deterioration of LCP film properties. Based on these results, the LCP with a total curing time of 65 minutes demonstrated optimal deformation, guiding subsequent experiments with the same curing duration.
 | ||
| Fig. 6 (a) Deformation angle variation of the LCP in response to laser power irradiation, depicting different total UV curing times, while maintaining a curing time ratio of 1:1 on the front and back sides, (b) variation of deformation angle resulting from laser irradiation on LCP samples prepared with varying curing time ratios on the front and back sides and (c) variation in deformation angle caused by laser irradiation on LCP with different curing times, including the 5:1 curing time ratio. | ||
Furthermore, this study investigated the influence of the UV curing time ratio between the front and back sides of the LC film on the photo-actuation characteristics of the LCP, as depicted in Fig. 6(b), while maintaining a constant total UV curing time of 65 minutes. The findings revealed that a lower curing time ratio adversely affected the photodeformation performance of the LCP, attributed to a reduced hardness difference between the front and back sides of the LCP film, resulting in a diminished bending angle when subjected to laser irradiation. Conversely, when the front-to-back curing time ratio exceeded 5:1, the photodeformation characteristics improved, and the photo-actuated deformation curves for LCP films prepared with ratios of 5:1, 6:1, and 8:1 appeared nearly identical. It was also noted that the LCP film produced with a curing ratio of 5:1 exhibited the most favorable structure. It was hypothesized that at a ratio of 8:1 and a total curing time of 65 minutes, the UV exposure time on the front was 58 minutes, while the exposure time on the back was only 7 minutes. This brief curing time may have been insufficient to fully cure the LC film, leading to the LCP adhering to the substrate surface when opening the upper substrate. Considering process convenience and film preparation yield, the conclusion drawn from this study was that, with a total curing time of 65 minutes, the optimal UV exposure parameters involved a curing time ratio of 5:1 on the front and back sides, representing the optimal process conditions for the LCP.
To evaluate whether the LCP film, fabricated with a front-to-back curing time ratio of 5:1 and a total curing time of 65 minutes, maintains its maximal photo-actuated deformation, an examination of the effects of five distinct total curing times on the photomechanical properties of the LCP film was conducted. The experimental findings depicted in Fig. 6(c) reveal that the photo-induced deformation of the LCP film, prepared with a total curing time of 65 minutes, surpasses the deformation induced by the other four varying laser powers. However, with an increase in the curing time to 70 or 100 minutes, the photomechanical response of the LCP diminishes, and the photo-response curve for a curing time of 100 minutes mirrors that of 25 and 50 minutes.
The decline in light-induced mechanical response observed in LC polymers with prolonged curing times is ascribed to the gradual augmentation in the crosslinking hardness of the coating film with extended UV light irradiation. LC films with higher hardness exhibit diminished susceptibility to structural deformation caused by external stimuli. Therefore, it is conclusively established that the LCP prepared with a total curing time of 65 minutes demonstrated optimal photo-induced deformation.
Additionally, an investigation was conducted to examine whether the deformation direction of the cured LCP film correlates with the duration of UV light exposure on the film surface. Table 2 presents the deformation direction of the LCP films obtained through various UV curing time ratios on the front and back sides of the sample. The experimental outcomes reveal that when subjected to laser stimulation, the LCP bends toward the surface exposed to UV light, particularly with a shorter curing time.
| Front/back side curing time (min) | Bending direction |
|---|---|
| 20/5 | Back side |
| 25/40 | Front side |
| 40/30 | Back side |
| 55/10 | Back side |
| 60/40 | Back side |
| 80/20 | Back side |
Based on our earlier experimental findings, it is evident that both the total UV curing time and the curing time ratio on the front and back sides of the substrate significantly influence the photomechanical response of the film under consideration. Furthermore, to explore the practical application of the light-actuated response exhibited by the developed LCP film, the prepared LCP was integrated with a PDMS film-based micropump system, as depicted in Fig. 2(c). Subsequently, a 60-mW green laser was utilized to actuate the LCP, and dynamic photo-mechanical analysis was conducted on the samples. Fig. 7 comprehensively illustrates how the LCP consistently propels the PDMS film downward when subjected to laser light. Remarkably, the PDMS film exhibits effective actuation, responding to the tensile behavior resulting from the light-induced downward movement of the LCP. Following laser stimulation, the LCP immediately deformed downward, compressing the PDMS coating and migrating close to the bottom surface. Upon turning off the laser, both the LCP and PDMS film swiftly revert to their initial positions, showcasing a reproducible and reversible photomechanical response. Laser activation induces noticeable compression of the PDMS micropillars within 2 seconds (Fig. 7(ii)–(vi)), while deactivation results in the LCP recovering to its original state in just 1 second (Fig. 7(vii)–(ix)). A Video S2 (ESI†) further illustrates the iterative contraction and recovery process upon exposure to laser light.
 | ||
| Fig. 7 (a) Schematic diagram of the PDMS microstructure (side view) and (b) the dynamic photomechanical response of the PDMS membrane under the influence of the light-powered LCP at different bending angles: (i) initial state (0°), (ii) laser ON (10°), (iii) laser ON (13°), (iv) laser ON (15°), (v) laser ON (18°), (vi) laser ON (20°), (vii) laser OFF (16°), (viii) laser OFF (8°), and (ix) laser OFF (0°). | ||
The characteristics observed in Fig. 7, depicting the response of the green laser-activated LCP and PDMS film, underwent a comprehensive quantitative analysis. The ensuing experimental findings are comprehensively presented in Table 3. The analysis highlights that the LCP can exert a downward force on the PDMS film, achieving a displacement of approximately 280 μm, almost reaching the bottom. Moreover, upon deactivation of the laser, both the LCP and PDMS film promptly and reversibly return to their original positions. The PDMS membrane demonstrated varying degrees of compression deformation corresponding to the bending angle of the LCP, illustrating a responsive photomechanical behavior when the laser was alternated between on and off states. Consequently, the prepared LCP actuator was found to be effective in serving as a remote light-responsive element, adeptly controlling the deformation of the PDMS film within the microsystem. This capability facilitated the micropump system in exhibiting switching characteristics, thereby overcoming the limitations associated with traditional electric-driven microsystems.
| Fig. i (off) | Fig. ii (on) | Fig. iii (on) | Fig. iv (on) | Fig. v (on) | Fig. vi (on) | Fig. vii (off) | Fig. viii (off) | Fig. i (off) | |
|---|---|---|---|---|---|---|---|---|---|
| LCP bending angle (°) | 0 | 10 | 13 | 15 | 18 | 20 | 16 | 8 | 0 |
| PDMS film downward compressed distance (μm) | 0 | 0 | 50 | 150 | 250 | 280 | 170 | 0 | 0 |
This study presents a photo-responsive LCP nanocomposite incorporating GO within a nematic LCP matrix. Taking advantage of the efficient thermal conductivity of GO, the nanocomposite absorbs light energy, converting it into heat and enabling actuation at lower laser powers. Experimental results highlight a significant enhancement in the mechanical performance of the LCP by incorporating GO into the nematic LC nanocomposites, with optimization achieved through careful adjustment of UV curing conditions. The prepared LCP nanocomposites exhibit a rapid (within seconds) and reversible generation of strain using light, characterized by consistent shape-change rates and substantially improved mechanical properties.
The light-actuated LCP demonstrates effective compression of the PDMS film in a designed microsystem, enhancing the practicality of LCP actuators in diverse biomedical applications, including photo-biomechanics research, optical biological cell sensors, and innovative cell culture methods. This work is anticipated to offer valuable insights into the advancement of intelligent materials for applications in biomedicine, soft micro-robotics, sustainable energy conservation, and beyond.
4. Conclusions
The light-induced actuation capabilities of the prepared LCP samples were systematically investigated. The improved photomechanical performance of LCP nanocomposites was found to be significantly influenced by the azo dye and GO dopant concentration, as well as photopolymerization conditions. This study utilizes the photoisomerization properties of azo molecules to produce macroscopic bending changes in the LCP shape. Considering factors such as photoactivation performance, reversibility, and structural stability of LCP, a methyl red doping concentration of 20% was found to be the optimal process parameter. In addition, the incorporation of GO into the LC material enhances the photomechanical response characteristics of LCP. Notably, higher concentrations of GO reduce the critical optical power required to induce deformation in the LCP. The maximum permissible doping concentration of GO solution limited to 10%. Furthermore, a series of experiments were conducted to explore the effect of UV curing time on the photo-actuation characteristics of the LCP. It was found that a total curing time of 65 minutes resulted in the greatest light-induced deformation of the LCP. As the ratio of the UV exposure time of the front and back sides of the LC substrate was 5:1, the best light-induced actuation characteristics were observed. This photo-actuation capability of LCP is significantly controlled by the manufacturing process conditions, and the prepared LCP successfully drives the PDMS micropillar to compress downwards to achieve the switching characteristics of the micropump system. Therefore, the approach reported here is very promising for further research on LCP components. Doping with various nanomaterials and controlling the photopolymerization conditions can further enhance the photo-actuation capability of LCPs. In the future, it is expected that LCPs will be used to advance the development of smart materials and biomedical technology through light-driven methods.
Author contributions
Guan-Ting Li: investigation, validation, data curation, formal analysis, and writing – original draft preparation. Chia-Hsien Hsu: conceptualization, supervision, visualization and validation. Bhupendra Pratap Singh: investigation, data curation, validation, writing – original draft preparation, and review & editing. Shug-June Hwang: conceptualization, resources, methodology, validation, formal analysis, supervision, project administration, funding acquisition, and writing – review & editing.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. All data generated or analyzed during this study are included in this published article [and its ESI†].Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to thank the National Science and Technology Council of Taiwan for financially supporting this research under contracts: (NSTC 111-2221-E-239-010-MY3) and (NSTC 111-2811-E-239-001-MY3).References
- S. M. Mirvakili and I. W. Hunter, Adv. Mater., 2018, 30, 1704407 CrossRef.
- Z. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS PubMed.
- G. M. Whitesides, Angew. Chem., Int. Ed., 2018, 57, 4258–4273 CrossRef CAS.
- L. Hines, K. Petersen, G. Z. Lum and M. Sitti, Adv. Mater., 2017, 29, 1603483 CrossRef PubMed.
- C. Wang, K. Sim, J. Chen, H. Kim, Z. Rao, Y. Li, W. Chen, J. Song, R. Verduzco and C. Yu, Adv. Mater., 2018, 30, 1706695 CrossRef.
- F. Rosso, G. Marino, A. Giordano, M. Barbarisi, D. Parmeggiani and A. Barbarisi, J. Cell. Physiol., 2005, 203, 465–470 CrossRef CAS.
- T. H. Ware, M. E. McConney, J. J. Wie, V. P. Tondiglia and T. J. White, Science, 2015, 347, 982–984 CrossRef CAS PubMed.
- D. Ha, B. G. Kim, T.-Y. Lin, Y. Ouyang, P. P. Irazoqui and W. J. Chappell, In 2010 IEEE MTT-S International Microwave Symposium Digest, 2010, pp. 612–615.
- J. Jeong, S. H. Bae, J.-M. Seo, H. Chung and S. J. Kim, J. Neural Eng., 2016, 13, 025004 CrossRef.
- F. Ge, X. Lu, J. Xiang, X. Tong and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 6126–6130 CrossRef CAS.
- D. D. Han, Y. L. Zhang, J. N. Ma, Y. Q. Liu, B. Han and H. B. Sun, Adv. Mater., 2016, 28, 8328–8343 CrossRef CAS.
- H. Zeng, P. Wasylczyk, D. S. Wiersma and A. Priimagi, Adv. Mater., 2018, 30, 1703554 CrossRef.
- T. J. White and D. J. Broer, Nat. Mater., 2015, 14, 1087–1098 CrossRef CAS.
- P. Beyer, E. M. Terentjev and R. Zentel, Macromol. Rapid Commun., 2007, 28, 1485–1490 CrossRef CAS.
- L. Guo, M. Liu, S. Sayed, B. Lin, P. Keller, X. Zhang, Y. Sun and H. Yang, Chem. Sci., 2016, 7, 4400–4406 RSC.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS.
- H. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149–2180 CrossRef CAS.
- X. Pang, J. a. Lv, C. Zhu, L. Qin and Y. Yu, Adv. Mater., 2019, 31, 1904224 CrossRef CAS.
- K. M. Lee, N. V. Tabiryan, T. J. Bunning and T. J. White, J. Mater. Chem., 2012, 22, 691–698 RSC.
- Y. Liu, B. Xu, S. Sun, J. Wei, L. Wu and Y. Yu, Adv. Mater., 2017, 29, 1604792 CrossRef.
- J. Wei and Y. Yu, Soft Matter, 2012, 8, 8050–8059 RSC.
- H. Jiang, C. Li and X. Huang, Nanoscale, 2013, 5, 5225–5240 RSC.
- T. Ube and T. Ikeda, Angew. Chem., Int. Ed., 2014, 53, 10290–10299 CrossRef CAS.
- W. Wang, X. Sun, W. Wu, H. Peng and Y. Yu, Angew. Chem., Int. Ed., 2012, 51, 4644–4647 CrossRef CAS.
- E. Pantuso, G. De Filpo and F. P. Nicoletta, Adv. Opt. Mater., 2019, 7, 1900252 CrossRef.
- L. Matějka, K. Dušek and M. Ilavský, Polym. Bull., 1979, 1, 659–664 CrossRef.
- L. Matějka, M. Ilavský, K. Dušek and O. Wichterle, Polymer, 1981, 22, 1511–1515 CrossRef.
- H. Finkelmann, E. Nishikawa, G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS.
- T. Ikeda, M. Nakano, Y. Yu, O. Tsutsumi and A. Kanazawa, Adv. Mater., 2003, 15, 201–205 CrossRef CAS.
- C. L. Van Oosten, C. W. Bastiaansen and D. J. Broer, Nat. Mater., 2009, 8, 677–682 CrossRef CAS.
- S. Iamsaard, S. J. Aßhoff, B. Matt, T. Kudernac, J. J. Cornelissen, S. P. Fletcher and N. Katsonis, Nat. Chem., 2014, 6, 229–235 CrossRef CAS.
- K. M. Lee, M. L. Smith, H. Koerner, N. Tabiryan, R. A. Vaia, T. J. Bunning and T. J. White, Adv. Funct. Mater., 2011, 21, 2913–2918 CrossRef CAS.
- K. M. Lee, T. J. Bunning and T. J. White, Adv. Mater., 2012, 24, 2839 CrossRef CAS.
- K. Kumar, C. Knie, D. Bléger, M. A. Peletier, H. Friedrich, S. Hecht, D. J. Broer, M. G. Debije and A. P. Schenning, Nat. Commun., 2016, 7, 11975 CrossRef CAS PubMed.
- Z. C. Jiang, Y. Y. Xiao and Y. Zhao, Adv. Opt. Mater., 2019, 7, 1900262 CrossRef.
- L. Qin, X. Liu and Y. Yu, Adv. Opt. Mater., 2021, 9, 2001743 CrossRef CAS.
- L. Yu, R. Peng, G. Rivers, C. Zhang, P. Si and B. Zhao, J. Mater. Chem. A, 2020, 8, 3390–3396 RSC.
- M. P. Da Cunha, E. A. van Thoor, M. G. Debije, D. J. Broer and A. P. Schenning, J. Mater. Chem. C, 2019, 7, 13502–13509 RSC.
- H. Shahsavan, A. Aghakhani, H. Zeng, Y. Guo, Z. S. Davidson, A. Priimagi and M. Sitti, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5125–5133 CrossRef CAS.
- W. Feng, Q. He and L. Zhang, Adv. Mater., 2024, 2, 2312313 CrossRef PubMed.
- C. Li, Y. Liu, C.-W. Lo and H. Jiang, Soft Matter, 2011, 7, 7511–7516 RSC.
- C. Li, Y. Liu, X. Huang and H. Jiang, Adv. Funct. Mater., 2012, 22, 5166–5174 CrossRef CAS.
- Y. Yang, Z. Pei, Z. Li, Y. Wei and Y. Ji, J. Am. Chem. Soc., 2016, 138, 2118–2121 CrossRef CAS.
- L. Yu, Z. Cheng, Z. Dong, Y. Zhang and H. Yu, J. Mater. Chem. C, 2014, 2, 8501–8506 RSC.
- Z. Cheng, T. Wang, X. Li, Y. Zhang and H. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 27494–27501 CrossRef CAS.
- R. Wei, Z. Wang, H. Zhang and X. Liu, Liq. Cryst., 2016, 43, 1009–1016 CrossRef CAS.
- Y. Yang, W. Zhan, R. Peng, C. He, X. Pang, D. Shi, T. Jiang and Z. Lin, Adv. Mater., 2015, 27, 6376–6381 CrossRef CAS PubMed.
- X. Lu, H. Zhang, G. Fei, B. Yu, X. Tong, H. Xia and Y. Zhao, Adv. Mater., 2018, 30, 1706597 CrossRef PubMed.
- A. H. Gelebart, G. Vantomme, E. Meijer and D. J. Broer, Adv. Mater., 2017, 29, 1606712 CrossRef.
- H. Zeng, O. M. Wani, P. Wasylczyk and A. Priimagi, Macromol. Rapid Commun., 2018, 39, 1700224 CrossRef.
- A. H. Gelebart, D. Jan Mulder, M. Varga, A. Konya, G. Vantomme, E. Meijer, R. L. Selinger and D. J. Broer, Nature, 2017, 546, 632–636 CrossRef CAS PubMed.
- W. Liu, L.-X. Guo, B.-P. Lin, X.-Q. Zhang, Y. Sun and H. Yang, Macromolecules, 2016, 49, 4023–4030 CrossRef CAS.
- M. Bu, T. Melvin, G. Ensell, J. S. Wilkinson and A. G. Evans, J. Micromech. Microeng., 2003, 13, S125 CrossRef.
- A. Shakeri, S. Khan and T. F. Didar, Lab Chip, 2021, 21, 3053–3075 RSC.
- J. Friend and L. Yeo, Biomicrofluidics, 2010, 4, 026502 CrossRef.
- E. I. Jull, R. J. Mandle, T. Raistrick, Z. Zhang, P. J. Hine and H. F. Gleeson, Macromolecules, 2022, 55, 4320–4330 CrossRef CAS.
- X. Du, Y. Liu, D. Zhao, H. F. Gleeson and D. Luo, Soft Matter, 2024, 20, 2562–2567 RSC.
- T. Beléndez, C. Neipp and A. Beléndez, Eur. J. Phys., 2002, 23, 371 CrossRef.
- T. Bailey and J. E. Hubbard Jr, J. Guid. Control Dyn., 1985, 8, 605–611 CrossRef.
- S. A. Mani, S. U. Hadkar, P. Jessy, S. Lal, P. Keller, S. Khosla, N. Sood and P. Sarawade, J. Inf. Disp., 2016, 17, 169–176 CrossRef.
- M. T. Brannum, A. M. Steele, M. C. Venetos, L. T. Korley, G. E. Wnek and T. J. White, Adv. Opt. Mater., 2019, 7, 1801683 CrossRef.
- J. M. Boothby, T. Van Volkenburg, N. Q. Le, K. Ohiri, M. Hagedon and Z. Xia, Multifunct. Mater., 2020, 3, 015002 CrossRef CAS.
- K. M. Lee and T. J. White, Macromolecules, 2012, 45, 7163–7170 CrossRef CAS.
- J. Wang, Y. Zheng, L. Li, E. Liu, C. Zong, J. Zhao, J. Xie, F. Xu, T. A. König and M. Grenzer Saphiannikova, ACS Appl. Mater. Interfaces, 2019, 11, 25595–25604 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc04063e |
| ‡ G. T. L. and B. P. S. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |