DOI:
10.1039/D4TC03867C
(Paper)
J. Mater. Chem. C, 2025,
13, 3886-3901
Mixed metal conductive MOFs constructed from Trypan blue linked metal nodes: characteristic features and electrochemical performance†
Received
9th September 2024
, Accepted 25th December 2024
First published on 30th December 2024
Abstract
Multifunctional ligands possess multiple coordination sites, enabling them to simultaneously connect with several metal nodes. In this study, we report the use of Trypan blue (Try) as a multifunctional organic linker to fabricate new three-dimensional (3D) 3d–3d and 3d–4f bimetallic metal–organic frameworks (MOFs). The multiple binding sites, including amino, hydroxyl, sulfonate, and azo groups, facilitated the coordination of metals (M = Cu, Co, Zn, Er, and Yb) with varying preferences and geometries. Depending on the metal interactions, these MOFs (Cu–Co–Try (P1), Cu–Zn–Try (P2), Cu–Er–Try (P3), Cu–Yb–Try (P4)) adopted empirical formulae such as Cu2M1C2O1.2N1/Cu1M2C2O1N1. They exhibit polymorphic crystal forms, predominantly featuring monoclinic symmetry in P21/c, P21/n, hexagonal symmetry in P3221, and triclinic symmetry in P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space groups. Electronic and vibrational spectroscopy results indicate that Trypan blue remained intact throughout the synthesis. The paste of these MOFs can serve as a stand-alone electroactive working electrode material without the need for carbon additives. They exhibited high peak current responses of 719.4 μA (P1), 637.5 μA (P2), 79.7 μA (P3), and 173.7 μA (P4) at a scan rate of 0.01 V s−1. The charge mobility values for the MOFs were determined as 1.08 × 10−7 (E), 1.25 × 10−3 (P1), 1.42 × 10−3 (P2), 1.58 × 10−4 (P3), and 1.51 × 10−4 (P4) cm2 V−1 s−1, suggesting that electron transfer occurs via hopping mechanisms. Additionally, the effective utilization of Trypan blue in MOF synthesis, without generating any waste, may reduce environmental impact and enhance economic benefits.
space groups. Electronic and vibrational spectroscopy results indicate that Trypan blue remained intact throughout the synthesis. The paste of these MOFs can serve as a stand-alone electroactive working electrode material without the need for carbon additives. They exhibited high peak current responses of 719.4 μA (P1), 637.5 μA (P2), 79.7 μA (P3), and 173.7 μA (P4) at a scan rate of 0.01 V s−1. The charge mobility values for the MOFs were determined as 1.08 × 10−7 (E), 1.25 × 10−3 (P1), 1.42 × 10−3 (P2), 1.58 × 10−4 (P3), and 1.51 × 10−4 (P4) cm2 V−1 s−1, suggesting that electron transfer occurs via hopping mechanisms. Additionally, the effective utilization of Trypan blue in MOF synthesis, without generating any waste, may reduce environmental impact and enhance economic benefits.
1. Introduction
Commonly used ligands for synthesizing conductive MOFs include tetrathiafulvalene (TTF), benzene-1,4-dithiolate (BDT), catechol derivatives, 2,3,6,7,10,11-hexaamino triphenylene (HITP), porphyrins and phthalocyanines, and carboxylates (e.g., 1,2,4,5-benzenetetracarboxylate, BTC).1–10 TTF is an electron-rich, π-conjugated molecule capable of donating electrons, making it a popular choice for conductive MOFs. BDT is known for its ability to coordinate with metal centers through sulfur atoms, promoting electron delocalization.4 Catechol groups provide strong coordination with metal ions and can facilitate electron transfer through redox-active behavior. HITP, porphyrins, and phthalocyanine derivatives are large, planar, π-conjugated molecules that easily coordinate with metal centers to create conductive frameworks with enhanced electron delocalization.8,9 However, TTF, HITP, porphyrin- or phthalocyanine, and BDT-based MOFs are sensitive to oxidative degradation, and their incorporation often requires multi-step synthetic procedures.11,12 Catechol- and carboxylate MOFs might require additional modification or doping as they may not inherently contribute to electrical conductivity. To address these challenges, other unexplored conjugated and multidentate commercially available organic molecules should be explored for designing conductive MOFs, thereby promoting their broad-spectrum use.
Furthermore, the electrochemical activity investigated in existing studies primarily relies on a single type of metal center.13 However, conductive MOFs with mixed metal centers can provide a higher density of active sites for electrochemical reactions. Different metals can possess varying electronic properties, such as distinct oxidation states or differing degrees of d-electron involvement.14 These variants can optimize the distribution of charge carriers within the MOF, leading to increased charge carrier mobility and overall electrical conductivity. Combining elements with diverse electronic properties, such as lanthanides (4f elements) and 3d transition metals, might induce multifunctionality in the resulting MOFs. The Lewis acidity of lanthanides can complement the redox activity of 3d metals.15–17 Together, they can introduce additional energy transfer pathways, enabling multi-wavelength emission. Lanthanides often form highly stable coordination environments due to their large ionic radii and coordination numbers. This can enhance the overall stability of the MOF, particularly under harsh conditions.14–17 However, achieving a uniform distribution may require precise control over synthesis conditions to prevent phase separation or preferential binding. 4f and 3d metals often have different coordination preferences and geometries, which can complicate the design of a MOF that accommodates both metal types in a uniform structure. In solution to this, a ligand with multiple binding sites can enhance coordination versatility, enabling strong metal–ligand interactions.15 Sulfonate groups, for example, form strong ionic bonds with metal ions, contributing to the overall stability of the MOF. Hydroxyl and amino groups can also form hydrogen bonds, further stabilizing the framework and increasing its resistance to thermal and chemical degradation. A ligand with both soft donor atoms (e.g., sulfur) and hard donor atoms (e.g., oxygen) can stabilize a heterometallic center by coordinating with a soft metal at one site and a hard metal at another, thus preventing the segregation of different metal ions into separate phases.18–20 Although 3D MOFs generally exhibit lower electrical conductivity compared to their 2D MOF counterparts, these materials exhibit desirable chemical functionality, chemical stability, electronic properties, proton conductivity, optical properties, and magnetic properties. 3D MOFs can be tailored using multifunctional organic linkers and multiple metal nodes, which allows for high selectivity in terms of chemical sensing, enhanced catalytic properties, or enhanced adsorption properties.13–15 Most of the 3D MOFs have greater structural rigidity and stability, especially under harsh conditions (e.g., high pressure or temperature). Via post-synthetic modifications or metal doping, the intrinsic conductivity of 3D MOFs can be improved without sacrificing their structural constitution. Incorporating metal nodes with mixed oxidation states facilitates electron hopping between metal centers and π-conjugated linkers, which can create extended pathways for electron delocalization, improving conductivity. Owing to this phenomenon, 3D MOFs excel in proton conductivity with accessible charge transport pathways, making them useful in fuel cells and proton-exchange membranes.14–18
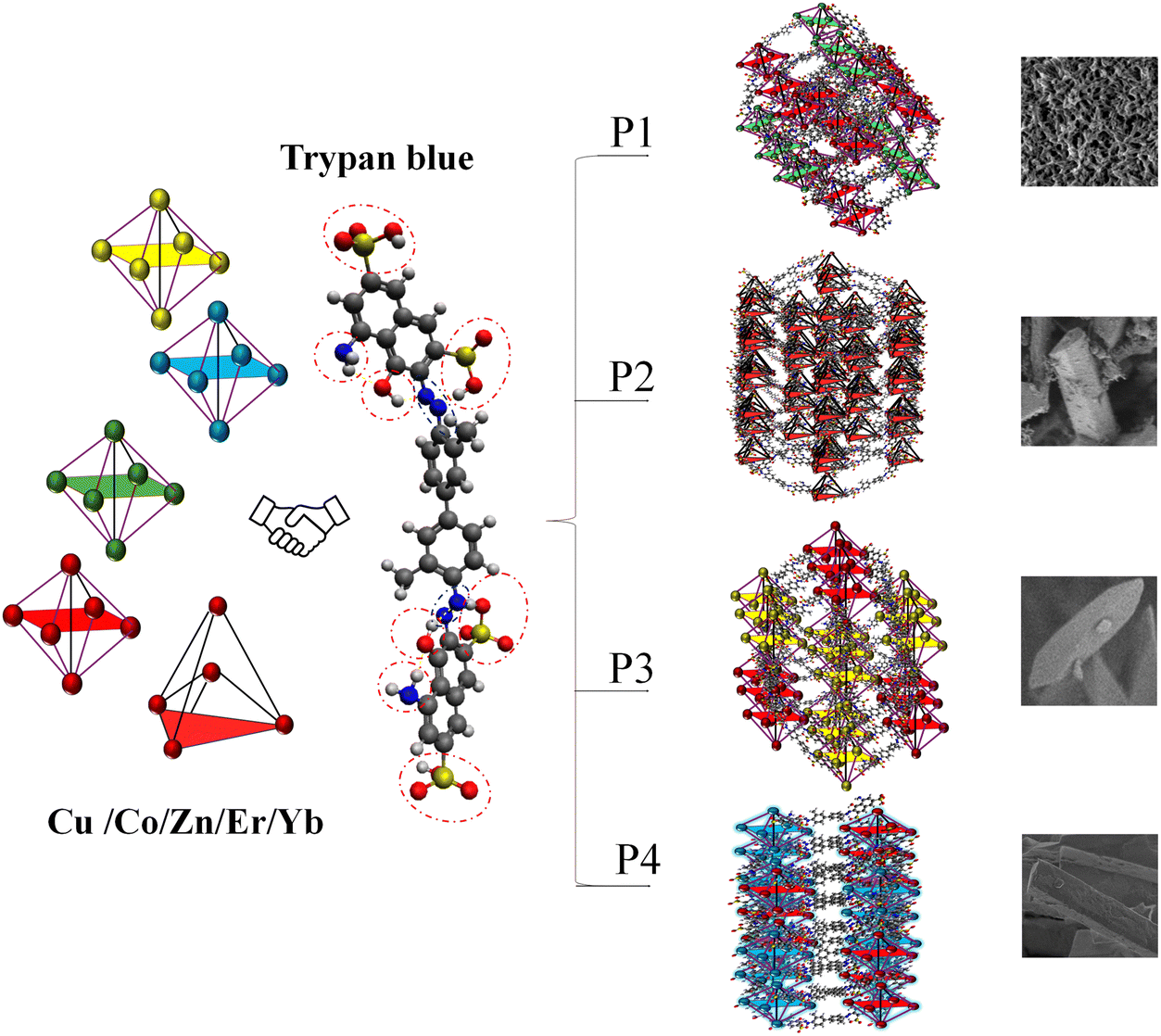
Herein, we report the fabrication of four unique three-dimensional (3D) multivariate MOFs (Cu–Co, Cu–Zn, Cu–Er, Cu–Yb MOFs) using Trypan blue [3,3′-[(3,3′-dimethyl(1,1′-biphenyl)-4,4′-diyl) bis(azo)] bis (5-amino-4-hydroxy-2,7-naphthalenedisulfonic acid)] as a multifunctional ligand (Fig. 1). The incorporation of Trypan blue (Try) as an organic linker in the design of these organometallic frameworks also serves as a mitigation measure against its potential toxicity. This innovative approach aids in developing diverse types of MOFs with different metal centers and binding sites while providing a sustainable solution for addressing Trypan blue toxicity. We used Cu, Co, Zn, Er, and Yb as metal centers and prepared four hybrid MOFs by combining the soft and electronegative Cu2+ ions with the relatively harder and more electropositive Co3+, Zn2+, Er3+, and Yb3+ ions. Given the multifunctionality of Trypan blue, harder metal centers might interact with the harder binding sites, while softer ones may preferably coordinate with the soft donor sites.18–20 The TGA curves of the reported MOFs P1, P2, P3, and P4 demonstrated superior thermal stability, remaining completely stable with no weight loss below 130 °C. After the degradation of solvents and organic components, approximately 30–45% of the material remained as residue upon heating to around 600 °C, corresponding to the formation of metal oxides. High-temperature treatment of MOFs led to the carbonization of organic linkers, resulting in the formation of conductive carbon frameworks and an increase in current response. The bimetallic compositions had a notable impact, with MOFs P1, P2, P3, and P4, containing Cu(II), Co(II), Zn(II), Er(III), and Yb(III), likely exhibiting strong charge transfer bands in the UV-visible range. Variations in metal composition and oxidation states caused shifts in the absorbance maxima to longer wavelengths for both transitions. Electrochemical experiments showed enhanced charge mobility and transfer kinetics for hybrid MOFs (P1, P2, P3, and P4) compared to single metal MOF systems. Linear sweep voltammetric experiments demonstrated enhanced HER performance for P3 and P4 compared to P1 and P2; P1 and P2 showed superior OER activity, achieving higher current densities. Electrochemical impedance spectroscopy (EIS) studies typically reveal a moderate charge transfer resistance for such MOFs, reflecting effective electron mobility across the framework.
 |
| | Fig. 1 Schematic view of formation of Cu–(Co/Zn/Er/Yb)–Trypan blue polymorphic MOFs. | |
2. Experimental
2.1. Materials
Copper nitrate hydrate.
Cu(NO3)2·H2O, cobalt acetylacetonate(II), zinc nitrate(II) hexahydrate, erbium chloride(III), ytterbium chloride(III), Trypan blue solution (0.4%), benzene tetra carboxylic acid (H4BTC), dimethyl formamide (DMF), phosphate buffer (PBS, pH 7.4), tetrathiafulvalene (TTF), hydrochloric acid (0.1) and other compounds and reagents were obtained commercially from Sigma Aldrich Chemical Co. USA (St. Louis, MO, USA).
2.2. Synthesis of MOFs
Synthesis of Cux (Try)x.
Trypan blue (Try) (100 mL) and Cu(NO3)2·H2O (100 mg) were dissolved in 4 mL of DMF, followed by 15 min sonication and then stirred at 110 °C for 24 h. The product was centrifuged from the mixture, washed repeatedly with ethanol, and then dried in a vacuum oven at a temperature of 100 °C for a period of 12 h.
Synthesis of Cux Coy (Try)x.
Trypan blue (Try) (100 μL) and Cu(NO3)2·H2O (100 mg), Co (NO3)2 (100 mg) was dissolved in 4 mL of DMF, followed by 15 min sonication and then heated at of 110 °C for a period of 24 h. The product was centrifuged from the mixture, washed repeatedly with ethanol, and then dried in a vacuum oven at a temperature of 100 °C for a period of 12 h.
Synthesis of Cux Zny (Try)x.
Trypan blue (Try) (100 μL) and Cu(NO3)2·H2O (100 mg), Zn(NO3)2·H2O (100 mg) was dissolved in 4 mL of DMF, followed by 15 min sonication and then heating at 110 °C for 24 h. The product was centrifuged from the mixture, washed repeatedly with ethanol to remove the residual unreacted precursors or ions, and then dried in a vacuum oven at a temperature of 100 °C for a period of 12 h.
Synthesis of Cux Ery (Try)x.
Trypan blue (Try) (100 μL) and Cu(NO3)2·H2O (100 mg), Er(Cl3)3 (100 mg) was dissolved in 4 mL of DMF, followed by 15 min sonication and heating at 110 °C for 24 h. The product was centrifuged from the mixture, washed repeatedly with ethanol, and then dried in a vacuum oven at a temperature of 100 °C for a period of 12 h.
Synthesis of Cux Yby (Try)x.
Trypan blue (Try) (100 μL) and Cu(NO3)2·H2O (100 mg), Yb(Cl3)3 (100 mg) was dissolved in 4 mL of DMF, followed by 15 min sonication and heating at 110 °C for 24 h. The product was centrifuged from the mixture, washed repeatedly with ethanol, and then dried in a vacuum oven at a temperature of 100 °C for a period of 12 h.
2.3. Instrumentation
Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra were acquired using a Thermo Fisher iS10 FTIR spectrometer (Waltham, MA). UV-Vis absorption spectra were acquired using Agilent Cary 300 & Agilent Cary 5000 UV-Vis-NIR spectrophotometers (USA). Scanning electron microscopy (SEM) data were collected using a Hitachi SU3900 variable pressure scanning electron microscope (VPSEM) (Tokyo, Japan). X-ray photoelectron spectroscopy (XPS) was utilized to evaluate the surface composition of MOFs. Measurements were undertaken using a Kratos XPS system with a monochromatic aluminum Kα (1.487 keV) source. The X-ray diffraction (XRD) studies were undertaken using a Panalytical Empyrean spinning stage X-ray diffractometer (Malvern Panalytical, Malvern, United Kingdom). Rietveld refinement was performed using X’pert High score software. BET and BJH studies were performed at Covalent Metrology Analytical Services (Sunnyvale, CA, USA).
2.4. Electrochemical measurements
The electrochemical behavior (CV and EIS) of the MOFs were evaluated with a DropSens SPELEC system (Metrohm AG, Herisau, Switzerland) and a PalmSens portable sensor bit potentiostat that contained a PSTrace Bluetooth user interface (EIS) in 0.1 N HCl solution. Both instruments contained a conventional three-electrode configuration. The final ground MOFs were converted into a paste with graphite powder (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and filled into a 3 mm well of graphite paste electrode (GPE). MOF-modified GPE was adopted as the working electrode, platinum wire was utilized as the counter electrode, and silver wire was used as the reference electrode. The CV cycles were recorded over a range of scan rates from 10–500 mV s−1, within the potential window of −0.15 to 0.6 V. The EIS studies were conducted with the initial voltage set to an open circuit voltage, a frequency of 0.1 Hz to 100 kHz, and an amplitude of 5 mV. Later, the electrode behavior was recorded at different fixed AC voltages (0.01–0.035) over changing frequency of 0.1 Hz to 100 kHz.
:1) and filled into a 3 mm well of graphite paste electrode (GPE). MOF-modified GPE was adopted as the working electrode, platinum wire was utilized as the counter electrode, and silver wire was used as the reference electrode. The CV cycles were recorded over a range of scan rates from 10–500 mV s−1, within the potential window of −0.15 to 0.6 V. The EIS studies were conducted with the initial voltage set to an open circuit voltage, a frequency of 0.1 Hz to 100 kHz, and an amplitude of 5 mV. Later, the electrode behavior was recorded at different fixed AC voltages (0.01–0.035) over changing frequency of 0.1 Hz to 100 kHz.
3. Results and discussion
3.1. Morphology and elemental analysis
Fig. 2 shows the SEM micrographs of MOFs (E, P1, P2, P3 & P4). Cu/Try (E) exhibits an irregular barrel-shaped clustered structural configuration (Fig. 2(a)). Cu–Co–Try (P1) has a distorted sheet-like morphology, appearing to form hexagonal/cubical structures joined by elongated chains (Fig. 2(b)). Cu–Zn–Try (P2) displays a rod-like morphology with flat ends, resembling mineral crystals like epidote, kyanite, or rhodonite, typical of monoclinic and triclinic crystal systems (Fig. 2(c)).21,22 Cu–Er–Try (P3) exhibits rod structures with pyramidal ends, giving them a monoclinic prism-like morphology (Fig. 2(d)). Cu–Yb–Try (P4) exhibits polygonal rod-like morphology with square flat ends (Fig. S1a, ESI†). The probable crystal configurations obtained via crystallographic refinement are discussed later in this text. The EDS profiles confirmed the presence of the elements Cu, Co, Zn, Er, Yb, C, O, and N. A stoichiometric ligand-to-metal feed ratio of 3:1 was used for the synthesis (Fig. 2(e)–(h) and Fig. S1b, ESI†). The high carbon content is attributed to the aromatic structure of the Try ligand. The potential compositional formulas were calculated as Cu1.45C4.5O2.5N (E), Cu2.1Co1.2C8O5.8N (P1), Cu0.4Zn0.9C4.6O2.7N (P2), and Cu0.06Er1.0C7.2O1.0N (P3), Cu0.1Yb1.1C9.7O3.9N (P4) (Fig. S1b, ESI†). Elemental maps shown in Fig. S2–S6 (ESI†) depict the distribution of elements (C, O, N, Cu, Co, Zn, Er, Yb) in the selected regions of corresponding electron micrographs.
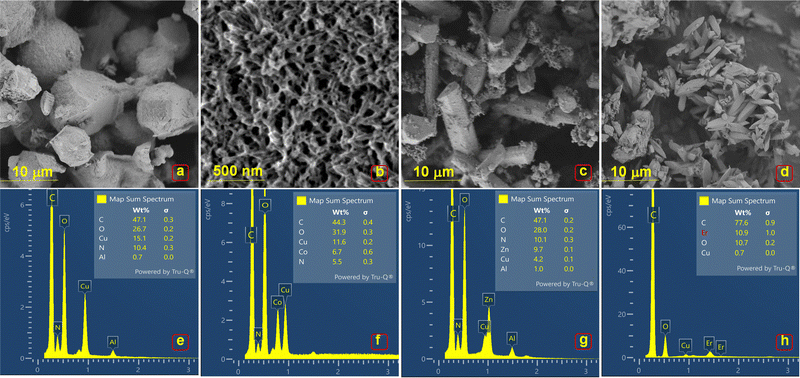 |
| | Fig. 2 SEM micrographs and corresponding EDS profiles of (a) and (b) E, (c) and (d) P1, (e) and (f) P2, (g) and (h) P3. | |
The surface and pore characteristics of the MOFs were evaluated using N2 adsorption–desorption isotherms (Fig. 3(a)–(d)). All MOFs exhibited type II isotherms, indicating non-porous or largely microporous behavior, with the inflection point corresponding to the end of monolayer adsorption. The large amount of adsorption near a relative pressure of 1 suggests the presence of macropores.23,24 E (12 m2 g−1) and P1 (15 m2 g−1) exhibited similar BET surface areas, with P2 (23.4 m2 g−1) having the largest surface area, followed by P3 (19 m2 g−1). Although the number of pores was small, MOFs E, P1, and P2 displayed pore sizes below the typical applicability limit of BJH (<3 nm). P3 had a significantly larger pore volume than the other MOFs, with BJH analysis showing a pore size of approximately 10 nm (Fig. 3(e) and (f)).
 |
| | Fig. 3 N2 adsorption curves of MOFs (a) E, (b) P1, (c) P2, (d) P3. BJH pore volume plots of MOFs (e) and (f). | |
3.2. Spectroscopic analyses
FTIR.
Trypan blue, a complex dye molecule and multifunctional organic linker, is reported to have several characteristic vibrations in the FTIR spectrum.25 Aromatic C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at ∼3100–3000, ∼1600–1500 cm−1 and C–H bending at 700–900 cm−1, SO, S–O stretching of sulfonate (SO3H) group at ∼1350–1250, ∼1500–1400 cm−1, NN (azo group) and C–N (amine group) stretching at ∼1550–1450, ∼1350–1250 cm−1, O–H, C–O stretching at ∼3500–3000, ∼1000–1300 cm−1, C–S stretching from 700–800 cm−1. Amine, sulfonate, and hydroxyl groups are the donor centers for MOF synthesis. Corresponding FTIR spectra of pristine Cu–Co–Try (P1), Cu–Zn–Try (P2), Cu–Er–Try (P3), Cu–Yb–Try (P4) MOFs are illustrated in Fig. 4(a)–(e) and are compared to Cu–Try MOF (E). Spectra of P1 exhibited a broad peak at 3296 cm−1 for O–H stretching vibrations, or possibly N–H stretching, sharp peaks at 1700 cm−1 associated with the carbonyl (CO) stretching vibration, 1620 cm−1 likely represents the CC and NN stretching vibrations (aromatic rings and azo groups), 1490 cm−1 and 1450 cm−1 for aromatic CC stretching which are core parts of dye structure, 1380 cm−1 and 1330 cm−1, correspond to symmetric SO3 and C–N stretching from aromatic amine groups, 1250 cm−1 of asymmetric SO3 stretching, 1140 cm−1 and 1110 cm−1 from C–O stretching and SO stretching, peaks at ∼1000 and 904 cm−1 may correspond to C–H in-plane and out of plane bending, 871, 821, and 679 cm−1 from C–H out-of-plane bending (aromatic), 724 cm−1 from C–S stretching, and 559 cm−1, 516 cm−1 likely from metal–oxygen (M–O) stretching, particularly Cu–O and Co–O. P2 possesses characteristic peaks at 3385 cm−1 (broad), 1666 cm−1, 1588 cm−1, 1493 cm−1, 1384 cm−1 and 1323 cm−1, 1140 cm−1 and 1117 cm−1, 1046 cm−1, 880 cm−1 and 843 cm−1, 814 cm−1, 770 cm−1, 688 cm−1, which corresponded to O–H stretching, CO stretching, CC as well as NN stretching, aromatic CC stretching, symmetric SO3 stretching as well as C–N stretching, C–O stretching as well as SO stretching, C–H In-plane bending, C–H out-of-plane bending (aromatic), C–H out-of-plane bending, C–S stretching, and metal–oxygen (M–O) stretching for Cu–O as well as Zn–O. Further, P3 spectra include band at 3300 cm−1, sharp peaks at 1689, 1646, 1618, 1568.8, 1494, 1381, 1298, 1135, 1117, 1061, 955, 879, 845, 809, 776, 746, 680, 660, 599, 512 cm−1, which may correspond to O–H stretching, CO stretching, CC and NN stretching, aromatic CC stretching, symmetric SO3 stretching and C–N stretching, C–O stretching and SO stretching, C–H in-plane bending, C–H out-of-plane bending (aromatic), C–H out-of-plane bending, C–S stretching, metal–oxygen (M–O), or metal–nitrogen (M–N) stretching vibrations. Similarly, P4 shows peaks at 3271 (broad), 1645, 1619, 1569, 1494, 1378, 1297, 1134, 1116, 954, 879, 840, 808, 776, 745, 680, 660, 597, 511 cm−1, most likely attribute to O–H stretching, CO stretching, CC and NN stretching, aromatic CC stretching, symmetric SO3 stretching and C–N stretching, C–O stretching and SO stretching, C–H in-plane bending, C–H out-of-plane bending (aromatic), C–H out-of-plane bending, C–S stretching, metal–oxygen (M–O), or metal–nitrogen (M–N) stretching vibrations. These spectra were compared to FTIR of E, which included peaks at 1617, 1491, 1445, 1386, 1334, 1138, 1107, 904, 870, 819, 678, 557, 512 cm−1 assigned to CO stretching, CC and NN stretching, aromatic CC stretching, symmetric SO3 stretching and C–N stretching, C–O stretching and SO stretching, C–H in-plane bending, C–H out-of-plane bending (aromatic), C–H out-of-plane bending, C–S stretching, metal–oxygen (M–O). The presence of stretching vibrations for azo groups, CC, and SO3 groups for all the MOFs indicates that these linkages remain intact throughout the synthesis, which means that Trypan blue did not undergo degradation. The appearance of distinct M–O or M–N stretching frequencies in the low wavenumber region (400–700 cm−1) indicates the linkage formation between different metal centers (Cu, Co, Zn, Er, or Yb).24–28 The presence of multiple peaks in this region suggests different metal–ligand coordination environments. Shifts in the M–O stretching frequency compared to a monometallic MOF could indicate changes in the bond strength due to the presence of a second metal.26–28 Symmetric stretching modes seem more pronounced than bending modes among the multiple peaks, showing the symmetrical coordination of the multifunctional ligand to two different metals.27 Additionally, well-defined peaks indicate a stable, crystalline framework for all the MOFs.29,30
C stretching at ∼3100–3000, ∼1600–1500 cm−1 and C–H bending at 700–900 cm−1, SO, S–O stretching of sulfonate (SO3H) group at ∼1350–1250, ∼1500–1400 cm−1, NN (azo group) and C–N (amine group) stretching at ∼1550–1450, ∼1350–1250 cm−1, O–H, C–O stretching at ∼3500–3000, ∼1000–1300 cm−1, C–S stretching from 700–800 cm−1. Amine, sulfonate, and hydroxyl groups are the donor centers for MOF synthesis. Corresponding FTIR spectra of pristine Cu–Co–Try (P1), Cu–Zn–Try (P2), Cu–Er–Try (P3), Cu–Yb–Try (P4) MOFs are illustrated in Fig. 4(a)–(e) and are compared to Cu–Try MOF (E). Spectra of P1 exhibited a broad peak at 3296 cm−1 for O–H stretching vibrations, or possibly N–H stretching, sharp peaks at 1700 cm−1 associated with the carbonyl (CO) stretching vibration, 1620 cm−1 likely represents the CC and NN stretching vibrations (aromatic rings and azo groups), 1490 cm−1 and 1450 cm−1 for aromatic CC stretching which are core parts of dye structure, 1380 cm−1 and 1330 cm−1, correspond to symmetric SO3 and C–N stretching from aromatic amine groups, 1250 cm−1 of asymmetric SO3 stretching, 1140 cm−1 and 1110 cm−1 from C–O stretching and SO stretching, peaks at ∼1000 and 904 cm−1 may correspond to C–H in-plane and out of plane bending, 871, 821, and 679 cm−1 from C–H out-of-plane bending (aromatic), 724 cm−1 from C–S stretching, and 559 cm−1, 516 cm−1 likely from metal–oxygen (M–O) stretching, particularly Cu–O and Co–O. P2 possesses characteristic peaks at 3385 cm−1 (broad), 1666 cm−1, 1588 cm−1, 1493 cm−1, 1384 cm−1 and 1323 cm−1, 1140 cm−1 and 1117 cm−1, 1046 cm−1, 880 cm−1 and 843 cm−1, 814 cm−1, 770 cm−1, 688 cm−1, which corresponded to O–H stretching, CO stretching, CC as well as NN stretching, aromatic CC stretching, symmetric SO3 stretching as well as C–N stretching, C–O stretching as well as SO stretching, C–H In-plane bending, C–H out-of-plane bending (aromatic), C–H out-of-plane bending, C–S stretching, and metal–oxygen (M–O) stretching for Cu–O as well as Zn–O. Further, P3 spectra include band at 3300 cm−1, sharp peaks at 1689, 1646, 1618, 1568.8, 1494, 1381, 1298, 1135, 1117, 1061, 955, 879, 845, 809, 776, 746, 680, 660, 599, 512 cm−1, which may correspond to O–H stretching, CO stretching, CC and NN stretching, aromatic CC stretching, symmetric SO3 stretching and C–N stretching, C–O stretching and SO stretching, C–H in-plane bending, C–H out-of-plane bending (aromatic), C–H out-of-plane bending, C–S stretching, metal–oxygen (M–O), or metal–nitrogen (M–N) stretching vibrations. Similarly, P4 shows peaks at 3271 (broad), 1645, 1619, 1569, 1494, 1378, 1297, 1134, 1116, 954, 879, 840, 808, 776, 745, 680, 660, 597, 511 cm−1, most likely attribute to O–H stretching, CO stretching, CC and NN stretching, aromatic CC stretching, symmetric SO3 stretching and C–N stretching, C–O stretching and SO stretching, C–H in-plane bending, C–H out-of-plane bending (aromatic), C–H out-of-plane bending, C–S stretching, metal–oxygen (M–O), or metal–nitrogen (M–N) stretching vibrations. These spectra were compared to FTIR of E, which included peaks at 1617, 1491, 1445, 1386, 1334, 1138, 1107, 904, 870, 819, 678, 557, 512 cm−1 assigned to CO stretching, CC and NN stretching, aromatic CC stretching, symmetric SO3 stretching and C–N stretching, C–O stretching and SO stretching, C–H in-plane bending, C–H out-of-plane bending (aromatic), C–H out-of-plane bending, C–S stretching, metal–oxygen (M–O). The presence of stretching vibrations for azo groups, CC, and SO3 groups for all the MOFs indicates that these linkages remain intact throughout the synthesis, which means that Trypan blue did not undergo degradation. The appearance of distinct M–O or M–N stretching frequencies in the low wavenumber region (400–700 cm−1) indicates the linkage formation between different metal centers (Cu, Co, Zn, Er, or Yb).24–28 The presence of multiple peaks in this region suggests different metal–ligand coordination environments. Shifts in the M–O stretching frequency compared to a monometallic MOF could indicate changes in the bond strength due to the presence of a second metal.26–28 Symmetric stretching modes seem more pronounced than bending modes among the multiple peaks, showing the symmetrical coordination of the multifunctional ligand to two different metals.27 Additionally, well-defined peaks indicate a stable, crystalline framework for all the MOFs.29,30
 |
| | Fig. 4 FTIR spectra of MOFs (a) E, (b) P1, (c) P2, (d) P3, (e) P4 with reference to trypan blue spectra in pink legend. (f) UV-Vis spectra of MOFs tracing the peaks of trypan blue (592, 318 nm). | |
UV-Visible spectroscopy.
Fig. 4(f) presents the UV-Vis spectra recorded from aliquots collected during the MOF synthesis process, showing absorbance maxima at 250–400 nm and 500–600 nm, which were analyzed to track the fate of Trypan blue during synthesis. Typical absorbance bands observed in bimetallic Trypan blue-linked MOFs include a π → π electronic transition around 200–300 nm, attributed to the conjugated aromatic rings of Trypan Blue, and a n → π transition around 400–500 nm, corresponding to the lone pairs on azo groups influenced by metal coordination. Additionally, a ligand-to-metal charge transfer (LMCT) band appears at ∼400–600 nm. The influence of bimetallic compositions led to noteworthy observations: MOFs P1, P2, P3, and P4, incorporating Cu(II), Co(II), Zn(II), Er(III), and Yb(III), likely exhibited strong charge transfer bands in the UV-visible range. Variations in the metal composition and oxidation states resulted in shifts of absorbance maxima to higher wavelengths for both transitions. Furthermore, differences in intensity were attributed to ligand coordination and MOF crystallinity. The MOFs can be ranked in the order of P2 > P3 > P4 > P1 based on band intensity, as shown in Fig. 4(f). As such, the extent of electron delocalization within the frameworks is expected to follow the same order. In agreement with the FTIR results, the bands corresponding to π → π* transition of the –NN– (azo group) at 315 nm and π → π* transition of an aromatic portion (596 nm) appear to remain intact.31,32 This result indicates that Trypan blue did not degrade or undergo significant chemical changes during synthesis at 110 °C, retaining its structural integrity. Table 1 shows a comparison of the key parameters of the reported MOFs obtained through various analysis approaches.
Table 1 Comparison of key parameters of the reported MOFs from various analyses
| MOFs |
FTIR stretching vibrations cm−1 |
| M–O |
C–S |
C–O |
C–N |
CO |
| Cu–Try B |
512 |
678 |
1138 |
1334 |
1617 |
| Cu–Co Try B |
597, 511 |
724 |
1140 |
1330 |
1700 |
| Cu–Zn Try B |
552, 510 |
770 |
1140 |
1323 |
1666 |
| Cu–Er Try B |
599, 512 |
746 |
1135 |
1298 |
1689 |
| Cu–Yb Try B |
559, 516 |
745 |
1134 |
1297 |
1645 |
| XPS profile/binding energy (eV) |
| MOFs |
C1s |
O1s |
N1s |
Cu 2p3/2 |
Cu 2p1/2 |
Cu Sat |
M2 Co/Zn/Er/Yb |
| Cu–Try B |
285.56 |
531.5 |
402.69 |
937.7 |
957.7 |
943.3 |
|
| 286.91 |
534.42 |
947.7 |
| 289.24 |
|
966.1 |
| Cu–Co Try B |
285.39 |
532.13 |
390.27 |
937.6 |
957.5 |
942.6 |
784.7 |
| 287.97 |
535.03 |
395.02 |
946.7 |
788.9 |
| 291.69 |
|
403.73 |
966.2 |
803.2 |
| Cu–Zn Try B |
285.94 |
533.01 |
393.98 |
937.9 |
957.8 |
943.38 |
1016.6 |
| 288.24 |
535.28 |
398.18 |
947.5 |
1025.9 |
| 292.21 |
|
|
965.1 |
1039.1 |
|
|
|
|
|
1049.04 |
| Cu–Er Try B |
286.01 |
532.39 |
395.85 |
936.5 |
956.38 |
946 |
201.59 |
| 287.71 |
534.79 |
400.01 |
962 |
173.16 |
| 291.91 |
|
|
|
189.12 |
|
|
|
|
|
201.59 |
| Cu–Yb Try B |
285.89 |
531.31 |
391.01 |
936.06 |
956.11 |
944.67 |
186.45 |
| 287.35 |
534.38 |
395.33 |
963.34 |
196.69 |
| 291.19 |
|
400.21 |
|
|
| Crystallographic profile |
| MOFs |
Space group – crystal system |
Ratios |
% Composition of polymorph |
| M1/M2 |
N/O |
| Cu–Try B |
P21/c – Monoclinic |
— |
— |
100 |
| Cu–Co Try B |
P3221 – Hexagonal |
2:1 |
2.5:1 |
77/11 |
|
Pm3m – Cubic |
|
C2/c – Monoclinic |
| Cu–Zn Try B |
P21/c1 – Monoclinic |
1:1 |
2:1 |
53.8/46.2 |
|
P – Triclinic |
| Cu–Er Try B |
P121/n1 – 2 Monoclinic systems |
3:2 |
1:6 |
52.6/47.4 |
X-Ray photoelectron analysis.
The chemical states and electronic interactions of MOFs (E, P1, P2, P3, P4) were analyzed using the XPS profiles. The survey spectrum (Fig. S7–S10, ESI†) demonstrated the presence of Cu, Co, Zn, Er, Yb, C, O, and N elements in the corresponding end products. C1s spectra revealed several peaks with varying FWHM values, each corresponding to various chemical states found in carbon (e.g., C–C, CC, C–O, CO). Different MOFs showed almost the same bands that can be assigned to C–O, C–N, and CO units at ∼285.37–285.9, 286.02, and 287.71–288.24 eV, respectively (Fig. 5(A)-a). The satellite peak indexed at 289.24–291.9 eV can be attributed to π–π* shake-up, indicating the presence of extended delocalized electron hopping in multivariate Cu–Tryp complexes.33,34 According to the calculated FWHM values, the C–C/C–H peaks are relatively narrow, indicating a uniform environment for these carbon atoms. The C–O peaks are broader, suggesting a more varied environment for carbon bonded to oxygen. The CO peaks are the broadest, indicating significant chemical and electronic diversity among the carbonyl groups. The O 1s regions of all products can be deconvoluted into two peaks (Fig. 5(A)-b). The corresponding peak positions showed a shift of approximately 0.9–1.2 eV. The two peaks are centered at 531.5 and 534.42 eV for E, at 532.14 and 534.94 eV for P1, at 533.30 and 535.33 eV for P2, at 532.81 and 534.88 eV for P3, and at 531.36 and 534.39 eV for P4. These peaks can be assigned to the oxygen atoms in the coordinated hydroxyl group (–OH) and coordinated carboxylate group (–CO, –C–O), respectively. The appearance of O1s spectra for all products was observed to be different. The lower binding energy (B.E.) peak was found to broaden and increase in height from E to P4. A shift to lower binding energies for the M–O component suggests strong metal–ligand interaction.33,34
 |
| | Fig. 5 (A) XPS spectra of (a) C1s, (b) O1s, and (c) N1s, in MOFs E, P1, P2, P3, and P4. (B) XPS spectra of (a) Cu in MOFs E, P1, P2, P3, and P4, (b) Co in P1, and (c) Zn in P2, (d) Er in P3, and (e) Yb in P4. | |
The N1s spectra of E show a single peak at 402.7 eV, suggesting that the nitrogen atoms are in a positively charged or protonated state (Fig. 5(A)-c). A peak around this binding energy typically indicates the presence of nitrogen atoms involved in protonated amine groups (–NH3+), possibly due to the coordination environment with metal centers or interactions with other functional groups in the MOF. P1 shows three distinct peaks in the N1s spectrum at 403.79 eV, 395.02 eV, and 390.27 eV. This suggests there are different chemical environments or bonding states for nitrogen in the material (Fig. 5(A)-c). 403.79 eV is possibly related to oxidized nitrogen or nitrogen in a protonated or complexed state due to interactions with the metal centers (Cu and Co). 395.02 eV likely corresponds to nitrogen in a basic or neutral state, such as the amine group of Try. 390.27 eV represents nitrogen in a more reduced state or directly coordinated with the metal centers (Cu and Co). Co exhibits redox flexibility (Co2+/Co3+), leading to varied nitrogen coordination environments. The low binding energy (390.27 eV) corresponds to strong charge transfer interactions with Co2+ or Cu+, while the higher energy (403.79 eV) indicates nitrogen is involved in protonation or hydrogen bonding. For P2, two N1s peaks were observed at 398.18 eV and 393.98 eV. The peak at 398.18 eV may correspond to nitrogen in the aromatic environment of Trypan blue. The lower binding energy peak might correspond to nitrogen in a more electron-rich or reduced state, around Cu/Zn in the MOF structure (Fig. 5(A)-c). Zn2+ is redox-inactive and primarily forms simple, stable coordination bonds; this results in narrower peaks and fewer distinct nitrogen environments. P3 shows two N1s peaks at 400.01 eV and 395.85 eV. The former one could correspond to nitrogen in the aromatic or relatively oxidized state (Fig. 5(A)-c). The latter one could indicate nitrogen in a more electron-rich or reduced state. Similarly, the N1s spectrum of P4 depicts three peaks at 400.21 eV, 395.85 eV, and 391.01 eV (Fig. 5(A)-c). The strong polarization effect of lanthanides (Er/Yb) shifts the N 1s peaks to higher binding energies (395.85–400.21 eV), indicating electron density redistribution around nitrogen. Er3+ and Yb3+ may polarize the ligand more strongly due to their high effective charge and larger ionic radii, creating distinct nitrogen environments. Additionally, they favor higher coordination numbers (8–10), which can create a broader range of nitrogen environments and can influence the chemical states.
The XPS profile of Cu 2p for Cu in E and all products showed the Cu 2p3/2 and Cu 2p1/2 bands at 937.36 and 957.35 eV for E, 937.8 and 957.41 eV for P1, 937.7 and 957.7 eV for P2, 936.5 and 956.38 eV for P3, and 936.06 and 956.11 eV for P4 (Fig. 5(B)-a). The obtained peaks were positioned 3–4 eV higher than the standard peak values. These two bands indicate the divalent chemical state of Cu in the products. The fitted bands in the range of 942.6–947.8 and 964.18–966.14 eV are the shake-up satellite peaks.35,36 Hybrid MOFs exhibited a noticeable enhancement in the shake-up peak energies, indicating a more paramagnetic state of Cu. The shift in binding energy suggests that the Cu is in a different chemical environment or oxidation state than the standard reference material, likely due to strong interactions with other elements, variations in coordination geometry, or multiple oxidation states. The high-resolution Co 2p XPS spectra of Cu–Co–Tryp (P1) exhibit two prominent bands at ∼787.4 and 803.03 eV, corresponding to spin–orbit splitting to Co 2p3/2 and Co 2p1/2, respectively. This result indicates that cobalt is mainly in its +3-oxidation state (Fig. 5(B)-b). Peak separation of 15.63 eV is consistent with what is observed for Co3+ species.37,38 The Zn 2p spectrum for Cu–Zn–Tryp (P2) reveals two prominent peaks at 1025.2 and 1049.01 eV, characteristic to Zn2+ and attributed to spin–orbit splitting to Zn 2p3/2 and Zn 2p1/2 components. The separation between the Zn 2p3/2 and Zn 2p1/2 peaks is about 23.8 eV, which is typical for Zn2+, confirming this oxidation state. The presence of Zn2+ suggests that zinc is coordinated with oxygen or nitrogen atoms in the Try ligand (Fig. 5(B)-c). Zn2+ ions do not appear to be in a highly distorted environment, consistent with typical coordination geometries such as tetrahedral or octahedral arrangements.39,40 The maintenance of typical +2 oxidation state in the MOF indicates minimal electronic interference from Cu. Er 3d spectra exhibited two peaks at units 173.16 and 189.12 eV with a separation of ∼16.5 eV, corresponding to spin–orbit split components Er 3d5/2 and Er 3d3/2 (Fig. 5(B)-d). The binding energies at 173.16 eV (3d5/2) and 189.12 eV (3d3/2) are characteristic of Er3+ in a coordination environment, confirming that erbium is in the +3-oxidation state within the MOF. The spin–orbit splitting of approximately 16.5 eV is consistent with the expected value for Er3+ ions, confirming the presence of this oxidation state. The appearance of satellite peaks at 164.39 and 201.59 eV suggests that the electronic environment around Er3+ in this MOF is complex, possibly due to strong interactions between erbium and the Trypan blue ligand or due to the mixed metal environment with copper. The separation of these satellite peaks from the main 3d peaks provides additional confirmation of the +3-oxidation state.41,42 XPS peaks in Yb 4f spectra were indexed at units 186.45 and 196.69 eV with a separation of ∼10.5 eV, corresponding to Yb 4f7/2 and Yb 4f5/2. The binding energy values, and the peak separation are characteristic of Yb3+ ions, suggesting that Yb is in a relatively standard coordination environment without significant distortion or interaction that would alter its electronic structure (Fig. 5(B)-e). Yb 4f spectra indicate that Yb maintains its +3-oxidation state, suggesting that it remains relatively unaffected by the presence of Cu and Zn in the framework.43,44 Corresponding shifts in B.E. compared to standard references probably indicate the changes in the electronic environment due to metal–ligand interactions or the presence of multiple metals. Previous studies suggest that 3d metals (Cu, Co, Zn) show complex multiple peaks splitting due to spin–orbit coupling. 4f metals (Er, Yb) exhibited characteristic sharp peaks with significant spin–orbit splitting.39–44 Multiple metal combinations (3d–3d and 3d–4f) can influence the binding energy of each metal core level due to differences in electronegativity, coordination environment, and electronic structure. The presence of a more electronegative metal (Cu) did increase the binding energy of neighboring 3d (Co, Zn) and 4f (Er, Yb) metal atoms.
Crystallographic analysis.
Powder X-ray diffraction (PXRD) profiles revealed the five MOFs (Fig. 6) with crystallinity ranging from high (E, P1, P2) to semi-crystalline (P3, P4). E displayed 12 sharp signals at 15.30, 17.46, 19.80, 22.71, 25.65, 26.61, 29.06, 33.94, 36.13, 39.68, 43.20°, which are indexed to (100), (110), ( 11), (111), (012), (121), (022), (040), (041), (221), (202) planes respectively (Fig. 6(a)). Further, P1 and P2 exhibited peaks at 9.76, 11.33, 12.76, 15.31, 17.59, 19.67, 20.48, 22.71, 25.68, 26.67, 29.14, 34.08, 36.21, 43.13° and at 8.36, 10.29, 10.83, 12.83, 16.19, 17.59, 17.96, 20.51, 22.61, 24.82, 25.29, 26.69, 28.82°. These correspond to the facets as (011), (012), (110), (111), (200), (210), (004), (204), (025), (300), (033), (713), (400), (![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 18) and (100), (1 1), (101), ( 20), (211), (220), (013), (311), ( 14), (203), (14), (
18) and (100), (1 1), (101), ( 20), (211), (220), (013), (311), ( 14), (203), (14), (![[5 with combining macron]](https://www.rsc.org/images/entities/char_0035_0304.gif) 13), respectively (Fig. 6(b)). Similarly, P3 and P4 were indexed with peaks at 2θ values of 8.69, 10.13, 15.77, 2.06, 27.78, 31.66, 33.84, 55.79, 66.55°; 9.89, 19.58, 28.80, 31.85, 40.22. They were assigned to the miller indices as (002), (110), (120), ( 23), (041), (331), (216), (615), (
13), respectively (Fig. 6(b)). Similarly, P3 and P4 were indexed with peaks at 2θ values of 8.69, 10.13, 15.77, 2.06, 27.78, 31.66, 33.84, 55.79, 66.55°; 9.89, 19.58, 28.80, 31.85, 40.22. They were assigned to the miller indices as (002), (110), (120), ( 23), (041), (331), (216), (615), (![[7 with combining macron]](https://www.rsc.org/images/entities/char_0037_0304.gif) 46), while P4 appears to be semi-crystalline (Fig. 6(c) and Fig. S11, ESI†). The Cu/Try-MOF structure appeared to possess monoclinic cells with crystallographic indices a = 5.26 Å, b = 9.92 Å, and c = 7.38 Å, and space group P21/c. The symmetry elements in this space group possibly impose specific constraints on the structural orientation and positioning of metal nodes and organic linkers, leading to unique pore structures and channels. Rietveld refinement could reveal the polymorphic nature of multivariate MOFs (P1, P2, P3 & P4). The quantification profile of the Cu/Co/Try-MOF (P2) complex shows ∼77% hexagonal (P3221), ∼11% cubic (Pm3m), and monoclinic (C2/c) components.45 The edges and angular dimensional parameters for hexagonal, cubic, and monoclinic include: [a = 10.96 Å, b = 10.96 Å, c = 22.45 Å; α = β = 90° and γ = 120°], [a = 9.85 Å, b = 9.85 Å, c = 9.85 Å; α = β = γ = 90°], and [a = 23.15 Å, b = 10.26 Å, c = 17.21 Å; α = γ = 90° and β = 96°]. The empirical arrangement of Cu and Co atoms is possibly 2:1, with a high carbon content (∼40 atoms) and a ratio of 2.5:1 for nitrogen/oxygen atoms. This suggests that two Cu atoms linked to aromatic Try ligands with heteroatoms (N, O) are bridged by a Co atom in each cell. The quantification profile of the Cu/Zn/Try-MOF (P2) complex is a mix of ∼53.8% monoclinic (P121/c1), and ∼46.2% triclinic (P) components. P means the polymorph has an inversion centre, making it centrosymmetric. The angular and dimensional parameters for the monoclinic and triclinic phases are as follows: [a = 16.24 Å, b = 10.56 Å, c = 20.85 Å; α = β = 90° and γ = 107.15°], and [a = 11 Å, b = 14.2 Å, c = 15.87 Å; α = 95°, β = 97°, γ = 90°]. The Cu and Zn atoms are present in a 1:1 ratio, with a high carbon content (∼40 atoms) and a ratio of 2:1 for nitrogen/oxygen atoms. This suggests that the equal metal centers (Zn/Cu) are bridged by multidentate Try ligand molecules in each cell. Cu/Er/Try-MOF (P3) complex is possibly a polymorphic mix (52.6/47.4%) of two similar monoclinic crystal systems (P121/n1). P means the polymorph has an inversion center, making it centrosymmetric.41–44 The angular and dimensional parameters for both monoclinic phases are as follows: [a = 11.65 Å, b = 12.87 Å, c = 19.15 Å; α = γ = 90° and β = 95°]. The Cu:Er atoms are present in a 3:2 ratio, and the refinement process shows n number of solvent (DMF) molecules included in the final model. Metal centers (Cu/Er) are bridged by multidentate Try ligand molecules, with an N:O ratio of 1:6 in each cell.
46), while P4 appears to be semi-crystalline (Fig. 6(c) and Fig. S11, ESI†). The Cu/Try-MOF structure appeared to possess monoclinic cells with crystallographic indices a = 5.26 Å, b = 9.92 Å, and c = 7.38 Å, and space group P21/c. The symmetry elements in this space group possibly impose specific constraints on the structural orientation and positioning of metal nodes and organic linkers, leading to unique pore structures and channels. Rietveld refinement could reveal the polymorphic nature of multivariate MOFs (P1, P2, P3 & P4). The quantification profile of the Cu/Co/Try-MOF (P2) complex shows ∼77% hexagonal (P3221), ∼11% cubic (Pm3m), and monoclinic (C2/c) components.45 The edges and angular dimensional parameters for hexagonal, cubic, and monoclinic include: [a = 10.96 Å, b = 10.96 Å, c = 22.45 Å; α = β = 90° and γ = 120°], [a = 9.85 Å, b = 9.85 Å, c = 9.85 Å; α = β = γ = 90°], and [a = 23.15 Å, b = 10.26 Å, c = 17.21 Å; α = γ = 90° and β = 96°]. The empirical arrangement of Cu and Co atoms is possibly 2:1, with a high carbon content (∼40 atoms) and a ratio of 2.5:1 for nitrogen/oxygen atoms. This suggests that two Cu atoms linked to aromatic Try ligands with heteroatoms (N, O) are bridged by a Co atom in each cell. The quantification profile of the Cu/Zn/Try-MOF (P2) complex is a mix of ∼53.8% monoclinic (P121/c1), and ∼46.2% triclinic (P) components. P means the polymorph has an inversion centre, making it centrosymmetric. The angular and dimensional parameters for the monoclinic and triclinic phases are as follows: [a = 16.24 Å, b = 10.56 Å, c = 20.85 Å; α = β = 90° and γ = 107.15°], and [a = 11 Å, b = 14.2 Å, c = 15.87 Å; α = 95°, β = 97°, γ = 90°]. The Cu and Zn atoms are present in a 1:1 ratio, with a high carbon content (∼40 atoms) and a ratio of 2:1 for nitrogen/oxygen atoms. This suggests that the equal metal centers (Zn/Cu) are bridged by multidentate Try ligand molecules in each cell. Cu/Er/Try-MOF (P3) complex is possibly a polymorphic mix (52.6/47.4%) of two similar monoclinic crystal systems (P121/n1). P means the polymorph has an inversion center, making it centrosymmetric.41–44 The angular and dimensional parameters for both monoclinic phases are as follows: [a = 11.65 Å, b = 12.87 Å, c = 19.15 Å; α = γ = 90° and β = 95°]. The Cu:Er atoms are present in a 3:2 ratio, and the refinement process shows n number of solvent (DMF) molecules included in the final model. Metal centers (Cu/Er) are bridged by multidentate Try ligand molecules, with an N:O ratio of 1:6 in each cell.
 |
| | Fig. 6 X-Ray diffraction patterns of MOFs (a) E, (b) P1, (c) P2, (d) P3, with corresponding refined curves and indexed miller indices. | |
3.3. Thermogravimetric analysis (TGA)
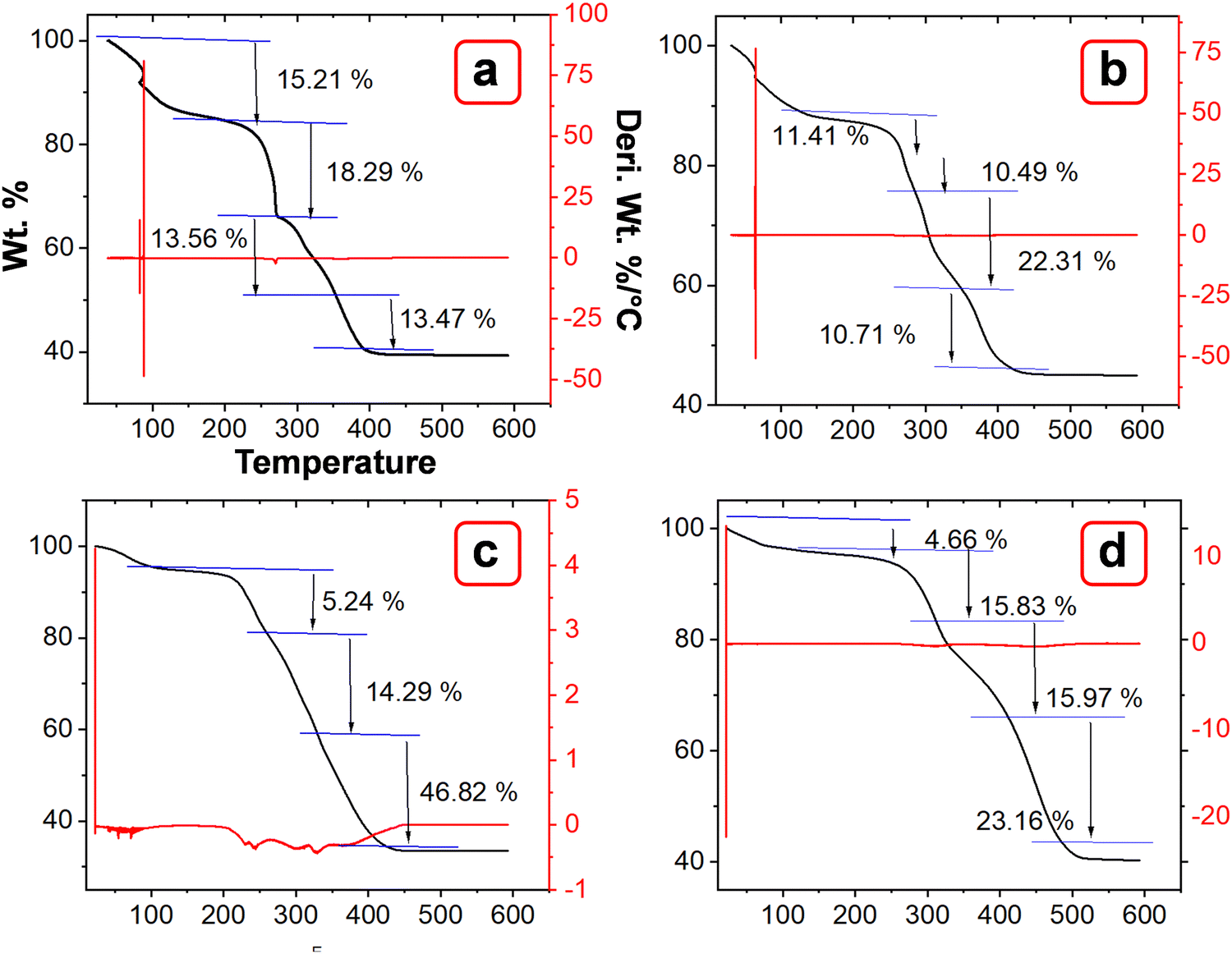
The TGA curves of MOFs P1, P2, P3, and P4 demonstrated superior thermal stability up to 400 °C (Fig. 7(a)–(d)). Table 2 summarizes the mass loss observed across specific temperature ranges. The materials showed no weight loss below 130 °C, indicating complete stability in this range. The TGA curves likely exhibited distinct stages of thermal degradation. Below 150 °C, desorption of physiosorbed and coordinated DMF may take place, as ligands may retain solvent molecules coordinated to the metal centers. Between 180–300 °C, partial decomposition of functional groups may occur, including the loss of water from hydroxyl groups (–OH) and the initial degradation of weakly bound sulfonate or azo groups. This stage may also involve the onset of oxidation of transition metals such as Cu and Co. Around 300–500 °C, framework decomposition may occur through aromatic backbone degradation, releasing CO, CO2, hydrocarbons, and nitrogenous gases due to azo bond cleavage (–NN–), while also forming mixed metal oxides such as CuO, ZnO, Er2O3, and Yb2O3. At temperatures exceeding 500 °C, complete degradation of the organic components of Trypan blue may occur, leading to the formation of stable residues and metal oxides. The higher residues observed (∼30–45%) are likely due to the presence of multiple metal oxides that are formed from the bimetallic centers.39–43
 |
| | Fig. 7 TGA and DTA profiles of (a) P1, (b) P2, (c) P3, and (d) P4. | |
Table 2 Summary of TGA parameters over temperature changes
| MOFs |
Temperature |
% Weight loss |
% Remaining |
| P1 |
194.14 |
15.2 |
84.719 |
| 271.90 |
18.29 |
66.428 |
| 346.93 |
13.56 |
52.859 |
| 447.27 |
13.473 |
39.386 |
|
|
| P2 |
135.53 |
11.414 |
88.56 |
| 277.46 |
10.496 |
78.090 |
| 367.43 |
22.316 |
55.774 |
| 501.69 |
10.706 |
45.068 |
|
|
| P3 |
173.50 |
4.66 |
95.340 |
| 325.64 |
15.838 |
79.502 |
| 420.90 |
15.972 |
65.530 |
| 567.69 |
23.169 |
40.361 |
|
|
| P4 |
139.69 |
5.249 |
94.751 |
| 253.06 |
14.295 |
80.456 |
| 442.23 |
46.821 |
33.610 |
3.4. Electrochemical behavior studies
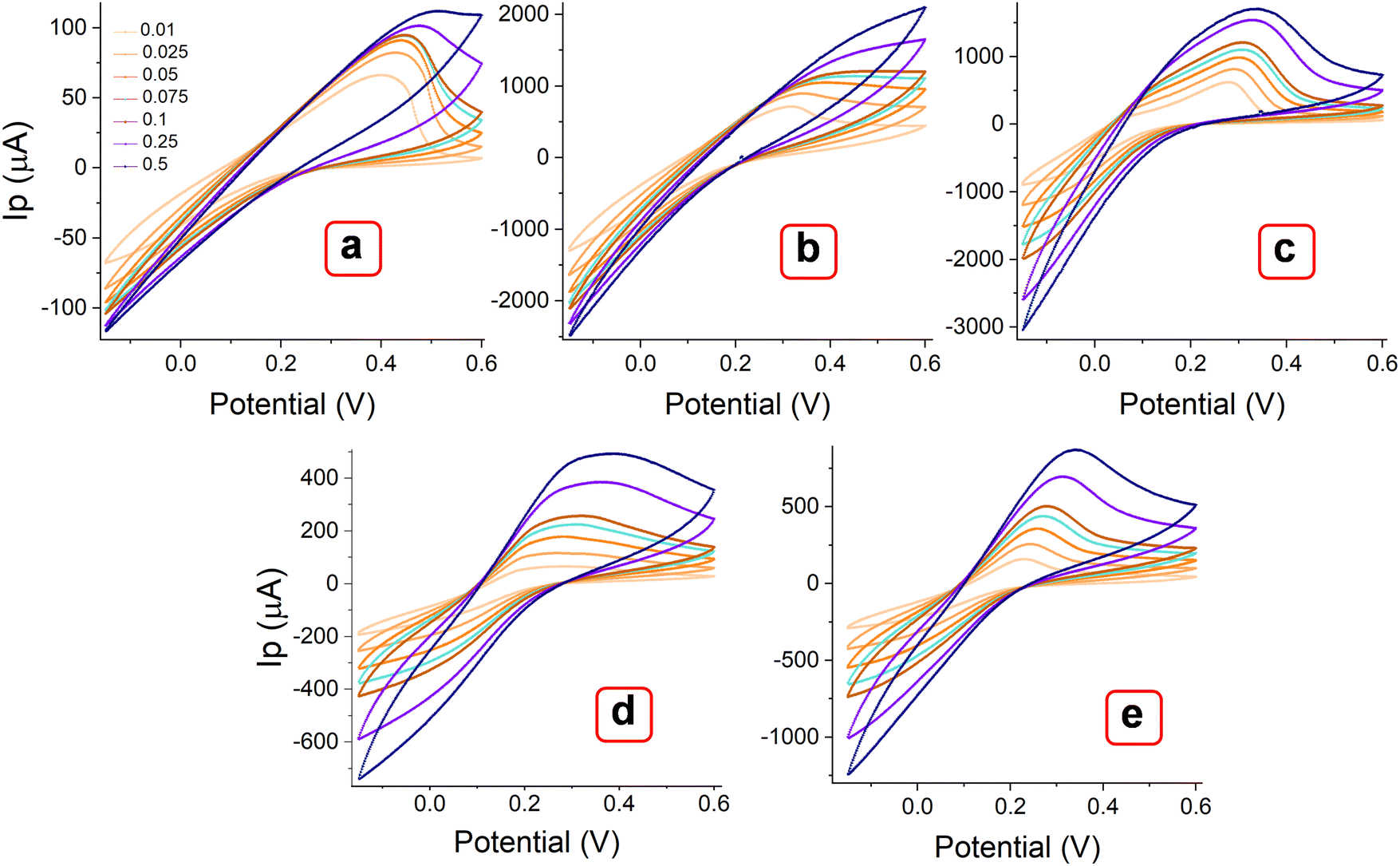
The MOF layer in the paste electrode could provide more electroactive sites and a smooth surface as a working electrode to ensure favorable electrochemical impacts.46 The effect of metal centers and ligand arrangement in mono and polymorphic crystal systems was recorded. Corresponding cyclic voltammetry (CV) measurements were conducted with the MOFs layer in the paste electrode. Fig. 8 illustrates the voltammograms of MOFs (E, P1, P2, P3 & P4) recorded within the potential window of −0.15 to 0.6 V over a range of scan rates (0.01, 0.025, 0.05, 0.075, 0.1, 0.25 and 0.5 V s−1). All the Try MOFs showed well-defined oxidation peaks, as shown in Fig. 8(a)–(e). The oxidation peak current responses of 62.7 (E), 719.4 (P1), 637.5 (P2), 79.7 (P3), 173.7 μA (P4), were obtained at 0.01 V s−1 sweep speed. Markedly, the P1 and P2 delivered substantially higher anodic currents than the P3, P4, and E variants. The higher cumulative current response from multi-metallic MOFs (P1, P2, P3 & P4) over E might be due to faster electron delocalization across the framework, along with enhanced structural stability in the 3d–3d metal MOFs over the 3d–4f variant.47,48
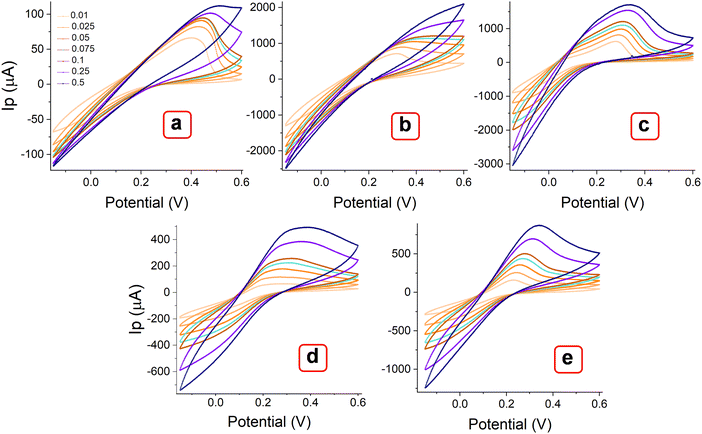 |
| | Fig. 8 CV curves of MOFs (a) E, (b) P1, (b) P2, (c) P3, and (d) P4, recorded at different scan rates from 0.01 to 0.5 V s−1. | |
Additionally, the polymorphic nature of these MOFs most likely led to the adoption of various coordination geometries and the formation of defect centers or structural distortions. This, in turn, may influence the electronic structure and increase the density of states near the Fermi level, contributing to a higher current response.49,50 With the increasing scanning rate from 10 to 500 mV s−1, the peak currents increased simultaneously. As observed, the oxidation peak shifted toward a higher potential, while the rudimentary reduction peak shifted toward a more negative potential. The log of peak current (logIp) versus the logarithm of the square root of the scan rate (log v1/2) plot (Fig. 9(a)–(e)) showed a linear relation at lower scan rate (below 100 mV s−1), while they are slightly nonlinear at higher scanning speeds. At lower scan rates, the process is possibly diffusion-controlled, following the equation below. As the scan rate increases, the process appears to be partially diffusion-controlled and partially governed by the kinetics of the electron transfer reaction. However, the coefficient of determination values (R2) obtained from the statistical analysis are significantly close to 1 [E (0.984), P1 (0.989), P2 (0.981), P3 (0.984), and P4 (0.957)]. This result indicates that the experimental data aligns well with the diffusion-controlled model, while the slight deviations suggest the influence of kinetic limitations.51,52
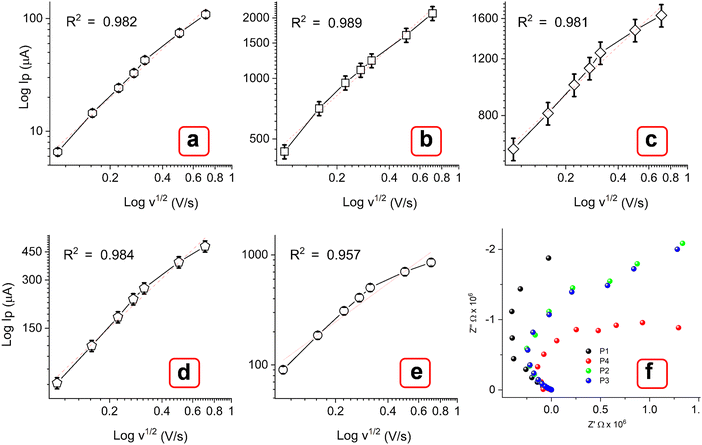 |
| | Fig. 9 Corresponding calibration plots (a)–(e) of log of peak current (Ip) vs. log of square root of scan rate (v1/2 V s−1). (f) Nyquist plots for P1, P2, P3, and P4. | |
The transport mechanism in these MOFs is analyzed from charge mobility (μ) values. It is calculated using the relation (eqn (1)):
| |  | (1) |
where
D is the diffusion coefficient at temperature
T,
n is the number of electrons transferred,
F is the Faraday constant (1.6 × 10
−19 C), and
kB is the Boltzmann constant (1.38 × 10
−23 J K
−1).
D is calculated using the Randles–Sevcik equation (
eqn (2)):
| | | Ip = 2.69 × 105n23AD21Cv21 | (2) |
The charge mobility values of MOFs were obtained as 1.08 × 10−7 (E), 1.25 × 10−3 (P1), 1.42 × 10−3 (P2), 1.58 × 10−4 (P3), and 1.51 × 10−4 (P4) cm2 V−1 s−1. The significantly lower values, below 0.1 cm2 V−1 s−1, indicate that charge transport is predominantly facilitated by electron hopping rather than band-like electron delocalization.53 Nyquist plots in Fig. 9(f) show a small semicircle at high frequencies and a nearly straight line at low frequencies in the order P1 > P2 ≈ P3 > P4. The small semicircle (arc) indicates a low charge transfer resistance (Rct), pointing to fast electron transfer kinetics. The straight line in the Warburg region signifies diffusion-controlled processes, which are less pronounced in multimetallic MOFs, likely due to enhanced ion transport.45,54 The lower value of Rct for P1 while higher Rct of P4 is probably due to the difference in electronic interactions. Typically, 3d–3d metal MOFs [P1 (Cu–Co), P2 (Cu–Zn)], have overlapping d-orbitals, which could provide continuous electron pathways and good mechanical stability, leading to lower Rct. Relatively, 4f metals (Er, Yb) have more localized f-orbitals that interact weakly with the surrounding ligands and other metal centers compared to 3d transition metals.45,54 This might often lead to less efficient charge transfer and higher Rct. Fig. 10 shows the dependence of electrochemical behavior based on thermal variations over 250–600 °C. The current response at different scan rates appeared to increase for thermally treated MOFs within the potential window of −0.1 to 1.2 V. This result may correspond to the carbonization of organic linkers in MOFs at high temperatures, resulting in conductive carbon frameworks. Moreover, the oxidation behavior of Trypan blue was less prominent in comparison to the untreated MOFs, with distinct peaks around 0.3 V. This result may be due to the onset of its decomposition at elevated temperatures, changes in the oxidation state of the metals, or the formation of mixed-valence systems.
 |
| | Fig. 10 Comparison of the electrochemical behavior of MOFs P1, P2, P3 and P4 using as synthesized (a)–(d), thermally treated 280 °C (a′–d′), and thermally treated 580 °C (a′–d′) materials, respectively. | |
The voltammograms of bimetal MOFs were compared with corresponding single metal MOFs (Co–Try, Zn–Try, Er–Try, and Yb–Try). Interestingly, their CV curves show quasi-reversible behavior (Fig. 11(a)–(e) and Fig. S12–S15, ESI†), characterized by weak anodic and cathodic peaks, along with non-faradaic processes contributing to the overall current response.55–58 The significant separation between the anodic and cathodic peak potentials may be attributed to a slow electron transfer rate, high charge transfer resistance at the electrode surface, restricted mass transport, and the capacitive behavior associated with electrode surface charging. In comparison, bimetal MOFs exhibit a well-defined oxidation peak with no sign of CV curve broadening.55,59 The polymorphic structures of these MOFs might facilitate efficient electron transfer, resulting in predominantly faradaic processes. Additionally, the choice of ligand, Trypan blue, may promote redox-active behavior by forming stable coordination environments around the metal centers, thus limiting the occurrence of non-faradaic capacitive behaviors.59 The increased structural stability in the oxidized form, complex electronic structures, and synergistic effects of multiple metals in multi-metallic MOFs may contribute to suppressing reduction while promoting a more kinetically favorable oxidation process. Further, these single metal–Trypan blue-based MOFs could be investigated for energy storage applications.
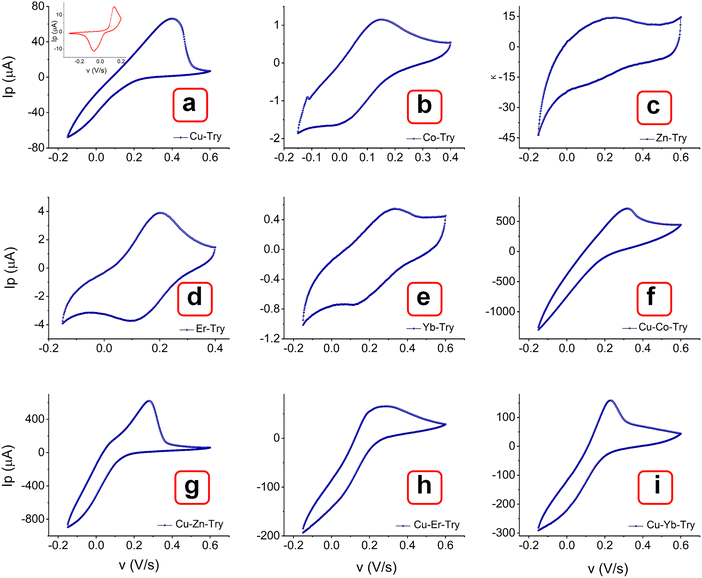 |
| | Fig. 11 Comparison of the voltammograms of single metal MOFs (a)–(e) with the multi centered MOFs (f)–(i) at a scan rate of 0.01 V s−1. Inset picture to graph (a) shows the CV curve of blank Trypan blue. | |
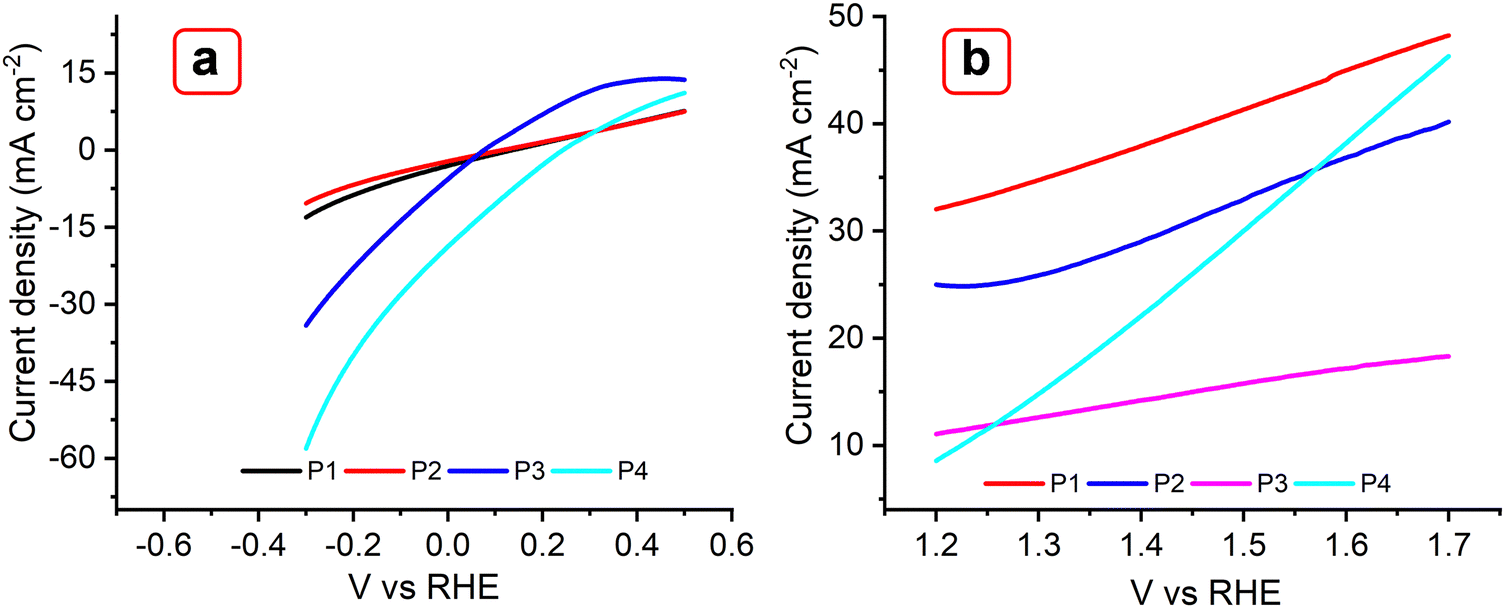
The electrochemical behavior of the reported MOFs was further investigated to understand their potential use as electrocatalysts for a water splitting reaction (Fig. 12). Linear sweep voltametric (LSV) curves of the MOFs P1, P2, P3, and P4, incorporating Cu(II), Co(II), Zn(II), Er(III), and Yb(III) metals were studied for the hydrogen evolution reaction (HER) at room temperature using a scan rate of 5 mV s−1 (Fig. 12(a)). In 1 M KOH, P3 and P4 show onset overpotentials of ∼107 mV; this value was 85 mV lower than that of P1 and P2 (140 mV). Achieving a current density of 10 mA cm−2 requires overpotentials of 160 mV for P1 and 190 mV for P2. For the oxygen evolution reaction (OER), P1 and P2 yielded a current density of ∼33 and 25 mA cm−2, respectively; while P3 and P4 exhibited a current density of 10 and 8 mA cm−2, respectively (Fig. 12(b)). These results suggest that the OER activities of the former catalysts are higher than those of the latter catalysts. These MOFs were also compared with the TTF-based MOF, as shown in Fig. S16a and b (ESI†). As the results suggest, the Trypan blue-based MOFs exhibit higher HER activity than the TTF MOF under ambient conditions. In agreement with previous studies, it is observed that TTF-based MOFs are primarily designed for electron transfer rather than catalytic applications, limiting their versatility. In contrast, the multifunctional character of Trypan blue enabled strong electrolytic activity.60–62
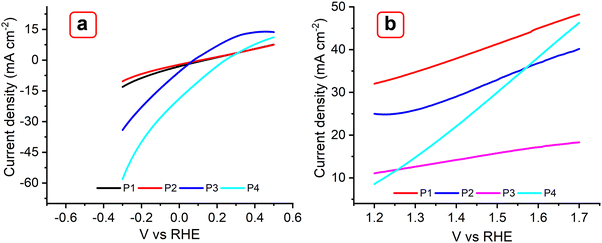 |
| | Fig. 12 LSV plots obtained with P1, P2, P3, and P4 MOFs for HER (a) and OER (b) at 5 mV s−1 in 0.1 M KOH. | |
4. Conclusions
We have demonstrated the fabrication of a series of chemically and thermally stable single and bimetal MOFs with a dye-based multifunctional macrocyclic ligand (Trypan blue) via a solvothermal route. Due to varied bonding interactions, metals and ligands arranged themselves to minimize energy and maximize stability, and thus these MOFs exhibited complex three-dimensional morphologies. Shifts in the M–O stretching frequency relative to a monometallic MOF may suggest changes in bond strength caused by the introduction of a second metal. Among the various peaks, symmetric stretching modes appear more prominent than bending modes, indicating the symmetrical coordination of the multifunctional ligand with two different metals. The detection of stretching vibrations for azo groups, CC bonds, and SO3 groups in all the MOFs suggests that these linkages remain intact throughout the synthesis, indicating that Trypan blue did not degrade. This finding contrasts with some reported Trypan blue-based MOF syntheses. The maintenance of the typical +2 and +3 oxidation states of Zn, Co, Er, and Yb in these MOFs indicates minimal electronic interference from Cu. The thermogravimetric analysis data revealed excellent thermal stability; no weight loss below 130 °C was noted. Upon heating to around 600 °C, the degradation of solvents and organic components left approximately 30–45% residue; this result was attributed to the formation of metal oxides. The observed increase in current response at various scan rates for thermally treated MOFs within the potential range of −0.1 to 1.2 V was attributed to the carbonization of organic linkers at elevated temperatures, resulting in the formation of conductive carbon frameworks. The difference in Rct values between 3d–3d and 3d–4f MOFs indicates that 3d–3d MOFs can achieve higher connectivity between metal centers and ligands, leading to a more conductive network compared to 3d–4f MOFs. The LSV analysis demonstrated enhanced HER performance for P3 and P4 as compared to P1 and P2. Conversely, P1 and P2 showed superior activity for OER, achieving higher current densities. We demonstrate the tunable electrochemical properties of Trypan blue-based single-metal and multi-metal MOFs to assess their efficiency for electron transport. The increased current contribution in the latter was attributed entirely to the faradaic process. In contrast, single-metal MOFs exhibited potential capacitive behavior, which could be further explored for energy storage applications in the future. The potential impacts of the study may include highly sensitive electrochemical sensors owing to enhanced electron transfer through multimetallic configurations, CO2 reduction catalysts and carbon capture materials based on electron-rich ligands (Trypan blue) and metal centers that can stabilize CO2 intermediates, as well as the stable redox chemistry of multi-metals that can enhance the performance of electrodes in lithium-ion or zinc-ion batteries.
Author contributions
S. S. was responsible for the project idea, writing of the original draft, data collection, interpretation, and calculations. N. J. procured SEM, EDS, and XPS data. S. K. was responsible for writing and reviewing the original draft. S. S. helped with BET and BJH data. R. J. N. was responsible for supervision, conceiving of the project, draft review, and funding.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
There are no acknowledgments to declare.
Notes and references
- Y. G. Weng, W. Y. Yin, M. Jiang, J. L. Hou, J. Shao, Q. Y. Zhu and J. Dai, ACS Appl. Mater. Interfaces, 2020, 12, 52615–52623 CrossRef CAS.
- D. Ukaj, H. Bunzen, J. Berger, G. Kieslich and R. A. Fischer, Chem. Mater., 2021, 33, 2532–2542 CrossRef CAS.
- L. J. Shen, X. F. Luo and X. Xiao, J. Solid State Chem., 2024, 339, 124933 CrossRef CAS.
- S. Adhikari, A. H. Sheikh, S. Kansız, N. Dege, N. Baildya, G. Mahmoudi, N. A. Choudhury, R. J. Butcher, W. Kaminsky, S. Talledo and E. M. Lopato, J. Mol. Struct., 2023, 1285, 135481 CrossRef CAS.
- C. A. Downes, A. J. Clough, K. Chen, J. W. Yoo and S. C. Marinescu, ACS Appl. Mater. Interfaces, 2018, 10, 1719–1727 CrossRef CAS PubMed.
- B. Yang, G. Zhang, Q. Niu, S. Liu, H. Li, S. Chen and K. Xu, J. Solid State Chem., 2023, 317, 123656 CrossRef CAS.
- B. Yang, Q. Niu, S. Liu, C. Wan, F. Zhao and K. Xu, J. Mol. Struct., 2022, 1261, 132943 CrossRef CAS.
- Y. Zhang, T. Qiu, F. Jiang, S. Amzil, Y. Wang, H. Fu, C. Yang, Z. Fang, J. Huang and G. Dai, Appl. Surf. Sci., 2021, 556, 149818 CrossRef CAS.
- N. Lv, Q. Li, H. Zhu, S. Mu, X. Luo, X. Ren, X. Liu, S. Li, C. Cheng and T. Ma, Adv. Sci., 2023, 10, 2206239 CrossRef CAS.
- M. Z. Iqbal, M. W. Khan, M. Shaheen, S. Siddique, S. Aftab, S. M. Wabaidur and S. Sharif, Mater. Today Sustain., 2023, 21, 100286 Search PubMed.
- H. V. Doan, H. Amer Hamzah, P. Karikkethu Prabhakaran, C. Petrillo and V. P. Ting, Nano-Micro Lett., 2019, 11, 1–33 CrossRef CAS.
- Y. Xie, J. Wan, Z. Yan, Y. Wang, T. Xiao, J. Hou and H. Chen, Chem. Eng. J., 2022, 429, 132237 CrossRef CAS.
- A. N. Yang, J. T. Lin and C. T. Li, ACS Appl. Mater. Interfaces, 2021, 13, 8435–8444 CrossRef CAS.
- S. Samira, J. C. A. Camayang, K. Patel, X. K. Gu and E. Nikolla, ACS Energy Lett., 2021, 6, 1065–1072 CrossRef CAS.
- H. Dong, X. Zhang, X. C. Yan, Y. X. Wang, X. Sun, G. Zhang, Y. Feng and F. M. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 45080–45086 CrossRef CAS.
- Z. J. Jiang, S. Cheng, H. Rong, Z. Jiang and J. Huang, J. Mater. Chem. A, 2017, 5, 23641–23650 RSC.
- L. X. You, B. B. Zhao, H. J. Liu, S. J. Wang, G. Xiong, Y. K. He, F. Ding, J. J. Joos, P. F. Smet and Y. G. Sun, CrystEngComm, 2018, 20, 615–623 RSC.
- K. Ma, H. R. Atapattu, Q. Zhao, Y. Gao, B. P. Finkenauer, K. Wang, K. Chen, S. M. Park, A. H. Coffey, C. Zhu and L. Huang, Adv. Mater., 2021, 33, 2100791 CrossRef CAS.
- P. Braunstein and A. A. Danopoulos, Chem. Rev., 2021, 121, 7346–7397 CrossRef CAS.
- H. Ryu, K. M. Park, M. Ikeda, Y. Habata and S. S. Lee, Inorg. Chem., 2014, 53, 4029–4038 CrossRef CAS.
- H. Jun, S. Oh, G. Lee and M. Oh, Sci. Rep., 2022, 12, 14735 CrossRef CAS PubMed.
- Y. Li, K. Zhang, Z. Xu, X. Zhao, Y. Wang, F. Zhang, X. Guo and Y. Zhang, J. Mater. Sci.: Mater. Electron., 2023, 34, 2229 CrossRef CAS.
- Y. Kim, K. Kim, H. H. Eom, X. Su and J. W. Lee, Chem. Eng. J., 2021, 421, 129765 CrossRef CAS.
- M. Yadav, R. Soni, M. K. Chauhan and N. Sandal, Biometals, 2021, 34, 351–363 CrossRef CAS PubMed.
- M. Maczka, A. Gagor, B. Macalik, A. Pikul, M. Ptak and J. Hanuza, Inorg. Chem., 2014, 53, 457–467 CrossRef CAS PubMed.
- M. H. Chisholm and A. M. Macintosh, Chem. Rev., 2005, 105, 2949–2976 CrossRef CAS.
- R. Jószai, I. Beszeda, A. C. Bényei, A. Fischer, M. Kovács, M. Maliarik, P. Nagy, A. Shchukarev and I. Tóth, Inorg. Chem., 2005, 44, 9643–9651 CrossRef.
- S. Tanaka, C. Dubs, A. Inagaki and M. Akita, Organometallics, 2004, 23, 317–319 CrossRef CAS.
- G. Brunet, D. A. Safin, I. Korobkov, A. Cognigni and M. Murugesu, Cryst. Growth Des., 2016, 16, 4043–4050 CrossRef CAS.
- Z. Chen, X. Li, C. Yang, K. Cheng, T. Tan, Y. Lv and Y. Liu, Adv. Sci., 2021, 8(20), 2101883 CrossRef CAS PubMed.
- G. Babakhanova, S. M. Zimmerman, L. T. Pierce, S. Sarkar, N. J. Schaub and C. G. Simon Jr., PLoS One, 2022, 17(1), e0262119 CrossRef CAS PubMed.
- R. Sasikala, K. Karthikeyan, D. Easwaramoorthy, I. M. Bilal and S. K. Rani, Environ. Nanotechnol., Monit. Manage, 2016, 6, 45–53 Search PubMed.
- T. Araújo, A. J. Parnell, G. Bernardo and A. Mendes, Carbon, 2023, 204, 398–410 CrossRef.
- B. B. Peterson, E. M. Andrews, F. Hung and J. C. Flake, J. Power Sources, 2021, 492, 229658 CrossRef CAS.
- K. Yang, Y. Yan, H. Wang, Z. Sun, W. Chen, H. Kang, Y. Han, W. Zahng, X. Sun and Z. Li, Nanoscale, 2018, 10(37), 17647–17655 RSC.
- A. S. Duke, E. A. Dolgopolova, R. P. Galhenage, S. C. Ammal, A. Heyden, M. D. Smith, D. A. Chen and N. B. Shustova, J. Phys. Chem. C, 2015, 119(49), 27457–27466 CrossRef CAS.
- X. Hu, H. Hu, C. Li, T. Li, X. Lou, Q. Chen and B. Hu, J. Solid State Chem., 2016, 242, 71–76 CrossRef CAS.
- Q. Zha, F. Yuan, G. Qin and Y. Ni, Inorg. Chem., 2020, 59(2), 1295–1305 CrossRef CAS PubMed.
- S. Kalhor, H. Sepehrmansourie, M. Zarei, M. A. Zolfigol and H. Shi, Inorg. Chem., 2024, 63(11), 4898–4914 CrossRef PubMed.
- D. K. Yadav, R. Gupta, V. Ganesan, P. K. Sonkar and M. Yadav, ChemElectroChem, 2018, 5(18), 2612–2619 CrossRef CAS.
- R. Shi, L. Wang, Z. Pi, L. Zhao, H. Xu, Z. Zhao, J. Liao, H. Ren, X. Tian, Q. Liu and X. Zhao, Electrochim. Acta, 2024, 483, 143993 CrossRef CAS.
- G. Shen, L. Zhong, Y. Bi, Y. Liu, J. Zhao, X. Wen, Y. Zhu, L. Feng, L. Geng, F. Yu and C. Hou, Sens. Actuators, B, 2024, 404, 135301 CrossRef CAS.
- Y. Li, C. Jiang, X. Chen, Y. Jiang and C. Yao, ACS Appl. Mater. Interfaces, 2022, 14(6), 8343–8352 CrossRef CAS PubMed.
- T. M. Pan, Y. S. Huang and J. L. Her, Sci. Rep., 2018, 8(1), 12902 CrossRef PubMed.
- P. P. Sun, Y. H. Zhang, H. Shi and F. N. Shi, Chem. Eng. J., 2022, 427, 130836 CrossRef CAS.
- M. A. Ramezani, M. Najafi and M. H. Karimi-Harandi, Food Chem., 2024, 458, 140296 CrossRef CAS.
- M. L. Aubrey, B. M. Wiers, S. C. Andrews, T. Sakurai, S. E. Reyes-Lillo, S. M. Hamed, C. J. Yu, L. E. Darago, J. A. Mason, J. O. Baeg and F. Grandjean, Nat. Mater., 2018, 17(7), 625–632 CrossRef CAS.
- J. G. Park, M. L. Aubrey, J. Oktawiec, K. Chakarawet, L. E. Darago, F. Grandjean, G. J. Long and J. R. Long, J. Am. Chem. Soc., 2018, 140(27), 8526–8534 CrossRef CAS PubMed.
- C. Avendano, Z. Zhang, A. Ota, H. Zhao and K. R. Dunbar, Angew. Chem., Int. Ed., 2011, 50(29), 6543–6547 CrossRef CAS.
- I. Huskić, N. Novendra, D. W. Lim, F. Topić, H. M. Titi, I. V. Pekov, S. V. Krivovichev, A. Navrotsky, H. Kitagawa and T. Friščić, Chem. Sci., 2019, 10(18), 4923–4929 RSC.
- P. Joshi, S. Shukla, S. Gupta, P. R. Riley, J. Narayan and R. Narayan, ACS Appl. Mater. Interfaces, 2022, 14(32), 37149–37160 CrossRef CAS.
- P. Joshi, S. Shukla, S. Gupta, N. Joshi, J. Narayan and R. Narayan, MRS Commun., 2023, 13(4), 554–560 CrossRef CAS.
- J. Duan, S. Goswami, S. Patwardhan and J. T. Hupp, J. Phys. Chem. C, 2022, 126(9), 4601–4611 CrossRef CAS.
- Y. Zhang, X. Bo, A. Nsabimana, C. Han, M. Li and L. Guo, J. Mater. Chem. A, 2015, 3(2), 732–738 RSC.
- J. Waelder and S. Maldonado, J. Electroanal. Chem., 2021, 878, 114665 Search PubMed; J. Waelder and S. Maldonado, Anal. Chem., 2021, 93(37), 12672–12681 CrossRef CAS PubMed.
- B. A. Brookes, G. Macfie and R. G. Compton, J. Phys. Chem. B, 2000, 104(24), 5784–5789 CrossRef CAS.
- S. Shukla, P. Joshi, P. Riley and R. J. Narayan, Biosens. Bioelectron., 2022, 216, 114592 CrossRef CAS PubMed.
- S. Shukla, S. Khanna, S. Sahoo, N. Joshi and R. Narayan, ACS Appl. Bio Mater., 2024, 7(1), 472–484 CrossRef CAS PubMed.
- S. Shukla, N. N. Joshi, S. Kadian and R. J. Narayan, ACS Appl. Bio Mater., 2024, 7(8), 5382–5396 CrossRef CAS PubMed.
- T. S. Renani, S. M. Khoshfetrat, J. Arjomandi, H. Shi and S. Khazalpour, J. Mater. Chem. A, 2021, 9, 12853–12869 RSC.
- C. Dietrich, D. A. Weber, S. J. Sedlmaier, S. Indris, S. P. Culver, D. Walter, J. Janek and W. G. Zeier, J. Mater. Chem. A, 2017, 5, 18111–18119 RSC.
- G. K. Shimizu, J. M. Taylor and S. Kim, Science, 2013, 341, 354–355 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Sachin
Kadian
a,
Siba Sundar
Sahoo
b,
Sachin
Kadian
a,
Siba Sundar
Sahoo