
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From simple delivery to multimodal systems: the critical role of macromolecular platforms in bioorthogonal drug synthesis
Tieze
van den Elsen
 and
Kevin
Neumann
*
and
Kevin
Neumann
*
Institute for Molecules and Materials, Radboud University, The Netherlands. E-mail: kevin.neumann@ru.nl
First published on 1st July 2025
Abstract
Targeted drug delivery strategies have emerged as promising solutions to overcome traditional challenges such as poor bioavailability and side effects associated with conventional drug delivery methods. Among these strategies, the use of prodrugs offers a viable approach by leveraging enzymatic or controlled chemical transformations in vivo to enhance drug efficacy and specificity. The advent of bioorthogonal chemistry has revolutionized prodrug activation, providing a multitude of activation strategies beyond conventional methods. This review explores the integration of bioorthogonal chemistry, particularly transition metal catalysis, into prodrug activation strategies, with a focus on the use of macromolecular scaffolds as platforms to enable and localize these chemistries in biological environments. Specifically, this review focuses on the growing field of in situ bond-forming synthesis mediated by transition metal catalysts, often enabled by the use of macromolecular platforms. By forming carbon–carbon or carbon–heteroatom bonds intra- or intermolecularly, this approach offers advantages over traditional uncaging strategies through the absence of the pharmacoactive motif in the prodrug. We emphasize the central role of macromolecular platforms in integrating bioorthogonal chemistries into multimodal systems that enable targeting strategies and stimuli-responsive behavior, both crucial for achieving site-specific activation and minimizing off-target effects. We conclude that the future of this field lies with the development of retrosynthetic prodrug design, and the use and development of multimodular macromolecular platforms to host and enable new bioorthogonal transition metal catalysis.
 Tieze van den Elsen | Tieze van den Elsen holds an MSc in Chemistry with expertise in organic, inorganic, and supramolecular chemistry. He earned both his Bachelor's and Master's degrees cum laude from Radboud University Nijmegen. During his Master's studies, he completed research internships with Professor R. J. M. Nolte at Radboud University, the Netherlands, and Professor R. A. Shenvi at The Scripps Research Institute in San Diego. From 2025, he pursues his further academic career as a PhD candidate in the group of Nobel Laureate Professor B. L. Feringa exploring smart surfaces and adaptive materials. |
 Kevin Neumann | Kevin Neumann holds a position as Assistant Professor at Radboud University in Nijmegen, the Netherlands. He joined the group of Prof. Mark Bradley at the University of Edinburgh for his PhD studies, focusing on synthetic approaches to drug activation within complex biological environments using polymeric platforms. From 2018 to 2021, Kevin worked as a postdoctoral fellow in the group of Prof. Jeffrey Bode at ETH Zurich, Switzerland, where he focused on developing new methodologies for peptide synthesis. Since 2021, Kevin has been an Assistant Professor in Nijmegen, the Netherlands, where he leads an interdisciplinary research group at the interface of organic and polymer chemistry. His work focuses on the development of next-generation drug delivery systems and the precision synthesis of therapeutically active biomolecule including protein–polymer conjugates. |
1. Introduction
Traditional drug delivery methods are ridden with various challenges. Besides drug efficacy, the most important challenges to overcome are those relating to bioavailability and reduction of side effects. The latter is especially important in cases such as traditional chemotherapy where severe side effects are often observed due to lack of tissue specificity.1 To address these challenges, targeted drug delivery strategies have emerged as promising solutions. These strategies aim to deliver drugs directly to the site of action, thereby minimizing off-target effects.2 The concept of ‘prodrugs’ is one of the strategies developed to minimize the undesirable properties of drugs. Prodrugs are inactive, derivatives of active drug molecules, that can be transformed in vivo via enzymatic or chemical ways to form the active parent drug. The use of prodrugs can improve bioavailability and distribution as well as improve selectivity for the desired target and thereby reduce side effects. Widely known examples of the first use of prodrugs include methenamine, a urinary track site selective prodrug for antibacterial formaldehyde, and aspirin, the acetylated and less irritating precursor of sodium salicylic acid.3 The most common method of prodrug activation is that of endogenous stimulation via microenvironmental factors such as pH and localized concentrations of biomolecules, or transformation via specific proteins. However, complete specificity and selectivity are difficult to obtain because of the limited differences between healthy and malignant tissues. Small amounts of transformation of the prodrug at off-target sites might still lead to significant side effects.4 Additionally, full spatiotemporal control is difficult to obtain due to the dependence on endogenous activation.A huge advancement in the field of medicinal chemistry was the introduction of ‘bioorthogonal chemistry’ by Bertozzi in 2003.5 This Nobel prize winning chemistry refers to chemical reactions that occur selectively within living systems without interfering with native biological processes. Early and most famous examples of bioorthogonal chemistry include the Staudinger ligation, and ‘click chemistry’ such as the copper-catalyzed azide–alkyne cycloaddition (CuAAC), the strain-promoted azide–alkyne cycloaddition (SPAAC), and the inverse-electron demand Diels–Alder (iEDDA) reaction. Many more reactions have been reported since.6 Nowadays, one of the main uses of bioorthogonal reactions, is the activation of prodrugs (Fig. 1). The main advantage of using bioorthogonal chemistry for prodrug activation is that it adds an extra layer of controlled administration over regular endogenous activation, resulting in higher prodrug selectivity and specificity. Many bioorthogonal chemistry mediated prodrug activation strategies are focused on uncaging/deprotection reactions. These reactions have the purpose of cleaving a bond, revealing the active drug molecule via unmasking, cleavage of linkers, or release of payload.7 For this purpose, bioorthogonal reactions often rely on the concept of ‘click-to-release’. These are modified ligation reactions with the formation of an unstable intermediate product, leading to dissociation of the drug molecule. Examples of this entail modified Staudinger ligations, iEDDA reactions with tetrazines, and modified SPAAC reactions with trans-cyclooctenes.7
 | ||
| Fig. 1 Schematic representation of transition metal catalyst (TMC) mediated in situ prodrug activation strategies. With the help of functionalized macromolecular platforms, the TMC accumulates at the site of the ailment. Then the non-active prodrug is distributed in the body and selectively activated near the ailment. This happens through bond formation catalyzed by the TMC. The activated drug only effectuates in the vicinity of the ailment only and over time cures it. | ||
The use of transition metal catalysts (TMCs) has become indispensable in all fields of synthesis over the last few decades, owing to their metal properties and versatile reactivity patterns in various transformations.8 The translation of TMCs into biorelevant environments for safe and selective catalysis in cellulo and in vivo to modulate cellular functions and introduce new-to-nature transformations to living systems, is a field of research with high potential for novel selective and efficient therapies and in vivo imaging. However, the translation towards bioorthogonal transition metal catalysis is faced with several challenges. Metal complexes often require water- and oxygen-free environments to prevent deactivation. In biological systems, they are confronted with many chelating and nucleophilic biomolecules such as amines and thiols, which can efficiently alter and deactivate the catalytic nature of the metal complexes and promote their excretion from the body. Additionally, some transition metals used in catalysis express toxicity towards cells.9 Despite these challenges, various bioorthogonal TMCs have been reported for in cellulo and in vivo catalyzed transformations, such as ligations, uncaging/deprotection reactions, oxidations, reductions and bond formations.10
While early efforts in bioorthogonal chemistry predominantly relied on small molecules or antibody–drug conjugates for prodrug activation, a limited number of pioneering studies have demonstrated the potential of macromolecular scaffolds to facilitate bioorthogonal transformations. Although, strictly not drug activation nor synthesis, in 2012, R. Weissleder and co-workers reported the use of biomacromolecular scaffolds in form of dextrans decorated with tetrazine residues for cellular resolution imaging and 18F labelling of biological target.11 In this study, pre-targeting was achieved by using anti-CD45 monoclonal antibodies labelled with dienophiles. The authors observed significantly enhanced in vivo labelling when polymer–tetrazine conjugates were employed instead of small molecule analogues. During the early phase of translating bioorthogonal chemistries to more complex environments, this study was among the first to employ a macromolecular scaffold in conjunction with bioorthogonal reactions and certainly inspired others to develop more functional macromolecular platforms that support bioorthogonal chemistry.
Since then, macromolecular platforms including polymeric nanoparticles, biomacromolecular networks, and protein conjugates, have evolved beyond passive delivery vehicles to incorporate intrinsic reactivity within their frameworks. Among other features, these systems offer compartmentalization and spatial resolution, enabling selective drug synthesis via bioorthogonal chemistry. The chemistry and architectures of such macromolecular platforms is vast, and includes besides some frequently used polymers such as polyethylene glycol, polyoxazoline and polysaccharides12 more tailored structures including poly(ylides),13,14 polymeric n-oxides15 and poly allyl glycolyl ether16 of which many are of interest for enabling new TM catalytic systems.
A major challenge in conventional therapy is the issue of site-selectivity which also applies to bioorthogonal drug synthesis. Besides acting as a synthetic platform for catalyst immobilisation, macromolecular scaffolds can be used to overcome site-selectivity issues. For example, one strategy to overcome this challenge involves the integration of exogenous trigger-responsive functionalities into macromolecular platforms. Unlike traditional small molecule-based systems, macromolecular drug delivery platforms such as polymeric nanomedicines offer inherent multimodality, enabling the incorporation of additional functional elements without the need to synthetically modify the bioactive payload.
When applied to prodrug activation strategies, most reported bioorthogonal TMCs are used in uncaging/deprotection reactions mainly focused on dealkylations such as ruthenium and palladium promoted deallylations, and palladium, gold and platinum mediated depropargylations.17 Using this strategy, bioactive functional groups (e.g. amines, hydroxides, or acids) are unmasked resulting in a bioactive molecule.
A more recently investigated method for prodrug activation is in situ bond-forming synthesis, where the focus lies on the formation of C–C or C–X (X = heteroatom) bonds resulting in the active drug rather than breaking bonds for unmasking. These reactions include various cycloadditions, cross-couplings, and metathesis reactions promoted by a variety of transition metals (Cu, Pd, Ru, Au, Rh, Ir). The TMC promoted reactions provide a wide range of possible chemical reactions, leading to a large amount of prodrug activation possibilities.10 Theoretically, this strategy has an additional advantage over decaging strategies, since the pharmacoactive motif does not yet exist in the prodrug molecule, resulting in a lower bioactivity of the prodrug compared to the bioactive drug, thereby significantly enhancing the therapeutic window. In addition, premature activation is prevented because of the lack of pre-existing drug scaffolds, and thus less side effects.18
In this review, the advances in the field of in situ TMC mediated bond-forming synthesis are discussed, from its conceptualization and first proofs of concept in 2016, to the most recent discoveries and applications in the present day. Furthermore, the crucial role of macromolecular platforms in enabling these strategies will become clear from the given examples (Table 1). In the first part of this review, we focus on the different chemistries and strategies for in cellulo and in vivo synthesis categorized by transition metal, and their applications towards in vivo therapy. This is followed by a brief overview of examples utilizing exogenous triggers to induce catalytic activity obtaining enhanced spatiotemporal control. Next, an overview is given of different modes of application of the most advanced examples in the field, where in situ catalysis is combined with targeting strategies to effectuate target specific catalysis and prevent off-target effects. Finally, the direction of this emerging field and promising new directions according to the authors are highlighted.
| Ref. | Macromolecular platform | Catalyst | Size of platform [nm] | Multimodality | Synthetic target |
|---|---|---|---|---|---|
| 19 | TentaGel resin | Cu | 16 × 104 | Injected delivery | Combretastatin A4 |
| 20 | Cross-linked lipoic acid NP | Cu | 97 | Increased activity by NP shape | Resveratrol analogue |
| 21 | Single-chain metal–organic NP | Cu | 7 | Dual role as ligand and crosslinker | Bisamidine scaffold |
| 22 | Polymeric NP | Cu | 100 | Affinity for localisation at cell membrane | Fluorophore |
| 23 | Zr MOF | Cu | 120 | Mitochondrial targeting | Fluorophore |
| 24 | Zn/Cu MOF | Cu | 120 | DNAzyme co-delivery | Resveratrol analogue |
| 25 | ZIF-8 copper chaperone mimics | Cu | 62 | FA-targeting and dual cis-platin delivery | Resveratrol analogue |
| 26 | CuAl-LDH nanosheets | Cu | Lateral size of 140–250 nm | CuAAC and Fenton-like reaction | resveratrol analogue |
| 27 | DNA–Cu-NP | Cu | ROS production and co-delivery of DNAzyme | Resveratrol analogue | |
| 28 | Polystyrene microsphere | Pd | 500 | Milestone as first reported system | Rhodamine 110 |
| 29 | Pd(0)-microsphere | Pd | Triggered endocytosis | PP-121 | |
| 30 | Hollow Si-microspheres | Pd | 500 | Encapsulation of metal catalysts | Fluorophore |
| 31 | PLGA–PEG polymeric NP | Pd | 57 | EPR and stealth effect | Fluorophore |
| 32 | Biotin–streptavidin complex | Ru | N/A | Increased activity by tailoring biomolecular environment | Fluorophore |
| 33 | Human serum albumin complex | Ru | N/A | Targeting residues | Umbelliprenin |
| 34 | Glycosylated albumin complex | Au | N/A | Targeting residues | Phenanthridinium |
| 35 | Glycosylated albumin complex | Au | N/A | Targeting residues | Dehydro-pyrrolizidine alkaloids |
2. Bioorthogonal in situ drug synthesis
2.1 Copper catalysis
Copper is a very versatile catalyst in organic synthesis, (co)catalyzing various reactions such as the Ullmann coupling,36 Sonogashira coupling,37 Buchwald–Hartwig amination,38 and Huisgen 1,3-dipolar cycloaddition reactions.39 Of these examples the best known Cu catalyzed reaction is the Huisgen 1,3-dipolar cycloaddition inspired Nobel prize-winning copper-catalyzed alkyne–azide 1,3-dipolar cycloaddition (CuAAC) or “click” reaction, pioneered by Sharpless in 2001.40 This reaction selectively forms 1,4-disubstituted 1,2,3-triazole products with high catalytic efficiency, and moreover in bioorthogonal fashion since the substrates are not naturally found in biological systems. Therefore, this reaction has high potential for in vivo applications.Although copper is considered an essential metal for living organisms as enzymatic cofactor, copper can exert high cytotoxic properties relating to the generation of radical oxygen species (ROS) inducing oxidative stress, and interference in biological pathways.41,42 To overcome the cytotoxicity and improve kinetics and efficiency, innovative approaches are required and many studies have been conducted using macromolecular platforms to protect the biological surrounding from undesired toxicity.43 Hence, macromolecular platforms are not only essential for enhancing catalytic efficiencies but also for providing safe and viable therapeutic platforms.
 | ||
| Fig. 2 Overview of reported in situ copper catalyzed click-reactions (A) by the respective copper polymeric enzyme mimics (B). (1) E-Cu-NP19 and Cu-cLANP20 catalyzed formation of combretastatin A4 (1). (2) Cu-MONP21 catalyzed formation of antimicrobial agent 2. (3) Cu-DSNP22 catalyzed formation of resveratrol analogue 3. Partly reproduced from ref. 20–22 with permission from the American Chemical Society. | ||
To increase catalyst selectivity, stability, and efficiency, transition metal catalysts have been incorporated in macromolecular scaffolds much like metalloenzymes. An early example of this was presented by Zimmerman in 2016 who employed a single chain linear polymer (SNCP) that was ‘folded’ with covalently bound Cu(II) atoms. The polymer was accessed via ring-opening metathesis and formed a water-soluble, copper-cross-linked single-chain metal–organic nanoparticles (MONP's).21 The Cu-MONPs were tested for their ability to catalyze CuAAC in water using sodium ascorbate to reduce the Cu(II) to Cu(I) in situ, resulting in a substrate conversion of >99% within 24 h using only 2.5 ppm of the Cu-MONPs in a model reaction. Various other substrates were also tested and resulted in isolated yields ranging from 83% to 98% at 5–30 ppm Cu-MONPs after 24 h at 50 °C. It was indicated that the Cu-MONPs were significantly less efficient towards hydrophilic substrates indicating the importance of hydrophobic binding. To show biocompatibility, the Cu-MONPs were successfully applied in an intracellular fluorescence study using fluorogenic coumarin precursors in live human cancer cells resulting in intense fluorescence. Additionally, the Cu-MONP was applied towards the synthesis of an antimicrobial agent 2 in E. coli bacteria effecting growth inhibition.
Further structure–activity studies using fluorogenic click reaction and dye uptake experiments conducted on the Cu-MONPs revealed the mechanism of its high catalytic efficiency and effectivity to involve an enzyme-like substrate binding process. It is primarily governed by the hydrophobic character of the substrate and the polymer and works best with a medium sized (19 kDa) MONPs.44 Although further research was conducted by Zimmerman on Cu-MONPs with differentiation towards surface protein binding catalysis45 and extracellular drug screening,46 no further application towards in vivo drug synthesis has yet been conducted.
Another example of a copper crosslinked polymeric nanoparticle was presented by Zhang in 2018.20 The Zhang group showed that an important element of nanocatalysts is not only reactivity and biocompatibility but also morphology. Nanocopper-doped cross-linked lipoic acid nanoparticles (Cu-cLANPs) of different sizes and shapes were synthesized. In a fluorescence study it was found that the intracellular CuAAC catalytic activity of the larger (∼97 nm) rugby-shaped nanoparticles was higher (up to 1.54×) than that of the smaller (47 and 11 nm) spherical nanoparticles. The same trend was found for the cellular uptake rate (up to 1.5×). A cell viability study of in situ generated resveratrol analogue 3via CuACC promoted click reaction was conducted with the Cu-cLANPs, resulting in cancer cell apoptosis (HeLa cells). Again, the rugby-shaped nanoparticle performing the best at every substrate concentration.
In a continuation on this study, Zhang presented a new application of the cLANPs in 2021.47 The group constructed Cu-NP and prodrug loaded nanocapsules from cross-linked lipoic acid (cLANCs), which could release their contents upon arrival in the tumor tissue due to glutathione (GSH) overexpression induced degradation. Besides the in situ CuAAC promoted CA4 prodrug activation, the cLANCs could be degraded into dihydrolipoic acid (DHLA) which showed a synergistic anticancer effect with the CA4 analogue. It was shown that the system displayed very little activity in a blood environment (1.4% drug yield) but strong activity in tumor environments (84% drug yield), due to the GSH induced release. It also showed good tumor accumulation (up to 11.3× higher than liver), and excellent tumor inhibitory rate (TIR, 83%) in an in vivo mice study, being much higher than directly administered CA4 derivative (TIR, 49%). Using the delivery vesicle in a synergistic antitumor therapy as done in this study shows potential.
Another polymer based macromolecular catalyst was reported by Bai in 2019.22 The group created a dense-shell nanoparticle (DSNP) scaffold consisting of a positively charged surface and hydrophobic interior with a core containing a high concentration of ligand moieties. This ligand core was able to readily coordinate Cu and Pd atoms and hereby the DSNP could contain and protect the catalytic function of these metal catalysts. The system showed enzyme-like catalysis through a bind-to-catalyze mechanism. It was found via fluorescence formation that the DSNP scaffold provided an inherent cell membrane targetability due its amphiphilic structure which caused it to attach to and reside within the lipid bilayer. A follow-up study from 2023 showed that because of this membrane affinity, the DSNP catalyst poses an ideal membrane-embedded catalyst (MEC).48 The MEC was useful for the synthesis and intracellular delivery of molecules with low cell permeability such as lipophilic or anionic substrates. This was shown via an in vitro cell viability study comparing the MEC promoted CuAAC click reaction of a proresveratrol analogue, and the extracellular addition of the preformed resveratrol analogue. This resulted in a lower cell viability (35% vs. 61% at 20 μM substrate and analogue, HeLa cells), indicating improved cellular uptake. Additionally, they show the MEC promoted ligation and delivery of a large, anionic cell autophagic agent with a 2-fold efficiency increase compared to the direct extracellular addition of the same agent. This approach is a promising example of the benefits of in situ drug synthesis promoting higher intracellular availability. However, because of non-specific membrane targeting a direct in vivo approach is still hard to imagine.
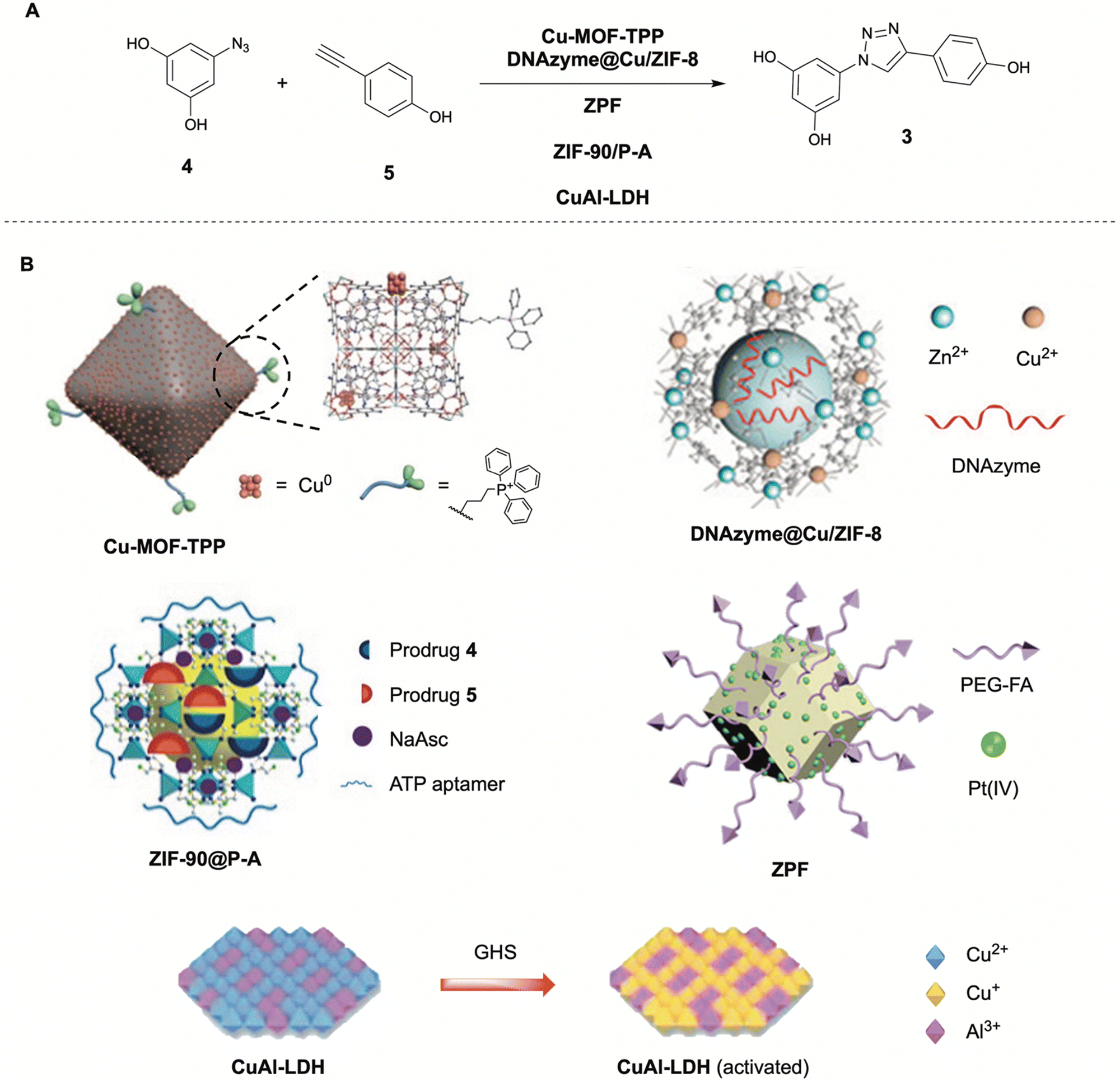 | ||
| Fig. 3 Reported in situ copper catalyzed click-reaction (A) and respective copper functionalized metal organic frameworks (MOFs) (B). (A) Formation of resveratrol analogue 3 from prodrug fragments 4 and 5via CuAAC by various MOFs (Cu-MOF-TPP,23DNAzyme@Cu/ZIF-8,24ZPF,25ZIF-90@P-A,50CuAl-LDH26). Partly reproduced from ref. 23–26, 50, 51 with permission from Wiley, ACS and Elsevier. | ||
Another application of the MOF-catalyzed CuAAC approach was shown by Qu in 2021.24 The group synthesized a bimetallic Zn/Cu MOF that, after uptake by cancer cells in lysosomes with acidic microenvironments, could release copper ions for the catalysis of the CuAAC click reaction. The acidic release properties were obtained from the zeolitic imidazolate framework-8 (ZIF-8) they employed as delivery vehicle. The MOF successfully promoted the CuAAC click reaction in cellulo to generate a resveratrol analogue (3) resulting in reduced cell viability (80%, 100 μM precursors, MCF-7 cells). This strategy was combined with the delivery of a human early growth response-1 (EGR-1) targeted DNAzyme with the corresponding Zn(II) cofactor, targeting metastasis-associated oncogenes to prevent proliferation and metastasis of the cancer cells. This chemotherapy–gene therapy nanoplatform (DNAzyme@Cu/ZIF-8) was reported to exert low cytotoxicity (up to 40 μg mL−1), good cellular uptake, and good synergistic anticancer effects in vitro (54% cell death, 75% cell proliferation inhibition). In an animal mice study the MOF showed tumor targetability due to enhanced permeability and retention (EPR) effect, and excellent tumor growth and metastasis inhibition upon intracellular synthesis of 3. The synergistic effect was clearly noticeable as the control experiments using only one of the two effective pathways showed significantly higher tumor growth and metastasis, indicating the effectiveness of the combined therapy method.
A third application of MOFs towards intracellular drug synthesis was presented by Qu in 2024.25 In this approach, the ZIF-8 MOF did not deliver exogenous copper atoms, but rather utilized endogenous copper to promote the CuAAC reaction. The amino-functionalized ZIF-8 MOF was covalently conjugated with a cis-platin prodrug (Pt(IV)) and modified with folic acid (FA) molecules for cancer cell targeting. The obtained ZIF-8–Pt(IV)–FA (ZPF) functioned as a Cu chaperone mimic, binding copper ions and transporting them to cancer cells through FA receptor-mediated endocytosis followed by acid induced release. The Pt(IV) prodrug was reduced in cellulo to Pt(II) by upregulated GSH, becoming functional inhibitors in the native copper ion transfer pathway, resulting in copper accumulation in the cancer cells. The ZPF was shown to be able to catalyze the CuAAC promoted formation of once again a resveratrol analogue using its endogenous copper accumulative properties, resulting in a significant apoptotic ratio of cancer cells in vitro (84%, B16F10 cells). An in vivo animal study on mice showed that the ZPF had good tumor targeting abilities, as well as significant mice survival rate promotion and cancer cell apoptosis and tissue damaging properties due to intracellular synthesis of 3.
This example was not the first using endogenous copper for the in situ CuAAC promoted formation of drugs. In 2019, the group already demonstrated the targeted synthesis of Cu-chelators using neurodegenerative Alzheimer's Disease (AD) related Cu accumulation in Aβ-plaques.51 After targeted delivery of the prodrug using H2O2-responsive mesoporous silica nanoparticles (MSN-IgG), the prodrug could be transformed into efficient Cu-chelators via CuAAC using endogenous copper. The Cu-chelators could extract Cu from Aβ-Cu aggregates and subsequently disassemble them, suppressing Aβ-mediated paralysis and restore locomotive function in an AD model C. elegans. Another study by Qu in 2023 utilizing endogenous copper, reported a MOF catalysis system which could be described as a ‘DIY’ package for self-destruction of cancer cells.50 The MOF ZIF-90 nanoparticle was functionalized with cancer cell ATP overexpressed targeted aptamers (ZIF-90@P-A). Upon endocytosis, a resveratrol derivative prodrug is released, together with the reducing agent sodium ascorbate to boost intracellular Cu(I) concentrations via reduction of Cu(II). The boosted Cu(I) could subsequently promote the CuAAC promoted formation of the resveratrol derivative 3. Its efficacy was shown in an animal mice study, significantly inhibiting tumor growth without exerting any side effects towards major organs. These examples show that using endogenous copper towards the in situ drug synthesis via CuAAC reaction is a promising strategy.
Comparable to the approach of using MOFs or inorganic nanoparticles is the approach presented by Chen in 2023.26 The group created a copper–aluminum layered double hydroxide (CuAl-LDH) nanosheet as a nanocatalyst for targeted in situ CuAAC drug synthesis for tumor therapy. Where many other Cu(I) promoted strategies rely on the in vivo reduction of Cu(II) with sodium ascorbate, Qu utilizes the upregulation of glutathione (GSH) in tumor microenvironments (TME) to reduce the CuAl-LDH Cu(II) to the catalytically active Cu(I).
Additionally, the Cu(I) atoms could not only promote the CuAAC formation of a resveratrol analogue 3in vitro and in vivo, it also showed cytotoxic effects through the generation of radical oxygen species (ROS) from increased hydrogen peroxide (H2O2) levels found in tumor microenvironments (TMEs). Although not explicitly stated, it is assumed that the nanosheets have passive tumor targeting abilities evident from the animal mice study showing significant tumor growth reduction as well as no observed tissue damage to major organs.
 | ||
| Fig. 4 (A) Formation of resveratrol analogue 3 from precursors 4 and 5via CuAAC and corresponding DNA-based catalysts DNA–Cu-NP,52 and DNAzyme–Cu-NP.27 (B) Schematic representation of the DNA-based catalysts. Secondary structures are not shown. Partly reproduced from ref. 52 with permission from Springer. | ||
The aptamer targeting approach was tested with two different aptamers which both showed excellent specific cancer cell type selectivity. The DNA–Cu-NP could readily promote the CuAAC click generation of resveratrol analogues in cellulo and in vivo, resulting in good to excellent tumor inhibitory effects. In the second paper the same DNA-templated Cu-NP approach was employed, however this time with an additional functionality. Instead of an aptamer used for targeting only, a hemin loaded peroxidase-mimicking DNAzyme aptamer was employed with retained cancer cell targeting ability (DNAzyme–Cu-NP). This was again linked to a polyT sequence functionalized with a Cu-NP. After targeted internalization in cancer cells, the DNAzyme aptamer part of the system could produce ROS using the high local H2O2 concentration in cancer cells. These active radical species could then oxidize the Cu-NP Cu(0) to Cu(I), enhancing its catalytic activity and enabling the in situ CuAAC generation of the anticancer resveratrol analogue. The resveratrol analogue and the cancer-killing ROS together, result in an effective synergistic anti-tumor combination.
2.2 Palladium catalysis
Palladium catalysts are extremely versatile in organic chemistry due to their capability of catalyzing various C–C bond forming cross-couplings leading to the awarding of the Nobel prize in chemistry in 2010. Examples include the Heck reaction, Buchwald–Hartwig reaction, Sonogashira coupling, and Suzuki–Miyaura coupling (Fig. 5).53 These reactions utilize substrates that are not commonly found in nature and therefore serve as an excellent recipe for bioorthogonal chemistry. Palladium exerts low toxicity unlike other transition metals such as copper and additionally can withstand oxygen and moisture. However, palladium catalysts have been shown to be poorly endocytosed, sometimes have low catalytic ability in water, and are readily inhibited by thiols such as glutathione (GSH), making their incorporation into nano-structured delivery tools essential.54 | ||
| Fig. 5 Overview of reported Pd catalyzed Suzuki–Miyaura cross-couplings (SMcc) (A) and respective Pd functionalized macromolecular platforms (B). (1) Formation of rhodamine 110 derivative (6) viaPd-PsMS catalyzed SMcc,28 (2) formation of cytotoxic agent PP-121 (7) viaPd-MPS29 and cRGD-Pd-NP55 catalyzed SMcc, (3) formation of Linifanib (8) viaPep-Pd catalyzed SMcc.56 Partly reproduced from ref. 28, 55 and 56 with permission from Wiley, ACS and Springer. | ||
The first example of in cellulo Pd mediated SMcc reaction was presented by Bradley in 2011.28 The group synthesized an amino-functionalized polystyrene microsphere (∼500 nm) and doped them with Pd(0) nanoparticles (Pd-PsMS), locked in place through extensive crosslinking of the amine moieties. The microspheres showed cellular uptake (HeLa cells) and good biological compatibility with low cytotoxicity (<9% cell death, 48 h, 0.38 × 1010 beads per mL). The microspheres capability for catalyzing the SMcc reaction was shown in a fluorescence study via the in cellulo synthesis of the fluorescent dye rhodamine 110 derivative (6) from triflate and boronate precursors. After 48 h clear fluorescence was observed around mitochondria due to TPP induced mitochondrial targeting of the formed dye.
Continuing this research, in 2016 the Bradley group presented the formation of an anticancer agent in the extracellular media using a similar SMcc strategy.29 This time the group did not employ endocytosed Pd(0) microspheres, but a large modular polymeric support functionalized with Pd(0) nanoparticles (Pd-MPS). This was then used in a cancer cell (PC-3 cells) viability study where cytotoxic agent PP-121 (7) was formed via SMcc from iodopyrazole and boronic ester precursors (2 and 10 μM respectively) in the extracellular media. This resulted in 50% cell death over 5 days.
In 2017, Bradley and co-workers expanded this research to Pd(0) functionalized microspheres for the intracellular catalysis of a tandem formation of two anticancer drugs via different chemistries.55 Here the original microspheres (∼208 nm) were again doped with Pd(0) nanoparticles and afterwards functionalized with cyclic-RGD cancer targeting moieties (cRGD-Pd-NP) selective for integrin (αvβ3)-positive cancer cells.
The cellular uptake was shown to be >95% within one hour in integrin-positive human glioblastoma cell line U87-MG as compared to <4% in integrin-negative human breast adenocarcinoma cell line MCF-7, indicating good targeting functionality. Afterwards the Pd(0) catalyst was employed in a cell viability study for the simultaneous deprotection of pro-5-fluorouracil (pro5FU) and the SMcc promoted formation of PP-121 (7), both known anticancer drugs. The synergistic effect of the dual therapy strategy was clearly shown with a cell viability as low as 22% over 5 days as compared to the separate strategies (66% and 44% respectively). This strategy has potential but shows relatively slow results as compared to for instance CuAAC strategies (5 days vs. 24 h).
A different application of the SMcc reaction in biologically relevant media was presented by Mascareñas in 2018.30 They employed hollow mesoporous silica nanoshell microspheres functionalized with an inner layer of Pd(0) nanoparticles (Pd-MSNs). The SMcc reaction yielded up to 86% conversion of starting materials over 24 h in water and PBS. However, where the depropargylation reaction of this system was successful, the SMcc reaction failed upon the presence of biorelevant thiols and did not proceed in cell lysates or in the presence of Vero cells.
An approach which will be more extensively visited later in this review, is the application of artificial metalloenzymes (ArMs). Palladium, however, has until now not yet been employed in this strategy. An approach closely resembling the ArM strategy, is the generation of metallopeptides as reported by Rappsilber in 2023.56 Rappsilber claims these might have a structural advantage over large metalloenzymes regarding delivery, no need for in situ assembly, and absence of immunogenicity. A methyl salicylate tagged hydrophilic peptide (LLEYLKR), chosen for its water soluble and hydrophobic pocket containing properties, was functionalized with a Pd(II) complex (Pep-Pd), and tested in a fluorescence study for its catalytic abilities towards depropargylation and SMcc reactions in PBS. The SMcc reaction was reported to be less efficient due to insufficient in situ reduction to Pd(0), the active catalyst for the SMcc reaction. The cytotoxicity of the metallopeptide was reported to be negligible up to 400 μM. A cell viability study (A549 lung cancer cells) showed that the in situ formation of two anticancer drugs (Paclitaxel (PTX) via depropargylation, Linifanib (LNF, 8) via SMcc) had a large synergistic effect evident from 28% cell viability over 5 days as compared to 67% and 61% of the separate methods. However, in this study the precursors for the SMcc reaction were applied in a 100× higher concentration than those of the depropargylation reaction because of the relatively low cytotoxic potency of LNF, rendering the bond-forming reaction less efficient. Although metallopeptides might not clearly fit in the description of ‘macromolecular platform’, they serve as a powerful example for the implementation of innovative strategies for catalyst shielding and delivery in a bioorthogonal manner.
Besides employing SMcc, other Pd catalyzed bond-forming reactions have been applied in biologically relevant media. An example of this is the application of a Heck coupling reaction as reported by Weissleder in 2017.31 They identified PdCl2(TFP)2 as an under physiological conditions active catalyst and trapped it into an FDA approved poly(lactic-co-glycolic acid)–polyethylene glycol (PLGA–PEG) scaffold to form nanoparticles (Fig. 6), ultimately providing a Pd-catalyst nanoparticle (PLGA–PEG–Pd). Although the Pd-nanoparticle was successfully applied towards in vivo prodrug decaging in antitumor therapy, the intramolecular Heck coupling reaction has only been applied in an in cellulo fluorescence study for the formation of a coumarin derivative.
 | ||
| Fig. 6 (A) Formation of coumarin derivative 9viaPLGA–PEG–Pd catalyzed Heck coupling. (B) Schematic structure of PLGA–PEG–Pd.31 Partly reproduced from ref. 31 with permission from Springer. | ||
If a suitable retrosynthetic Heck coupling anticancer compound could be identified, this strategy could be very interesting for further studies towards in situ bond-forming drug synthesis.
2.3 Ruthenium catalysis
Ruthenium (Ru) has gained significant attention for catalysis in organic chemistry due to its high versatility, catalyzing a wide variety of reactions, such as hydrogenation, hydrogen transfer, metathesis, oxidation and cross-coupling reactions.57 Ru catalyzed metathesis and cross-coupling reactions are both bond-forming reactions, of which Grubbs olefin metathesis is the most famous example. This reaction has been used for different kinds of olefin metathesis such as cross metathesis (CM), ring-closing metathesis (RCM), ring-opening metathesis polymerization (ROMP), enyne metathesis, and cross-coupling metathesis (CMC).58Artificial metalloenzymes (ArMs) are a combination of proteins with organometallic catalysts, anchored through covalent, dative, or supramolecular interactions.59 Ruthenium complexes are highly toxic,60 and therefore benefit from being shielded and prevented from leaking by ArMs if applied to in vivo catalysis. Embedding the catalysts in a biomacromolecular scaffold, protects the Ru catalyst from inhibition by proteins and metabolites and promotes enzyme-like catalysis.
One of the earliest examples of ruthenium promoted metathesis reactions using the ArM approach was presented by Ward in 2016 (Fig. 7).32 The group employed biotin–streptavidin technology by anchoring a biotin functionalized second generation Hoveyda–Grubbs Ru catalyst into a streptavidin protein, shielding it from biological inhibition of the catalyst (SAV-biot-Ru). They then used and evolved this ArM (SAV-biot-Ru) for the bioorthogonal olefin ring-closing metathesis (RCM) reaction. It was shown in a fluorescence study that this system was able to catalyze the RCM reaction forming umbelliferone (10) in the periplasm of Eschericia coli bacteria. The Ru-ArM was not affected by the glutathione disulfide (GSSG) present here but was inhibited by glutathione (GSH) present in the cytoplasm resulting in reduced catalytic efficiency. Upon saturation mutagenesis optimization screening, an ArM was found with improved in vitro and in vivo activity towards various substrates (TON: up to 90), outperforming two commercially available metathesis catalysts (second-generation Hoveyda–Grubbs and AquaMet).
 | ||
| Fig. 7 Overview of reported ruthenium catalyzed metathesis reactions (A) and respective ruthenium artificial metalloenzymes (Ru ArMs) (B). (1) Formation of umbelliferone (10) viaSAV-biot-Ru catalyzed ring closing metathesis (RCM),32 (2) formation of umbelliprenin (11) viaRu-GArM catalyzed RCM,33 (3) formation of pivalate combretastatin A4 (13) from precursor 12viaRu-GArM33 and cRGD-ArM-Ru-I61 catalyzed RCM. Partly reproduced from ref. 33 and 61 with permission from RSC and Springer. | ||
Similar to this approach is that presented by Tanaka in 2019, as a continuation on his earlier endeavors focusing on in vivo gold catalysis in 2017 (vide infra34).33 The group developed a human serum albumin (HSA)-based ArM that could protect the Ru RCM catalytic ability of a second generation Hoveyda–Grubbs Ruthenium catalyst in the presence of up to 20 mM GSH, rendering it biocompatible. The Ru catalyst was anchored in the HSA using a coumarin moiety attached with a PEG spacer. To extend this system towards in vivo anti-cancer therapy, a glycosylated albumin targeting strategy was employed. After functionalization of the Ru-ArM with α(2,3)-linked sialic acid terminated N-glycans (Ru-GArM), good targetability of cancer cells (SW620 > HeLa > A549) was shown in an RCM activated fluorescence study. To assess the possibility of prodrug therapy, the Ru-GArM was employed in a cell cytotoxicity study with the SMcc prodrug activation of the anti-cancer drug umbelliprenin (11). This resulted in cell growth inhibition of up to >95% (32 μM precursor, 25 μM Ru-GArM), showing significant difference compared to the non-glycosylated ArM (∼10% cell growth inhibition), indicating the importance of targeted therapy strategies.
In a continuation on these studies, Tanaka focused on a concept which he called ‘retrosynthetic prodrug design’, taking existing drugs and retrosynthetically disassembling them for prodrug design, focusing on optimization of the prodrug rather than the optimization of the biocatalyst.18 The key advantage of retrosynthetic prodrug design is the large increase in possibilities towards the optimization of reactivity, catalytic efficiency, bioavailability and difference in bioactivity between the prodrug and drug resulting in higher side effect suppression.
Using this concept, rather than improving the biocatalyst (Ru-GArM), it was attempted to optimize the prodrug, improving cascade reactivity (via enhanced aromatization), activity with the biocatalyst, hydrolytic stability in blood, and reducing intrinsic prodrug bioactivity. As previously seen, it was found that substrate hydrophobicity played an important role in the substrate/ArM interaction. Multiple CA4 derivatives were identified as suitable retrosynthetic prodrug candidates. A prodrug designed with a citronellate moiety was found to exert low tubulin affinity compared to the drug (−1.15 vs. −9.2 kcal mol−1), favorably increasing the bioactivity difference. However, out of the tested substrates a prodrug containing a pivalate moiety (12) instead, showed the highest catalytic conversion rate (kcat/KM = 457.9 vs. 84.1 & 108.7 M−1 s−1) as well as largest hydrolytic stability in blood (9.8% vs. 22.3 & 31.5% hydrolysis, 2 h), and was therefore selected as the most suitable candidate. Upon cytostatic analysis potential in a HeLa cell line, the pivalate prodrug/Ru-GArM combination proved highly effective, achieving a 50% cell growth reduction already at 4 μM (12) and 130 nM (Ru-GArM). Upon conducting an in vivo mice study via intravenous (i.v.) injection, the system was shown to exert a significant although not complete tumor growth inhibition property.
Since the prodrug/Ru-GArM system was previously injected intraperitoneally instead of intravenously, the difference between in vitro and in vivo application, was attributed to blood (cells and metabolites) induced quenching of the reaction.
To combat the blood induced quenching of the Ru-ArM promoted RCM, Tanaka improved the Ru-ArM in a recent study in 2023.61 After exchanging the Ru–Cl anchored catalyst for a Ru–I catalyst, the yield of a model RCM reaction in blood increased significantly (88% vs. 19% over 3 h) at a catalyst loading of the albumin of only 2.5%. It even continued to show RCM yields (21%) after 24 h incubation in blood. Besides RCM it was shown that the newly obtained Ru–I-ArM was also able to efficiently catalyze enyne RCM and olefin cross metathesis in blood environment. After functionalization of the Ru–I-ArM with a cyclic-Arg-Gly-Asp (cRGD) pentapeptide (cRGD-ArM-Ru-I), selective for cancer cell integrin overexpression, an in vivo tumor growth inhibition study with i.v. administration was performed on SW620-xenografted mice. At a loading of only 20–40 mg kg−1 of the cRGD-ArM-Ru-I, a significant dose dependent tumor growth inhibition effect was observed due to formation of pivalate combretastatin A4 (13), whereas none was observed whatsoever using the cRGD-ArM-Ru-Cl, indicating the high efficiency and blood stability of the new Ru-ArM. Additionally, the cRGD targeting efficacy was clearly noticeable as the system exerted a higher tumor growth suppression than the direct administration of the post RCM CA4 drug.
2.4 Gold catalysis
Traditionally, gold (Au) was considered inert and inactive in general chemistry. However, in modern-day chemistry, Au nanoparticles and complexes are highly effective catalysts due to their electronic properties and ability to activate a wide range of substrates. Gold catalysis is used for a diverse array of transformations including hydroamination, hydroarylation, hydroalkoxylation, and C–H activation. The most common pattern in Au-catalysis is the activation of alkynes allenes and olefins for nucleophilic attack, efficiently creating carbon–heteroatom and carbon–carbon bonds. Au-catalysts tolerate both oxygen, water, and acidic environment and are compared to other transition metal catalysts for similar reactions significantly more active.62Au-catalysis in biological systems suffers from side reactivity with biomolecules and intracellular reducing agents, such as GSH, which inhibit the catalytic activity. To overcome this, various strategies have been employed to protect and enhance the catalytic activity in intracellular environments. Additionally, since Au activates substrates for nucleophilic attack, intramolecular cyclization reactions are preferred over intermolecular reactions for the purpose of intracellular drug synthesis because of the presence of many nucleophiles in biological environments.63
The first application of gold mediated intracellular catalysis using an ArM approach, was reported by Tanaka in 2017, a preliminary study to Tanaka's Ru-ArM studies previously described (vide supra).34 The group developed a glycosylated albumin Au(III) ArM, which enabled in vivo localized propargyl ester amidation, resulting in organ specific fluorescence in mice studies. The Au-GArM consisted of a coumarin functionalized cyclometallated Au(III) catalyst anchored inside the organ targeting glycosylated albumin. Upon localized Au-activation of the fluorescent probe, surface amino groups could ligate to it resulting in fluorescence accumulation.
In a continued study in 2021, Tanaka applied the albumin Au-ArM approach towards the in situ synthesis of anticancer phenanthridinium derivatives via Au(I) promoted hydroamination (Fig. 8).64 The group identified a new carbene functionalized Au(I)–Cl catalyst that performed the hydroamination reaction on various substrates with high TONs (>200) and good yields (50–99%, 3 h). The unmodified Au-catalyst was applied in a cell viability study (A549 cells) to form a phenanthridinium derivative (17) via Au-catalyzed hydroamination. This resulted in a decrease in cell growth of >50% already at a prodrug and catalyst concentration of 8 and 2.5 μM respectively (4 d). To be able to employ this system in vivo and shield it from biological GSH, the catalyst was anchored in an albumin scaffold, forming a new Au-ArM. This Au-ArM resulted in sustained catalytic activity even upon addition of 1200 μM GSH (TON > 100). The new Au-ArM was applied in a new cell viability study, resulting in even better results compared to the unmodified Au-catalyst (50% cell growth inhibition at 0.63 μM catalyst, 4 d).
 | ||
| Fig. 8 Overview of reported gold catalyzed reactions (A) and respective albumin based gold artificial metalloenzymes (Au-ArMs) (B). (1) Formation of phenanthridinium derivative (17) viaAu-ArM catalyzed hydroamination64. (2) Formation of dehydropyrrolizidine alkaloid 18viaAu-GArM catalyzed hydroamination.35 Partly reproduced from ref. 35 with permission from Wiley. | ||
In 2022, Tanaka and Yokoshima demonstrated that the same albumin-Au-ArM with SW620 cancer cell-targeted glycosylation (Au-GArM) could also be employed for the synthesis of dehydro-pyrrolizidine alkaloids (DHPs, 18) via a gold catalyzed hydroamination cyclization of the precursor molecule.35 In a targeted cancer cell viability study (HeLa, PC3, A549, SW620 cells), a cell growth inhibition of >50% was achieved with prodrug and Au-GArM concentrations of 64 and 2.5 μM respectively. These albumin-based targeted Au-GArM approaches shows great potential for in vivo anti-tumor therapy strategies.
2.5 Iridium catalysis
Iridium (Ir) is a versatile transition metal for catalysis in organic chemistry. Due to its wide range of oxidation states (−3 to +9), Ir exerts various unique properties for a broad area of applications. It is known to catalyze reactions such as cycloadditions, allylic substitutions, C–H functionalization, hydrogenation, hydrogen transfers and photo-redox catalysis.65 Iridium's low intrinsic cytotoxicity and its high tolerance for water, oxygen and bio-additives indicate biocompatibility.66 However, despite its versatility, it is still highly underrepresented in bioorthogonal chemistry when compared to metals such as palladium and copper.The difficulty of the translation of iridium chemistry into biological systems is the general insolubility of iridium compounds as well as catalytic inhibition due to metabolites and enzymes that bind iridium. A few examples of in vivo Ir-catalyzed reactions have been reported although limited to hydrogen transfer, uncaging and photo-redox reactions.66–68
Iridium catalyzed bond-forming reactions have scarcely been used towards potential intracellular catalysis. One known example was shown by Hartwig in 2021 and elaborated upon in 2022 (Fig. 9).69,70 The group reported an iridium porphyrin cofactor (Ir-Por) that complexes in cellulo (E. coli) with a cytochrome P119 to form an iridium ArM (Ir-Por-ArM) that catalyzes stereoselective carbene insertions into benzylic C–H bonds. Although these studies are focused on synthetic biology, they display the potential for Ir-catalyzed reactions in vivo.
 | ||
| Fig. 9 (A) Formation of 19 and 20via intracellular Ir-Por-ArM catalyzed carbene insertion reactions. (B) Intracellular formation of Ir-Por-ArM from Ir-Por cofactor and hemoprotein cytochrome P119.69,70 Partly reproduced69 with permission from ACS. | ||
Other examples of cyclopropanation through cofactor insertion in native and engineered proteins have been reported for iridium71 and iron (heme cofactor,72–79). However, since these studies are focused on synthetic biology and not in vivo catalyzed drug formation, they are considered outside of the scope of this review and will not be further discussed.
2.6 Rhodium catalysis
Rhodium (Rh) catalysis has been around in organic chemistry a little over 25 years, significantly shorter than other transition metal catalysis such as Pd-catalysis.80 With respect to C–C bond forming reactions, however, rhodium has displayed interesting catalysis, coupling common reagents in ways not demonstrated before by other transition metal catalysts, enriching the possibilities within organic chemistry. Examples include cycloadditions, hydroacylation reactions, and allylic functionalizations.80 Rhodium catalysts also show great potential for environmentally benign reactions since water can often be used as cosolvent or sole solvent. However, in vivo applications are limited due to rhodium's high toxicity and carcinogenicity due to its DNA binding preferences and are therefore still highly underrepresented in this field.81The first and, to the best of the authors knowledge, only report of in vivo Rh-catalysis was presented by Ward in 201882. The group employed biotin–streptavadin technology to develop a dirhodium ArM (SAV-biot-Rh) capable of catalyzing carbene transfer reactions inside living bacteria. With this SAV-biot-Rh it was possible to promote intermolecular cyclopropanation (diazo + olefin precursors) and C–H carbene insertion reactions. Although carbene transfer reactions catalyzed by dirhodium complexes prove to be biocompatible, the application towards in vivo drug synthesis might be difficult. Diazo compounds have been shown to be useful tools in chemical biology,83 but many reactive components found in biological environments serve as good substrates for C–H insertion (e.g. amino acids84). An interesting approach to overcome this in future studies, might be the design of an intramolecular prodrug scaffold.
3. Multimodality: the need for macromolecular platforms
Site and time specific (spatiotemporal) control over in situ synthesis can be obtained through external triggers. By using drug delivery tools including polymeric or biomacromolecular nanocarrier both active and passive targeted drug delivery have been realized. In comparison to non-specific therapeutic treatments, prodrug activation strategies give promising results and thereby greatly reduce off-target effects. However, because these strategies are based on endogenous factors which lack perfect selectivity, non-specific delivery or activation might still occur leading to unwanted toxicity. One approach to overcome this challenge is the addition of an exogenous trigger responsive functionalization. Compared to traditional small molecule-based drug delivery platforms, polymeric nanomedicines offer the advantage of multimodality, allowing the incorporation of additional functions – often without synthetically altering the bioactive payload. Exogenous triggers such as light, magnetic fields, heat, or ultrasound, can be applied with high precision and – importantly – spatiotemporal control to selectively activate drug formation at the desired location, further preventing off-target effects (Fig. 10).85,86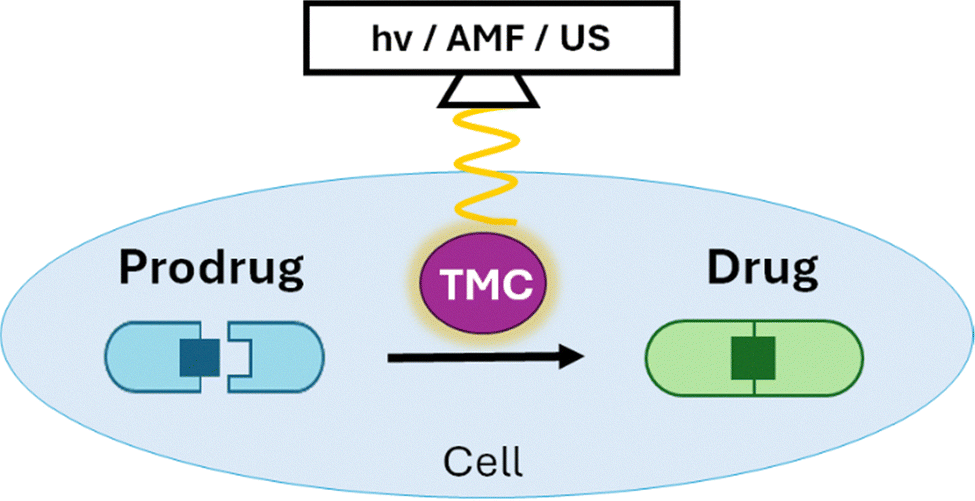 | ||
| Fig. 10 Bond-forming in situ prodrug activation through external trigger-controlled catalysis. | ||
In this section representative examples are given of exogenous control applied to in situ bond-forming drug synthesis, mainly showing its potential and efficacy. Throughout the given examples it will become clear that the implementation of nanostructures is vital to the incorporation of secondary functionalities, from incorporation of responsive materials to shielding of catalytic functionality and accommodation of nanoreactors. For further reading on stimuli-responsive systems, we refer to specialized reviews.87–90
3.1 Light triggered in situ catalysis
The application of light is an effective and easy way of administering energy to a system, activating and accelerating chemical reactions.91 Light of different wavelengths carry different energies (small wavelengths have high energy) and have different effects on biological tissues. UV-light (<400 nm) has high energy, can cause severe photo-damage to tissue, and has very low tissue penetration depth making it invaluable for many in vivo applications. Visible light (400–700 nm) and near-infrared light (NIR-I: 700–900 nm, NIR-II: 900–2000 nm) cause significantly less photodamage and have higher penetration depts up to 10 cm.92,93 Even though light is already extensively used in multimodular nanomedicine strategies for drug delivery, (uncaging) prodrug activation, and targeting,94 its application towards in situ drug synthesis is novel and underexplored.A light-promoted palladium catalyzed SMcc approach for in situ synthesis was reported by Qu as early as 2018 (Fig. 11).95 The group presented a microporous silica–Pd(0) nanoparticle functionalized with azobenzene and shielded with β-cyclodextrin (CD) through host–guest interactions (CASP). Here, usage of the microporous silica nanoparticle is vital for the accommodation of the light responsive azobenzene. The CD shielding of the Pd-nanoparticles caused catalyst inhibition through obstruction of the catalytic sites. However, upon illumination with UV-light, isomerization of the azobenzene induced release of the CD blocker, restoring the catalytic function of the Pd. The CASP showed good cellular uptake, low cytotoxicity, also in combination with low-dose UV-irradiation. CASP displayed good intracellular light activated catalytic activity in a fluorescence study through the formation of rhodamine 110 (6) via cleavage and a rhodamine derivative via SMcc reaction. This study presents a promising approach towards light-activated bioorthogonal catalysis. However, UV-light is not the ideal choice for in vivo applications because of its intrinsic biological interference.
 | ||
| Fig. 11 Overview of reported catalyzed reactions (A) and respective light activated catalysts/catalytic systems (B). (1) Formation of rhodamine 110 derivative (6) viaCASP catalyzed Suzuki–Miyaura cross-coupling after activation with NIR-1 light.95 (2) Formation of resveratrol derivative 3via CuAAC catalyzed by MCNs-Cu after NIR-I light activation,91 and CuS@PDA/Pd after NIR-II light activation.96 Partly reproduced from ref. 91, 95 and 96 with permission from ACS and Springer. | ||
The first copper based light-promoted in vitro and in vivo catalysis approach was presented by Qu in 2020.91 For this purpose, the group constructed a mesoporous carbon nanosphere functionalized with copper nanoparticles (MCNs-Cu). Upon irradiation with near infrared light (NIR, λem = 808 nm) the carbon nanostructure heats up (again showing nanostructure necessity) and the MCNs-Cu can form reactive oxygen species (ROS) which were able to convert Cu(0) into CuAAC active Cu(I). Additionally, the MCNs-Cu could convert the NIR irradiation efficiently into heat (η = 51%), increasing local temperature and accelerating the catalytic process significantly (5-fold fluorescence increase in vial). In a cell viability study, only small effects (85% cell viability) were observed for NIR irradiation of intracellular MCNs-Cu (up to 100 μg mL−1) at a power density of 1.0 W cm−2 for 5 minutes. Addition of resveratrol derivative precursors (10 μM) resulted in significantly higher cell death (75%) with NIR irradiation than without (40%), which was attributed to the more efficient in situ resveratrol (3) formation as well as the increase in oxidative stress. After toxicological analysis, the system was applied in vivo in a mice tumor model, showing great antitumor potential with minimal side effects over 14 days.
To further improve on their previous strategy, Qu reported a new approach for light promoted antitumor therapy in 202296. Instead of using NIR-I light (λem = 650–950 nm), they employed NIR-II light (λem = 1000–1700 nm) for its deeper tissue penetration and higher maximum permissible exposure.
The 1064 nm laser was used to catalyze CuAAC as well as Pd decaging reactions via activation of functionalized nanocatalysts. The nanocatalysts consisted of CuS nanoplatelets (nanostructure with secondary functionality) coated with polydopamine and functionalized with Pd nanoparticles (CuS@PDA/Pd). The catalyst displayed great photothermal properties increasing temperature to 52 °C within 10 minutes of irradiation (η = 58%), thereby increasing the Pd catalyzed decaging rate by 6-fold as determined by fluorescence. However, the CuAAC reaction showed no acceleration upon NIR-II irradiation. In a cell viability study, it was shown that the NIR-II light was indeed less invasive than the NIR-I light reported in the previous study (only 82% cell viability over 30 min, 1.0 W cm−2). Also, the combined anticancer strategy (Pd decaging and CuAAC) of in situ formation of 5FU and a resveratrol derivative (3) upon NIR-II irradiation, showed good synergistic effects as compared to the separate strategies (cell viability 27% vs. 54% and 43%, 3 d). In an in vivo mice study, the system showed highest efficacy using the dual therapy strategy with NIR-II light irradiation compared to the separate strategies or the dual strategies with NIR-I light irradiation. This indicates the utility of dual drug anticancer therapies as well as the applicability of NIR-II irradiation in in vivo synthesis. However, it remains a question whether NIR-II could also be applied to deep tissue therapy.
3.2 Magnetic field induced catalysis
The application of magnetic fields as an external trigger for in situ drug synthesis has great intrinsic potential. Because of the deep tissue penetration, controllability, and low energy transfer to the bioenvironment, magnetic fields provide a great option for energy stimulation. This has been shown by Lee in 2020, who employed a silica-confined magnetothermia-induced nanoreactor for intracellular Pd catalyzed carbocyclization (Fig. 12).97 The nanoreactors (MAG-NER) were created through amino–silica coating of iron-oxide nanoparticles, followed by hollowing of the silicon shell through hydrolysis and subsequent Pd nanoparticle growth on the iron-oxide core. Notably, the iron-oxide core functions as an alternating magnetic field (AMF)-nanoheater, transferring energy to the Pd nanoparticle activating it towards catalysis. The silicon shell nanoparticle accommodates the nanoreactor shielding it from deactivation. The MAG-NER showed high catalytic ability in various biologically relevant media (55–70% yield, 1 h, PBS, BSA, DMEM, cell lysates) upon AMF induction. The cellular uptake of the nanoreactor was shown to be high with 65% after 4 h, and the cytotoxicity low (89% cell viability, 500 μg mL−1, 48 h) even under AMF exposure (90% cell viability, 30 min). Finally, in a fluorescence experiment, procoumarin was converted into coumarin derivative (16) via Pd promoted carbocyclization reaction, resulting in clear strong fluorescence after AMF application for 30 minutes, compared to no fluorescence without AMF application. This study clearly shows the excellent applicability of magnetism activated in situ synthesis of molecules, showing great potential for future in vivo studies. | ||
| Fig. 12 (A) Formation of coumarin derivative 16viaMAG-NER catalyzed carbocyclization after catalyst activation using an alternating magnetic field (AMF). (B) Schematic representation of MAG-NER and its AMF activation mechanism.97 Partly reproduced from ref. 97 with permission from ACS. | ||
3.3 Ultrasound-triggered catalysis
An external trigger that has not often been combined with in situ catalyst activation, is the use of ultrasound (US). US, just as magnetic fields, is intrinsically biocompatible and can be applied with high spatiotemporal control. In 2022, Chen reported a copper based polymeric nanocomplex (Cu@PAA-NC), able to achieve US promoted CuAAC catalysis (Fig. 13).98Via exogenous ultrasound irradiation the interconversion between CuAAC inactive Cu(II) and the CuAAC active Cu(I) in the Cu@PAA-NC could be reversely regulated. | ||
| Fig. 13 (A) Formation of resveratrol analogue 3viaCu@PAA-NC catalyzed CuAAC after catalyst activation with ultrasound (US). (B) Schematic representation of Cu@PAA-NC and its activation mechanism using US.98 Partly reproduced from ref. 98 with permission from Wiley. | ||
The reverse transformation to Cu(II) was caused by oxygen or H2O2 exposure, generating ROS in the process. The Cu@PAA-NC proved to successfully catalyze the CuAAC reaction under US irradiation in cellulo, forming a triazole fluorophore.
In a cell viability study a resveratrol derivative (3) was formed in situ with high yield (84%, 12 h) resulting in 36.8% cell viability (80 μg mL−1 Cu-NC), indicating Cu-NC to act as an effective sonosensitizer. In an in vivo mice study, the Cu@PAA-NC displayed fast passive tumor accumulation, efficient renal metabolism mediated clearance, and no toxicity effects. Additionally, the system displayed excellent synergistic tumor growth suppression in 4T1 and B16-F10 melanoma models: 66% and 68% suppression with Cu@PAA-NC + US irradiation only (ROS induced cell death), and 79% and 94% suppression with Cu@PAA-NC + US irradiation + resveratrol derivative precursors respectively, without any observed off-target effects. This highly effective novel method of in situ catalyst activation thus shows great potential for tumor therapy with high spatiotemporal control.
4. Modes of application
So far, it has been demonstrated that there are a lot of diverse strategies for bioorthogonal in situ bond-forming synthesis of various molecules, fluorophores, and drugs, ranging from in vial to in vivo examples. However, towards clinical applications, targeted delivery of the therapeutic system towards lesion sites is of vital importance to prevent off-target effects. Various strategies have been employed for targeted therapy, especially in cancer research, where adverse effects are often severe. The different targeting strategies can be categorized in two groups: passive and active targeting. Passive targeting in cancer therapy, is possible because of tumor microenvironments (TMEs), which differ from healthy tissue. Active targeting on the other hand, involves the functionalization of surface moieties of the catalyst/drug nanocarrier with ligands, antibodies or other molecular functionalities, to identify and attach to specific cells, cellular receptors, or tissues.2 An essential requirement for both passive and active targeting is that nanocarriers are not cleared too rapidly from the bloodstream, thereby providing adequate circulation time. This is typically achieved by applying so-called stealth coatings, which form a hydrated, neutrally charged polymer layer such as PEG or zwitterionic brushes on the particle surface to resist protein adsorption and evade recognition and uptake by the mononuclear phagocyte system.99 Because of the increasing risk of PEG antigenicity by the immune system, alternative scaffolds to PEG are investigated.100,101 For example, our group recently reported the decoration of the PEG-termini with charge neutral ylide residue and have shown that recognition by anti-PEG antibodies is diminished.102,103Here, a brief overview is presented how some examples discussed before are equipped with targeting functionalization of the enabling macromolecular scaffold, again highlighting the importance of their use in multimodular strategies.
4.1 Passive targeting
As observed, most studies discussed in this review that have been extended to in vivo applications, are focused on cancer therapy research. In cancer therapy a targeting strategy often applied is passive targeting using properties of the TME that are vastly different from healthy tissue. TMEs are acidic environments and express high glutathione (GSH), high hydrogen peroxide (H2O2), and low oxygen (hypoxia) contents (Fig. 14).104 The complex TME can form great barriers towards drug delivery to tumors. However, because of its differences with healthy tissue it can also be utilized for targeted drug therapy.105,106 One property of the TME that is often utilized is the leaky vasculature and impaired lymphatic drainage. This property leads to the enhanced permeability and retention (EPR) effect, first observed by Maeda and Matsumura in the 1980s.107,108 In some cancers, this effect causes macromolecules (liposomes, nanoparticles, macromolecules) to preferentially accumulate in tumor tissue as compared to healthy tissue, to sustain and stimulate the rapid growth of cancer cells. This effect can subsequently be abused to passively accumulate drug nanocarriers, nanozymes, or in our case, nanoparticle bioorthogonal catalysts at tumor sites, resulting in concentrations high enough for therapeutic efficacy.109 EPR targeted anti-cancer therapy through in situ mediated bond-forming drug synthesis has been shown for polymer based Pd-catalyst NPs,31 Pd-functionalized CuS nanoplatelets,96 Cu-NP functionalized mesoporous carbon nanospheres91 and polymer based copper nanocomplexes.98 In cases of the latter three examples tumor targeting control using the EPR effect was combined with external stimuli for additional spatiotemporal control using NIR and US irradiation (vide supra). Although this targeting strategy seems promising, it must be noted that there are significant physiological differences between rodents and humans and its success can not directly be assumed in human clinical trials.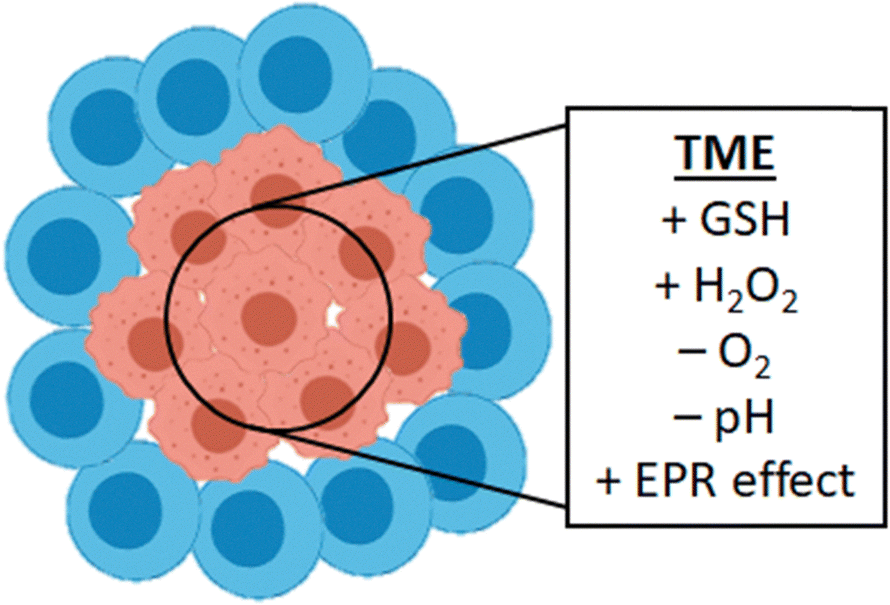 | ||
| Fig. 14 Tumor tissue is different from healthy tissue and forms a so-called tumor micro-environment (TME). This TME often shows high GSH and hydrogen peroxide concentrations and low oxygen and pH. It also accommodates for the observed EPR effect. | ||
Some studies utilize specific factors from the TME to selectively activate their catalyst. Studies that use the GSH, and H2O2 overexpression in TMEs to target catalyst delivery and activation have been presented in this review.
Biocompatible CuAl-LDH nanosheets utilize both EPR and TME for targeting. The nanosheets are first accumulated in tumor tissue via the EPR effect and consequently reduced via high GSH concentration to form Cu(I) for CuAAC promoted prodrug activation and the generation of ROS because of the presence of high H2O2 content.26 The application of cross-linked lipoic acid nanocapsules (cLANCs) also uses the TME for targeted catalyst release. Upon coming into contact with tumoral GSH, the cLANCs structure was disrupted resulting in catalyst and prodrug release and consequent in situ drug activation.47 Bimetallic Cu/Zn MOF based on zeolitic imidazolate framework-8 (ZIF-8) also uses both EPR and TME for its targeted activation. After EPR induced accumulation at the tumor, the release is effectuated by the acidity of the TME, dissolving the ZIF-8 and releasing the Zn and Cu ions to work their magic.24
One similar approach, not targeted to TMEs but Alzheimer's Disease (AD) generated microenvironments, utilizes elevated H2O2 due to amyloid-β (Aβ) aggregation and copper accumulation. Prodrug carrying mesoporous silica nanoparticles can penetrate the blood–brain barrier and disassemble under the AD elevated H2O2, releasing its contents, followed by the endogenous copper mediated prodrug activation and subsequent Aβ-plaque dissociation.51
4.2 Active targeting
Active targeting utilizes ligands, antibodies, or other molecular functionalities to specifically target, identify and attach specific cells, cellular receptors, or tissues. Different tissues and cell types often display different receptor expressions because of their different bodily functions. Diseased tissues such as cancer also differ from normal tissues in their receptor expression, because of their hypermetabolism.110 By modifying NPs with ligands for molecular recognition of these overexpressed receptors, cellular uptake is increased and thereby the therapy efficiency enhanced. Various types of ligands can be employed for this purpose, such as proteins, polysaccharides, nucleic acids, peptides, and small molecules.111 Active targeting has been applied in in vivo drug synthesis studies of which a small selection of examples is discussed in this review. The rapid development of new click reactions for the decoration of complex molecules has significantly advanced the field112 and many bioconjugation approaches are frequently used to decorate macromolecular scaffolds with targeting residues (Fig. 15).113 | ||
| Fig. 15 Various active targeting strategies have been reported in combination with in situ bond forming drug synthesis including the use of targeting glycans, aptamers, peptides, small molecules, and targeted injection. M = Macromolecular scaffold with embedded catalyst. | ||
Additionally, glycans have been shown to be able effectuate tumor specific accumulation as well.118 This strategy has been employed for in situ drug synthesis to selectively accumulate glycoalbumin based Au-GArMs in liver and intestine for in vivo Au-mediated fluorescence labeling and ref. 34, Ru-GArMs in SW620 colon adenocarcinoma cells and in HeLa tumors in mice for metathesis mediated in situ and in vivo drug synthesis,18,33 and Au-GArMs in SW620 cells for hydroamination cyclization mediated in situ drug synthesis.35
5. Conclusions and outlook
In situ bioorthogonal synthesis of drug molecules opens new doors in the fields of cell biology and biomedicine. With the introduction of many new-to-nature reactions to in vivo systems, possibilities arise for the invention of new techniques for the interrogation and alteration of cells and their metabolism, but also new therapy strategies, enhancing efficiency, specificity, and selectivity ultimately leading to higher efficacy and the reduction of side effects.Currently, the research in the field of in situ synthesis is mainly focused on the development of new bioorthogonal chemistries and application strategies. Hereby, a significant move is being made towards the optimization of physiological stability of the catalytic systems, and target specificity through smart combination with known targeting strategies for in vivo application. Currently, studies towards new bioorthogonal bond-forming chemistries are rapidly emerging. These chemistries have been shown to be possible under aqueous and/or biologically relevant media in vitro and therefore show potential use for in situ synthesis for in vivo applications. For example, the authors consider a range of reactions including copper catalyzed Diels–Alder cycloaddition,121 Friedel–Crafts alkylation,122 and nitrene transfer sulfimidation,123 palladium-only catalyzed Sonogashira coupling,124–126 ruthenium catalyzed atroposelective metathesis,127 alkyne–alkene coupling,128,129 and (light-induced) deboronative, and decarboxylative alkynylation,130,131 and azide–thioalkyne cycloaddition,132,133 and iridium catalyzed 1,3-dipolar cycloadditions of azides with ynamides, internal alkynes, and internal disulfanyl alkynes.134–136 The recent past has shown that improved control obtained from bioorthogonal methods is greatly enhanced through the addition of TMC promoted reactions. Notably, the addition of new functionalities for spatiotemporal control through exogenous stimulation may result in even higher specificity and efficacy. As highlighted, the need for nano-carriers with secondary functionality is essential for both targeting functionalization and exogenous stimulation as well as for shielding of catalyst functionality. The research presented in this review shows immense potential for future clinical applications, especially evident from some highly effective cancer therapy strategies in mice studies. Synergistic multi-modal therapy strategies are in this case most prominent, especially for diseases like cancer with multifactorial origins.
One aspect of in situ synthesis that so far has remained underexplored is the concept of ‘retrosynthetic prodrug design’ as introduced by Tanaka.18 The advantage of in situ synthesis, is the possibility to design the ideal prodrug utilizing all possible bioorthogonal chemistries. To design the ideal prodrug, optimization should be pursued in creating the largest difference in bioactivity between drug and prodrug resulting in higher side effect suppression, but also in reactivity, catalytic efficiency, and bioavailability.
As the field of nanomedicine involving macromolecular scaffolds continues to emerge, regulatory agencies are still adapting their frameworks, and several key challenges remain to be addressed. One critical issue is batch-to-batch consistency, which is essential for ensuring safety, regulatory approval, and the feasibility of large-scale manufacturing.137 In situ-forming scaffolds including injectable drug depots, pose additional challenges, particularly when self-administered by patients, and require rigorous monitoring for obtaining safety profiles. Another growing concern is the immunogenicity associated with nanomedicines.138,139 For example, anti-PEG antibodies, whether pre-existing or induced by repeated administration of PEGylated drugs or vaccines, can compromise both the safety and efficacy of nanomedicines. These antibodies may affect biodistribution of nanocarriers, trigger unwanted inflammatory responses, destabilize polymer drug formulations, and in some cases result in hypersensitivity reactions.
Artificial intelligence (AI) holds great promise for advancing the design of macromolecular platforms in nanomedicine. By analyzing complex datasets, AI may help to optimize drug combinations, release profiles, and scaffold architectures to enhance therapeutic outcomes.140,141 This is particularly valuable for combination therapies, where drug synergy is time-, dose-, and patient-dependent. Unlike fixed-dose approaches, AI may help to guide the development of adaptive nanocarriers tailored to individual needs, enabling more effective and personalized treatments. In context of drug synthesis using metal catalysts, such computational power would be of high value to determine most effective concentrations of drug fragments, taking into account stability and circulation time.
In summary, the introduction of transition metal promoted synthesis in biological systems with the help of macromolecular platforms is primed for remarkable advancements in the coming years.
Author contributions
TvE revised literature. TvE and KN wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
We like to acknowledge Radboud University for providing access to literature.References
- W. M. van den Boogaard, D. S. Komninos and W. P. Vermeij, Chemotherapy side-effects: not all DNA damage is equal, Cancers, 2022, 14(3), 627 CrossRef CAS PubMed.
- J. Li, et al., Recent advances in targeted drug delivery strategy for enhancing oncotherapy, Pharmaceutics, 2023, 15(9), 2233 CrossRef CAS PubMed.
- K. M. Huttunen, H. Raunio and J. Rautio, Prodrugs—from serendipity to rational design, Pharmacol. Rev., 2011, 63(3), 750–771 CrossRef CAS PubMed.
- Q. Fu, et al., Bioorthogonal chemistry for prodrug activation in vivo, Chem. Soc. Rev., 2023, 52, 7737–7772 RSC.
- H. C. Hang, et al., A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(25), 14846–14851 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, Finding the right (bioorthogonal) chemistry, ACS Chem. Biol., 2014, 9(3), 592–605 CrossRef CAS PubMed.
- J. Tu, M. Xu and R. M. Franzini, Dissociative bioorthogonal reactions, ChemBioChem, 2019, 20(13), 1615–1627 Search PubMed.
- C. Zhong and X. Shi, When organocatalysis meets transition-metal catalysis, Eur. J. Org. Chem., 2010,(16), 2999–3025 CrossRef CAS.
- H. Madec, et al., Metal complexes for catalytic and photocatalytic reactions in living cells and organisms, Chem. Sci., 2023, 14(3), 409–442 Search PubMed.
- D. P. Nguyen, H. T. Nguyen and L. H. Do, Tools and methods for investigating synthetic metal-catalyzed reactions in living cells, ACS Catal., 2021, 11(9), 5148–5165 CrossRef CAS PubMed.
- N. K. Devaraj, et al., Reactive polymer enables efficient in vivo bioorthogonal chemistry, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(13), 4762–4767 Search PubMed.
- J. M. Mejia Oneto, et al., In vivo bioorthogonal chemistry enables local hydrogel and systemic pro-drug to treat soft tissue sarcoma, ACS Cent. Sci., 2016, 2(7), 476–482 CrossRef CAS PubMed.
- D. Karagrigoriou, et al., Unveiling the Antifouling Potential of Stabilized Poly(phosphorus ylides), ACS Macro Lett., 2023, 12(12), 1608–1613 Search PubMed.
- K. Neumann, The case for poly(ylides) as a class of charge-neutral, hydrophilic polymers with applications in biomaterials science, Biomater. Sci., 2024, 12(21), 5481–5490 RSC.
- B. Li, et al., Trimethylamine N-oxide–derived zwitterionic polymers: A new class of ultralow fouling bioinspired materials, Sci. Adv., 2019, 5(6), eaaw9562 CrossRef CAS PubMed.
- K. Neumann, S. Jain, J. Geng and M. Bradley, Nanoparticle “switch-on” by tetrazine triggering, Chem. Commun., 2016, 52, 11223 RSC.
- M. O. van de L'Isle, M. C. Ortega-Liebana and A. Unciti-Broceta, Transition metal catalysts for the bioorthogonal synthesis of bioactive agents, Curr. Opin. Chem. Biol., 2021, 61, 32–42 CrossRef PubMed.
- I. Nasibullin, et al., Synthetic prodrug design enables biocatalytic activation in mice to elicit tumor growth suppression, Nat. Commun., 2022, 13(1), 39 CrossRef CAS PubMed.
- J. Clavadetscher, et al., Copper catalysis in living systems and in situ drug synthesis, Angew. Chem., Int. Ed., 2016, 128(50), 15891–15895 CrossRef.
- J. Huang, et al., Nanocopper-doped cross-linked lipoic acid nanoparticles for morphology-dependent intracellular catalysis, ACS Catal., 2018, 8(7), 5941–5946 CrossRef CAS.
- Y. Bai, et al., A highly efficient single-chain metal–organic nanoparticle catalyst for alkyne–azide “click” reactions in water and in cells, J. Am. Chem. Soc., 2016, 138(35), 11077–11080 CrossRef CAS PubMed.
- Q. Lu, et al., A dense-shell macromolecular scaffold for catalyst-or substrate-guided catalysis in a cellular environment, ACS Mater. Lett., 2019, 2(1), 89–94 CrossRef.
- F. Wang, et al., A biocompatible heterogeneous MOF–Cu catalyst for in vivo drug synthesis in targeted subcellular organelles, Angew. Chem., Int. Ed., 2019, 58(21), 6987–6992 CrossRef CAS PubMed.
- Z. Wang, et al., A bimetallic metal–organic framework encapsulated with DNAzyme for intracellular drug synthesis and self-sufficient gene therapy, Angew. Chem., 2021, 133(22), 12539–12545 CrossRef.
- Y. Wei, et al., MOFs Modulate Copper Trafficking in Tumor Cells for Bioorthogonal Therapy, Nano Lett., 2024, 24(4), 1341–1350 CrossRef CAS PubMed.
- S. Wang, et al., In situ activation of CuAl-LDH nanosheets to catalyze bioorthogonal chemistry in vivo in tumor microenvironment for precise chemotherapy and chemodynamic therapy, Chem. Eng. J., 2023, 457, 141186 CrossRef CAS.
- Y. You, et al., A DNAzyme-augmented bioorthogonal catalysis system for synergistic cancer therapy, Chem. Sci., 2022, 13(26), 7829–7836 Search PubMed.
- R. M. Yusop, et al., Palladium-mediated intracellular chemistry, Nat. Chem., 2011, 3(3), 239–243 CrossRef CAS PubMed.
- E. Indrigo, et al., Palladium-mediated in situ synthesis of an anticancer agent, Chem. Commun., 2016, 52(99), 14212–14214 RSC.
- P. Destito, et al., Hollow nanoreactors for Pd-catalyzed Suzuki–Miyaura coupling and O-propargyl cleavage reactions in bio-relevant aqueous media, Chem. Sci., 2019, 10(9), 2598–2603 RSC.
- M. A. Miller, et al., Nano-palladium is a cellular catalyst for in vivo chemistry, Nat. Commun., 2017, 8(1), 15906 CrossRef CAS PubMed.
- M. Jeschek, et al., Directed evolution of artificial metalloenzymes for in vivo metathesis, Nature, 2016, 537(7622), 661–665 CrossRef CAS PubMed.
- S. Eda, et al., Biocompatibility and therapeutic potential of glycosylated albumin artificial metalloenzymes, Nat. Catal., 2019, 2(9), 780–792 CrossRef CAS.
- K. Tsubokura, et al., In vivo gold complex catalysis within live mice, Angew. Chem., Int. Ed., 2017, 56(13), 3579–3584 CrossRef CAS PubMed.
- M. Kurimoto, et al., Anticancer approach inspired by the hepatotoxic mechanism of pyrrolizidine alkaloids with glycosylated artificial metalloenzymes, Angew. Chem., Int. Ed., 2022, 61(43), e202205541 CrossRef CAS PubMed.
- C. Sambiagio, et al., Copper catalysed Ullmann type chemistry: from mechanistic aspects to modern development, Chem. Soc. Rev., 2014, 43(10), 3525–3550 RSC.
- A. M. Thomas, A. Sujatha and G. Anilkumar, Recent advances and perspectives in copper-catalyzed Sonogashira coupling reactions, RSC Adv., 2014, 4(42), 21688–21698 RSC.
- M. M. Heravi, et al., Buchwald-Hartwig reaction: an overview, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS.
- M. Breugst and H. U. Reissig, The Huisgen reaction: Milestones of the 1,3-dipolar cycloaddition, Angew. Chem., Int. Ed., 2020, 59(30), 12293–12307 CrossRef CAS PubMed.
- H. C. Kolb, M. Finn and K. B. Sharpless, Click chemistry: diverse chemical function from a few good reactions, Angew. Chem., Int. Ed., 2001, 40(11), 2004–2021 CrossRef CAS PubMed.
- S. Shaligram and A. Campbell, Toxicity of copper salts is dependent on solubility profile and cell type tested, Toxicol. In Vitro, 2013, 27(2), 844–851 CrossRef CAS PubMed.
- L. M. Gaetke and C. K. Chow, Copper toxicity, oxidative stress, and antioxidant nutrients, Toxicology, 2003, 189(1–2), 147–163 CrossRef CAS PubMed.
- L. Li and Z. Zhang, Development and applications of the copper-catalyzed azide-alkyne cycloaddition (CuAAC) as a bioorthogonal reaction, Molecules, 2016, 21(10), 1393 CrossRef PubMed.
- J. Chen, et al., Enzyme-like click catalysis by a copper-containing single-chain nanoparticle, J. Am. Chem. Soc., 2018, 140(42), 13695–13702 CrossRef CAS PubMed.
- J. Chen, et al., Polymeric “clickase” accelerates the copper click reaction of small molecules, proteins, and cells, J. Am. Chem. Soc., 2019, 141(24), 9693–9700 CrossRef CAS PubMed.
- J. Chen, et al., A bioorthogonal small molecule selective polymeric “clickase”, J. Am. Chem. Soc., 2020, 142(32), 13966–13973 CrossRef CAS PubMed.
- L. Wang, et al., “One-stitch” bioorthogonal prodrug activation based on cross-linked lipoic acid nanocapsules, Biomaterials, 2021, 273, 120823 CrossRef CAS PubMed.
- Y. Deng, et al., A membrane-embedded macromolecular catalyst with substrate selectivity in live cells, J. Am. Chem. Soc., 2022, 145(2), 1262–1272 CrossRef PubMed.
- H. Furukawa, et al., The chemistry and applications of metal-organic frameworks, Science, 2013, 341(6149), 1230444 CrossRef PubMed.
- J. Zhu, et al., Boosting Endogenous Copper(I) for Biologically Safe and Efficient Bioorthogonal Catalysis via Self-Adaptive Metal–Organic Frameworks, J. Am. Chem. Soc., 2023, 145(3), 1955–1963 CrossRef CAS PubMed.
- Z. Du, et al., Self-triggered click reaction in an Alzheimer's disease model: in situ bifunctional drug synthesis catalyzed by neurotoxic copper accumulated in amyloid-β plaques, Chem. Sci., 2019, 10(44), 10343–10350 RSC.
- Y. You, et al., DNA-based platform for efficient and precisely targeted bioorthogonal catalysis in living systems, Nat. Commun., 2022, 13(1), 1459 CrossRef CAS PubMed.
- A. Biffis, et al., Pd metal catalysts for cross-couplings and related reactions in the 21st century: a critical review, Chem. Rev., 2018, 118(4), 2249–2295 CrossRef CAS PubMed.
- J. G. Rebelein and T. R. Ward, In vivo catalyzed new-to-nature reactions, Curr. Opin. Biotechnol, 2018, 53, 106–114 Search PubMed.
- J. Clavadetscher, et al., In-Cell Dual Drug Synthesis by Cancer-Targeting Palladium Catalysts, Angew. Chem., 2017, 129(24), 6968–6972 CrossRef.
- A. M. Pérez-López, et al., Dual-bioorthogonal catalysis by a palladium peptide complex, J. Med. Chem., 2023, 66(5), 3301–3311 CrossRef PubMed.
- J. Hafeez, et al., Synthesis of ruthenium complexes and their catalytic applications: A review, Arabian J. Chem., 2022, 15(11), 104165 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts, Chem. Rev., 2010, 110(3), 1746–1787 CrossRef CAS PubMed.
- T. R. Ward, Artificial metalloenzymes based on the biotin− avidin technology: enantioselective catalysis and beyond, Acc. Chem. Res., 2011, 44(1), 47–57 CrossRef CAS PubMed.
- H. Kruszyna, et al., Toxicology and pharmacology of some ruthenium compounds: Vascular smooth muscle relaxation by nitrosyl derivatives of ruthenium and iridium, J. Toxicol. Environ. Health, Part A, 1980, 6(4), 757–773 CAS.
- I. Nasibullin, et al., Catalytic olefin metathesis in blood, Chem. Sci., 2023, 14(40), 11033–11039 RSC.
- A. S. K. Hashmi, Gold-catalyzed organic reactions, Chem. Rev., 2007, 107(7), 3180–3211 CrossRef CAS PubMed.
- S. R. Thomas and A. Casini, Gold compounds for catalysis and metal-mediated transformations in biological systems, Curr. Opin. Chem. Biol., 2020, 55, 103–110 CrossRef CAS PubMed.
- T. C. Chang, et al., Prodrug Activation by Gold Artificial Metalloenzyme-Catalyzed Synthesis of Phenanthridinium Derivatives via Hydroamination, Angew. Chem., Int. Ed., 2021, 133(22), 12554–12562 CrossRef.
- S. B. Singh, Iridium chemistry and its catalytic applications: A brief, Green Chem. Technol. Lett., 2016, 2(206), 456–518 Search PubMed.
- Y. Okamoto and T. R. Ward, Transfer hydrogenation catalyzed by organometallic complexes using NADH as a reductant in a biochemical context, Biochemistry, 2017, 56(40), 5223–5224 CrossRef CAS PubMed.
- N. Singh, et al., Iridium-triggered allylcarbamate uncaging in living cells, Inorg. Chem., 2021, 60(17), 12644–12650 CrossRef CAS PubMed.
- C. Huang, et al., In vitro and in vivo photocatalytic cancer therapy with biocompatible iridium(III) photocatalysts, Angew. Chem., 2021, 133(17), 9560–9565 CrossRef.
- Z. Liu,
et al., Assembly and Evolution of Artificial Metalloenzymes within E. coli Nissle 1917 for Enantioselective and Site-Selective Functionalization of C–H and C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C Bonds, J. Am. Chem. Soc., 2022, 144(2), 883–890 CrossRef CAS PubMed.
C Bonds, J. Am. Chem. Soc., 2022, 144(2), 883–890 CrossRef CAS PubMed. - J. Huang, et al., Unnatural biosynthesis by an engineered microorganism with heterologously expressed natural enzymes and an artificial metalloenzyme, Nat. Chem., 2021, 13(12), 1186–1191 CrossRef CAS PubMed.
- H. M. Key, et al., Abiological catalysis by artificial haem proteins containing noble metals in place of iron, Nature, 2016, 534(7608), 534–537 CrossRef CAS PubMed.
- S. Wallace and E. P. Balskus, Interfacing microbial styrene production with a biocompatible cyclopropanation reaction, Angew. Chem., Int. Ed., 2015, 54(24), 7106–7109 CrossRef CAS PubMed.
- P. S. Coelho, et al., A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo, Nat. Chem. Biol., 2013, 9(8), 485–487 CrossRef CAS PubMed.
- S. J. Kan, et al., Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life, Science, 2016, 354(6315), 1048–1051 CrossRef CAS PubMed.
- K. Chen, et al., Enzymatic construction of highly strained carbocycles, Science, 2018, 360(6384), 71–75 CrossRef CAS PubMed.
- M. Garcia-Borràs, et al., Origin and control of chemoselectivity in cytochrome c catalyzed carbene transfer into Si–H and N–H bonds, J. Am. Chem. Soc., 2021, 143(18), 7114–7123 CrossRef PubMed.
- Z. Liu, et al., Dual-function enzyme catalysis for enantioselective carbon–nitrogen bond formation, Nat. Chem., 2021, 13(12), 1166–1172 CrossRef CAS.
- A. L. Chandgude, X. Ren and R. Fasan, Stereodivergent intramolecular cyclopropanation enabled by engineered carbene transferases, J. Am. Chem. Soc., 2019, 141(23), 9145–9150 CrossRef CAS PubMed.
- X. Ren, A. L. Chandgude and R. Fasan, Highly stereoselective synthesis of fused cyclopropane-γ-lactams via biocatalytic iron-catalyzed intramolecular cyclopropanation, ACS Catal., 2020, 10(3), 2308–2313 CrossRef CAS PubMed.
- K. Fagnou and M. Lautens, Rhodium-catalyzed carbon− carbon bond forming reactions of organometallic compounds, Chem. Rev., 2003, 103(1), 169–196 CrossRef CAS PubMed.
- H. T. Chifotides and K. R. Dunbar, Interactions of metal− metal-bonded antitumor active complexes with DNA fragments and DNA, Acc. Chem. Res., 2005, 38(2), 146–156 CrossRef CAS PubMed.
- J. Zhao, et al., An artificial metalloenzyme for carbene transfer based on a biotinylated dirhodium anchored within streptavidin, Catal. Sci. Technol., 2018, 8(9), 2294–2298 RSC.
- K. A. Mix, M. R. Aronoff and R. T. Raines, Diazo compounds: versatile tools for chemical biology, ACS Chem. Biol., 2016, 11(12), 3233–3244 CrossRef CAS PubMed.
- Z. T. Ball, Molecular recognition in protein modification with rhodium metallopeptides, Curr. Opin. Chem. Biol., 2015, 25, 98–102 CrossRef CAS PubMed.
- Z. Zhao, et al., Dual stimulus-triggered bioorthogonal nanosystem for spatiotemporally controlled prodrug activation and near-infrared fluorescence imaging, Chem. Commun., 2023, 59(26), 3878–3881 RSC.
- Y. Zhang, et al., Nanoparticulation of prodrug into medicines for cancer therapy, Adv. Sci., 2021, 8(18), 2101454 CrossRef CAS.
- K. Neumann, A. Lilienkampf and M. Bradley, Responsive polymeric nanoparticles for controlled drug delivery, Polym. Int., 2017, 66(12), 1756–1764 CrossRef CAS.
- N. Mamidi, et al., Carbonaceous nanomaterials incorporated biomaterials: The present and future of the flourishing field, Composites, Part B, 2022, 243, 110150 CrossRef CAS.
- C. I. Crucho, Stimuli-responsive polymeric nanoparticles for nanomedicine, ChemMedChem, 2015, 10(1), 24–38 CrossRef CAS PubMed.
- N. Mamidi, F. F. De Silva and A. O. M. Salehi, Advanced disease therapeutics using engineered living drug delivery systems, Nanoscale, 2025, 17, 7673–7696 RSC.
- Y. You, et al., Near-infrared light dual-promoted heterogeneous copper nanocatalyst for highly efficient bioorthogonal chemistry in vivo, ACS Nano, 2020, 14(4), 4178–4187 CrossRef CAS PubMed.
- P. A. Shaw, et al., Two-photon absorption: an open door to the NIR-II biological window?, Front. Chem., 2022, 10, 921354 CrossRef CAS PubMed.
- R. Weissleder, A clearer vision for in vivo imaging, Nat. Biotechnol., 2001, 19(4), 316–317 CrossRef CAS PubMed.
- R. C. Remmers and K. Neumann, Reaching new lights: a review on photo-controlled nanomedicines and their in vivo evaluation, Biomater. Sci., 2023, 11(5), 1607–1624 RSC.
- F. Wang, et al., Designed heterogeneous palladium catalysts for reversible light-controlled bioorthogonal catalysis in living cells, Nat. Commun., 2018, 9(1), 1209 CrossRef PubMed.
- H. Zhao, et al., NIR-II light leveraged dual drug synthesis for orthotopic combination therapy, ACS Nano, 2022, 16(12), 20353–20363 CrossRef CAS PubMed.
- J. Lee, et al., Magnetothermia-induced catalytic hollow nanoreactor for bioorthogonal organic synthesis in living cells, Nano Lett., 2020, 20(10), 6981–6988 CrossRef CAS PubMed.
- L. Xia, et al., Spatiotemporal ultrasound-driven bioorthogonal catalytic therapy, Adv. Mater., 2023, 35(7), 2209179 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, PEG conjugates in clinical development or use as anticancer agents: an overview, Adv. Drug Delivery Rev., 2009, 61(13), 1177–1188 CrossRef CAS PubMed.
- A. S. A. Lila, H. Kiwada and T. Ishida, The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage, J. Controlled Release, 2013, 172(1), 38–47 CrossRef PubMed.
- J. J. Verhoef, et al., Potential induction of anti-PEG antibodies and complement activation toward PEGylated therapeutics, Drug Discovery Today, 2014, 19(12), 1945–1952 CrossRef CAS PubMed.
- G. Poulladofonou and K. Neumann, Poly(sulfur ylides): a new class of zwitterionic polymers with distinct thermal and solution behaviour, Polym. Chem., 2022, 13(30), 4416–4420 RSC.
- D. M. Sánchez-Cerrillo, et al., Introducing Ylides as Charge-Neutral Termini for Poly(Ethylene Glycol) with Applications in Nanomedicine. 2025.
- E. Henke, R. Nandigama and S. Ergün, Extracellular matrix in the tumor microenvironment and its impact on cancer therapy, Front. Mol. Biosci., 2020, 6, 160 CrossRef PubMed.
- R. Zhang, et al., Tumor microenvironment-responsive BSA nanocarriers for combined chemo/chemodynamic cancer therapy, J. Nanobiotechnol., 2022, 20(1), 223 Search PubMed.
- Y. Zhou, et al., Overcoming the biological barriers in the tumor microenvironment for improving drug delivery and efficacy, J. Mater. Chem. B, 2020, 8(31), 6765–6781 RSC.
- Y. Matsumura and H. Maeda, A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs, Cancer Res., 1986, 46(12_Part_1), 6387–6392 CAS.
- H. Maeda and Y. Matsumura, Tumoritropic and lymphotropic principles of macromolecular drugs, Crit. Rev. Ther. Drug Carrier Syst., 1989, 6(3), 193–210 CAS.
- R. Bazak, et al., Passive targeting of nanoparticles to cancer: A comprehensive review of the literature, Mol. Clin. Oncol., 2014, 2(6), 904–908 CrossRef PubMed.
- F. S. Anarjan, Active targeting drug delivery nanocarriers: Ligands, Nano-Struct. Nano-Objects, 2019, 19, 100370 CrossRef.
- J. Yoo, et al., Active targeting strategies using biological ligands for nanoparticle drug delivery systems, Cancers, 2019, 11(5), 640 CrossRef CAS PubMed.
- K. Gavriel, et al., Click’n lock: rapid exchange between unsymmetric tetrazines and thiols for reversible, chemoselective functionalisation of biomolecules with on-demand bioorthogonal locking, RSC Chem. Biol., 2023, 4(9), 685–691 RSC.
- W. Yi, et al., Recent advances in developing active targeting and multi-functional drug delivery systems via bioorthogonal chemistry, Signal Transduction Targeted Ther., 2022, 7(1), 386 CrossRef CAS PubMed.
- Y. Cheng and Y. Ji, RGD-modified polymer and liposome nanovehicles: Recent research progress for drug delivery in cancer therapeutics, Eur. J. Pharm. Sci., 2019, 128, 8–17 CrossRef CAS PubMed.
- X. Ni, et al., Nucleic acid aptamers: clinical applications and promising new horizons, Curr. Med. Chem., 2011, 18(27), 4206–4214 CrossRef CAS PubMed.
- M. Brayman, A. Thathiah and D. D. Carson, MUC1: a multifunctional cell surface component of reproductive tissue epithelia, Reprod. Biol. Endocrinol., 2004, 2, 1–9 CrossRef PubMed.
- A. Ogura, et al., Visualizing trimming dependence of biodistribution and kinetics with homo-and heterogeneous N-glycoclusters on fluorescent albumin, Sci. Rep., 2016, 6(1), 21797 CrossRef CAS PubMed.
- A. Ogura, et al., Glycan multivalency effects toward albumin enable N-glycan-dependent tumor targeting, Bioorg. Med. Chem. Lett., 2016, 26(9), 2251–2254 CrossRef CAS PubMed.
- K. Urbiola, et al., Efficient serum-resistant lipopolyplexes targeted to the folate receptor, Eur. J. Pharm. Biopharm., 2013, 83(3), 358–363 CrossRef CAS PubMed.
- B. A. Gruner and S. D. Weitman, The folate receptor as a potential therapeutic anticancer target, Invest. New Drugs, 1998, 16, 205–219 CrossRef CAS PubMed.
- W. Ghattas, et al., Receptor-based artificial metalloenzymes on living human cells, J. Am. Chem. Soc., 2018, 140(28), 8756–8762 CrossRef CAS PubMed.
- S. Chordia, et al., In vivo assembly of artificial metalloenzymes and application in whole-cell biocatalysis, Angew. Chem., Int. Ed., 2021, 60(11), 5913–5920 CrossRef CAS PubMed.
- E. J. Meeus, et al., Copper-Catalyzed Sulfimidation in Aqueous Media: a Fast, Chemoselective and Biomolecule-Compatible Reaction, Chem. – Eur. J., 2024, 30(14), e202303939 CrossRef CAS PubMed.
- N. Li, et al., Copper-free Sonogashira cross-coupling for functionalization of alkyne-encoded proteins in aqueous medium and in bacterial cells, J. Am. Chem. Soc., 2011, 133(39), 15316–15319 CrossRef CAS PubMed.
- J. Li, et al., Ligand-free palladium-mediated site-specific protein labeling inside Gram-negative bacterial pathogens, J. Am. Chem. Soc., 2013, 135(19), 7330–7338 CrossRef CAS PubMed.
- N. Li, et al., A genetically encoded alkyne directs palladium-mediated protein labeling on live mammalian cell surface, ACS Chem. Biol., 2015, 10(2), 379–384 CrossRef CAS PubMed.
- T. Vornholt, et al., An Artificial Metalloenzyme for Atroposelective Metathesis, ChemCatChem, 2023, 15(23), e202301113 CrossRef CAS.
- L. Adriaenssens, et al., Bio-and air-tolerant carbon–carbon bond formations via organometallic ruthenium catalysis, Collect. Czech. Chem. Commun., 2009, 74(7), 1023–1034 CrossRef CAS.
- A. Gutiérrez-González, et al., Ruthenium-catalyzed intermolecular alkene–alkyne couplings in biologically relevant media, Chem. Sci., 2023, 14(23), 6408–6413 RSC.
- H. Huang, et al., Visible-light-induced chemoselective deboronative alkynylation under biomolecule-compatible conditions, J. Am. Chem. Soc., 2014, 136(6), 2280–2283 CrossRef CAS PubMed.
- J. Yang, et al., Visible-light-induced chemoselective reductive decarboxylative alkynylation under biomolecule-compatible conditions, Chem. Commun., 2015, 51(25), 5275–5278 RSC.
- P. Destito, et al., Ruthenium-Catalyzed Azide–Thioalkyne Cycloadditions in Aqueous Media: A Mild, Orthogonal, and Biocompatible Chemical Ligation, Angew. Chem., Int. Ed., 2017, 56(36), 10766–10770 CrossRef CAS PubMed.
- A. Gutiérrez-González, et al., Bioorthogonal Azide–Thioalkyne Cycloaddition Catalyzed by Photoactivatable Ruthenium(II) Complexes, Angew. Chem., Int. Ed., 2021, 60(29), 16059–16066 CrossRef PubMed.
- W. Song and N. Zheng, Iridium-catalyzed highly regioselective azide–ynamide cycloaddition to access 5-amido Fully substituted 1, 2, 3-triazoles under mild, air, aqueous, and bioorthogonal conditions, Org. Lett., 2017, 19(22), 6200–6203 CrossRef CAS PubMed.
- R. Chen, et al., Iridium-Catalyzed Hydroxyl-Enabled Cycloaddition of Azides and Alkynes, Adv. Synth. Catal., 2019, 361(5), 989–994 CrossRef CAS.
- M. Li, et al., Iridium-catalyzed orthogonal and regioselective synthesis of triazole disulfides in aqueous media under mild conditions, Green Chem., 2020, 22(8), 2394–2398 RSC.
- N. Mamidi, et al., Innovative hydrogel-based delivery systems for immunotherapy: A review of pre-clinical progress, Nano Res., 2024, 17(10), 9031–9043 CrossRef CAS.
- Y. Chen, Y.-C. Su and S. R. Roffler, Polyethylene glycol immunogenicity in nanomedicine, Nat. Rev. Bioeng., 2025, 1–19 Search PubMed.
- B.-M. Chen, T.-L. Cheng and S. R. Roffler, Polyethylene glycol immunogenicity: theoretical, clinical, and practical aspects of anti-polyethylene glycol antibodies, ACS Nano, 2021, 15(9), 14022–14048 CrossRef CAS PubMed.
- D. Ho, P. Wang and T. Kee, Artificial intelligence in nanomedicine, Nanoscale Horiz., 2019, 4(2), 365–377 RSC.
- N. Serov and V. Vinogradov, Artificial intelligence to bring nanomedicine to life, Adv. Drug Delivery Rev., 2022, 184, 114194 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |