
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the effect of hydrothermal carbonisation reaction times on the photoluminescence of bio-oil-derived carbon polymer dots†
Lawrence A.
Bruce
 a,
Liam
Desmond
b,
Abigail A.
Seddon
a,
Leon
Bowen
c,
Greg A.
Mutch
b,
Anh N.
Phan
*b and
Elizabeth A.
Gibson
*a
a,
Liam
Desmond
b,
Abigail A.
Seddon
a,
Leon
Bowen
c,
Greg A.
Mutch
b,
Anh N.
Phan
*b and
Elizabeth A.
Gibson
*a
aEnergy Materials Laboratory, Bedson Building, Newcastle University, Edward's Walk, Newcastle upon Tyne NE1 8QB, UK. E-mail: anh.phan@newcastle.ac.uk; elizabeth.gibson@newcastle.ac.uk
bSchool of Engineering, Merz Court, University, Newcastle upon Tyne NE1 7RU, UK
cDurham University, Department of Physics, South Road, Co Durham, DH1 3LE, UK
First published on 3rd December 2024
Abstract
Carbon dots (CDs) have favourable properties such as broad spectral absorption, strong photoluminescence, low toxicity, and high specific surface area. Here, carbon polymer dots (CPDs), which are CDs with a carbon core and polymeric surface, were synthesised from chitin-derived bio-oil, supporting the transition away from fossil- based feedstocks in carbon nanomaterial synthesis. The influence of hydrothermal carbonisation (HTC) reaction times on the structural, morphological, and optical properties were studied. Structural analysis revealed the formation of nitrogen-doped CPDs with a cross-linked polymeric shell surrounding a crystalline graphitic carbon core, with the size, structure, and surface composition all influenced by HTC reaction time. All CPD samples showed the same excitation dependent emission, with a λmax of 394 nm. Deconvolution of the photoluminescence showed multiple components with contributions identified from both molecular luminophores and the carbon core. Overall, this work investigates the mechanism of photoluminescence in CPDs, providing insights that could promote application in areas such as bioimaging, sensing, and optoelectronics.
1. Introduction
Carbon dots (CDs) are an emergent class of photoluminescent carbon nanomaterial, discovered in 2004 by Xu et al. during the purification of single-walled carbon nanotubes fabricated by arc-discharge from soot.1 Their favourable properties such as broad spectral absorption,2 strong photoluminescence,3 tunable emission,4–6 low toxicity,7,8 conductivity,9 and high specific surface area10 have facilitated their application in biomedical sciences and as energy materials.11–13 However, in the literature, the term “CDs” is often used to refer broadly to a range of carbon nanomaterials, which can vary significantly in structure and properties. The classification of CDs in the literature contains some discrepancies, CDs can be loosely classified into four categories (Table 1). More research is needed to elucidate mechanisms of formation and structure–activity relationships.6 In particular, the influence of synthesis methodology on the size, structure, and optical properties of CDs is poorly understood.14| Classification | Size (nm) | Structure | Photoluminescence |
|---|---|---|---|
| Graphene quantum dot (GQD)15 | 1–20 | Crystalline, graphene lattice with sp2 hybridised carbon domains. Functionalised groups on the edges and basal plane. | Quantum confinement16–18 |
| Edge effects17 | |||
| Surface states19,20 | |||
| Defects21 | |||
| Molecular fluorophores22 | |||
| Carbon quantum dot (CQD)23 | 2–10 | Quasi-spherical nanoparticles, crystalline carbon core and chemical groups on the surface (core–shell). | Quantum confinement |
| Core–shell structure24 | |||
| Surface states/defects | |||
| Molecular fluorophores | |||
| Carbon nanodots (CNDs or C-dots)20 | 2–10 | Amorphous nanoparticles highly carbonised sp3 hybridised graphitic/turbostratic carbon core with groups on the surface (core–shell). | Quantum confinement |
| Core–shell structure24 | |||
| Surface states/defects | |||
| Molecular fluorophores | |||
| Carbonised polymer dots (CPDs)25 | <100 | Carbon core (CQD or CND) with surface cross-linked or aggregated functional groups. | Core–shell structure |
| Surface states/defects | |||
| Cross-link enhanced emission26 | |||
| Conjugated π-domain | |||
| Molecular fluorophores |
In this introduction, the term CD will be used collectively to refer to the broad range of carbon nanomaterials, including graphene quantum dots, carbon quantum dots, carbon nanodots, and carbon polymer dots, as classified in Table 1, to discuss the general trends observed in their structure, morphology, and properties.
Doping CDs with heteroatoms effectively tunes their electronic and optical properties. This can be achieved through in-plane substitution, intercalation, or surface replacement. Various heteroatoms are used such as boron, phosphorus, silicon, and metals.19,27 For example, Zhai et al. (2012) found that increasing nitrogen content in CDs via microwave-assisted pyrolysis boosted the fluroscence quantum yield (QY) from 2.2% to 30.2%.28 While Qian et al. (2014) observed that the incorporation of nitrogen atoms significantly boosted the QY of N-CQDs, achieving a maximum emission efficiency of 36.3%.29
Biomass (waste and residues) is abundant, low-cost and naturally incorporates dopants, making it a cost-effective method for synthesising doped CDs.30–32 Improvements in photoluminescence have been observed where the enhanced nitrogen content contributes to increased QY and tunable optical properties by further functionalisation.33 Chitin, a by-product of the fishing industry processed from prawn and crab shell waste, was selected for its high nitrogen content (∼7 wt%).28 Previously, Briscoe et al. (2017) synthesised a series of biomass derived CDs by hydrothermal carbonisation (HTC) at 200 °C for 6 h from glucose, chitin, and chitosan (the deacylated form of chitin) with corresponding quantum yield (QY) of 1.4%, 11.6% and 13.4%. The results demonstrate a notable improvement in QY with nitrogen-doped CDs.34 Jiang et al. (2022) achieved an impressive QY of 54% from spheroidal CDs derived from chitin, synthesised via HTC at 240 °C for 10 h. These CDs had an average diameter of 4.2 nm. The X-ray diffraction (XRD) pattern displayed a broad peak at 23° (d002 = 0.34 nm), indicating the presence of amorphous carbon. The photoluminescence was excitation-dependent, accredited to quantum confinement and surface states.35 Chitin-CQDs have also been synthesised directly from prawn shells by (i) deproteinisation with NaOH (ii) demineralisation with HCl (iii) then HTC at 200 °C for 8 h. Transmission electron microscopy (TEM) and XRD analysis confirmed the presence of homogeneous spherical nanoparticles composed of amorphous carbon. Blue excitation dependent emission was recorded with a QY of 25.8%.36 A literature review of CDs produced from chitin, chitosan, and combinations such as chitosan with p-phenylenediamine or κ-carrageenan by HTC was conducted (Table S1, ESI†). The operating conditions ranged from 180–220 °C with reaction times between 5–24 h. The size of the resulting CDs was between 0.5 nm and 20 nm. For the majority of the CDs, photoluminescence was attributed to surface emission sites and carbon cores. The emission wavelengths (λmax) ranged from 400–to 533 nm, with QY up to 59%. Lifetimes, reported in nanoseconds, reflected complex decay profiles, indicating multiple emissive states. More research is required to understand the structure–property relationships of CDs; particularly the mechanisms underlying their photoluminescence, which could lead to enhanced control over their optical properties for optoelectronic applications.6,37 Challenges persist in the synthesis of biomass-derived carbon CDs as the varying composition of different biomass sources results in inconsistencies in their properties. Determining the optimal synthetic conditions, such as temperature, reaction time, pH, and precursor concentration is crucial. Additionally, the mechanism of CD formation from biomass remains unclear, highlighting the need for methods to control and modify CD properties more effectively. In this work, nitrogen doped carbon polymer dots (N-CPDs) were synthesised from chitin.38 N-CPDs were synthesised from the crude bio-oil derived from the pyrolysis of chitin, followed by HTC. The synthesised dots were classified as carbon polymere dots (CPDs) due to their size (17–45 nm) and morphology. The reaction time of the hydrothermal treatment was varied to examine how changes in morphology, chemical composition, and structure affected the photoluminescence. The proposed formation mechanism involves pyrolysis degrading chitin into a mixture of nitrogen (N) & oxygen (O) containing heterocyclics and small molecules, with high temperature (700 °C) being sufficient to induce graphitization.4 During the HTC, these fragments serve as nucleation sites for cross-linking polymerisation, where dehydration and condensation reactions lead to the formation of a cross-linked polymeric shell around the carbon core.39–42
Photoluminescence was found to have contributions from both the crystalline carbon core and a luminescent molecule; namely 3-aminopyridene.43–45 Overall, our work demonstrates that bio-oil derived from chitin can serve as an efficient feedstock for producing nanomaterials. This work introduces a methodology that can be extended to other bio-oil streams, offering a versatile approach for producing nanomaterials from various bio-oil sources (including waste). This approach provides benefits such as resource efficiency, carbon sequestration, and reduced environmental impact.46 Utilising biomass waste aligns with the circular economy, which prioritises resource reuse and waste minimisation according to the waste hierarchy.47,48
2. Results
2.1. Synthesis of N-CPDs
Chitin was pyrolysed at 700 °C in an inert atmosphere to produce a biochar (∼35%), liquid known as bio-oil (∼50%), and gas (∼15%). This temperature was selected to ensure complete conversion of the biomass and achieve a sufficiently high bio-oil yield.49 A temperature of 700 °C was found to give a higher yield of bio-oil than 450 °C (∼36%) (Table S2, ESI†). Analysis of the bio-oil produced at 700 °C by gas chromatography mass spectrometry (GC-MS) showed that acetic acid and acetamide were the dominant products formed (Fig. S1, ESI†). The production of large amounts of acetic acid was expected due to the hydrolysis of the acetyl amide side chain. A large mixture of O and N-heterocyclics were observed (Table S3, ESI†), which is consistent with other work.50The bio-oil was further processed by HTC at 200 °C over various holding times to understand the effect of reaction time on the product's structure, morphology, surface chemistry, and photoluminescence. Typical HTC temperatures for chitosan/chitin CD synthesis range from 180–220 °C.32 The temperature of 200 °C was chosen to ensure the efficient formation of the N-CPDs while avoiding excessive thermal degradation. A midpoint of 200 °C facilitated the comparison with previously synthesised chitin/chitosan derived CDs. The reaction time was varied from 2, to 4, and 8 h, with resulting samples labelled 2H-CPD, 4H-CPD, and 8H-CPD, respectively. After the HTC reaction, the N-CPDs were purified by filtration, followed by centrifugation, and solvent extraction with ether to remove impurities. Previous experiments incorporated an additional step of dialysis; however, it had no discernible effect on the purification process. The aqueous fraction containing the N-CPDs was freeze-dried and afterwards washed and sonicated with ether again until the solid N-CPDs crashed out of solution. This protocol was developed to ensure that no impurities or residual solvents are adsorbed onto the surface or trapped within the structure of the N-CPDs during freeze-drying. Vigorous purification, including centrifugation, filtration, and solvent extraction is essential to ensure accurate characterisation of the luminescence as it eliminates byproducts that can otherwise distort the true emission properties.51
During the purification of the N-CPDs, the organic fraction (ether) containing impurities was isolated from the solvent extraction for each sample (2H-CPD, 4H-CPD, and 8H-CPD). The fractions were analysed by GC-MS, and a mixture of ketone, phenol and pyridine derivatives were observed. 3-Acetamidopyridine was present in all the fractions and bio-oil (Table S4: 2H-CPD, Table S5: 4H-CPD, and Table S6: 8H-CPD, ESI†). The maximum yield from bio-oil to N-CPDs (8.8%) occurred at 8 h HTC time (Table S7, ESI†). The overall yield of chitin to N-CPDs was 4.5%. The N-CPDs were hygroscopic, which was attributed to there abundant hydrophilic surface groups, small size with a high surface area, and a potential crosslink-enhanced effect that allows them to effectively bind and retain water molecules.52 All the samples were found to be hydrophilic; easily dispersing in water and with a zeta potential of −15 mV.
2.2. Morphology and structure of N-CPDs
TEM images of N-CPDs synthesised with HTC reaction times of 2, 4, and 8 h are shown in Fig. 1. The TEM results confirmed the synthesis of nanoscale N-CPDs by pyrolysis followed by HTC. The images collected show the overall size of the N-CPDs, including both the crystalline carbon core and the amorphous cross-linked polymeric shell. Due to the size and the structure, the synthesied nanomaterials were classified as CPDs. Fig. 1a shows the N-CPDs had a uniform spherical morphology and were well dispersed, with some agglomeration in the 2H-CPD sample. Additionally, the TEM images show an increase in the average particle size as the reaction time increased; 2H-CPD: (17 ± 7) nm, 4H-CPD: (33 ± 8) nm, and 8H-CPD: (45 ± 6) nm. | ||
| Fig. 1 N-CPD samples were prepared for TEM by dispersion in IPA and sonication, before deposition on a carbon grid. (a) TEM image of 2H-CPD and (inset) magnification showing the crystal lattice grating, and (b) size-distribution from TEM analysis of 2H-CPD. TEM image and size distribution for (c) 4H-CPD and (d) 8H-CPD. The size distributions were determined from averaging all imaged N-CPDs. | ||
In Fig. 1a, a crystal lattice spacing of 0.23 nm was calculated corresponding to the reported (100) crystal facet of graphite.53,54 The crystal lattice was not observed by TEM for 4H-CPD or 8H-CPDs, consistent with the Raman spectroscopy in Fig. 2. CPDs synthesised from HTC by bottom-up processes typically exhibit lattice spacings incomparable with the graphite/graphene lattice due to the polymeric or ordered polymer/carbon hybrid structure.26,42 Luo et al. (2022)55 demonstrated that CDs synthesised via HTC at temperatures above 350 °C under an inert atmosphere led to structural instability, resulting in carbonisation. The process resulted in the removal of weakly bonded oxygen-containing functional groups and the formation of the conjugated sp2 carbon to generate graphitic planes.55 It was reasoned that the crystalline core formed during the pyrolysis step, as the pyrolysis temperature of 700 °C was sufficient to induce the crystallisation observed in the TEM images (Fig. 1a). Small fragments of graphitic carbon present in the bio-oil likely acted as nucleation sites during HTC, where their growth was facilitated by increased hydrothermal cross-linking polymerisation. At extended reaction times, the dehydration of functional groups in the precursors, driven by intramolecular collisions, promoted the formation and extension of polymer chains with cross-links. This process led to the development of a polymeric cross-linked shell that enveloped the pre-formed crystalline carbon core.42 It was rationalised that the increase in size was due to more extensive hydrothermal cross-linking reactions occurring at longer reaction times, leading to the growth of a larger cross-linked polymeric structure.
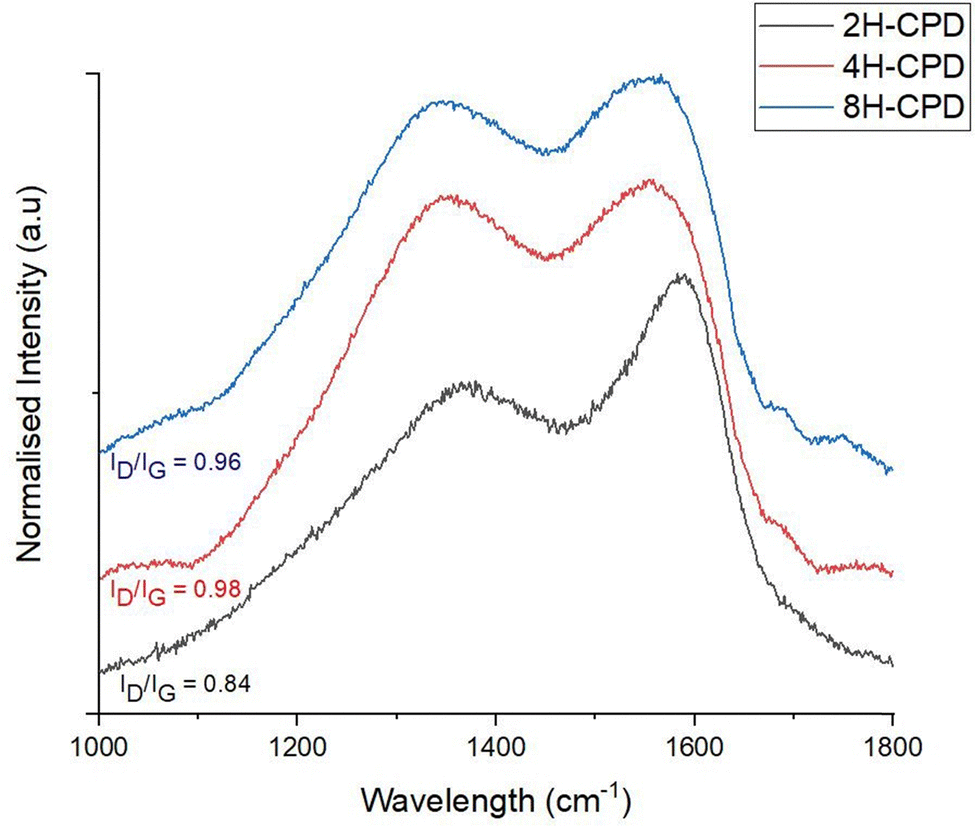 | ||
| Fig. 2 Analysis of Raman spectroscopy data related to the D-band and G-bands; data was processed by a 2-point baseline correction then normalised. The excitation wavelength was at 532 nm with a grating 600 g mm−1. | ||
The collection of XRD data was limited by the need for a larger material quantity, and the hygroscopic nature also complicated handling and preparation of dry samples. Additionally, the N-CPDs contained a mixture of amorphous and crystalline regions, with limited crystalline content that is often too small or disordered to produce distinct XRD peaks. This is consistent with similar studies, where broad, weak peaks are observed in XRD for amorphous materials, contributing little beyond the information from Raman spectroscopy and TEM.35,56,57
2.3. Vibrational spectroscopy
Raman spectroscopy was used to further investigate the effect of reaction time on structural properties (Fig. 2). Raman scattering is sensitive to highly symmetric covalent bonding with no or little natural dipole moments. Therefore, it is an insightful technique to use for carbon–carbon bonds in nanomaterials as slight changes in the structure can be discerned. Of interest is the D-band (disordered band ≈ 1350 cm−1), originating from the hybridised vibrational mode associated with dangling bonds, edge effects and structural defects (i.e., disordered carbon).58 The G-band (1580 cm−1) is attributed to the in plane vibrational mode of the of sp2-bonded carbon atoms in the hexagonal lattice, associated with graphene and graphite. The relative intensity ratio of the D- and G bands (ID/IG) is used to measure the relative defect density. 2H-CPD showed a D-band at 1369 cm−1 and a G-band at 1586 cm−1. As the reaction time increased, a discernible change in structure was signalled by a downward shift of the G-band to lower wavenumbers, observed in samples 4H-CPD: 1556 cm−1 and 8H-CPD: 1555 cm−1. The ID/IG of 0.84 for 2H-CPD is consistent with crystalline character with a degree of disorder (Fig. 2).59 The breadth of the D band also indicates disorder in the aromatic cluster. As the reaction time was increased, the intensity of the D band increased for both 4H-CPD and 8H-CPD. The ID/IG of 0.98 for 4H-CPD and 0.96 for 8H-CPD is consistent with a decrease in the crystallinity as the reaction time increased. Previous studies have shown that longer HTC reaction times lead to increased crystallinity.39,60 Ehrat et al. (2017) synthesised CDs from citric acid and ethylenediamine by HTC from 15 min to 10 h. As the reaction time increased, the initially formed molecular fluorophores acted as nucleation seeds for the growth of aromatic domains, resulting in enhanced crystallinity.60 However, in this study as the HTC reaction time increased, the amount of graphitic character decreased 2H-CPD: ID/IG = 0.84, 4H-CPD: ID/IG = 0.98 and 8H-CPD ID/IG = 0.96 (Fig. 2). This was attributed to by the increasing size of the CPD from 2H-CPD: (17 ± 7) nm to 8H-CPD: (45 ± 6) nm (Fig. 1) and the increased size of the disordered cross-linked polymeric shell that grew during the HTC by hydrothermal cross-linking polymerisation.The Fourier transform infrared FT-IR spectrum (Fig. 3a) confirmed the transformation of the starting material chitin to N-CPDs. The N-CPD samples (Fig. 3b) all showed a strong and broad O–H stretch from 3600–3200 cm−1. Additionally, the N–H stretch (3200–3180 cm−1), N–H bending (1560 cm−1) and C–N bending (1310 cm−1) were retained. The sharp amide carbonyl stretching (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) mode at ∼1660 cm−1 was also retained, however, at longer HTC reaction time, the amide peak became weaker from 2H-CPD to 8H-CPD (Fig. 3b). This was consistent with the X-ray photoelectron spectroscopy (XPS) analysis of the surface composition of the CPDs (Fig. 4).
O) mode at ∼1660 cm−1 was also retained, however, at longer HTC reaction time, the amide peak became weaker from 2H-CPD to 8H-CPD (Fig. 3b). This was consistent with the X-ray photoelectron spectroscopy (XPS) analysis of the surface composition of the CPDs (Fig. 4).
 | ||
| Fig. 3 (a) FT-IR spectra of chitin, bio-oil derived from the pyrolysis of chitin at 700 °C, and nitrogen doped CPDs synthesised from HTC at 200 °C (2H-CPD) (b) FT-IR spectra of nitrogen doped CPDs synthesised at 200 °C HTC at 2 h (2H-CPD), 4 h (4H-CPD), and 8 h (8H-CPD). | ||
 | ||
| Fig. 4 Row (a) 2H-CPDs, from left elemental composition, C 1s fitted XPS spectrum, and N 1s fitted spectrum. Row (b) 4H-CPDs, from left elemental composition, C 1s fitted XPS spectrum, and N 1s fitted spectrum. Row (c) 8H-CPDs, from left elemental composition, C 1s fitted XPS spectrum, and N 1s fitted spectrum. | ||
2.4. X-ray photoelectron spectroscopy
The elemental composition of carbon and nitrogen for the N-CPDs samples was found to remain consistent, with a nitrogen composition (7.5–8.6%) larger than the nitrogen content found from CHN analysis of the starting reagent chitin (6.57%). The oxygen composition increased with longer reaction times (2H-CPD: 14.5%, 4H-CPD: 15.0%, and 8H-CPD: 15.8%).For 2H-CPD, three peaks were observed in the C 1s region (Table 2) at 284.8, 265.6, and 287.9 eV which were assigned to CC, C–O–H/C–O–C, and C–N respectively.46,53 In the N 1s region there is a peak at 399.8 eV which was assigned to OC–N/H–N–H, and is characteristic of a pyridone/amine group.61–63 Of the total N 1s peak area, the contribution from OC–N/H–N–H functional groups was calculated to be ∼83%. The remaining ∼17% was assigned to H–N–H protonated amine at 401.1 eV.53,62,64,65 This was confirmed by the N–H stretch (3200–3180 cm−1), N–H bending (1560 cm−1) and C–N bending (1310 cm−1) in the FT-IR (Fig. 3). For 4H-CPD, the C 1s spectrum closely resembled that of 2H-CPD. However, in the N 1s spectrum, a distinct peak at 400.4 eV was observed, which was attributed to pyrrolic nitrogen.61 Additionally, a shift of the pyridone/amine peak to 399.5 eV was observed, along with a decrease in the intensity of the amide (CO) mode around 1660 cm−1 in the FT-IR (Fig. 3). The peak showed a more significant amine contribution.61,66 For 8H-CPD the C 1s peak area of the CC functional group significantly increased to 59.0% from 32.3% in 4H-CPD. The N 1s remained consistent in assignment, position, and distribution of peak area with 4H-CPD, with a further decrease observed in the sharpness of the amide (CO) mode. After the 8 h HTC the cross-linked polymeric structure is still intact, and full carbonisation hasn’t yet occurred as confirmed by XPS (Table 2). The C 1s peak area at 2 h and 4 h was predominantly composed of C–O–H/C–O–C bonds, accounting for 59.1% and 58.4%, respectively. Interestingly, after 8 h HTC it appears as though the process of cross-linked polymer to amorphous carbon is starting to occur. From 2 h to 8 h there is an increase in the amount of CC bonds on the surface from 30.3% to 59.0%.
| Sample | C 1s | N 1s | ||||
|---|---|---|---|---|---|---|
| Peak | Binding energy (eV) | Area (%) | Peak | Binding energy (eV) | Area (%) | |
| 2H-CPD | CC |
284.8 | 30.3 | OCNC/H–N–H |
399.8 | 83.44 |
| C–O–H/C–O–C | 265.6 | 59.1 | C–NH+/Hydrogen | 401.1 | 16.56 | |
| C–N | 288.0 | 10.5 | Bonded | |||
| 4H-CPD | CC |
284.8 | 32.3 | OC–N–C/H–N–H |
399.5 | 53.2 |
| C–O–H/C–O–C | 285.6 | 58.4 | C–(NH)–C | 400.4 | 46.8 | |
| C–N | 287.9 | 9.3 | ||||
| 8H-CPD | CC |
284.9 | 59.0 | OC–N–C/H–N–H |
399.5 | 48.5 |
| C–O–H/C–O–C | 285.1 | 30.6 | C–(NH)–C | 400.7 | 51.5 | |
| C–N | 287.7 | 10.4 | ||||
2.5. Optical properties
The UV-visible spectra of all three N-CPD samples (Fig. 5a and Fig. S5, ESI†) contained two broad peaks at approximately 280 nm corresponding to the π → π* transition of the aromatic sp2 domains,67 and at 320 nm corresponding to a n → π* transition (CO).68,69 The absorption occurring at longer wavelengths was assigned to the energy level transitions from the conjugated π-structure.69 Strong photoluminescence was recorded in the blue and visible regions of the emission spectrum, and photoluminescence was excitation wavelength dependent, with a red shift as the excitation wavelength increased. The maximum emission wavelength (λmax) was 394 nm at an excitation wavelength of 320 nm (Fig. 5b and Table 3). Excitation spectra were measured to examine the absorption characteristics of the emissive state. The consistent structure of the peaks suggests the excitation of a similar emissive state (Fig. 5c and Fig. S5, ESI†). The photoluminescence decay was fitted to two exponential components, which were within the timescale (∼1–10 ns) (Table 3). This is consistent with previously reported CDs (Table S7, ESI†).25,70 The optical properties of all N-CPDs samples were identical (Fig. S2, ESI†).
 | ||
| Fig. 5 (a) absorbance, (b) excitation-dependent photoluminescence and (c) excitation spectra for 2H-CPD. 2H-CPDs were synthesised from the 2H HTC of chitin-derived bio-oil, they dispersed in deionised water and were sonicated for 20 minutes prior to measurements. (d) absorbance, (e) photoluminescence and (f) excitation spectra for the impurity fraction isolated after the 2H HTC. The fraction was dispersed in deionised water and was sonicated for 20 minutes prior to measurements. | ||
| Sample | λ max (nm) | QY (%) | τ 1 (ns) | τ 2 (ns) |
|---|---|---|---|---|
| 2H-CPD | 394@320 | 6 | 4.44 ± 0.03 (61%) | 12.44 ± 0.08 (28%) |
| 4H-CPD | 394@320 | 7 | 4.65 ± 0.04 (62%) | 12.64 ± 0.09 (28%) |
| 8H-CPD | 394@320 | 8 | 4.90 ± 0.04 (62%) | 12.94 ± 0.09 (28%) |
To investigate further whether an independent fluorophore contributed to the photoluminescence, the optical properties of the organic fraction containing impurities from the purification step was recorded and compared to that of the N-CPDs (Fig. 5). The optical properties of the organic fraction containing impurities remained consistent irrespective of the duration of the HTC reaction (Fig. S3, ESI†). The UV-vis spectra contained two peaks, at 250 nm and 280 nm, with a small tail extending into the visible spectrum. The maximum emission wavelength (λmax) was 394 nm at an excitation wavelength of 310 nm. From an excitation wavelength of 310 nm to 350 nm, the emission is excitation-independent with a λmax of 394 nm. However, from 360 nm excitation-dependent emission is observed corresponding with the beginning of the tail in the absorption spectra (Fig. 6d). Excitation spectra were recorded from an emission wavelength of 390–450 nm; the spectra are the same suggesting a similar emissive state. However, at an emission wavelength of 360 nm the excitation spectrum looks more like the absorption observed in Fig. 6d with peaks at 250 nm and 280 nm (Fig. 6f).
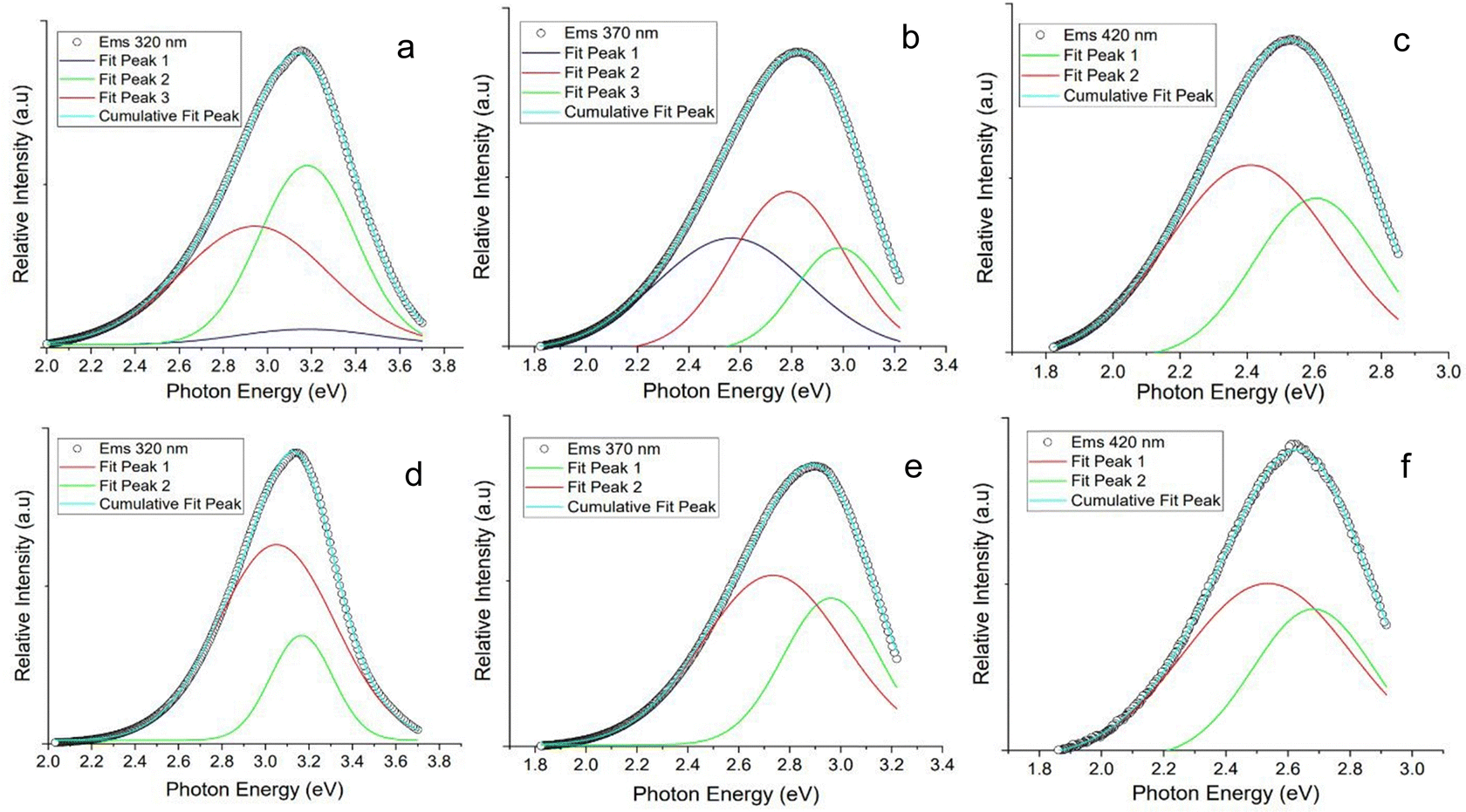 | ||
| Fig. 6 Gaussian peak fitting of photoluminescence data of 2H-CPDs synthesised from the 2H HTC of chitin-derived bio-oil, with excitation wavelengths of (a) 320 nm, (b) 370 nm, and (c) 420 nm. Gaussian peak fitting of photoluminescence data of the impurity fraction isolated after the 2 h HTC, with excitation wavelengths of (d) 320 nm, (e) 370 nm, and (f) 420 nm. | ||
Gaussian deconvolution of the photoluminescence for both the N-CPDs and the isolated impurity was performed to identify the components contributing to the overall photoluminescence (Fig. 6). A large contribution to the total photoluminescence at an excitation wavelength 320 nm for the N-CPD shared a considerable overlap with the emission of the isolated impurity at the same wavelength. An additional small and broad component was identified in the N-CPD that was attributed to the contribution from the N-CPD. At an excitation wavelength of 370 nm, the contribution from a molecular luminophore in the isolated impurity remained, contributing strongly to the overall photoluminescence of the N-CPD. An additional component was observed at 2.5 eV (495 nm). At an excitation wavelength of 420 nm the Gaussian deconvolution of both the N-CPD and isolated impurity appeared identical.
GC-MS analysis of the organic fraction containing impurities consistently revealed the presence of 3-aminopyridine. The photoluminescence of 3-aminopyridine exhibited optical properties similar to the synthesised N-CPDs. The photoluminescence of 3-amino pyridine was characterised in deionised water with absorption λmax of 215, 233 nm and at 290 nm, and a λmax emission of 390 nm when excited at 320 nm (Fig. S4, ESI†).43,45
Photoluminescence in CPDs has been attributed to several factors including interactions from the core–shell structure, surface states, defects, cross-link enhanced emission, conjugated π-domains and from molecular fluorophores.26,42 A contribution from a surface state was neglected; we rationalised that an observable change in the photoluminescence would be seen corresponding to the changing surface composition observed in the XPS (Fig. 4). The deconvolution of the photoluminescence (Fig. 6) revealed the presence of multiple peaks, confirming the existence of numerous photoluminescent centres. A large contribution to the overall photoluminescence of the N-CPDs from a molecular luminophore was observed. Previously, Song et al. (2015) identified multiple photoluminescent centres for CDs; IPCA (imidazo[1,2-a]pyridine-7-carboxylic acid, 1,2,3,5-tetrahydro-5-oxo-), a bright blue fluorophore, and its derivatives were found to contribute to a molecular state photoluminescence in the CDss. In addition, they found a weaker contribution originated from carbon core, consisting of nanosized carbon cores and polymeric clusters.71
Emission occurring from the crystalline carbon core has been characterised as having intrinsic and extrinsic emission. Intrinsic emission has strong blue emission at 400 nm when excited near 325 nm. It originates from the minimally oxidised and low defect isolated sp2 hexagonal carbon domains.72,73 Yoon et al.(2016) investigated the origin of this intrinsic luminescence by investigating the subdomain formation. They found the that the preferential size of the sub-domain was a cluster of four to seven carbon hexagons isolated by oxygen containing functional groups forming stable formations of subdomains. Extrinsic emission has been described as emission occurring at 500 nm when excitation at 325 nm and above, resulting from excessive defects, including oxygen functional groups.72
A λmax of 394 nm for emission at an excitation wavelength 320 nm was observed for all N-CPD samples, which is close to the value reported for intrinsic emission. However, the deconvolution of the photoluminescence and comparison with the isolated impurity would suggest that this is coming from a molecular luminophore as opposed to the carbon core itself. A smaller and broader component was attributed to the contribution from the carbon core (Fig. 6). An explanation for the excitation-dependent emission observed in the N-CPD was attributed to the quantum confinement of differently sized π-conjugated sub-domains within the crystalline core. Size dependent photoluminescence would be expected due to the energy gap transitions from the quantisation of the π-conjugated system, with a red-shift observed as larger sub-domains form. The broadness of the observed peak can be explained by a large distribution of sub-domains.74,75 The predominant source of the overall photoluminescence in the N-CPDs was primarily associated with a molecular luminophore, 3-aminopyridine, alongside a lesser contribution from sub-domains within the crystalline carbon core. However, XPS analysis did not confirm the presence of 3-aminopyridine on the surface (Table 2), as no peak corresponding to the pyridine (CN–C) bond was observed in the 397.8–398.8 eV range.61 The absence of this peak suggests that either the concentration of 3-aminopyridine on the surface is too low to be detected, or that it is embedded within the cross-linked polymeric matrix. The rigorous purification protocol significantly reduces the likelihood that the observed photoluminescence is due to surface contamination. Emphasising the need for vigorous purification to accurately characterise the true mechanism of photoluminescence.
3. Conclusion
Overall, the findings of this study underscore the potential of utilising bio-oil for the sustainable synthesis of carbon nanomaterials and contributes to a deeper understanding of their structure–property relationships. N-CPDs were synthesised from pyrolysis of chitin followed by hydrothermal treatment, introducing the potential of using a waste stream, bio-oil, for the synthesis of N-CPDs. The effect of HTC reaction time on the size, structure, surface composition, and optical properties was explored. Longer reaction times led to the formation of larger N-CPDs with a polymeric cross-linked shell surrounding a crystalline graphitic carbon core. The proposed formation mechanism involves pyrolysis degrading chitin into smaller fragments, with the temperature being sufficient to induce graphitization. These fragments then serve as nucleation sites for hydrothermal cross-linking polymerization, resulting in the creation of a cross-linked polymeric structure. At longer HTC reaction times, this structure gradually carbonises. Interestingly, although the size, structure, and surface composition were noted to change as the reaction time increased, the observed optical properties remained consistent. The optical properties of N-CPDs were thoroughly characterised, revealing strong photoluminescence in the blue and visible regions of the spectrum. Excitation wavelength-dependent emission and multi-component photoluminescence spectra were observed, indicating the presence of diverse photoluminescent centres. Gaussian deconvolution analysis suggested contributions from both a molecular luminophore (3-amino pyridine) and the carbon core to the overall photoluminescence. Future work could investigate alternative bio-oil sources and how surface modifications of N-CPDs influences their photophysical properties. Nuclear magnetic resonance spectroscopy, 1H NMR and 13C were not preformed due to the complex surface chemistry of the N-CPDs, with mixed functional groups and cross-linked structures, would likely lead to convoluted 1H NMR and 13C NMR spectra, making interpretation difficult. By enhancing or tailoring the optical properties, CPDs could become more suitable for applications in energy materials. The consistent photoluminscent properties and reasonable QY provide a solid foundation for further improvements and potential advancements in this field.4. Experimental methodology
4.1. Materials
Chitin, diethyl ether, and acetonitrile (≥99.9%, gradient grade, suitable for HPLC) were purchased from Merck. 2-Acetamidophenol, 3-amino pyridine, 1-octaonol, isoamyl ether, and methyl laurate were purchased from Sigma Aldrich. 3-Acetamidopyridine was purchased from Combi-blocks. 0.22 μm PTFE hydrophilic filter was purchased from Fisher Scientific.4.2. Method
N-CPDs were synthesised from chitin by pyrolysis at 700 °C (heating rate ∼10 °C min−1) for 2 h under a N2 atmosphere to produce bio-oil and bio-char. Bio-oil (10 mL) was transferred to a Teflon-lined autoclave for hydrothermal treatment, water (30 mL, ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 biomass:water). The autoclave was heated in an oven at 200 °C, and the reaction times were 2, 4, and 8 h. The reaction mixture was filtered through a Buckner funnel to collect a black precipitate. The resulting filtrate was then centrifuged at 4000 rpm for 15 minutes then filtered through a 0.22 μm PTFE hydrophilic filter. The solution was then extensively washed with ether. The total volume of the solution was reduced by rotary evaporation and then freeze dried for 48 h. A brown viscous oily solid was obtained. This solid was washed with ether and sonicated until a brown powder crashed out of solution. The N-CPDs were noted to be hygroscopic, with little contact with water formed a viscous oil.
:3 biomass:water). The autoclave was heated in an oven at 200 °C, and the reaction times were 2, 4, and 8 h. The reaction mixture was filtered through a Buckner funnel to collect a black precipitate. The resulting filtrate was then centrifuged at 4000 rpm for 15 minutes then filtered through a 0.22 μm PTFE hydrophilic filter. The solution was then extensively washed with ether. The total volume of the solution was reduced by rotary evaporation and then freeze dried for 48 h. A brown viscous oily solid was obtained. This solid was washed with ether and sonicated until a brown powder crashed out of solution. The N-CPDs were noted to be hygroscopic, with little contact with water formed a viscous oil.
4.3. Characterisation
Analysis using gas chromatography mass spectrometry (GC-MS) was conducted utilising an Agilent 8890 Chromatograph, which was fitted with an HP-5 capillary column (30 m × 0.25 mm, 0.25 um film thickness), and a 5977 MSD mass spectrometer. Transmission electron microscopy was performed using a JEOL 2100F operated at 200 keV. For the Raman spectra, a Horiba LabRAM HR Evolution spectrometer was used with an exciting line wavelength λ = 532 nm, source power 455 mW. To attenuate the power at the sample, a non-dispersive filter was used reducing it to around ∼1% of the laser power. An Olympus LMPLFLN20X magnification objective was used to examine the samples. FT-IR was recorded on a PerkinElmer spectrum two FT-IR spectrometer. The XPS analysis was conducted using a Thermo NEXSA system equipped with a monochromated Al Kα X-ray source (1486.7 eV), with a spherical sector analyser, and three multichannel resistive plate detectors, each with a 128-channel delay line. All data were recorded at 19.2 W with an X-ray beam size of 400 × 200 μm. Absorption spectra were recorded on a Shimadzu UV-1800 spectrophotometer UV-vis and luminescence spectra were recorded on a Shimadzu RF-6000 spectrofluorophotometer. Luminescence spectra and excited-state lifetimes were measured using an Edinburgh FLS980 photoluminescence spectrometer, equipped with a 450 W Xenon arc lamp, Czerny Turner excitation and emission monochromators (1.8 nm mm−1 dispersion; 1800 grooves per mm), time-correlated single photon counting (TCSPC) module and a Hamamatsu R928P photomultiplier tube (in a fan assisted TE cooled housing, operating temperature −20 °C). For lifetime measurements, samples were excited with an EPL-375 (370.8 nm; 61.1 ps pulse width) and an EPL-475 (471.8 nm; 61.1 ps pulse width) picosecond pulsed diode lasers and data analysis was performed on the F980 software with numerical data. A LECO CHN628 combustion elemental carbon, hydrogen, and nitrogen analyser was used to determine the composition of the samples. The Zeta potential was measured with a Zetasizer Nano ZS Instrument (Malvern Panalytical Ltd).Author contributions
Lawrence Bruce: conceptualisation (ideas), methodology, investigation, writing – original draft, visualisation. Liam Desmond: conceptualisation (ideas), methodology, investigation. Abagail Seddon: methodology. Leon Bowen: resources, investigation Greg. A. Mutch: funding acquisition, resources, writing – review & editing Anh Phan: conceptualisation (of project), funding acquisition, supervision, writing – review & editing Elizabeth A. Gibson: conceptualisation (of project), funding acquisition, supervision of LB and AS, writing – review & editing.Data availability
The data supporting the findings of this study are available upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering & Physical Sciences Research Council [grant numbers EP/V047078/1, EP/W03395X/1, EP/Y034961/1, EP/S023836/1, EP/R51309X/1]. GAM was supported by the Royal Academy of Engineering under the Research Fellowship scheme.References
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736 CrossRef CAS.
- L. Li, G. Wu, G. Yang, J. Peng, J. Zhao and J.-J. Zhu, Nanoscale, 2013, 5, 4015 RSC.
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. Zhang, D. Li, H. Tan, Z. Zhao, Z. Xie and Z. Sun, Nanoscale, 2013, 5, 12272 RSC.
- H. Luo, N. Papaioannou, E. Salvadori, M. M. Roessler, G. Ploenes, E. R. H. van Eck, L. C. Tanase, J. Feng, Y. Sun, Y. Yang, M. Danaie, A. Belen Jorge, A. Sapelkin, J. Durrant, S. D. Dimitrov and M. M. Titirici, ChemSusChem, 2019, 12, 4432–4441 CrossRef CAS.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Adv. Mater., 2018, 30, 1704740 CrossRef PubMed.
- J. Yu, X. Yong, Z. Tang, B. Yang and S. Lu, J. Phys. Chem. Lett., 2021, 12, 7671–7687 CrossRef CAS PubMed.
- B. Mehrdad-Vahdati, S. Pourhashem, M. Sedghi, Z. Vaezi, B. Shojaedin-Givi, A. Rashidi and H. Naderi-Manesh, Toxicol. In Vitro, 2019, 61, 104649 CrossRef CAS.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- Y. Liu, S. Roy, S. Sarkar, J. Xu, Y. Zhao and J. Zhang, Carbon Energy, 2021, 3, 795–826 CrossRef CAS.
- Z. Xu, Z. Chen, T. Ji, D. Jv and P. Guan, Mater. Lett., 2022, 309, 131273 CrossRef CAS.
- A. Abbas, L. T. Mariana and A. N. Phan, Carbon, 2018, 140, 77–99 CrossRef CAS.
- R. Wang, K. Q. Lu, Z. R. Tang and Y. J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- P. Namdari, B. Negahdari and A. Eatemadi, Biomed. Pharmacother., 2017, 87, 209–222 CrossRef CAS.
- Y. Liu, H. Huang, W. Cao, B. Mao, Y. Liu and Z. Kang, Mater. Chem. Front., 2020, 4, 1586–1613 RSC.
- P. Tian, L. Tang, K. S. Teng and S. P. Lau, Mater. Today Chem., 2018, 10, 221–258 CrossRef CAS.
- Z. Ji, E. Dervishi, S. K. Doorn and M. Sykora, J. Phys. Chem. Lett., 2019, 10, 953–959 CrossRef CAS.
- S. Zhu, Y. Song, J. Wang, H. Wan, Y. Zhang, Y. Ning and B. Yang, Nano Today, 2017, 13, 10–14 CrossRef CAS.
- A. Bhattacharya, S. Chatterjee, R. Prajapati and T. Kanti Mukherjee, Phys. Chem. Chem. Phys., 2015, 12833 RSC.
- H. Ding, X.-H. Li, X.-B. Chen, J.-S. Wei, X.-B. Li and H.-M. Xiong, J. Appl. Phys., 2020, 127, 231101 CrossRef CAS.
- A. Sciortino, A. Cannizzo and F. Messina, C, 2018, 4, 67 CAS.
- W. Liu, Y. Han, M. Liu, L. Chen and J. Xu, RSC Adv., 2023, 13, 16232 RSC.
- M. Langer, L. Zdražil, M. Medveď and M. Otyepka, Nanoscale, 2023, 15, 4022–4032 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- K. J. Mintz, Y. Zhou and R. M. Leblanc, Nanoscale, 2019, 11, 4634–4652 RSC.
- S. Tao, T. Feng, C. Zheng, S. Zhu and B. Yang, J. Phys. Chem. Lett., 2019, 10, 5182–5188 CrossRef CAS.
- S. Tao, C. Zhou, C. Kang, S. Zhu, T. Feng, S. T. Zhang, Z. Ding, C. Zheng, C. Xia and B. Yang, Light: Sci. Appl., 2022, 11, 56–66 CrossRef CAS PubMed.
- X. Wang, G. Sun, P. Routh, D.-H. Kim, W. Huang and P. Chen, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- X. Zhai, P. Zhang, C. Liu, T. Bai, W. Li, L. Dai and W. Liu, Chem. Commun., 2012, 48, 7955–7957 RSC.
- Z. Qian, J. Ma, X. Shan, H. Feng, L. Shao and J. Chen, Chem. – Eur. J., 2014, 20, 2254–2263 CrossRef CAS PubMed.
- T. C. Wareing, P. Gentile and A. N. Phan, ACS Nano, 2021, 15, 15471 CrossRef CAS PubMed.
- N. Tejwan, S. K. Saha and J. Das, Adv. Colloid Interface Sci., 2020, 275, 102046 CrossRef CAS PubMed.
- H. Ababneh and B. H. Hameed, Int. J. Biol. Macromol., 2021, 186, 314–327 CrossRef CAS PubMed.
- F. Yan, Y. Jiang, X. Sun, Z. Bai, Y. Zhang and X. Zhou, Microchim. Acta, 2018, 185, 424–458 CrossRef PubMed.
- A. Marinovic, L. S. Kiat, S. Dunn, M. M. Titirici and J. Briscoe, ChemSusChem, 2017, 10, 1004–1013 CrossRef CAS.
- Q. Jiang, Y. Jing, Y. Ni, R. Gao and P. Zhou, Microchem. J., 2020, 157, 105111 CrossRef CAS.
- G. Gedda, C.-Y. Lee, Y.-C. Lin and H. Wu, Sens. Actuators, B, 2016, 224, 396–403 CrossRef CAS.
- L. Cao, K. A. Shiral Fernando, W. Liang, A. Seilkop, L. Monica Veca, Y.-P. Sun and C. E. Bunker, J. Appl. Phys., 2019, 125, 220903 CrossRef.
- V. P. Santos, N. S. S. Marques, P. C. S. V. Maia, M. A. B. de Lima, L. de, O. Franco and G. M. de Campos-Takaki, Int. J. Mol. Sci., 2020, 21, 1–17 Search PubMed.
- N. Papaioannou, M. M. Titirici and A. Sapelkin, ACS Omega, 2019, 4, 21658 CrossRef CAS PubMed.
- C. Xia, J. Zhong, X. Han, S. Zhu, Y. Li, H. Liu and B. Yang, Angew. Chem., Int. Ed., 2024, 63, e202410519 CrossRef CAS.
- F. Rigodanza, M. Burian, F. Arcudi, L. Đorđević, H. Amenitsch and M. Prato, Nat. Commun., 2021, 12, 2640 CrossRef CAS.
- C. Xia, S. Zhu, T. Feng, M. Yang and B. Yang, Adv. Sci., 2019, 6, 1901316 CrossRef CAS.
- A. Weisstuch and A. C. Testa, J. Phys. Chem., 1968, 72, 1982–1987 CrossRef CAS.
- B. Y. Yubin Song, S. Zhu, S. Zhang, Y. Fu, L. Wang and X. Zhao, Mater. Chem. C, 2015, 3, 5976 RSC.
- H. Huang, K. Nishi, H. J. Tsai and B. D. Hammock, Anal. Biochem., 2007, 363, 12–21 CrossRef CAS.
- M. Royle, B. Chachuat, B. Xu and E. A. Gibson, RSC Sustainability, 2024, 2, 1337–1349 RSC.
- A. S. Nizami, M. Rehan, M. Waqas, M. Naqvi, O. K. M. Ouda, K. Shahzad, R. Miandad, M. Z. Khan, M. Syamsiro, I. M. I. Ismail and D. Pant, Bioresour. Technol., 2017, 241, 1101–1117 CrossRef CAS PubMed.
- J. Sherwood, Bioresour. Technol., 2020, 300, 122755 CrossRef CAS.
- R. E. Guedes, A. S. Luna and A. R. Torres, J. Anal. Appl. Pyrolysis, 2018, 129, 134–149 CrossRef CAS.
- X. Gao, X. Chen, J. Zhang, W. Guo, F. Jin and N. Yan, ACS Sustainable Chem. Eng., 2016, 4, 3912–3920 CrossRef CAS.
- N. Ullal, R. Mehta and D. Sunil, Analyst, 2024, 149, 1680–1700 RSC.
- C. Dong, M. Xu, S. Wang, M. Ma, O. U. Akakuru, H. Ding, A. Wu, Z. Zha, X. Wang and H. Bi, J. Nanobiotechnol., 2021, 19, 1–8 CrossRef.
- C. D. Yaqian Feng, R. Li and P. Zhou, Microchem. J., 2022, 180, 107627 CrossRef.
- M. Jia, L. Peng, M. Yang, H. Wei, M. Zhang and Y. Wang, Carbon, 2021, 182, 42–50 CrossRef CAS.
- H. Luo, L. Lari, H. Kim, S. Herou, L. C. Tanase, V. K. Lazarov and M. M. Titirici, Nanoscale, 2022, 14, 910–918 RSC.
- J. Zhan, R. Peng, S. Wei, J. Chen, X. Peng and B. Xiao, ACS Omega, 2019, 4, 22574 CrossRef CAS PubMed.
- M. Feng, Y. Wang, B. He, X. Chen and J. Sun, ACS Appl. Nano Mater., 2022, 5, 7502–7511 CrossRef CAS.
- C. Klinke, R. Kurt, J. M. Bonard and K. Kern, J. Phys. Chem. B, 2002, 106, 11191 CrossRef CAS.
- S. Lu, L. Sui, M. Wu, S. Zhu, X. Yong and B. Yang, Adv. Sci., 2019, 6, 1801192 CrossRef PubMed.
- F. Ehrat, S. Bhattacharyya, J. Schneider, A. Löf, R. Wyrwich, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2017, 17, 7710–7716 CrossRef CAS PubMed.
- M. Ayiania, M. Smith, A. J. R. Hensley, L. Scudiero, J. S. McEwen and M. Garcia-Perez, Carbon, 2020, 162, 528–544 CrossRef CAS.
- J. Briscoe, A. Marinovic, M. Sevilla, S. Dunn and M. Titirici, Angew. Chem., Int. Ed., 2015, 54, 4463–4468 CrossRef CAS PubMed.
- L. Zhao, Y. Wang, X. Zhao, Y. Deng and Y. Xia, Polymers, 2019, 11, 1731–1743 CrossRef CAS.
- S. Liao, X. Zhao, F. Zhu, M. Chen, Z. Wu, X. Song, H. Yang and X. Chen, Talanta, 2018, 180, 300–308 CrossRef CAS PubMed.
- Y. Ni, P. Zhou, Q. Jiang, Q. Zhang, X. Huang and Y. Jing, Dyes Pigm., 2022, 197, 109923 CrossRef CAS.
- Y. Chen, C. Zhao, Y. Wang, H. Rao, Z. Lu, C. Lu, Z. Shan, B. Ren, W. Wu and X. Wang, Mater. Sci. Eng., C, 2020, 117, 111264 CrossRef CAS PubMed.
- Y. Xiong, J. Schneider, C. J. Reckmeier, H. Huang, P. Kasák and A. L. Rogach, Nanoscale, 2017, 9, 11730 RSC.
- M. Sudolská, M. Dubecký, S. Sarkar, C. J. Reckmeier, R. Zbořil, A. L. Rogach and M. Otyepka, J. Phys. Chem. C, 2015, 119, 13369 CrossRef.
- W. Kwon, S. Do, J. H. Kim, M. Seok Jeong and S. W. Rhee, Sci. Rep., 2015, 5, 12604 CrossRef CAS PubMed.
- Z. Liang, M. Kang, G. F. Payne, X. Wang and R. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 17478 CrossRef CAS.
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, J. Mater. Chem. C, 2015, 3, 5976–5984 RSC.
- H. Yoon, Y. H. Chang, S. H. Song, E. S. Lee, S. H. Jin, C. Park, J. Lee, B. H. Kim, H. J. Kang, Y. H. Kim and S. Jeon, Adv. Mater., 2016, 28, 5255–5261 CrossRef CAS PubMed.
- S. H. Song, M. Jang, J. Chung, S. H. Jin, B. H. Kim, S. Hur, S. Yoo, Y. Cho and S. Jeon, Adv. Opt. Mater., 2014, 2, 1016–1023 CrossRef CAS.
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. Tan, A. Chen, M. Jin and S. Yang, Nat. Commun., 2018, 9, 2249–2260 CrossRef PubMed.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. Tan, L. Fan and S. Yang, Adv. Mater., 2024, 36, 2312060–2312063 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03858d |
| This journal is © The Royal Society of Chemistry 2025 |