
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrasonic activation of polymer–drug conjugates for targeted and combinational pancreatic cancer therapy†
Dimitra
Toumpa
 a,
Athina
Angelopoulou
a,
Konstantinos
Avgoustakis
b and
George
Pasparakis
*a
a,
Athina
Angelopoulou
a,
Konstantinos
Avgoustakis
b and
George
Pasparakis
*a
aDepartment of Chemical Engineering, University of Patras, Patra 26504, Greece. E-mail: gpasp@chemeng.upatras.gr
bDepartment of, Pharmacy, University of Patras, Patra 26504, Greece
First published on 13th June 2025
Abstract
In this work, we present a series of polymer–drug conjugates (PDCs) incorporating gemcitabine (GEM) and camptothecin (CPT), linked to polymethacrylate backbones via ester and disulfide linkers. Using monomeric prodrug precursors, we employed reversible addition–fragmentation chain transfer (RAFT) polymerization to synthesize colloidally stable PDCs. Upon ultrasound irradiation, these PDCs exhibited accelerated drug release, which was further enhanced by the presence of a sonosensitizer due to reactive oxygen species (ROS) generation. Systematic in vitro testing across different treatment modalities revealed formulations capable of outperforming the IC50 values of the parent drugs by up to five orders of magnitude. Our findings highlight how the interplay between the PDC structure (e.g., drug combinations and linkers) and ultrasound-triggered activation in the presence of a sonosensitizer significantly enhances the therapeutic potency of these nanomedicines.
1. Introduction
In recent years, remotely activated therapeutic modalities (RATMs) have garnered attention for their ability to enhance the precision and efficacy of cancer drug delivery owing to minimal off-target toxicity and the ability to spatiotemporally modulate drug release at tumor sites.1–3 RATMs comprise systems that are primarily triggered by magnetic fields, radiofrequencies, light irradiation and ultrasound (US). The complexity of the formulations responding to these external cues can vary from simple administration of drugs in their parent form to more functionally complex micellar vehicles, solid lipid nanoparticles, and mesoporous nanomaterials.4Light activated systems constitute an interesting sub-class of RATMs as they can achieve facile activation by irradiation with red and near infrared wavelengths enabling deep tissue penetration. For example, photodynamic therapy (PDT) utilizes photosensitizers (PS) as prodrugs (usually porphyrin molecules) that can generate cytotoxic reactive oxygen species (ROS) selectively only upon light irradiation.5–7 PDT can be further enhanced with the co-administration of anticancer drug molecules, which act in a synergistic manner. A variation of this method is photochemical internalization (PCI) where the light dose delivered is of low, sublethal intensity but sufficient enough to induce extensive membrane/organelle photooxidation to increase cell membrane permeability and ultimately cell apoptosis by the increased drug uptake.8 PCI is far superior compared to PDT in that it can significantly reduce the IC50 of the co-administered drug and at the same time enhance the apoptotic to necrotic death ratio, which is beneficial for secondary immune response in vivo. Similar therapeutic modalities can be achieved by replacing the light source with an US probe to elicit sonodynamic-type effects. Sonodynamic therapy (SDT) can also be delivered with PSs (now known as sonosensitizers, SSs) and drug molecules for combinational therapies.9–11 SDT offers the advantage that US probes can potentially cover larger tissue areas; they exert higher versatility in terms of energy that can be delivered and can also be combined with clinically established US imaging (i.e., for image guided drug delivery purposes).
We and others have reported PCI therapeutics with various front-line oncology drugs including vinca alkaloids, taxanes, nucleosides and biologics either in their parent form or formulated as nanomedicines (i.e., drug loaded nanoparticles, polymer drug conjugates etc.).12–16 The exact prerequisites that enhance the IC50 of a drug molecule in PCI are relatively elusive as one cannot quantitatively predict the extent of cytotoxicity enhancement of a given drug molecular structure; however, it seems that the interplay of PS/drug confinement, drug lipophilicity, and irradiation timing and dose plays a critical role. These factors can be adjusted by employing nanosized formulations that can co-carry PSs and anticancer drugs in a confined manner.15
In this study we build on previous findings to address key issues that affect the degree of enhancement of IC50 of two potent anticancer drugs, namely gemcitabine (GEM) and camptothecin (CPT), in the form of polymer–drug conjugates (PDCs). Although confinement strategies have been reported with physically entrapped micelles or similar systems, the utilization of polymer–drug conjugates as a key RATM component has not been extensively explored despite the distinct formulation advantages that they convey:17,18 (1) covalent drug attachment that prevents burst release events while allowing for a controlled rate of drug liberation via suitable linker chemistries, (2) precise and repeatable drug loading that can be fine-tuned early at the synthesis stage, and (3) the possibility to confine multiple drug molecules and PSs under one polymer scaffold and (4) harness passive targeting capabilities via prolonged drug circulation19 followed by activation only at the site of activation.
Our hypothesis stems from previous findings obtained using PCI protocols where nanoformulation strategies lead to enhanced therapeutic effects. Therefore, it is reasonable to expect similar potency with SDT protocols. By exploring the interplay between different drug and linker combinations and US we perform mechanistic studies on the potency of PDCs against a model pancreatic cell line.
Starting from polymerizable monomer–drug precursors, we employ reversible addition fragmentation chain transfer (RAFT) polymerization from polyethylene glycol chain transfer agents to generate PDCs of different drug molecules attached with two types of linkers, a slow (ester) and a fast (disulfide) hydrolyzing one. The resulting PDCs can be co-activated by an US probe to elicit combinational therapeutic action by confined drug liberation and sensitization. This approach allows for the alteration of drug combinations, linker composition, irradiation dose, and drug loading to optimize the formulation towards IC50 minimization. Optimization of the delivery regime, i.e., by adjusting the timing of cells’ exposure to sonosensitizers, PDCs and US irradiation, led to the discovery of formulations that either match or even outperform the cytotoxicity of their native-drug counterparts even by several orders of magnitude (Scheme 1).
 | ||
| Scheme 1 Our proposed concept of prodrug monomer synthesis that leads to samples with different drug combinations and linker hydrolysis rates with augmented IC50 under ultrasound irradiation. | ||
2. Experimental part
2.1. Materials
4,4′-Azobis(4-cyanovaleric acid) (ACVA), aq. hydrochloride (HCl), magnesium sulfate (MgSO4), sodium bicarbonate (NaHCO3), the macro-chain transfer agent poly(ethylene glycol)-4-cyano-4-(phenylcarbonothioylthio)pentanoate (macro PEG-CTA, Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), pyridine, phthalocyanine (Pc), 1,3-diphenylisobenzofuran (DPBF), zinc acetate dihydrate, tributylamine, terephthalic acid (TA), 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB) and dansyl chloride were purchased from Merck. 3,3′-Dithiodipropionic acid, CPT, dimethyl formamide (DMF), dimethylaminopyridine (DMAP), 1-(3-dimethylaminopropyl)3-ethylcarbodiimide hydrochloride (EDCI·HCl), gemcitabine hydrochloride (GEM·HCl), 1-hydroxybenzotriazole (HOBt) formic acid, pyrene and succinic anhydride were purchased from Fluorochem. Dichloromethane (DCM), ethyl acetate (EtOAc), hexane (Hex), 2-hydroxypropyl methacrylate (HPMA), methanol (MeOH), tetrahydrofuran (THF), toluene (PhMe) and n-pentanol were supplied by ThermoFisher Scientific. Deuterated chloroform (CDCl3) and deuterated methanol (MeOH-d4) were purchased from Merck. Ultra-pure three-distilled water (3D-H2O) was obtained by means of an ELGA Medica R7/15 device. Flash column chromatography (FCC) was performed on Merck silica gel 60 (240–400 mesh, Darmstadt, Germany), and analytical thin layer chromatography (TLC) was performed on Macherey silica gel-F254 pre-coated aluminum foils (0.2 mm film).
000 g mol−1), pyridine, phthalocyanine (Pc), 1,3-diphenylisobenzofuran (DPBF), zinc acetate dihydrate, tributylamine, terephthalic acid (TA), 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB) and dansyl chloride were purchased from Merck. 3,3′-Dithiodipropionic acid, CPT, dimethyl formamide (DMF), dimethylaminopyridine (DMAP), 1-(3-dimethylaminopropyl)3-ethylcarbodiimide hydrochloride (EDCI·HCl), gemcitabine hydrochloride (GEM·HCl), 1-hydroxybenzotriazole (HOBt) formic acid, pyrene and succinic anhydride were purchased from Fluorochem. Dichloromethane (DCM), ethyl acetate (EtOAc), hexane (Hex), 2-hydroxypropyl methacrylate (HPMA), methanol (MeOH), tetrahydrofuran (THF), toluene (PhMe) and n-pentanol were supplied by ThermoFisher Scientific. Deuterated chloroform (CDCl3) and deuterated methanol (MeOH-d4) were purchased from Merck. Ultra-pure three-distilled water (3D-H2O) was obtained by means of an ELGA Medica R7/15 device. Flash column chromatography (FCC) was performed on Merck silica gel 60 (240–400 mesh, Darmstadt, Germany), and analytical thin layer chromatography (TLC) was performed on Macherey silica gel-F254 pre-coated aluminum foils (0.2 mm film).
For the cellular studies in this work, Dulbecco Modified Eagle Medium (DMEM, 1×, Gibco) supplemented with 4.5 g L−1D-glucose, L-glutamine, and pyruvate was used. Phosphate buffered saline, PBS, pH 7.4 (1×, Gibco) and trypsin 0.25%–EDTA in HBSS (biosera) were obtained. 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) and dimethyl sulfoxide (DMSO) for cell culture were purchased from Merck.
2.2. Analytical methods
000 g mol−1). This allowed the determination of the number-average molar mass (Mn), the weight-average molar mass (Mw) and the dispersity index (Đ = Mw/Mn). All samples were filtered through a nylon membrane with 0.22 μm pore size before injection. Data were collected and processed with Clarity software.
2.3. Synthesis procedures
:4 as the eluent, to give a pure product as a white solid (90%); Rf (Hex/EtOAc 6:4): 0.30; 1H NMR (CDCl3, 600 MHz) δ 1.23 (d, J = 6.54 Hz, 3H), 1.88 (s, 3H), 2.54–2.64 (m, 4H), 4.07–4.13 (m, 1H), 4.19 (dt, J = 3.54 & 4.56 Hz, 1H), 5.12–5.20 (m, 1H), 5.53 (dt, J = 1.5 & 5.46 Hz, 1H), 6.05 (d, J = 6.9 Hz, 1H), 9.83 (br s, 1H) ppm; 13C NMR (CDCl3, 151 MHz) δ 16.33, 18.13, 28.83, 28.97, 66.17, 68.55, 126.06, 135.82, 167.07, 171.47, 177.82 ppm.
:8 as the eluent, to give pure product as a yellow solid (80.9%); Rf (Hex/EtOAc 2:8): 0.23; 1H NMR (CDCl3, 600 MHz) δ 0.98 (t, J = 7.44 Hz, 3H), 1.13–1.26 (m, 3H), 1.87 (t, J = 13.86 Hz, 3H), 2.11–2.17 (m, 1H), 2.22–2.29 (m, 1H), 2.59–2.68 (m, 2H), 2.79–2.90 (m, 2H), 3.98–4.21 (m, 2H), 5.04–5.18 (m, 1H), 5.26 (d, J = 5.34 Hz, 2H), 5.38 (d, J = 17.1 Hz, 1H), 5.51 (t, J = 21.84 Hz, 1H), 5.66 (dd, J = 3 & 14.04 Hz, 1H), 6.05 (dd, J = 16.68 & 6.72 Hz, 1H), 7.30–7.33 (m, 1H), 7.66 (t, J = 7.5 Hz, 1H), 7.83 (t, J = 7.98Hz, 1H), 7.93 (d, J = 8.1 Hz, 1H), 8.24 (d, J = 8.52 Hz, 1H), 8.39 (s, 1H) ppm; 13C NMR (CDCl3, 151 MHz) δ 7.50, 16.30, 18.20, 28.83, 31.73, 49.92, 65.97, 66.99, 68.69, 76.26, 96.55, 120.18, 126.01, 128.08, 128.22, 128.53, 128.57, 129.33, 130.77, 131.40, 135.77, 145.93, 145.98, 148.51, 152.19, 157.31, 166.91, 167.31, 171.05, 171.22 ppm; ESI-MS (30 eV) m/z: [M + H]+ calcd for C31H29N2O9+ 574.20, found 575.32, [M + Na]+ calcd for C31H30N2NaO9+ 597.18, found 597.30, [M + K]+ calcd for C31H30KN2O9+ 613.16, found 613.62.
:1 as the eluent, to give pure product as a white solid (39.2%); Rf (DCM/MeOH 15:1): 0.11; 1H NMR (CDCl3, 600 MHz) δ 1.27 (d, J = 6.48 Hz, 3H), 1.90 (s, 3H), 2.68 (s, 3H), 2.83–2.92 (m, 2H), 3.89 (d, J = 10.8 Hz, 1H), 3.99–4.04 (m, 2H), 4.11–4.15 (m, 1H), 4.19–4.23 (m, 1H), 4.61 (s, 1H), 5.15–5.19 (m, 1H), 5.55 (d, J = 6.42 Hz, 1H), 6.07 (d, J = 6.6 Hz, 1H), 6.11–6.14 (m, 1H), 7.40 (s, 1H), 8.13 (s, 1H) ppm; 13C NMR (CDCl3, 151 MHz) δ 16.40, 18.18, 28.08, 31.98, 59.72, 66.13, 68.89, 87.71, 97.68, 122.51, 125.94, 126.23, 135.82, 136.17, 167.13, 172.27, 172.60 ppm; ESI-MS (30 eV) m/z: [M + H]+ calcd for C20H25F2N3O9+ 490.16, found 490.40, [M + Na]+ calcd for C20H25F2N3NaO9+ 512.14, found 512.30, [M + K]+ calcd for C20H25F2KN3O9+ 528.12, found 528.40.
:2 as the eluent, to give pure product as a colorless oil (49.2%); Rf (PhMe/EtOAc 8:2): 0.24; 1H NMR (CDCl3, 600 MHz) δ 1.29 (d, J = 6.54 Hz, 3H), 1.94 (s, 3H), 2.72–2.80 (m, 4H), 2.91–2.93 (m, 4H), 4.12–4.17 (m, 1H), 4.22–4.26 (m, 1H), 5.19–5.25 (m, 1H), 5.59 (dt, J = 1.5 & 8.34 Hz, 1H), 6.11 (d, J = 7.14 Hz, 1H) ppm; 13C NMR (CDCl3, 151 MHz) δ 16.62, 18.39, 32.88, 33.25, 33.94, 34.35, 66.32, 68.90, 126.29, 136.02, 167.18, 171.16, 177.27 ppm.
:8 as the eluent, to give pure product as a yellow oil (46%); Rf (Hex/EtOAc 2:8): 0.26; 1H NMR (CDCl3, 600 MHz) δ 0.99 (t, J = 7.44 Hz, 3H), 1.24 (q, J = 6.06 Hz, 3H), 1.91 (s, 3H), 2.13–2.19 (m, 1H), 2.26–2.32 (m, 1H), 2.69–2.72 (m, 2H), 2.87–2.96 (m, 6H), 4.06–4.20 (m, 2H), 5.12–5.20 (m, 1H), 5.28 (s, 2H), 5.40 (d, J = 14.04 Hz, 1H), 5.54–5.56 (m, 1H), 5.67 (d, J = 17.1 Hz, 1H), 6.08 (d, J = 6.6 Hz, 1H), 7.28 (s, 1H), 7.67 (td, J = 1.02 & 6 Hz, 1H), 7.83 (td, J = 1.32 & 5.52 Hz, 1H), 7.94 (d, J = 7.92 Hz, 1H), 8.23 (d, J = 8.25 Hz, 1H), 8.40 (s, 1H) ppm; 13C NMR (CDCl3, 151 MHz) δ 7.58, 16.44, 18.24, 31.78, 32.42, 33.10, 33.77, 34.18, 49.96, 66.12, 67.09, 68.63, 76.31, 96.29, 120.28, 126.07, 128.09, 128.20, 128.51, 129.51, 130.76, 131.31, 135.86, 145.67, 146.15, 148.69, 152.22, 157.31, 166.93, 167.31, 170.72, 170.92 ppm; ESI-MS (30 eV) m/z: [M + H]+ calcd for C33H35N2O9S2+ 667.18, found 667.21, [M + Na]+ calcd for C33H34N2NaO9S2+ 689.16, found 689.50, [M + K]+ calcd for C33H34KN2O9S2+ 705.13, found 705.43 [M + K]+.
:1 as the eluent, to give pure product as a white solid (66.6%); Rf (DCM/MeOH 15:1): 0.07; 1H NMR (CDCl3, 600 MHz) δ 1.29 (d, J = 6.42 Hz, 3H), 1.93 (s, 3H), 2.72–2.76 (m, 2H), 2.91–2.99 (m, 6H), 3.93 (d, J = 11.34 Hz, 1H), 4.03 (dd, J = 4.44 & 19.02 Hz, 2H), 4.12–4.16 (m, 1H), 4.21–4.25 (m, 1H), 4.49 (s, 1H), 5.19–5.22 (m, 1H), 5.59 (d, J = 8.34 Hz, 1H), 6.10 (d, J = 6.66 Hz, 1H), 6.18 (s, 1H), 7.46 (s, 1H), 8.49 (s, 1H) ppm; 13C NMR (CDCl3, 151 MHz) δ 16.50, 18.25, 32.13, 32.99, 33.87, 34.16, 59.11, 66.12, 68.82, 82.20, 85.49, 96.99, 120.66, 122.39, 125.96, 126.25, 135.85, 136.20, 168.88, 167.10, 171.23, 171.56 ppm; ESI-MS (30 eV) m/z: [M + H]+ calcd for C22H30F2N3O9S2+ 582.14, found 582.31, [M + Na]+ calcd for C22H29F2N3NaO9S2+ 604.12, found 604.29, [M + K]+ calcd for C22H29F2KN3O9S2+ 620.09, found 620.16 [M + K]+.
000 g mol−1 (25 mg, 0.0015 mmol, 1 eq.) and ACVA (0.1 mg, 0.000375 mmol, 0.25 eq.) were dissolved in anhydrous DMF (0.425 mL). The flask was sealed with a rubber septum and purged using N2 for 15 min. The flask was heated at 70 °C for 24 h under magnetic stirring. The reaction was stopped by exposing the solution to open air and the polymer/monomer mixture was precipitated using diethyl ether. The residual monomer was washed away repeating this procedure two more times and the polymer was dried under vacuum. All purified polymers were analyzed with SEC (Fig. S5, ESI†) and NMR (Fig. S1–S4, ESI†).
2.4. Formulation
:1 v/v) in a sample vial with a total volume of 2 mL and the organic phase was evaporated in a controlled manner using a rotor evaporator. The vial was immersed in a water bath at 40 °C and was rotated at a constant speed of 100 rpm under a pressure of 100 mbar, until constant weight was achieved. The nanoparticles were characterized using DLS and TEM. Data analysis was performed using Matlab software. The same protocol was followed to form the PDC combinations by mixing different polymers at 1:1 (i.e., GEM: CPT) drug molar stoichiometry.
2.5. US treatment
For the US treatment we used the SONIDEL SP300 sonoporator. To expose the samples to US, water was added to a Petri dish containing the sample vials or cell well-plates, serving as the medium for ultrasound wave transmission. The US exposure parameters used are as follows: 1 MHz frequency, 25% duty cycle, and a pulse repetition frequency of 100 Hz. The intensities and exposure times ranged from 1.00 to 4.00 W cm−2 for 30 sec to 5 min, respectively.2.6. Evaluation of US-mediated ROS production
:1 solvent system. The solution was then irradiated with US emitting at a frequency of 1 MHz, using a power density of 4 W cm−2 for 60 min and a 25% duty cycle at a pulse repetition frequency of 100 Hz. This procedure was repeated for the DPBF solution in the absence of photosensitizer and for a deoxygenated DPBF solution containing the photosensitizer. Aliquots were taken ever 5 min and with a UV-vis spectrometer the DPBF absorbance at 410 nm was recorded. Measurements were taken in triplicate and data were analyzed using Matlab software.
2.7. Drug release studies
:1:0,01, and the released drug was quantified by LC-MS/MS. Chromatographic analysis was performed on a Waters HPLC system (Alliance HT 2795) equipped with a temperature-controlled autosampler and a degasser. Chromatographic separation was achieved using a Waters BEH C18 column (2.1 mm × 50 mm, 1.7 μm, Waters Corp., MA, USA), with a flow rate of 0.3 mL min−1 for a mobile phase consisting of H2O (solvent A) and acetonitrile (solvent B). The column temperature was maintained at 40 °C throughout all the experiments, while the sample temperature was kept at 10 °C in the autosampler tray. The sample (50 μL) was injected for analysis, and the gradient started immediately after the injection. The elution program was as follows: solvent B was initially equilibrated at 10%, linearly increased from 10% to 50% within 2 min, linearly increased from 50% to 90% in 5 min, kept stable at 90% for 1 min, then decreased at 10% in 1 min, and kept stable at 10% for 4 min. The total data acquisition duration was 13 min.27 The HPLC system was coupled with a Micromass Quattro Micro tandem MS system equipped with a quadrupole analyzer and an electrospray ion source that operated in positive ion mode. The MS parameters were optimized as follows: source temperature, 100 °C; desolvation temperature, 400 °C; desolvation gas flow, 500 L h−1; and argon gas flow 50 L h−1 was used as the collision gas. The capillary voltage was set at 3.5 kV, the multiplier was set at 650 V, while the cone voltage values for cotinine and IS were 41 and 22 V, respectively. Cotinine and IS were both detected using the multiple reaction mode (MRM) scan, with product ions m/z 349.34 > 305.05 > 249.00, respectively. Measurements were taken in triplicate and data were processed using the MassLynx v.4.0 software.
2.8. Biological studies
:1 drug ratio in μM). The concentration range for free GEM and GEM NPs was 1–500 μM, for CPT and CPT NPs was 1.5–290 μM, and for the combinations a window range of 1.5–150 μM was selected. After predetermined time periods of 48 and 72 hours of incubation at 37 °C, the supernatant was completely removed, and the cells were washed with PBS pH 7.4. Then, 180 μL of DMEM and 20 μL of MTT solution (stock 12 mM in PBS pH 7.4) were added in each well, followed by further incubation for 3 h at 37 °C. Then, the medium was completely removed, 150 μL of DMSO were added in each well to dissolve the formazan crystals, and the absorbance was measured at 490 nm (absorbance at a second wavelength of 630 nm was measured also to subtract background noise). The experiments were performed in triplicates and the IC50 values were extracted from the dose–effect curves of viability (%) vs. concentration (on a log scale).
The same protocol was repeated in order to evaluate the cytotoxicity of all the samples under the effect of US. Three types of treatments were examined: (1) treatment 1: cells incubated first for 24 h in ZnPc rich DMEM (2.8 μM), then DMEM was replaced with fresh ZnPc free medium and the cells were treated with US 22 hours later. 2 hours after US exposure, NPs were added followed by cell viability monitoring at 48 and 72 hours, (2) treatment 2: cells were first cultured for 24 hours followed by simultaneous exposure to ZnPc and NPs for 22 hours; 2 hours later (without changing the medium) they were treated with US followed by cell viability monitoring as previously described and (3) treatment 3: cells were first cultured for 24 hours followed by exposure to ZnPc and NPs for one hour followed by US treatment (again without changing the medium). Each well-plate was treated with US, upon slight immersion in a water bath using a frequency of 1 MHz, a duty cycle of 25% (pulse frequency = 100 Hz) for 3 min and an ultrasound power density of 4 W cm−2. The group of the samples and their concentrations were the same as mentioned above and the experiments were performed in triplicates.
The optical density was assayed in a TECAN infinite F50 spectrophotometer with Gen5 software (Gen5™ Microplate Data Collection & Analysis software, BioTek® Instruments Inc.).
 | (1) |
 | (2) |
In order to test the synergistic effect of US, ZnPc, and the NPs, a three-parameter equation was used.
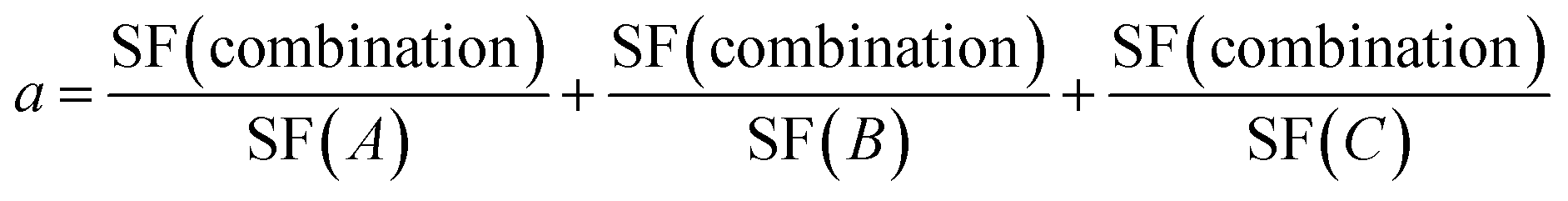 | (3) |
3. Results and discussion
3.1. Polymer prodrug conjugate synthesis
Starting from the commercially available HPMA through esterification we added succinic anhydride to afford a monomer with a slow-reducible linker (HPMA-Suc),31 due to the ester functional group (Fig. 1). The successful synthesis of the modified monomer was confirmed by 1H NMR, with characteristic peaks observed at 2.64–2.66 and 1.88 ppm, corresponding to the hydrogens of the succinic group and to the methyl group of HPMA, respectively (Fig. S1, ESI†). Additionally, 13C NMR showed a distinct peak at 171.47 ppm, attributed to the carbonyl of the ester bond between HPMA and the succinic group (Fig. S2, ESI†). HPMA was selected due to its lower toxicity compared to hydroxyl-group-containing monomers; the cytotoxicity ranking is as follows: 2-hydroxyethyl acrylate (HEEA) > 2-hydroxypropyl acrylate (HPAA) > HPMA > 2-hydroxyethyl methacrylate (HEMA). Additionally, when comparing the cytotoxicity of HPMA and HEMA with their corresponding alkyl esters, the HPMA alkyl ester exhibits slightly lower toxicity32 and hence HPMA constitutes the cornerstone starting monomer synthon throughout the study. Then, we used the carboxylic moiety of the modified monomer to attach GEM via the amide bond (HPMA-Suc-GEM) and CPT via an ester bond (HPMA-Suc-CPT) with acceptable yields of 40% and 81%, respectively (Fig. 1(ii) and (iii), respectively).33,34 Both compounds were confirmed by 1H NMR, which showed characteristic aromatic hydrogen signals for each drug (peaks at 7.80 and 7.48 ppm for GEM, and 8.39–7.30 ppm for CPT, Fig. S3 and S5, ESI†). 13C NMR revealed the characteristic peaks of HPMA-Suc and the drugs at 172.58 ppm for HPMA-Suc-GEM, corresponding to the carbonyl of the amide bond, and at 171.22 ppm for HPMA-Suc-CPT (Fig. S4 and S6, ESI†). These NMR findings were further supported by ESI-MS analysis confirming the successful synthesis. | ||
| Fig. 1 Synthetic procedure for the monomer prodrugs. Reagents and reaction conditions: (i) succinic anhydride, DMAP, DCM, 30 °C, o/n, under N2, 49.2%; (ii) GEM, EDCI·HCl, pyridine, DMF, rt, 72 h, under N2, 39.2%; (iii) CPT, EDCI·HCl, DMAP, DCM, rt, 24 h, under N2, 80.9%; (iv) DTDP, EDCI·HCl, DMAP, DCM, rt, o/n, under N2, 49.2%; (v) CPT, EDCI·HCl, DMAP, DCM, rt, 24 h, under N2, 46%; and (vi) GEM, EDCI·HCl, HOBt, pyridine, DMF, rt, 24 h, under N2, 47.5%. | ||
A key issue in our strategy is the choice of the linker, which at later phases will play a critical role during the in vitro testing. Disulfide bonds can be easily hydrolyzed in the cell by enzymes and mildly acidic pH (i.e., pH 5.4). Free thiol helps in the hydrolysis of the amide/or ester bond and the subsequent release of the drug.35 To this end, the prodrug monomers were synthesized in 2 steps, DTDP was first reacted with HPMA to produce an acid functional monomer with a reducible disulfide bond (HPMA-SS) (Fig. 1(iv)).36 The synthesis of HPMA-SS was confirmed both by 1H NMR and 13C NMR. 1H NMR confirmed the characteristic peaks at 2.93–2.72 and 1.94 ppm that correspond to DTDP and HPMA, respectively (Fig. S7, ESI†). 13C NMR showed a distinct peak at 171.16 ppm, attributed to the carbonyl of the ester bond between HPMA and DTDP (Fig. S8, ESI†). The acid side chain of this monomer was in turn coupled to the hydroxy group of CPT (HPMA-SS-CPT) or the free amine of GEM (HPMA-SS-GEM) to form polymerizable prodrug monomers (Fig. 1(v) and (vi), respectively).33,34 In particular for CPT, the presence of the hydrophobic moiety at the 20S position improves the stability of the lactone towards hydrolysis and decreases the activity under non reducing conditions.37 Both compounds were confirmed by 1H NMR, which showed characteristic aromatic hydrogen signals for each drug (peaks at 8.49 and 7.46 ppm for GEM, and 8.40–7.28 ppm for CPT, Fig. S9 and S11, ESI†). 13C NMR revealed the characteristic peaks of HPMA-Suc and the drugs at 171.23 ppm for HPMA-SS-GEM, corresponding to the carbonyl of the amide bond, and at 170.72 ppm for HPMA-SS-CPT (Fig. S10 and S12, ESI†). These NMR findings were further supported by ESI-MS analysis.
The polymerizable methacrylate prodrugs were subsequently used as co-monomers in RAFT polymerization. RAFT allows control of the molecular weight and produces well defined polymers with very narrow polydispersity indices (Đ < 1.2). Therefore, we used macroPEG-CTA (Mn = 10000 g mol−1) in RAFT polymerization with our methacrylate monomer prodrugs in anhydrous DMF at 70 °C for 24 h, in the presence of ACVA as a radical initiator (Fig. 2).
 | ||
| Fig. 2 RAFT polymerization that led to the PDCs. Reagents and general reaction conditions: methacrylate prodrug, ACVA DMF, 70 °C, 24 h, under N2, (i) 62%; (ii) 60.5%; (iii) 93.6%; and (iv) 60.5%. | ||
A degree of polymerization (DP) of 50 was chosen as sufficient to induce robust colloidal stability via self-assembly and to afford high drug loading with the total mass of the polymer chain for each PDC. The successful synthesis of the PDCs was confirmed by 1H NMR and SEC (Fig. S13–S17, ESI†). It was possible to assign peaks corresponding to the drug molecules (i.e., from 7.89 and 8.35 ppm for GEM and 8.80 to 7.39 ppm for CPT); in addition, the diminishing of the vinyl protons of the methacrylate moieties could be tracked, which confirmed the successful polymerization. The Mn for CPT-rich PDCs was calculated based on integration of 8.80–7.39 ppm peaks and for GEM-rich samples, it was calculated based on 5.26–5.19 ppm peaks (see ESI,† for Fig. S13–S16 for more details). In terms of macromolecular characteristics, Mn values were in the 11000–13400 g mol−1 range with low dispersities (Đ = 1.1–1.2), suggesting the formation of well-defined samples with good control of the polymerization (Fig. S17, ESI†). Some discrepancy between the Mn values obtained from NMR and SEC is expected as SEC analysis measures the hydrodynamic volume based on the hydrodynamic volume of the calibration standards used, which may be distorted in the mobile phase of the GPC due to the block-copolymer architecture. Hence, for greater accuracy, the DP for all the PDCs was calculated from 1H NMR data (Table 1).
| Ref | Polymer | M n,SEC a | Đ a | M n,NMR b | DPb | CAC (mg mL−1) |
|---|---|---|---|---|---|---|
| P1 | PEG-(HPMA-Suc-CPT)10 | 10800 |
1.1 | 15700 |
10 | 0.09 |
| P2 | PEG-(HPMA-SS-CPT)13 | 13400 |
1.2 | 18700 |
13 | 0.21 |
| P3 | PEG-(HPMA-Suc-GEM)25 | 12700 |
1.15 | 22200 |
25 | 0.12 |
| P4 | PEG-(HPMA-SS-GEM)19 | 12000 |
1.2 | 21000 |
19 | 0.22 |
3.2. Nanoparticle formation
The synthesized PDCs could form core–shell types of nanoparticles (NPs) in aqueous media resulting from the block copolymer structure, that is, the hydrophilic PEG and the more hydrophobic drug-rich moieties. NPs were formulated by nanoprecipitation by the drop addition of a CHCl3 solution of PDCs in water followed by the removal of the organic solvent by evaporation. Effective removal of the organic solvent is important for controlling the size of the emergent nanostructures as its presence reduces the stability of the nanoparticles due to Ostwald ripening,38 in which smaller structures dissociate in order to feed the formation of larger ones.39 In order to probe the effect of slow and fast drug release we tested different PDC combinations. The formation of colloidally stable self-assembled NPs was confirmed by DLS (Fig. S20–S22, ESI†) and TEM (Fig. S23, ESI†), which were relatively in good agreement (Table 2).| NP | D h (nm)a | D h (nm)b | D h (after 15 days) (nm)a | % Dh changea | Zeta potential (mV)a |
|---|---|---|---|---|---|
| P1 | 312.6 ± 4.8 | 236.8 ± 30 | 286 ± 1.5 | 8.4 | 0.03 ± 0.07 |
| P2 | 549 ± 2.5 | 360.7 ± 41 | 516.3 ± 8.6 | 6 | 0.022 ± 0.005 |
| P3 | 207 ± 2.6 | 294.8 ± 76 | 187.9 ± 2.9 | 9.2 | 0.07 ± 0.08 |
| P4 | 215 ± 2.3 | 182.13 ± 62 | 200.8 ± 0.9 | 6.6 | −0.08 ± 0.01 |
| P1 & P2 | 265 ± 4.8 | 213 ± 86 | 196.8 ± 1.7 | 25.7 | 0.002 ± 0.027 |
| P3 & P4 | 226.6 ± 2.6 | 212.7 ± 55 | 186.8 ± 3.5 | 17.6 | −0.03 ± 0.05 |
| P1 & P4 | 593.3 ± 2.3 | 348.4 ± 49 | 532.6 ± 2.4 | 10.2 | 0.05 ± 0.02 |
| P2 & P3 | 123.6 ± 2.5 | 159.8 ± 37 | 93.5 ± 2.5 | 24.4 | −0.05 ± 0.07 |
The PDCs generally formed spherical homogenous NP populations with some samples being affected by the presence of large particles, which shifted the overall size distribution towards larger particles (Fig. S22, ESI†). Large NPs could also be traced in the TEM images corroborating the observed size distribution in DLS results. The small difference in diameter values is attributed to the drying effect of the NPs onto the TEM grid. Overall, the particle sizes of the NPs are all relatively large, which can be explained in some formulations (i.e., P2 and P1 & P4) from the distribution of the DLS graphs (Fig. S20 and S22, ESI†), which tend to move Dh to higher numbers. Nevertheless, the sizes of the NPs may be suitable for direct intratumoral injection (i.e., via transarterial chemoembolization or catheterization), which could potentially achieve better confinement with a sonosensitizer in a clinical scenario. Furthermore, the stability of the NPs was investigated over time. After 15 days, the Dh values were measured using DLS and were found to be similar to the starting Dh, which is indicative of their good colloidal stability. Interestingly the least stable samples were those made by combining different PDCs (i.e., P1 & P2, P3 & P4, P1 & P4, P2 & P3); still the overall stability was adequate with less than 25% change of size at the tested timeframe.
Also, the zeta potential of all the samples is virtually zero, which is expected due to the absence of charged moieties in the molecular structure, implying that the overall stability is solely attributed to the steric effect of the PEG coronae.
3.3. Evaluation of the US effect
First, we conducted preliminary experiments with the PDCs that contain CPT to adjust the time and power of the ultrasound probe. The drug release profiles were found to be non-dependent on US irradiation dose within the window of 30 seconds up to 5 minutes, for a probe power of up to 4 W cm−2 (Fig. S24–S27, ESI†).The release of CPT was performed in neutral (pH 7.4) and acidic (pH 5) aqueous media at 37 °C and monitored by LC-MS/MS. P1 and P2 exhibited similar, limited release behavior at pH 7.4, with both showing less than 8% release (Fig. 3a and b). However, under acidic conditions, the release was moderately increased, particularly for P2, due to the faster hydrolysis of the disulfide.35 The presence of Pc was also found to have no effect in the release events for this sample. Overall, the US appears to have an effect under the various conditions evaluated, but the results are unclear because the percentages of the released drug are similar and not distinct enough.
 | ||
| Fig. 3 (a) CPT release of the NPs P1 and (b) CPT release of the NPs P2. | ||
Based on the initial release data, we hypothesized that perhaps the ROS yield from the Pc was relatively low to have an effect on the release profile and we sought to insert a metal center in the Pc ring to enhance spin–orbit coupling in favor of higher quantum yield in ROS production (Fig. 4).22,40 It should be noted that Zn was chosen owing to its proven effect in ROS quantum yield enhancement and its previous use in PDT.41
 | ||
| Fig. 4 Metalation reaction of Pc to produce ZnPc. | ||
1O2 is the main component of ROS with biological relevance as it is the main photooxidation product of phototherapies and can be monitored by the DPBF assay. It was confirmed that the metalated ZnPc could generate significantly more 1O2 than the non-metalated under the tested conditions (Fig. S29–S30, ESI†). ZnPc could also generate hydroxyl radicals (˙OH) by US irradiation as evidenced by the TA assay, which acts as a selective ˙OH radical trap producing the UV-vis measurable product ΗΤΑ (Fig. 5a).42–45
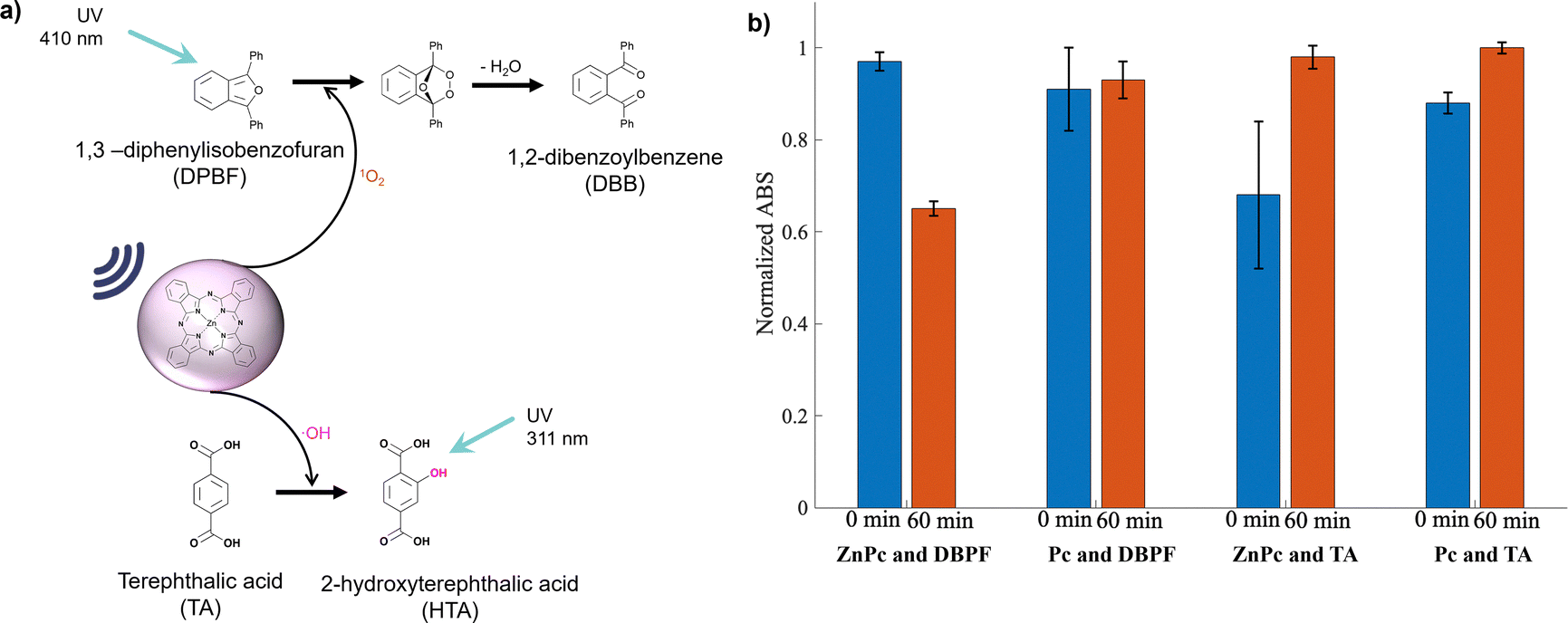 | ||
| Fig. 5 (a) Photo-oxidation of DPBF by 1O2 and the formation of HTA via the reaction of TA and ˙OH and (b) quenching of DPBF and generation of HTA by US irradiation in the presence of Pc or ZnPc. | ||
Fig. 5b compares the ability of Pc and ZnPc to generate ROS, including ˙OH, with ABS normalized to the maximum value. In the DBPF assay, the absorbance of DBPF decreases as 1O2 is produced (Fig. 5a). The change in absorbance (ΔABS = ABS at 0 min – ABS at 60 min) for Pc is only 2%, compared to 32% for ZnPc, demonstrating a significantly enhanced ROS generation capacity for ZnPc. In the TA assay, absorbance increases as ˙OH generates more HTA. To evaluate the ˙OH production, the ΔABS (ABS at 60 min – ABS at 0 min) was calculated, yielding 30% for ZnPc and 12% for Pc. Overall, ZnPc exhibits a superior ability to generate ROS, which translates to enhanced drug release in release experiments. While Pc slightly enhances drug release, ZnPc significantly increases the release rate (see release profiles in Fig. 7).
It is well known that US can induce the formation of ˙OH in water, in a similar way as ionizing radiation. Since ˙OH is a major free-radical intermediate and an important precursor for many products formed by the action of US in aqueous solutions, ˙OH can also have a critical role in the breakage of the disulfide bond (Fig. 6a).43
 | ||
| Fig. 6 (a) Hydrolysis of the disulfide bond, (b) Ellman's assay for P2, and (c) Ellman's assay for P4. | ||
The effect of US and the use of Pc or ZnPc was verified using Ellman's assay with the normalized ABS with the highest ABS values (Fig. 6b and c). In the presence of free thiols, an absorbance at 412 nm is observed due to the chromogenic 5-nitro-2-thiobenzoic acid (TNB). Both P2 and P4 contain a disulfide linker with either CPT or GEM, respectively. US promotes the breakage of this disulfide bond, as confirmed by the release studies (Fig. 3a and b). The presence of Pc further enhances this effect, with an even greater enhancement observed with ZnPc. To investigate the role of ˙OH, experiments were conducted in the presence of TA, a scavenger used to trap ˙OH. In these experiments, no absorbance was recorded at 412 nm, indicating that the disulfide bond did not break and highlighting the critical role of ˙OH in bond cleavage. Furthermore, Fig. 5b shows that Pc produces less 1O2 and ˙OH compared to ZnPc, which explains the larger TNB peak at 412 nm with ZnPc.
As ZnPc has enhanced production of ROS, release experiments were conducted for all PDCs in the presence of ZnPc at a fixed US dose. Generally, the presence of Zn seems to enhance the rate of release across the samples. At pH 7.4, CPT release from P1 exhibited similar rates in the presence and absence of Pc (Fig. 3a and 7a). The presence of ZnPc did not significantly improve the drug release even at acidic pH, which remained at 35%. However, at acidic pH, for P2 a clear difference was observed between experiments with Pc and ZnPc. With Pc, the maximum release rate of CPT reached 30%, whereas with ZnPc, the total release of CPT was 80% (Fig. 7b).
 | ||
| Fig. 7 (a) Release profile of the NPs P1, (b) release profile of the NPs P2, (c) release profile of the NPs P3 and (d) release profile of the NPs P4. | ||
At pH 7.4, the GEM release was consistently low (<10%) across all conditions. However, at pH 5, GEM release increased in the presence of ZnPc, reaching nearly 20%, whereas without ZnPc, GEM release remained similar to the levels observed at pH 7.4 (Fig. 7c). Notably, at pH 5, the highest GEM release was achieved when ultrasound was applied, following a similar trend as in CPT. To assess the effect of US on the disulfide linker, we repeated release experiments with P4 with and without the presence of ZnPc (Fig. S27a and b, ESI†). As expected, at pH 7.4 the release was very low; the highest rate was 10%, which was reached at 4 W cm−2 of US (Fig. 7c). At acidic pH the release was significantly higher with US (ca. 40%) and increased by 25% with ZnPc (Fig. 7d). From this set of experiments, we conclude that US combined with ZnPc has a significant impact on the release events at acidic pH across all samples. The release seems to be affected in the first 6–7 hours followed by steady state release later as monitored up to 24 hours.
A notable distinction lies in the extent of drug release: while CPT achieves near-complete release under the tested conditions; GEM exhibits a controlled release profile. This attenuated release behavior suggests enhanced modulation of drug delivery, potentially attributable to differences in chemical bond stability. Specifically, CPT is conjugated via an ester bond, which is comparatively labile under hydrolytic conditions, facilitating rapid cleavage and full release.46 In contrast, GEM is linked through a more stable amide bond, which resists hydrolysis and delays drug liberation.46 The enhanced stability of the amide bond likely prolongs retention of GEM within the nanocarrier, enabling sustained, controlled release. This mechanistic distinction underscores the importance of bond selection in tailoring drug delivery profiles for therapeutic precision.
3.4. In vitro cell studies
 | ||
| Fig. 8 Different treatment modalities. | ||
:GEM molar ratio of 1:1 were also evaluated. Cytotoxicity was evaluated at 48 (Fig. S32, ESI†) and 72 h (Fig. 9), which generally showed a similar trend across the samples. However, the overall cytotoxic effect was more pronounced at 72 h, as GEM and CPT reach their maximum therapeutic potential over time. This delay occurs because CPT must penetrate the nucleus, while GEM, as a prodrug, requires activation via phosphorylation pathways to exert its effects.47
 | ||
| Fig. 9 IC50 values of (a) free CPT, NPs P1 and P2, (b) free GEM, NPs P3 and P4, and (c) their combinations: GEM + CPT, PEG-Suc-CPT & PEG-SS-CPT (P1 & P2), PEG-Suc-GEM & PEG-SS-GEM (P3 & P4), PEG-Suc-CPT & PEG-SS-GEM (P1 & P4), and PEG-Suc-GEM & PEG-SS-CPT (P3 & P2). | ||
The IC50 values of free CPT and GEM were 154 ± 4 μM and 254 ± 4 μM, respectively (Fig. 9a and b), which were considerably higher than those reported in the literature (107.6 nM for CPT48 and 10 nM for GEM49). This suggests that the cell line used in our study exhibits resistance to both frontline chemotherapeutics GEM and CPT, while common drug resistant PANC-1 cells exhibit an IC50 value of 50 μM for GEM.50 An interesting observation is that while GEM PDCs (P3 and P4) exhibited higher IC50 values (631 ± 4 μM for P3 and 1013 ± 3 μM for P4), expectedly indicating reduced cytotoxicity, CPT NPs (P1 and P2) showed lower IC50 values compared to the parent drug (142 ± 4 μM for P1 and 17 ± 3 μM for P2). This suggests that the CPT free drug may have undergone hydrolysis of the lactone ring prematurely within the cells, leading to a reduced cytotoxic effect;51 the polymer scaffold could possibly have a protective effect of premature hydrolysis events in this scenario. In comparison with previous studies,52 where low IC50 values have been reported in absence of any additional treatment, our GEM and the CPT-ester NPs seem to exhibit reduced cytotoxic efficacy, as reflected by their higher IC50 values. This implies that they exert a safer (i.e., less toxic) therapeutic profile if not activated, that is if US is not applied. Interestingly, the presence of ZnPc under treatment 2 protocol resulted in the most favorable IC50 values (5 ± 0.2 μM for P1, 2 ± 0.7 μM for P2, 8.11 ± 2 μM for P3, and 0.41 ± 0.12 μM for P4), outperforming all other treatments (Fig. 9). In contrast, treatment 1 showed reduced cytotoxicity, likely because the effect was primarily driven by radicals produced from ZnPc, which were not expected to enhance cytotoxicity significantly as shown in control experiments (Fig. S34, ESI†). Meanwhile, treatment 3 exhibited significantly lower IC50 values compared to treatment 1, but the shorter time of cells exposur to PDCs before US application was insufficient for sufficient cellular uptake, unlike treatment 2. Treatment 2 exhibited the most potent IC50 values, likely due to the adequate time allowed for ZnPc and the PDCs to internalize into the cells and, with the co-assistance of US, showed a significantly more potent therapeutic effect.
In order to evaluate the role of the different linkers, four PDC combinations were tested: combinations with the same drug (CPT or GEM) but different linkers and combinations with mixed linkers and drugs. These combinations were selected to investigate how linker chemistry and drug pairings influence the release kinetics and therapeutic efficacy of the PDCs, providing insights into optimizing drug delivery strategies. The most potent combination was P2 & P3, with the IC50 value being 5 ± 1 μM (Fig. 9c). Remarkably, upon application of US in the presence of ZnPc (treatment 2 protocol) a 71-fold decrease in cell viability was observed, with an IC50 value of 0.07 ± 0.1 μM (Fig. 9c) far exceeding the potency of the parent drugs and their combinations, by 5 orders of magnitude. Presumably this result is the outcome of the quick response of CPT (disulfide linked) and the slower but systemic exposure of the cells with GEM (ester linked), leading to an overall enhanced therapeutic effect at 72 hours. This pattern is significantly augmented by the co-treatment with ZnPc.
In our previous work, where we evaluated the toxicity of GEM-nanomedicines activated by laser rather than US, an IC50 of 0.5 μM was achieved in the MiaPaCa-2 cell line.15 This is significantly higher than the lowest IC50 observed in this study (0.07 ± 0.1 μM for P2 & P3), highlighting the enhanced potency of our drug-delivery system. Moreover, it is important to note that the PANC-1 cell line used in this study is resistant, further underscoring the therapeutic potential of our approach.
To evaluate the synergistic effect of the combination of US, ZnPc, and the PDCs, we determined the synergy parameter for treatment 2 (eqn (3)),30 which showed the best IC50 values out of all treatments. At low concentrations, all PDCs (Fig. 10a) and their combinations (Fig. 10b) demonstrated a synergistic effect. However, at higher concentrations, the effect shifted toward a borderline antagonistic and additive effect. This trend at higher concentrations can be attributed to the antagonistic effects observed in the absence of US and ZnPc. If the drugs alone were synergistic, the synergy effect would probably have been further enhanced. Moreover, given that the applied US intensity and ZnPc alone were not toxic, and the PDCs had greater IC50 values than the parent drugs, implies that their combination appears to yield an enhanced therapeutic outcome—consistent with the IC50 values obtained.
 | ||
| Fig. 10 Synergy parameter for treatment 2: (a) NPs from a single polymer type and (b) NPs from a combination of two polymers. | ||
Although synergism studies are of limited phenomenological interpretation of the PDC combinations under the different treatment protocols, they can provide effective guiding in the optimization of potent therapeutic modalities that consist of multiple parameters (i.e., drug cocktails, external triggering mechanisms, etc.). For example, CIs as feedback readouts may provide guiding in focusing parameters (US intensity, ZnPc dose, or treatment duration, drug combinations, etc.) that will clearly impact the in vitro response and lead to optimization of the treatment modalities in future studies.
Finally, to investigate the efficacy of the treatments, we compared the Emax value, which is indicative of a treatment's efficacy and represents the value of cell viability at the maximum tested drug concentration (here, 1000 μM). The key treatment effects, indicative of treatment potency and efficacy (IC50; Emax), were identified from the chemotherapy (that is, without US) and the sonochemical (that is, ZnPc with US) cytotoxicity profiles for each drug and their combinations and are summarized in Table 3 (for chemotherapy and treatment 2) and Table S1 (ESI†) (for US, treatment 1, and treatment 3). Chemotherapy and sonochemical therapy effects were also compared with each other, and the corresponding changes in potency and efficacy are also listed. The most potent sonochemical therapy is treatment 2, with P1, P1 & P2, and P1 & P4 being the most efficacious, exhibiting a 6.7, 2.3, and 3.1-fold increase compared to sole chemotherapy, respectively. Notably, P4 demonstrates a remarkable ∼2470-fold improvement in its IC50 value (0.41 μM in treatment 2 compared to 1013 μM in chemotherapy). In contrast, while the combination of P2 & P3 achieves the lowest IC50 value (0.07 μM in treatment 2 vs. 5 μM in chemotherapy), its fold improvement is less pronounced due to the already low IC50 in chemotherapy.
| Drug | Treatment | IC50 (μM) | Fold improvement | E max (% control) [±SE] | Efficacy change (fold) |
|---|---|---|---|---|---|
| CPT | Chemotherapy | 154 | 42 (±6) | ||
| P1 | 142 | 40 (±7) | |||
| P2 | 17 | 43 (±9) | |||
| GEM | 254 | 40 (±2) | |||
| P3 | 631 | 50 (±4) | |||
| P4 | 1013 | 50 (±6) | |||
| GEM & CPT | 174 | 32 (±2) | |||
| P1 & P2 | 152 | 41 (±6) | |||
| P3 & P4 | 291 | 46 (±1) | |||
| P1 & P4 | 52 | 25 (±5) | |||
| P2 & P3 | 5 | 31 (±5) | |||
| CPT | Treatment 2 | 10 | +15.4 | 39 (±1) | +1.1 |
| P1 | 0.4 | +355 | 6 (±14) | +6.7 | |
| P2 | 0.25 | +68 | 28 (±3) | +1.5 | |
| GEM | 1.74 | +146 | 17 (±7) | +2.3 | |
| P3 | 8.11 | +77.8 | 33 (±3) | +1.5 | |
| P4 | 0.41 | +2471 | 36 (±9) | +1.4 | |
| GEM & CPT | 4 | +43.5 | 12 (±11) | +2.7 | |
| P1 & P2 | 15 | +10.1 | 18 (±4) | +2.3 | |
| P3 & P4 | 1 | +294 | 26 (±2) | +1.8 | |
| P1 & P4 | 9 | +5.8 | 8 (±2) | +3.1 | |
| P2 & P3 | 0.07 | +71.4 | 43 (±2) | −0.7 |
To address the feasibility of translating this platform to clinical settings and the safety profile of ZnPc and PDCs, several factors must be considered. First of all, the feasibility to access pancreatic tumors with ultrasound probes has been shown in clinical studies.54–58 Potentially, US treatment could be accompanied by the co-delivery of PDCs by direct injection to achieve effective accumulation of drug molecules at the site of treatment. This is critical if one considers the presence of a sonosensitizer to fully harness the benefit of US in drug release events as shown in this study. Confinement of the different drug molecules is another critical factor that must be fulfilled to maximize the therapeutic effect as seen by the combination indexes. Co-formulation strategies with spatially controlled US treatment may be a potent approach that exerts aggressive cytotoxicity while minimizing side effects in non-irradiated areas. This is clearly shown in Fig. S34 (ESI†) where ZnPc and PDCs exhibit minimal cytotoxicity in the absence of US activation, indicating a potentially safer therapeutic profile when not triggered. This can be further enhanced by direct delivery of PDCs at the tumor sites, which eliminates the exposure of healthy tissues to PDCs.
4. Conclusions
This study demonstrated the successful synthesis and characterization of PDCs designed for pancreatic tumor-targeted drug delivery. By employing tailored linker chemistries, we achieved controlled and stimuli-responsive drug release profiles, emphasizing the importance of disulfide bonds in enhancing the release kinetics under acidic conditions and in response to US application. The incorporation of ZnPc significantly improved ROS production, further enhancing the release efficacy of CPT and GEM from the formulations. To evaluate the role of different linkers, four PDC combinations were tested, combinations with of the same drug, CPT or GEM, but different linkers and combinations with mixed linkers and drugs, to investigate how linker chemistry and drug pairings influence release kinetics and therapeutic efficacy, thereby optimizing drug delivery strategies. These findings underline the interplay between linker chemistry, formulation parameters (e.g., drug combinations), and US treatment protocols; particularly, PDC pretreatment timing critically influences cytotoxicity against resistant PANC-1 cell lines, with IC50 values as low as 0.07 ± 0.1 μM for optimized combinations (e.g., P2 & P3 under the treatment 2 protocol). This represents a key result of our study for in vitro data evaluation in that NP pretreatment can support a critical cascade of biological events promoting anticancer activity. This is a key message not just for our study but for any study that reports in vitro data, in that incubation and US treatment timing play a significant role in biological events and it is always worth exploring possible blind spots that can be revealed by harnessing subtle experimental variations while testing the disease protocol. Finally, in porphyrin activated therapeutics (be they of photochemical or sonodynamic nature), the choice of the porphyrin molecule is absolutely key to fully exploit the potential of any RATM; in this study, we used the simplest phthalocyanine, which already showed enhanced ROS generation via metalation; however, the overall effect could be further enhanced by increasing its amphiphilic character as we have shown in our previous studies.15 All in all, it is clear that sonochemical RATMs utilizing PDC combinations constitute a potent concept that, if carefully used, can lead to highly cytotoxic outcomes with extremely high ΔIC50 (that is, fold-improvement) at the activated areas.Author contributions
Dimitra Toumpa: conceptualization, writing – original draft, validation, methodology, investigation, formal analysis, experimental design, and data collection and curation; Athina Angelopoulou: reviewing and editing, methodology and in vitro data analysis; Konstantinos Avgoustakis: reviewing and editing, resources, methodology; and George Pasparakis: writing – reviewing and editing, writing – original draft, validation, supervision, resources, project administration, methodology, investigation, funding acquisition, formal analysis, data curation, conceptualization.Conflicts of interest
There are no conflicts to declare.Data availability
The data of the manuscript are available through the University of Patras repository at the websites https://www.polylab.gr and https://www.upatras.gr. All data are also available at OSFHOME under the project “ultrasound nanomedicines” at the URL: https://osf.io/6rbhj/.Acknowledgements
This work was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I., project ULTRAMED No. 00234). We thank Alexandros Kyriakos Bikogiannakis and Prof. Georgios Kyriakou (Department of Chemical Engineering, University of Patras) for the XPS measurements and analysis. We thank Prof. Michael Kornaros (Department of Chemical Engineering, University of Patras) for the access to the UV-vis spectrometer, Prof. Theodoros Christopoulos (Department of Chemistry, University of Patras) for providing access to the spectrofluorometer and Prof. Theodore Tselios (Department of Chemistry, University of Patras) for the access to ESI-MS. We thank Dr Mohamed A. El Mubarak and Prof. Gregory B. Sivolapenko (Department of Pharmacy, University of Patras) for conducting the LC-MS/MS measurements. The Instrumental Analysis Laboratory (School of Natural Sciences, University of Patras) is gratefully acknowledged for carrying out the NMR analysis. The Laboratory of Electron Microscopy and Microanalysis (Department of Biology, University of Patras) for imaging is kindly acknowledged.References
- H. Hu, P. Busa, Y. Zhao and C. Zhao, Smart Mater. Med., 2024, 5, 386–408 CrossRef.
- D. V. Voronin, A. A. Abalymov, Y. I. Svenskaya and M. V. Lomova, Int. J. Mol. Sci., 2021, 22, 9149 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS.
- A. Zhang, K. Jung, A. Li, J. Liu and C. Boyer, Prog. Polym. Sci., 2019, 99, 101164 CrossRef CAS.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- G. Gunaydin, M. E. Gedik and S. Ayan, Front. Chem., 2021, 9, 691697 CrossRef CAS PubMed.
- J. H. Correia, J. A. Rodrigues, S. Pimenta, T. Dong and Z. Yang, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed.
- W. Jerjes, T. A. Theodossiou, H. Hirschberg, A. Høgset, A. Weyergang, P. K. Selbo, Z. Hamdoon, C. Hopper and K. Berg, J. Clin. Med., 2020, 9, 528 CrossRef CAS PubMed.
- D. Costley, C. Mc Ewan, C. Fowley, A. P. McHale, J. Atchison, N. Nomikou and J. F. Callan, Int. J. Hyperthermia, 2015, 31, 107–117 CrossRef CAS.
- I. Rosenthal, J. Z. Sostaric and P. Riesz, Ultrason. Sonochem., 2004, 11, 349–363 CrossRef CAS.
- Y. Zhang, X. Zhang, H. Yang, L. Yu, Y. Xu, A. Sharma, P. Yin, X. Li, J. S. Kim and Y. Sun, Chem. Soc. Rev., 2021, 50, 11227–11248 RSC.
- M. H.-C. Lin, L.-C. Chang, C.-Y. Chung, W.-C. Huang, M.-H. Lee, K.-T. Chen, P.-S. Lai and J.-T. Yang, Pharmaceutics, 2021, 13, 1877 CrossRef CAS PubMed.
- H. Maemoto, R. Suzaki, K. Watanabe, K. Itaka and T. Ohtsuki, Eur. J. Med. Chem. Rep., 2025, 13, 100242 CAS.
- H.-L. Lu, W.-J. Syu, N. Nishiyama, K. Kataoka and P.-S. Lai, J. Controlled Release, 2011, 155, 458–464 CrossRef CAS.
- C. Barnett, F. Joubert, A. Iliopoulou, R. S. Álvarez and G. Pasparakis, Mol. Pharmaceutics, 2023, 20, 1818–1841 CrossRef CAS.
- G. Pasparakis, T. Manouras, M. Vamvakaki and P. Argitis, Nat. Commun., 2014, 5, 3623 CrossRef.
- P. Thakor, V. Bhavana, R. Sharma, S. Srivastava, S. B. Singh and N. K. Mehra, Drug Discovery Today, 2020, 25, 1718–1726 CrossRef CAS.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- J. Wu, J. Pers. Med., 2021, 11, 771 CrossRef PubMed.
- S. Tsatsos, S. Ladas and G. Kyriakou, J. Phys. Chem. C, 2020, 124, 26268–26278 CrossRef CAS.
- C. D. Wagner, L. E. Davis, M. V. Zeller, J. A. Taylor, R. H. Raymond and L. H. Gale, Surf. Interface Anal., 1981, 3, 211–225 CrossRef CAS.
- I. M. Denekamp, F. L. P. Veenstra, P. Jungbacker and G. Rothenberg, Appl. Organomet. Chem., 2019, 33, e4872 CrossRef.
- A. Singh, S. Malhotra, D. Bimal, L. M. Bouchet, S. Wedepohl, M. Calderón and A. K. Prasad, ACS Omega, 2021, 6, 103–112 CrossRef CAS PubMed.
- S. O. McDonnell, M. J. Hall, L. T. Allen, A. Byrne, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2005, 127, 16360–16361 CrossRef CAS.
- B. Tang, L. Zhang and Y. Geng, Talanta, 2005, 65, 769–775 CrossRef CAS.
- A. Habeeb, Methods in enzymology, Elsevier, 1972, vol. 25, pp. 457–464 Search PubMed.
- Z. Wang, C. Shao, Z. Hu, J. Zheng, Q. Zhang, X. Kou, Z. Wang, T. Yu, Z. Cao and Y. Wang, J. Pharm. Biomed. Anal., 2020, 179, 112963 CrossRef CAS.
- W. R. Gray, Methods Enzymol., 1967, 11, 139–151 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- K. J. Takafumi Nakayama, FUJFILM Corporation, Tokyo (JP), 2020, US 2020/0115316 A1, 28.
- E. Yoshii, J. Biomed. Mater. Res., 1997, 37, 517–524 CrossRef CAS PubMed.
- L. Xiao, Y. Zhou, X. Zhang, Y. Ding and Q. Li, Chem. Pharm. Bull., 2019, 67, 1082–1087 CrossRef CAS.
- F. Joubert and G. Pasparakis, J. Mater. Chem. B, 2018, 6, 1095–1104 RSC.
- Z. Shi, J. Wu, Q. Song, R. Göstl and A. Herrmann, J. Am. Chem. Soc., 2020, 142, 14725–14732 CrossRef CAS.
- Z. T. W. H. L. X. B. H. W. Jiafeng, CN202110428510 20210421, 2021.
- M.-J. Li, C. Jiang, M.-Z. Li and T.-P. You, J. Mol. Struct. THEOCHEM, 2005, 723, 165–170 CrossRef CAS.
- V. Kumar and R. Prud’homme, Chem. Eng. Sci., 2009, 64, 1358–1361 CrossRef CAS.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Chem. Soc. Rev., 2017, 46, 6638–6663 RSC.
- J. Karolczak, D. Kowalska, A. Lukaszewicz, A. Maciejewski and R. P. Steer, J. Phys. Chem. A, 2004, 108, 4570–4575 CrossRef CAS.
- T. D. de Souza, F. I. Ziembowicz, D. F. Müller, S. C. Lauermann, C. L. Kloster, R. C. Santos, L. Q. Lopes, A. F. Ourique, G. Machado and M. A. Villetti, Eur. J. Pharm. Sci., 2016, 83, 88–98 CrossRef PubMed.
- J. C. Barreto, G. S. Smith, N. H. P. Strobel, P. A. McQuillin and T. A. Miller, Life Sci., 1994, 56, PL89–PL96 CrossRef PubMed.
- X. Fang, G. Mark and C. von Sonntag, Ultrason. Sonochem., 1996, 3, 57–63 CrossRef CAS.
- S. E. Page, W. A. Arnold and K. McNeill, J. Environ. Monit., 2010, 12, 1658–1665 RSC.
- D. H. Gonzalez, X. M. Kuang, J. A. Scott, G. O. Rocha and S. E. Paulson, Anal. Lett., 2018, 51, 2488–2497 CrossRef CAS.
- A. Dal Corso, L. Pignataro, L. Belvisi and C. Gennari, Chemistry, 2019, 25, 14740–14757 CrossRef CAS.
- E. Moysan, G. Bastiat and J.-P. Benoit, Mol. Pharmaceutics, 2013, 10, 430–444 CrossRef CAS.
- S. Park, J. Sung, E. Kim and N. Chung, Braz. J. Med. Biol. Res., 2014, 48, 111–119 CrossRef.
- D. P. Jeansonne, G. Y. Koh, F. Zhang, H. Kirk-Ballard, L. Wolff, D. Liu, K. Eilertsen and Z. Liu, Oncol. Rep., 2011, 25, 1473–1480 CAS.
- H. Kakwere, E. S. Ingham, S. K. Tumbale and K. W. Ferrara, Mater. Sci. Eng., C, 2020, 117, 111251 CrossRef CAS.
- M. Ghanbari-Movahed, T. Kaceli, A. Mondal, M. H. Farzaei and A. Bishayee, Biomedicines, 2021, 9, 480 CrossRef CAS PubMed.
- K. Kaliya, N. Bhardwaj, Ruchika and A. Saneja, ChemMedChem, 2025, 9, 8 CAS.
- T.-C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed.
- T. Inui, H. Amitani, K. Kubo, D. Kuchiike, Y. Uto, T. Nishikata and M. Mette, Anticancer Res., 2016, 36, 3767–3770 CAS.
- T. Inui, K. Makita, H. Miura, A. Matsuda, D. Kuchiike, K. Kubo, M. Mette, Y. Uto, T. Nishikata and H. Hori, Anticancer Res., 2014, 34, 4589–4593 Search PubMed.
- Y. Jiang, J. Fan, Y. Li, G. Wu, Y. Wang, J. Yang, M. Wang, Z. Cao, Q. Li and H. Wang, Int. J. Cardiol., 2021, 325, 132–139 CrossRef PubMed.
- A. Bunevicius, S. Pikis, F. Padilla, F. Prada and J. Sheehan, J. Neuro-Oncol., 2022, 156, 1–10 CrossRef PubMed.
- A. Sofuni and T. Itoi, J. Med. Ultrason., 2022 DOI:10.1007/s10396-022-01263-x.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb01250c |
| This journal is © The Royal Society of Chemistry 2025 |