DOI:
10.1039/D5TB02247A
(Paper)
J. Mater. Chem. B, 2025,
13, 15556-15564
Photosensitizer-pendant biotinylated polyester as a nanocarrier for targeted photodynamic therapy
Received
6th October 2025
, Accepted 25th October 2025
First published on 29th October 2025
Abstract
Combinatorial cancer therapy that combines photodynamic therapy with chemotherapy has gained tremendous importance in recent times. Reactive oxygen species (ROS) generation in cancer cells via photosensitizer-loaded polymeric nanoparticles represents one of the non-invasive methods for cancer treatment. As a proof of concept, herein, we demonstrate a molecular design based on a fully degradable polyester scaffold featuring a photosensitizer for targeted ROS generation in cancer cells. An enzymatically degradable, amphiphilic polyester was synthesized by organocatalyzed step-growth polymerization via a transesterification reaction between an activated diester and functional diols, incorporating a phenothiazine dye as the photosensitizer and biotin as the targeting ligand, since biotin receptors are known to be overexpressed in cancer cells. The polymer self-assembled into nanoaggregates in water, exhibiting selective uptake in cancer cells (HeLa and MCF7) with ROS-generating ability upon light irradiation, which caused significant cytotoxic effects. In addition, the hydrophobic core within the nanoaggregates exhibits the ability to encapsulate a chemotherapeutic drug, doxorubicin, and selectively release it in cancer cells.
1. Introduction
Integrating photodynamic therapy (PDT) with various other modes of treatment such as chemotherapy, photothermal therapy (PTT), immunotherapy, and more has emerged as a powerful combinatorial cancer therapy approach to address the limitations of single-mode treatments, especially in combating drug-resistant cancer cells.1 In PDT,2 upon exposure to visible or near-infrared light irradiation, the non-cytotoxic photosensitizer is excited to a higher energy state, where it can transfer its energy or electrons to molecular oxygen, generating reactive oxygen species (ROS) such as hydrogen peroxide, superoxide anions, oxygen radicals, and others. These generated ROS ultimately induce tumor cell death. Despite the therapeutic efficiency of many photosensitizers, including porphyrin-based organic π-systems and heavy-atom-free photosensitizers, explored predominantly for their high singlet oxygen quantum yields, they exhibit poor solubility along with dark cytotoxicity beyond a specific threshold dosage.3 Furthermore, the passive uptake of the photosensitizer causes non-tissue-specific internalization, resulting in ROS-mediated phototoxicity also in normal cells along with cancer cells. To overcome some of these critical challenges, active transport of the photosensitizer has become increasingly important for tissue-specific delivery. Notably, organic dyes with sulfur-containing heteroatoms display significant potential as photosensitizers.4 For example, phenothiazines, featuring nitrogen- and sulfur-substituted tricyclic derivatives, have been known for over 100 years for their wide range of biological and chemical properties.5 Phenothiazinium-based polar dyes such as methylene blue (MB) and toluidine blue (TBO) are frequently used photosensitizers; however, due to their hydrophilic nature, they exhibit poor penetration properties across lipid membranes that significantly impede the cellular uptake of these compounds.6a By increasing the hydrophobic alkyl content in methylene blue (MB), the lipophilicity of the molecule can be enhanced, but it comes with other challenges, for example, long alkyl chains can render MB derivatives water-insoluble and resistant to degradation, raising concerns over their bioaccumulation and environmental or toxicological hazards. As the lipophilicity of a pharmaceutical compound is critical for its cellular uptake and intracellular localization, an appropriate balance between hydrophobicity and hydrophilicity is essential.6b We envisaged that photosensitizer-functionalized biodegradable polymers featuring cell-targeting motifs may provide advantages of both cell-specific uptake and enzyme-triggered faster clearance from the body. To test this possibility, here we propose a novel molecular design based on a cancer cell-targeting degradable biotinylated polyester scaffold, with a phenothiazine derivative incorporated as a pendant group functioning as a photosensitizer (Scheme 1). The amphiphilic polymer P1 self-assembles into nanoaggregates in water and exhibits dual photosensitizing and hydrophobic dye loading properties. As a proof of concept, we demonstrated the loading of a model anticancer drug, doxorubicin (DOX), and its selective release in cancer cells combined with light-triggered ROS-induced cytotoxicity. The combinatorial effect of ROS- and DOX-mediated cancer cell killing, achieved through a targeted design approach based on a completely degradable polyester backbone, remains underexplored.
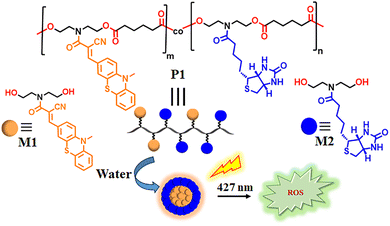 |
| | Scheme 1 Structure of the amphiphilic polyester P1 and schematic representation of its aggregation in water with ROS generating properties upon irradiation with 427 nm light. | |
2. Experimental
2.1. Materials and methods
All reagents were obtained from commercial suppliers and used without further purification unless otherwise mentioned. Adipoyl chloride, diethanolamine, pentafluorophenol, biotin, phenothiazine, and 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) were purchased from TCI Chemicals. 4-Dimethylaminopyridine (DMAP) and EDC·HCl were purchased from Avra Synthesis Pvt. Ltd. Doxorubicin·HCl was purchased from BLD Pharmatech Pvt. Ltd. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from HiMedia Laboratories. All the solvents were dried properly following standard procedures before setting up the reactions. Dried solvents for polymerization were purchased from Sigma-Aldrich. 1H-NMR and 19F-NMR spectra were measured on Bruker 400 MHz and 500 MHz NMR spectrometers using CDCl3 and DMSO-d6 as solvents from Eurisotop. Chemical shifts (δ) are reported in ppm units with TMS as the internal standard. The coupling constants (J) are reported in hertz (Hz). HRMS was conducted using a XEVO G2-XS QTof and a Micromass Q-Tof Micro machine. The number average molecular weight (Mn) and dispersity (Đ) of the polymers were measured by size exclusion chromatography (SEC) with N,N-dimethylformamide (DMF) as an eluent at 60 °C with a flow rate of 0.5 mL min−1 using a GPC instrument containing a Waters 515 HPLC pump, a Waters 2414 refractive index (RI) detector, one PolarGel-M guard column (5037.5 mm) and two Polar Gel-M analytical columns (30037.5 mm). FT-IR spectra were recorded using a PerkinElmer Spectrum 100 FT-IR spectrometer. Column chromatography was carried out on silica gel (100–200 mesh). Spectroscopic grade solvents were used for UV-vis studies. UV-vis spectra were recorded using a JASCO V-750 spectrophotometer. Fluorescence spectra were recorded in a FluoroMax-3 spectrophotometer from Horiba Jobin Yvon. Dynamic light scattering (DLS) measurements were conducted using a Malvern instrument. A Leica DMIL LED fluorescence microscope was used for fluorescence imaging. The absorbance of the MTT assay at 570 nm was monitored using a microplate reader (VARIOSKAN, Thermo Fisher). Transmission electron microscopy (TEM) was performed using a Jeol 2100 LaB6 transmission electron microscope operating at 200 kV voltage. All images were analyzed using ImageJ software. Fluorescence-activated cell sorting (FACS) analysis was performed using a BD FACSAria III. EPR data were obtained using a JEOL FA200 spectrophotometer with the following parameters: modulation width, 20 G; amplitude, 2; time constant, 30 ms; power, 3 mW; and frequency, 9.09 GHz.
2.2. Sample preparation and self-assembly studies
Concentrated stock solutions of P1 were prepared in HPLC grade MeOH (“good” solvent). A measured quantity of the polymer solution was transferred into a glass vial. MeOH was evaporated by gently heating the solution to prepare a thin film in the vial. Then a measured amount of Milli Q water was added and the dispersion was subjected to heating, followed by cooling at room temperature to ensure proper dissolution of the film. This aqueous solution was then used for spectroscopic analysis (UV-vis and PL), dynamic light scattering (DLS), and transmission electron microscopy (TEM). For TEM analysis, an aqueous solution of P1 (C = 0.1 mg mL−1) was drop-cast onto a carbon coated Cu grid. After 5 min, the excess surface solvent on the grid was removed by tapping a filter paper on it. Then the grid was kept overnight for air drying before the TEM experiment. For the DLS experiment, an aqueous solution of P1 (C = 0.1 mg mL−1) was used. The stock solution of M1 was prepared in CHCl3 and thin films were obtained by slow evaporation of the solvent. The aqueous solution of M1 at the desired concentration (equivalent to the dye concentration in 0.1 mg mL−1 of P1) was prepared following the same procedure as mentioned for P1, and all the above-mentioned experiments were performed with M1 for direct comparison with P1.
2.3. FT-IR studies
These studies were performed with activated adipoyl ester monomer A1 and P1 crude in transmittance mode. The spectral signatures were measured using the following parameters: scan range = 4000–1000 cm−1, resolution = 1.0 cm−1, number of scans = 64, and T = 25 °C.
2.4. Degradation study
For the degradation study, an aqueous solution of P1 (C = 1 mg mL−1) was added with 500 µL of an enzymatic solution of lipase B from Pseudomonas cepacia (6.6 mg mL−1) in phosphate buffer solution (pH = 7.4, 38.6 U mg−1) incubated at 37 °C for 12 h and 24 h with continuous stirring at 200 rpm. After incubation, the sample was lyophilized and changes in the molecular mass were characterized by size exclusion chromatography (SEC) in DMF solvent.7
2.5. Determination of the critical aggregation concentration (CAC)
Measured amounts of the polymer stock solutions in MeOH were taken in different vials assigned for different concentrations of the polymers. A stock solution of pyrene (Py) (C = 1 × 10−4 M) was prepared in CHCl3 and a measured aliquot of the solution was taken in all the polymer containing vials of different concentrations. After drying off the organic solvent, the final Py concentration was adjusted to 10−6 M by dispersing the polymer films in a total volume of 1000 µL water. The individual solutions were then subjected to heating for 5 minutes, cooled at room temperature and equilibrated for 60 minutes before recording their emission at an excitation wavelength of 337 nm.
2.6. Doxorubicin encapsulation and release study
The commercially available DOX·HCl was neutralized by treatment with excess triethylamine solution and the neutralized hydrophobic DOX was extracted from the organic part during workup using a CHCl3/water mixture. A measured quantity (0.5 mg or 0.9 µM) of hydrophobic DOX in THF was added into a solution of polymer P1 in MeOH (C = 2 mg mL−1). The organic solvent was then evaporated to form a film, which was then dispersed in water and then vortexed for 10 min, followed by sonication for 2 min. The solution was then subjected to dialysis in water for 24 h in a dialysis bag with a 3500 Mw cutoff. The remaining solution was considered as stock and preserved for further use. Before any experiment, the same solution was again filtered through a 0.45 µm syringe filter (nylon). UV-vis and emission spectra of the DOX-loaded polymers confirmed drug encapsulation. The drug loading efficiency (DLE) and drug loading content (DLC) were estimated using the standard calibration curve obtained from the concentration-dependent UV-vis absorbance spectra of DOX and using the following formula:8
| DLC (%) = [weight of loaded DOX/weight of DOX loaded nanoaggregates] × 100% |
| DLE (%) = [weight of loaded DOX/weight of DOX infeed] × 100% |
2.7. Cell culture conditions
Human cervical cancer (HeLa), human breast cancer (MCF 7), and mouse fibroblast (NIH 3T3) cell lines were used for the cellular uptake studies described in this work. All the cells were seeded in a high glucose Dulbecco's Modified Eagle Medium (DMEM), which was supplemented with 10% fetal bovine serum (FBS) and 1% L-penicillin–glutamine–streptomycin (PSG) to prepare the complete media. Cells were maintained by passaging them at ∼80% confluency at 37 °C in the presence of 5% CO2 in an incubator with a humidified environment.
2.8. Cell viability study by the MTT assay
The cell viability of the HeLa cells was investigated in the presence of P1 using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Approximately, 104 cells per well were seeded in a 96-well plate in complete medium and left overnight for the cells to adhere. The next day, the spent medium was replaced with fresh complete DMEM containing polymers at various concentrations (0.1, 0.2, 0.3, 0.4 and 0.5 mg mL−1) and incubated for another 24 h. After 24 h, the medium containing polymers was again removed and 100 µL of the fresh medium was added, followed by 50 µL of 5 mg mL−1 MTT salt per well, and incubated for another 4 h at 37 °C. After 4 h, the medium was carefully removed without disturbing the formazan crystals formed at the base of each well. Dimethyl sulfoxide (DMSO) (100 µL per well) was added to dissolve the purple formazan crystals and cells were incubated for another 30 min before the absorbance of these treated cells was recorded at 570 nm using a plate reader (VARIOSKAN, Thermo Fisher). MTT added to the untreated cells was considered as the positive control.9 The cell viability percentage of cell death was calculated as follows:
| [OD of the polymer treated cells/OD of the untreated cells] × 100, |
where OD stands for optical density.
2.9. Fluorescence-activated cell sorting (FACS) analysis
Approximately 105 cells were seeded in 35 × 10 mm2 cell culture plates in complete media and incubated overnight for the cells to adhere. The next day, the spent medium was removed, and fresh complete medium containing either the aqueous solution of the polymer (C = 0.4 mg mL−1) or DOX-encapsulated polymer solution was added. The cells were further incubated at 37 °C for a predetermined time (24 h) period to achieve effective cellular internalization. To study the effect of biotin receptor blocking on cellular internalization, the adherent cells were first incubated with commercially available free biotin (5 µM) for 1 h at 37 °C. Subsequently, after the completion of incubation, the cell culture medium was replaced with the polymer solution (C = 0.4 mg mL−1) or DOX-encapsulated polymer solution in fresh complete medium, followed by incubation for another 24 h. After this, the medium was removed, cells were treated with trypsin EDTA, and cell pellets were collected after centrifugation. After the removal of the supernatant, the cells were re-suspended in complete media and transferred to a FACS tube (12 × 75 mm2 polystyrene round bottom style). Data for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events of live cells were collected and analyzed using a BD FACSAria™ III.
000 events of live cells were collected and analyzed using a BD FACSAria™ III.
For the ROS generation assay, both HeLa and MCF7 cells were incubated with P1 (0.4 mg mL−1) for 24 h. Subsequently, they were washed with PBS buffer (pH = 7.4) and treated with 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, 20 µM) in complete media for 30 min. Cells were again washed with phosphate buffer and irradiated for 10 minutes (427 nm LED bulb), followed by incubation for another 24 h. After this, the medium was removed, cells were treated with trypsin EDTA, and cell pellets were collected after centrifugation. After the removal of the supernatant, the cells were re-suspended in complete media and transferred to a FACS tube (12 × 75 mm2 polystyrene round bottom style). Data for 10000 events of live cells were collected and analyzed using a BD FACSAria™ III.
2.10. ROS generation
The solutions for P1 (C = 0.4 mg mL−1) and M1 (C = 28 µM) were prepared following the procedure described earlier for aqueous sample preparation. The total ROS generation efficiency of aqueous P1 solution as well as M1 was determined using 2′,7′-dichlorofluorescein diacetate (DCFH-DA) as a probe. Initially, DCFH-DA was activated by treating with aqueous NaOH solution (0.01 N) to produce DCFH (200 µM). After that, 1.0 mL of 0.4 mg mL−1P1 solution was mixed with 175 µL of 200 µM DCFH solution (the final concentration of DCFH was 35 µM) and fluorescence spectra were measured in the dark as well as after light irradiation (λ = 427 nm) at 5-min intervals for a total of 30 minutes. The time-dependent evolution of the fluorescent DCF dye via oxidation of DCFH by in situ generated ROS was monitored both with and without light irradiation of the P1 polymer. The same process was repeated for the 0.4 mg mL−1P1 in water sample under hypoxic conditions (inert atmosphere). To achieve hypoxic conditions, the sample was purged with dry argon for 2 h.
2.11. Fluorescence imaging of cells
105 cells were seeded in confocal imaging dishes (35 × 10 mm2, purchased from Genetix, Biotech Asia Pvt. Ltd.) and incubated overnight for their adhesion to the dish. The next day, the medium was removed and replaced with the fresh complete medium containing the polymer (C = 0.4 mg mL−1), followed by incubation for 24 hours before washing again with complete medium and then incubation with Hoechst 33342 for 10 min and DCFH-DA solution for 10 min, and the cells were imaged in the green channel (λ = 488 nm), blue channel (λ = 405 nm), and orange channel (λ = 488 nm). Similarly, the cells were treated with the DOX-encapsulated polymer solution or propidium iodide (PI) and incubated for an hour, followed by incubation with Hoechst 33342 for 10 min and washing thrice with complete media, and were then imaged in the excitation of the red channel (λ = 543 nm), green channel and blue channel. Finally, live cell microscopy imaging was performed in the presence of complete media. Images were captured using an Eclipse Ti-E confocal laser-scanning microscope equipped with a Plan Apochromat VC 60×/1.4 oil objective and a Digital Sight DS-Qi1MC monochromatic camera with NIS-AR software (Nikon, Tokyo, Japan) and a Leica DMIL LED fluorescence microscope.
3. Results and discussion
3.1. Polymer synthesis, characterization and kinetics
With the aim to impart the ROS-generating ability to a degradable polyester backbone (Scheme 1), herein we synthesized a diol monomer, M1, functionalized with a fluorescent phenothiazine photosensitizer10 (Scheme S1), which imparts luminescent character to P1, enabling monitoring of its intracellular uptake by fluorescence imaging. The aqueous dispersibility was acquired from the biotinylated diol monomer M2,11a which enables the newly synthesized polyester P1 to exhibit cancer cell targeting ability through its overexpressed biotin receptors.12 Together with these two monomers in a feed ratio of 1:4 (M1:M2), polyester P1 was synthesized following our earlier established methodology13 through an organocatalyzed polycondensation reaction with the pentafluorophenyl ester of adipic acid (A1) (Scheme S2). Near-complete conversion of P1 was realized after 22 h evidenced by its crude 19F NMR spectrum through the disappearance of the peaks corresponding to the A1 monomer with the appearance of additional shielded peaks corresponding to that of the released pentafluorophenol (Fig. S1a), corroborating our previous observations.9c,11,13 Furthermore, the FT-IR spectra showed complete disappearance of the activated ester peak at 1788 cm−1 (Fig. S1b), accompanied by the appearance of a new prominent signal at 1672 cm−1 along with shoulder peaks between 1700 and 1750 cm−1 corresponding to the carbonyl stretching of the newly formed backbone ester and the other two incorporated monomers. The 1H NMR spectrum of purified P1 showed the signature of the aromatic phenothiazine protons, suggesting its successful incorporation. However, the integration ratio of Hc and Hm protons concluded 2% attachment of M1 with respect to M2 (Fig. S2). P1 showed an absorption band at ∼450 nm similar to M1, suggesting successful attachment of the photosensitizer to the polymer chain (Fig. S3a). Poor incorporation of M1 can be attributed to its lower reactivity with respect to M2, which was found to increase when the same polymerization reaction was extended for 48 h keeping the feed ratio constant, to obtain the P2 polymer with 10% incorporation of M1 (Fig. S4). The 1H NMR stack plot revealed that the proton signals from A1 (δ = 2.88 and 1.78 ppm) got shielded to δ = 2.31 and 1.5 ppm due to the release of the highly electronegative PFP moiety, whereas the peaks corresponding to the diethanolamine counterparts  of M1 and M2 at δ = 3.81–3.62 ppm and 3.56–3.51 ppm, respectively, got deshielded to δ = 4.06 and 4.2 ppm due to their higher proximity to the electron withdrawing ester groups in P1 (Fig. S5). To further elucidate the lower incorporation of M1 with respect to M2, we investigated the polymerization ability of M1 with only A1 (P3 polymer) in the absence of M2. The progress of the polymerization of P3 was monitored by time-dependent 1H NMR spectroscopy. The emergence of a new downfield shifted peak at δ = 4.2–4.0 ppm for the methylene proton (Ha) adjacent to the ester group in the crude P3 polymer and concomitant disappearance of the methylene proton
of M1 and M2 at δ = 3.81–3.62 ppm and 3.56–3.51 ppm, respectively, got deshielded to δ = 4.06 and 4.2 ppm due to their higher proximity to the electron withdrawing ester groups in P1 (Fig. S5). To further elucidate the lower incorporation of M1 with respect to M2, we investigated the polymerization ability of M1 with only A1 (P3 polymer) in the absence of M2. The progress of the polymerization of P3 was monitored by time-dependent 1H NMR spectroscopy. The emergence of a new downfield shifted peak at δ = 4.2–4.0 ppm for the methylene proton (Ha) adjacent to the ester group in the crude P3 polymer and concomitant disappearance of the methylene proton  adjacent to the hydroxy (–OH) group in free M1 at δ = 3.82–3.62 ppm were monitored at different time intervals (Fig. S6a). The integration ratio of the newly emerged peak with respect to the disappearing peak revealed near-quantitative incorporation of M1 after 4 h in P3 (Fig. S6b), which indicates efficient uptake of M1 by this methodology, when there is no other competing monomer. A stack plot of polymers P3 and P4 with P2 indeed shows the presence of signature peaks of both phenothiazine and biotin in copolymer P2 (Fig. S7), further suggesting the successful inclusion of the individual monomers in P2. The two polymers were further characterized by size exclusion chromatography (SEC), which yielded broad peaks for P1 and P2 corresponding to molecular weights (Mn) of 23000 g mol−1 (Đ = 1.52) and 26500 g mol−1 (Đ = 1.07), respectively, with respect to the polymethyl methacrylate (PMMA) standard and dimethyl formamide (DMF) as the eluent (Fig. S3b).
adjacent to the hydroxy (–OH) group in free M1 at δ = 3.82–3.62 ppm were monitored at different time intervals (Fig. S6a). The integration ratio of the newly emerged peak with respect to the disappearing peak revealed near-quantitative incorporation of M1 after 4 h in P3 (Fig. S6b), which indicates efficient uptake of M1 by this methodology, when there is no other competing monomer. A stack plot of polymers P3 and P4 with P2 indeed shows the presence of signature peaks of both phenothiazine and biotin in copolymer P2 (Fig. S7), further suggesting the successful inclusion of the individual monomers in P2. The two polymers were further characterized by size exclusion chromatography (SEC), which yielded broad peaks for P1 and P2 corresponding to molecular weights (Mn) of 23000 g mol−1 (Đ = 1.52) and 26500 g mol−1 (Đ = 1.07), respectively, with respect to the polymethyl methacrylate (PMMA) standard and dimethyl formamide (DMF) as the eluent (Fig. S3b).
3.2. Self-assembly and degradation studies
Next, we investigated the self-assembly behavior and the ROS generation ability of P1 with a lower loading content of the hydrophobic phenothiazine dye. The UV-vis spectra showed a 37 nm blue shift in the aggregated polymer in water compared to its molecularly dispersed state in CHCl3, suggesting that the aromatic phenothiazine moiety at the core is strongly π-stacked (Fig. S8). The container property of the core was verified through the encapsulation of a hydrophobic fluorescent probe (pyrene), yielding a critical aggregation concentration (CAC) of 37.8 µg mL−1 (Fig. S9). The transmission electron microscopy (TEM) image of P1 in water revealed spherical nanoaggregates with an average size of ∼200 nm (Fig. 1a), which closely matched with the hydrodynamic diameter (Dh) of 150 nm obtained from the dynamic light scattering (DLS) data (Fig. 1c). The water dispersibility of P1 can be attributed to the polar M2 motif, which renders its homopolymer P4 with amphiphilic character, resulting in a spherical aggregated structure, as can be observed from the TEM images (Fig. S10). The nanoaggregates of P1 were found to retain their thermal stability at high temperatures (Fig. S11), as evident from the temperature-dependent DLS studies. The enzymatic degradability of the ester backbone was verified by treating P1 with the lipase B enzyme from Pseudomonas cepacia.11a After 12 h and 24 h of incubation at 37 °C, partial degradation was observed as indicated by the increase in the retention time compared to that of the pristine polymer P1 from the SEC data (Fig. S12),11a yet this type of amphiphilic polyester-based nanoaggregates maintain their structural integrity in cell culture media (DMEM) as we observed earlier.9c Furthermore, the amphiphilic nature of M1 arising due to the presence of both the polar hydroxyl groups and the hydrophobic phenothiazine dye enabled us to study its self-assembling ability in water for comparison with the P1 polymer. TEM images of M1 in water also revealed nanoaggregates with an average diameter of ∼120 nm (Dh), slightly smaller than that of P1 (Fig. 1b and c). This facilitated the evaluation of the ROS generating properties of the polymer with respect to the monomer, as discussed in the following section.
 |
| | Fig. 1 TEM images of (a) P1 (C = 0.1 mg mL−1) and (b) M1 (C = 7 µM) in water; (c) DLS data of P1 (C = 0.1 mg mL−1) and M1 (C = 7 µM) in water; (d) a schematic representation of the DCFH-DA assay for monitoring in situ ROS generation from the evolution of DCF fluorescence; (e) plot of DCF emission at 525 nm at various time intervals upon irradiation of DCFH-treated P1 (C = 0.4 mg mL−1) and M1 (C = 28 µM) maintaining an equal sensitizer concentration (0.1 mg mL−1P1 is equivalent to 7 µM of M1 (Fig. S3a); (f) EPR spectra taken after exposure to light (λ = 427 nm) for 5 min of P1 (C = 0.4 mg mL−1) and M1 (C = 28 µM) in water (frozen state). | |
3.3. ROS generation studies
The ROS generation ability of phenothiazine containing M1 and P1 was evaluated through the DCFDA assay,14 where a commercially available dye, DCFH-DA (2′,7′-dichlorofluorescein diacetate) was pretreated with NaOH to produce active DCFH, a weak fluorophore. In the presence of ROS, DCFH oxidizes to DCF, generating a green emission at 525 nm (Fig. 1d).14 Thus, time-dependent emission of DCF was monitored for probing the ROS generation ability, which yielded a 26-fold increase in the fluorescence intensity within just 5 min of LED light exposure (Fig. 1e and Fig. S13a, b) for both P1 and M1. To further assess the generation of ROS, 2,2,6,6-tetramethylpiperidine (TEMP) was employed as the singlet oxygen capturing agent, and an electron paramagnetic resonance (EPR) experiment was performed after 5 min of irradiation with an LED bulb (12 W, λ = 427 nm) in the frozen state. Similar EPR signals (Fig. 1f) were observed for both M1 and P1 in water, signifying the singlet oxygen (1O2) generation ability of M1 that was inherited by P1 without any alteration.15 1,3-Diphenylisobenzofuran (DPBF) is used as a typical 1O2 UV-vis probe, as it undergoes an irreversible reaction with 1O2 to generate colorless 1,2-dibenzoyl benzene with a decrease of absorbance at 414 nm.15 But due to the overlap of this absorbance region with that of the photosensitizers M1 and P1, the DPBF assay could not be performed by UV-vis spectroscopy. However, this assay was proved to be successful with gas chromatography–mass spectrometry (GC–MS) (Fig. S13c), since upon irradiation of P1 by 427 nm light for 5 min in the presence of DPBF, a prominent mass peak at 286.186 corresponding to 1,2-dibenzoyl benzene was obtained, reiterating the 1O2 generation ability of P1. Furthermore, polymer P1 was also able to exhibit ROS generation properties, though to a lesser extent, even under limited oxygen conditions (Fig. S13d), compared to normoxic conditions (aerial), suggesting its efficacy to operate under hypoxic conditions (inert atmosphere) as well.15c
Since the P1 polymer showed excellent ROS generating ability, we pursued further cellular studies with the P1 polymer. The light-induced intracellular ROS-triggered cytotoxicity of P1 and M1 was investigated by the MTT assay. The polymer itself was found to be negligibly toxic against cancerous HeLa cells, with around ∼90% cell viability even at a concentration of 0.5 mg mL−1, which was 13 times higher than the CAC. Also, more than 90% cell viability was observed at 0.4 mg mL−1 upon 24 h of incubation (Fig. 2a). This was followed by time-dependent photo-irradiation (λ = 427 nm) of the P1-treated cells. The cell viability significantly reduced to ∼40% and ∼30% upon light irradiation for 10 and 15 min, respectively, compared to more than 90% viability in non-light-irradiated cells, while only light exposure without the polymer showed no toxicity. This firmly establishes the cytotoxic effect explicitly arising due to the light-induced intracellular ROS generation by P1 (Fig. 2a). The IC50 of P1 was calculated to be 0.38 mg mL−1 (23.7 µM) and 0.34 mg mL−1 (26.6 µM) for the HeLa cell line upon 10 and 15 min of irradiation, respectively (Fig. 2b and Fig. S14). The ROS-mediated cell killing was also obtained with another cancer cell line, MCF7, under identical experimental conditions (Fig. 2a). To further validate the light-induced cytotoxicity of P1 arising due to its intracellular ROS generation ability, the DCFDA assay was performed with P1-treated cancer cells (HeLa and MCF7), and the results were analysed via fluorescence-activated cell sorting (FACS) studies. Upon light irradiation, green fluorescence from DCF evolved, probing cellular uptake of P1 and its ROS generation ability (Fig. 2c and Fig. S15a and b). Interestingly, much diminished green fluorescence of DCF in the non-cancerous NIH 3T3 cell line indicates cancer cell-selective internalization of the biotinylated polyester P1, which was attributed to the biotin receptor-mediated endocytosis due to the overexpression of biotin receptors in the cancer cells as compared to non-cancerous cells. Noteworthy, although free M1 exhibits identical ROS generation ability to P1 at the same photosensitizer concentration (28 µM M1 ≈ 0.4 mg mL−1P1), it showed minimal uptake by passive diffusion across all three tested cell lines (Fig. 2c and Fig. S15c), indicating its lack of cancer cell targeting ability. Furthermore, the cancer cell-selective uptake of P1 was confirmed by the reduced emission of DCF in the MCF7 cell line pretreated with excess free biotin, blocking the biotin receptor-mediated endocytosis (Fig. 2d) and establishing P1's biotin-based targeted molecular design. Next, the visual confirmation of the cancer cell-selective uptake of P1 was realized by fluorescence microscopy imaging in HeLa cells (Fig. 3), where the intrinsic orange emission of P1 showed an overlap with the green emission of DCF generated inside the cytosol upon light irradiation, demonstrating the intracellular ROS generating potential of P1. A similar result was also observed in another cancerous cell line, MCF7, additionally treated with propidium iodide (PI). Along with the emission from P1 and DCF, additional red fluorescence from PI (Fig. 4) was observed due to its known ability to selectively penetrate membrane-compromised dead cells.14 The absence of orange or green fluorescence in light-irradiated non-cancerous NIH 3T3 cells further proved the selective uptake of P1 in cancer cells (Fig. 3).
 |
| | Fig. 2 (a) MTT assay of P1 with and without light irradiation (λ = 427 nm) in HeLa cells and MCF7 cells; + L denotes the presence of only light, P1 means with only the polymer, and P1 + L denotes with the polymer in the presence of light with different irradiation times [data are shown as the mean ± SD of three experimental replicates]; (b) calculation of IC50 in P1 incubated HeLa cells after irradiation for 10 and 15 min; (c) mean DCF intensity data obtained from FACS analysis of P1 and M1 in different cancerous (MCF7 and HeLa) and non-cancerous (NIH 3T3) cell lines after incubation for 24 h at 37 °C, followed by irradiation for 10 min [data are shown as the mean ± SD of three experimental replicates]; (d) FACS analysis showing the relative fluorescence intensity of DCF in the MCF7 cell line with and without pretreatment with excess free biotin; the left shift of the chromatograms suggests decreased intracellular ROS generation for the pretreated cell lines with free biotin. | |
 |
| | Fig. 3 Fluorescence imaging of HeLa cells and NIH 3T3 cells after incubation with P1 for 24 hours at 37 °C; Images from top to bottom rows: differential interference contrast (DIC), blue-channel emission for the nuclei stained with Hoechst 33342, green-channel emission for the DCF dye, orange emission for P1, and their overlay in the merged images. | |
 |
| | Fig. 4 Fluorescence microscopy images of MCF7 cells after incubation with P1 for 24 h at 37 °C followed by 10 minutes of light irradiation (λ = 427 nm). Left to right images: differential interference contrast (DIC); blue-channel emission for the nuclei stained with Hoechst 33342; orange-channel emission for the P1 polymer; green-channel emission for the DCF dye; and red-channel emission for the dead cell assay propidium iodide (PI) dye. | |
3.4. DOX encapsulation and combinatorial therapy
Next, successful encapsulation of the anti-cancerous drug doxorubicin (DOX) within the hydrophobic pocket of the P1 nanoaggregates was confirmed by the red shift16 in the UV-vis spectra of DOX-encapsulated P1 [abbreviated as P1(DOX)] with respect to free DOX (Fig. S16a), coupled with significant quenching in the absorption-normalized emission band of DOX within the encapsulated polymer (Fig. S16b).16 The swelling behavior of P1(DOX) was observed from the DLS data, showing an increase in the particle size from 150 to 220 nm (Fig. S16c). The prominent red fluorescence from DOX-encapsulated P1 was observed in MCF7 cells with a mean uptake of ∼48%, indirectly quantifying P1's cellular internalization (Fig. 5a). However, the uptake of P1 was significantly reduced to only ∼9% upon pretreatment of the MCF7 cells with excess free biotin (Fig. 5a), confirming its internalization via a biotin receptor pathway. The fluorescence microscopy image of the P1(DOX)-treated MCF7 cell line showed prominent colocalization of the intrinsic orange fluorescence of P1 and the red fluorescence of DOX, further demonstrating the dye encapsulation properties of the nanoaggregate (Fig. S17a). The loss of morphological integrity of the cancerous cells upon treatment with P1(DOX) was clearly visualized from the optical microscopy images (Fig. S17b), which can be attributed to the possible polyester degradation mediated by the acidic microenvironment of cancer cells17 and endogenous enzymes, triggering the release of the encapsulated drug.18 This effect was found to be more pronounced by the combinatorial effect of DOX release and light-triggered ROS generation, which was also evident from the MTT assay, where P1(DOX) showed an additional ∼20% decrease in cell viability post irradiation with 427 nm light (Fig. 5b).
 |
| | Fig. 5 (a) FACS analysis showing the relative fluorescence intensity of P1(DOX) in the MCF7 cell line with and without pretreatment with excess free biotin; (b) MTT assay of P1 and P1(DOX) before and after irradiation with 427 nm light (+ L) in MCF7 cells [data are shown as the mean ± SD of three experimental replicates]. | |
4. Conclusions
In summary, degradable polyesters (P1 and P2) incorporating a cancer cell-targeting biotin motif and a phenothiazine-based photosensitizer for reactive oxygen species (ROS) generation were successfully synthesized by organocatalyzed step-growth polymerization via a transesterification reaction between an activated diester monomer and functional diols. In water, P1 self-assembled into nanoaggregates with surface decorated biotin motifs, which enabled its cellular uptake and ROS-producing ability selectively in cancer cells (HeLa and MCF7) compared to non-cancer cells (NIH 3T3). This selectivity caused a significant decrease in the cell viability of cancer cells upon light-triggered ROS generation, while the polymer itself showed negligible cytotoxicity in the absence of light irradiation. Additionally, self-assembled P1 exhibited hydrophobic container properties, enabling efficient encapsulation of a model anticancer drug, doxorubicin, and its selective release in cancer cells. As a proof of concept, P1 demonstrates the potential for synergistic combination of photodynamic and chemotherapeutic strategies for enhanced anticancer therapy. The fundamental findings of this work may inspire new design principles for targeted combinatorial anticancer therapy from a completely degradable polyester scaffold.
Author contributions
These authors contributed equally.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: 10.1039/d5tb02247a.
Acknowledgements
C. C. and S. B. thank NET-UGC, New Delhi and IACS Kolkata, respectively, for fellowships. A. D. thanks ANRF-SERB, India (grant no. CRG/2022/003069) and TRC, IACS for funding.
Notes and references
-
(a) S. Mignani, M. Bryszewska, B. K. Maculewicz, M. Zablocka and J. P. Majoral, Biomacromolecules, 2015, 16, 1–27 CrossRef CAS PubMed;
(b) A. Kanojiya, J. Terglane, V. Gerke and B. J. Ravoo, Soft Matter, 2025, 21, 1639 RSC;
(c) Z. Xie, T. Fan, J. An, W. Choi, Y. Duo, Y. Ge, B. Zhang, G. Nie, N. Xie, T. Zheng, Y. Chen, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2020, 49, 8065 RSC;
(d) J. Yi, L. Liu, W. Gao, J. Zeng, Y. Chen, E. Pang, M. Lan and C. Yu, J. Mater. Chem. B, 2024, 12, 6285 RSC;
(e) M. Overchuk, R. A. Weersink, B. C. Wilson and G. Zheng, ACS Nano, 2023, 17, 7979 CrossRef CAS PubMed;
(f) N. Kwon, H. Kim, X. Li and J. Yoon, Chem. Sci., 2021, 12, 7248 RSC;
(g) L. Gong, L. Yan, R. Zhou, J. Xie, W. Wu and Z. Gu, J. Mater. Chem. B, 2017, 5, 1873 RSC;
(h) Z. Xie, T. Fan, J. An, W. Choi, Y. Duo, Y. Ge, B. Zhang, G. Nie, N. Xie, T. Zheng, Y. Chen, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2020, 49, 8065 RSC.
-
(a) B. M. Luby, C. D. Walsh and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 2558 CrossRef CAS;
(b) G. S. Attar, M. Kumar and V. Bhalla, Chem. Commun., 2024, 60, 11610 RSC;
(c) Y. Liu, S. Pujals, P. J. M. Stals, T. Paulohrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423 CrossRef CAS PubMed;
(d) X. Wang, J. Peng, C. Meng and F. Feng, Chem. Sci., 2024, 15, 12234 RSC;
(e) T. C. Pham, V. N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454 CrossRef CAS PubMed;
(f) C. Zhang, W. Chen, T. Zhang, X. Jiang and Y. Hu, J. Mater. Chem. B, 2020, 8, 4726 RSC.
-
(a) A. Gorman, J. Killoran, C. O’Shea, T. Kenna, W. M. Gallagher and D. F. O’Shea, J. Am. Chem. Soc., 2004, 126, 10619 CrossRef CAS;
(b) C. Wang, Y. Xiu, Y. Zhang, Y. Wang, J. Xu, W. Yu and D. Xing, Nanoscale, 2025, 17, 1812 RSC;
(c) W. X. Ren, J. Han, S. Uhm, Y. J. Jang, C. Kang, J. H. Kim and J. S. Kim, Chem. Commun., 2015, 51, 10403 RSC;
(d) G. M. N. Neubi, Y. O. Damoah, X. Gu, Y. Han, J. Zhou and Y. Ding, Biomater. Sci., 2018, 6, 958 RSC;
(e) S. F. Karkan, S. Sargazi, S. Shojaei, B. F. Far, S. Mirinejad, M. Cordani, A. Khosravi, A. Zarrabi and S. Ghavami, Nanoscale, 2024, 16, 12750 RSC.
-
(a) J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Chem. Sci., 2020, 11, 6701 RSC;
(b) L. A. Ortiz-Rodríguez and C. E. Crespo-Hernández, Chem. Sci., 2020, 11, 11113 RSC.
- M. Wainwright, Photodiagn. Photodyn. Ther., 2005, 2, 263 CrossRef CAS PubMed.
-
(a) G. Viola and F. Dall’Acqua, Curr. Drug Targets, 2006, 7, 1135 CrossRef CAS;
(b) F. Calik, A. Degirmenci, M. Eceoglu, A. Sanyal and R. Sanyal, Bioconjugate Chem., 2019, 30, 1087 CrossRef CAS PubMed.
-
(a) J. Montané, E. Armelin, L. Asín, A. Rodríguez-Galán and J. Puiggalí, J. Appl. Polym. Sci., 2002, 85, 1815 CrossRef;
(b) Y. Tokiwa and T. Suzuki, Nature, 1977, 270, 76 CrossRef CAS PubMed.
-
(a) P. S. Pramod, R. Shah and M. Jayakannan, Nanoscale, 2015, 7, 6636 RSC;
(b) S. Bera, R. Barman and S. Ghosh, Polym. Chem., 2022, 13, 5188 RSC.
-
(a) C. L. Wang, L. Kong, D.-Q. Zhang, L. Ye, S.-C. Nao, D. S. H. Chan, X. Li, Y. Peng, L. Yang, C. Y. Wong, V. K. W. Wong, W. Wang, H. Chao and C. H. Leung, J. Am. Chem. Soc., 2025, 147, 14824 CrossRef PubMed;
(b) A. Narayanan, S. Kaur, C. Peng, D. Debnath, K. Mishra, Q. Liu, A. Dhinojwala and A. Joy, Biomacromolecules, 2019, 20, 2577 CrossRef CAS PubMed;
(c) S. Biswas, P. Rajdev, A. Banerjee and A. Das, Nanoscale, 2025, 17, 5732 RSC.
- C. Chakraborty, A. Rajak and A. Das, Nanoscale, 2024, 16, 13019 RSC.
-
(a) S. Biswas, P. Rajdev, A. Banerjee and A. Das, Biomacromolecules, 2025, 26, 4661 CrossRef CAS PubMed;
(b) S. Biswas, R. Barman, M. Biswas, A. Banerjee and A. Das, Polym. Chem., 2024, 15, 2753 RSC.
-
(a) N. U. Deshpande and M. Jayakannan, Biomacromolecules, 2018, 19, 3572 CrossRef CAS PubMed;
(b) S. Bag, M. P. Gadpayle, D. Ghosh, S. Maiti and P. De, Biomacromolecules, 2024, 25, 4233 CrossRef CAS PubMed.
- S. Biswas and A. Das, Chem. – Eur. J., 2023, 29, e202203849 CrossRef CAS PubMed.
- T. Banerjee, K. Dan and S. Ghosh, Nanoscale, 2024, 16, 19756 RSC.
-
(a) T. X. Luan, L. Du, J. R. Wang, K. Li, Q. Zhang, P. Z. Li and Y. Zhao, ACS Nano, 2022, 16, 21565 CrossRef CAS;
(b) S. Sriwastav, A. R. Sarkar, A. Datta, N. R. Jana and S. Malik, ACS Appl. Nano Mater., 2024, 7, 21674 CrossRef CAS;
(c) L. Hao, Y. Wang, S. Zeng, Z. Yang, S. Long, W. Sun, J. Wang, X. Peng and H. Li, Chem. Commun., 2025, 61, 10981 RSC.
- R. Bej, K. Achazi, R. Haag and S. Ghosh, Biomacromolecules, 2020, 21, 3353 CrossRef CAS.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289 RSC.
-
(a) R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836 CrossRef CAS;
(b) H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61 CrossRef CAS;
(c) P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps, Chem. Soc. Rev., 2013, 42, 7055 RSC;
(d) N. R. B. Boase, E. R. Gillies, R. Goh, R. E. Kieltyka, J. B. Matson, F. Meng, A. Sanyal and O. Sedláček, Biomacromolecules, 2024, 25, 5417 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*

 of M1 and M2 at δ = 3.81–3.62 ppm and 3.56–3.51 ppm, respectively, got deshielded to δ = 4.06 and 4.2 ppm due to their higher proximity to the electron withdrawing ester groups in P1 (Fig. S5). To further elucidate the lower incorporation of M1 with respect to M2, we investigated the polymerization ability of M1 with only A1 (P3 polymer) in the absence of M2. The progress of the polymerization of P3 was monitored by time-dependent 1H NMR spectroscopy. The emergence of a new downfield shifted peak at δ = 4.2–4.0 ppm for the methylene proton (Ha) adjacent to the ester group in the crude P3 polymer and concomitant disappearance of the methylene proton
of M1 and M2 at δ = 3.81–3.62 ppm and 3.56–3.51 ppm, respectively, got deshielded to δ = 4.06 and 4.2 ppm due to their higher proximity to the electron withdrawing ester groups in P1 (Fig. S5). To further elucidate the lower incorporation of M1 with respect to M2, we investigated the polymerization ability of M1 with only A1 (P3 polymer) in the absence of M2. The progress of the polymerization of P3 was monitored by time-dependent 1H NMR spectroscopy. The emergence of a new downfield shifted peak at δ = 4.2–4.0 ppm for the methylene proton (Ha) adjacent to the ester group in the crude P3 polymer and concomitant disappearance of the methylene proton  adjacent to the hydroxy (–OH) group in free M1 at δ = 3.82–3.62 ppm were monitored at different time intervals (Fig. S6a). The integration ratio of the newly emerged peak with respect to the disappearing peak revealed near-quantitative incorporation of M1 after 4 h in P3 (Fig. S6b), which indicates efficient uptake of M1 by this methodology, when there is no other competing monomer. A stack plot of polymers P3 and P4 with P2 indeed shows the presence of signature peaks of both phenothiazine and biotin in copolymer P2 (Fig. S7), further suggesting the successful inclusion of the individual monomers in P2. The two polymers were further characterized by size exclusion chromatography (SEC), which yielded broad peaks for P1 and P2 corresponding to molecular weights (Mn) of 23
adjacent to the hydroxy (–OH) group in free M1 at δ = 3.82–3.62 ppm were monitored at different time intervals (Fig. S6a). The integration ratio of the newly emerged peak with respect to the disappearing peak revealed near-quantitative incorporation of M1 after 4 h in P3 (Fig. S6b), which indicates efficient uptake of M1 by this methodology, when there is no other competing monomer. A stack plot of polymers P3 and P4 with P2 indeed shows the presence of signature peaks of both phenothiazine and biotin in copolymer P2 (Fig. S7), further suggesting the successful inclusion of the individual monomers in P2. The two polymers were further characterized by size exclusion chromatography (SEC), which yielded broad peaks for P1 and P2 corresponding to molecular weights (Mn) of 23



