
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A buoyant plasmonic microbubble-based SERS sensing platform for amyloid-beta protein detection in Alzheimer's disease†
Willis Kwun Hei
Ho‡
a,
Qin
Zhang‡
*ab,
Fariza
Zhorabe
e,
Jiaxiang
Yan
a,
Yutian
Gu
a,
Shujun
Wang
a,
Changqing
Yi
 f,
Yu
Zhang
g and
Mo
Yang
*abcd
f,
Yu
Zhang
g and
Mo
Yang
*abcd
aDepartment of Biomedical Engineering, The Hong Kong Polytechnic University, Hong Kong 999077, China. E-mail: qin7zhang@polyu.edu.hk; mo.yang@polyu.edu.hk
bThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen 518000, China
cResearch Center for Nanoscience and Nanotechnology, The Hong Kong Polytechnic University, Kowloon, Hong Kong 999077, China
dJoint Research Center of Biosensing and Precision Theranostics, The Hong Kong Polytechnic University, Kowloon, Hong Kong 999077, China
eDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Hong Kong 999077, China
fKey Laboratory of Sensing Technology and Biomedical Instruments (Guangdong Province), School of Biomedical Engineering, Sun Yat-Sen University, Guangzhou 510000, China
gDepartment of Mechanical and Automotive Engineering, Royal Melbourne Institute of Technology, Melbourne, VIC 3000, Australia
First published on 24th June 2025
Abstract
Amyloid-β (Aβ) plaques are a key pathological hallmark of Alzheimer's disease (AD), highlighting the need for highly sensitive bioassays for Aβ detection to enable AD diagnosis. Here, we synthesized a buoyant plasmonic substrate composed of polyvinyl alcohol microbubbles (MBs) decorated with in situ-reduced gold nanoparticles (Au NPs). Benefiting from its inherent buoyancy and near-infrared plasmonic properties, the Au/MB substrate serves as an ideal platform for biomolecular sensing via the surface-enhanced Raman spectroscopy (SERS) technique. Compared to conventional flat SERS substrates, the three-dimensional (3D) curved surface of the Au/MB substrate significantly increases the effective sensing area, while its inherent buoyancy facilitates the efficient removal of unbound targets, thereby enhancing detection specificity. By functionalizing Au/MB substrates with copper ions (Cu2+) and 4-mercaptobenzoic acid (4-MBA), we achieved sensitive detection of AD-related Aβ proteins. In the presence of the target analyte, the interaction between Aβ proteins and Cu2+ induces molecular deformation and orientation changes in 4-MBA, leading to distinct spectral changes in the SERS signals. The results demonstrate that the developed Au/MB-based SERS sensor enables sensitive detection of Aβ1–40 oligomers with a sensitivity as low as 10−9 M. Therefore, this work not only establishes a foundational framework for designing buoyant plasmonic substrate-based SERS sensing platform but also paves the way for the quantitative detection of disease-associated protein biomarkers, contributing to advancements in AD diagnostics.
1. Introduction
Alzheimer's disease (AD) is one of the progressive neurodegenerative diseases afflicting individuals all over the world, with toxic amyloid-β (Aβ) protein fibrils as the main pathological feature.1–3 The abnormal oligomerization and aggregation processing of Aβ contributes to the accumulation of neurotoxic plaques and bundle fibrils in the brain, which subsequently lead to neurodegenerative symptoms such as memory loss and cognitive impairment.4–6 Current clinal monitoring methods such as positron-emission tomography (PET) brain imaging are complicated, high-cost, and invasive, thus limiting the large-scale preclinical screening to meet the substantial public need.7,8 In contrast, detection of biomarkers in bodily fluids offers a more economical and convenient alternative for monitoring AD progression. Recent studies have identified Aβ oligomers as efficient biomarkers for predicting AD.9,10 Therefore, developing a sensitive method for Aβ detection is crucial for assessing AD progression during the preclinical stages.Raman signals provide molecular fingerprint spectra of target analytes, enabling precise characterization of biochemical systems.11 As a result, Raman-based techniques have been widely integrated into various biosensing platforms. However, due to the inherently low Raman scattering cross-section of most molecules, the native Raman signals are often too weak for effective detection and analysis.12 To overcome this limitation, plasmonic nanoparticles are frequently employed as substrates to enhance Raman signals. When localized surface plasmon resonance (LSPR) is excited, the incident light energy becomes confined within a sub-wavelength volume, generating intense electromagnetic near fields that amplify Raman signals by several orders of magnitude. This amplification phenomenon, known as surface-enhanced Raman spectroscopy (SERS), has revolutionized the sensitivity of Raman-based sensing techniques.13,14 However, the success of SERS relies heavily on the development of robust substrates capable of providing uniform and reproducible signal enhancement. Unfortunately, many existing SERS substrates are fabricated using complex and expensive techniques, such as electron beam lithography, which significantly limit their scalability and practical applicability.15 In contrast, buoyant plasmonic substrates have emerged as a promising alternative due to their unique structural and functional advantages.16 These substrates are typically composed of plasmonic nanoparticles immobilized on lightweight, floating microstructures. By concentrating analytes at the liquid–air interface, buoyant plasmonic substrates facilitate enhanced signal uniformity through self-assembly and improve light–matter interactions by providing greater accessibility to excitation light.17 These features not only simplify the fabrication process but also make buoyant plasmonic substrates highly attractive for scalable, cost-effective, and high-performance SERS applications.
Amid recent developments in nanotechnology and plasmonic materials, microbubbles (MBs) have garnered significant attention as innovative platforms for SERS due to their unique structural and functional properties.18 In contrast to traditional solid-state SERS substrates, which are often fabricated using complex and expensive techniques, MBs provide a scalable, cost-effective, and versatile alternative.19 The 3D curved surface of the MB substrate significantly increases the effective sensing area. Composed of lightweight, gas-filled structures typically stabilized by polymeric shells, MBs can serve as buoyant platforms for the immobilization of plasmonic nanoparticles, such as gold nanostructures.19,20 These hybrid plasmonic microbubbles exhibit distinct advantages, making them highly promising for a wide range of SERS-based applications. Recently, SERS-based sensing strategies have been developed for protein quantification.21,22 These strategies work by capturing target proteins on functionalized surfaces, where mechanical stress induced by protein binding deforms stress-sensitive nanostructures or molecules, subsequently converting these deformations into measurable optical or spectroscopic signals, thereby effectively reporting the presence and concentration of proteins. Therefore, it is reasonable to assume that a SERS platform combining mechanical force-driven stress sensing transduction with the high sensitivity and specificity of SERS could enable precise and label-free detection of disease-related proteins in complex biological environments. Furthermore, the SERS sensing platform holds great promise for diagnosing AD by enabling the sensitive and non-invasive detection of Aβ protein at early stages, thereby facilitating timely diagnosis and intervention.
Herein, we developed a buoyant plasmonic SERS substrate by in situ reduction of gold nanoparticles (Au NPs) on the surface of polyvinyl alcohol microbubbles (PVA-MBs). The resulting Au/MB substrate exhibits two key features that make it an excellent SERS platform for biomolecule detection: (1) the inherent buoyancy of the MBs and (2) the extended plasmonic properties in the near-infrared (NIR) region of Au NPs. To enable the detection of Aβ protein, the Au/MB substrate was further functionalized with 4-mercaptobenzoic acid (4-MBA) as a SERS reporter molecule (Fig. 1). The 4-MBA molecules bind to the Au nanoclusters via their thiol terminal groups, leaving the carboxyl terminal groups available for further coordination with copper(II) ions (Cu2+), thereby enabling selective detection of Aβ proteins, as Aβ contains several amino acids that serve as coordination sites for Cu2+.23,24 The Aβ1–40 protein specifically interacts with the Cu2+ complex, inducing molecular deformation and orientation changes in 4-MBA, which lead to intensity variations and distinct peak shifts in the Raman spectrum (Fig. 1). These spectroscopic changes in the SERS signals enable the quantitative detection of Aβ1–40 protein at varying concentrations and provide insights into the aggregation state of Aβ. Our results demonstrate that this Au/MB SERS sensor achieves a detection sensitivity as low as 10−9 M for Aβ1–40 protein. Thus, the Au/MB-based SERS platform provides a powerful tool for detecting AD-related biomarkers, contributing to the broader application of SERS in biomedical diagnostics.
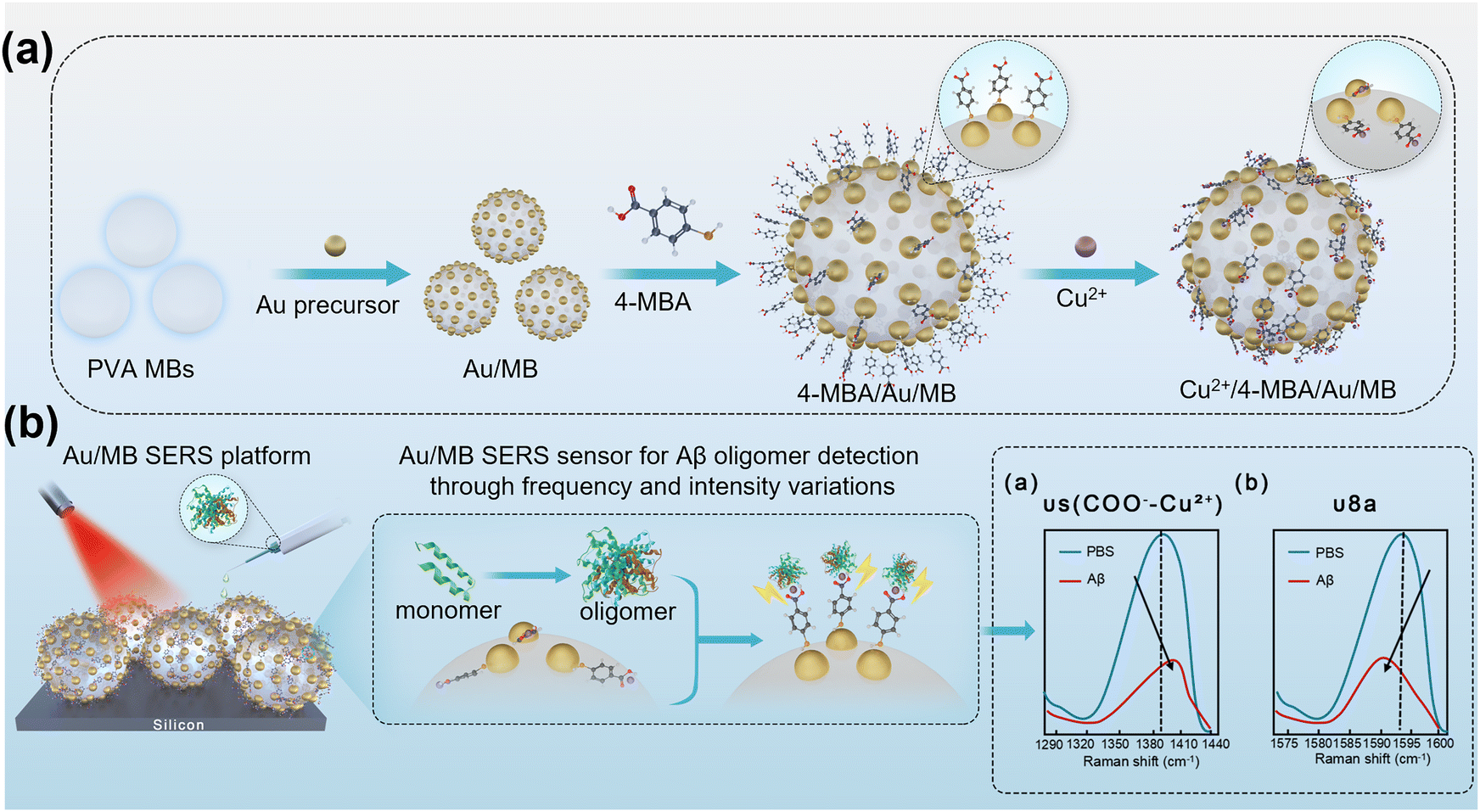 | ||
| Fig. 1 (a) Schematic diagram illustrating the preparation of Cu2+ and 4-MBA functionalized Au/MB SERS sensing substrate. (b) Schematic illustration of the application of buoyant plasmonic Au/MB SERS sensing platform for the detection of oligomerized Aβ protein. | ||
2. Materials and methods
2.1. Materials and equipment
Unless otherwise noted, all reagents were obtained from commercial suppliers and used without further purification. Polyvinyl alcohol (99%), sodium periodate (NaIO4, 99%), Au(III) chloride hydrate (HAuCl4, 99%), L-ascorbic acid (AA, 99%), 4-mercaptobenzoic acid (4-MBA, 99%), and copper(II) chloride (99%) were purchased from Sigma Aldrich. Dimethyl sulfoxide (DMSO), hexafluoro-2-propanol (HFIP) were purchased from Aladdin Co. Ltd. Tris-buffered saline (TBS), bovine serum albumin (BSA), human serum albumin (HSA) and rana grylio virus (RGV) glycol protein were purchased from Thermofisher. Aβ1–40 was purchased from Sango Biotech. The size distribution and zeta potential were determined by a dynamic light scattering (DLS) analyzer (Malvern Zetasizer Nano ZS90). Scanning electron microscopy (SEM) and element mapping images were obtained in a JEOL field emission SEM microscope. ultraviolet-visible (UV-vis) absorbance was measured by Ultrospec 2100 Pro spectrophotometer (Amersham Biosciences). Zeta potential was measured using a Malvern ZEN 3600 Zetasizer. Optical darkfield images were taken from optical system (Olympics BX-150). All the SERS spectra were taken from Renishaw Micro-Raman Spectroscopy System.2.2. Preparation of polyvinyl alcohol air-filled microbubbles (PVA-MBs)
Air-filled PVA-MBs were synthesized by crosslinking telechelic PVA polymers in accordance with previously reported methods.25,26 First, 2% PVA aqueous solution was prepared under hot plate stirring (70 °C, 900 rpm). The condenser was to prevent evaporation of the solvent during heating. Once a homogeneous solution was obtained, 800 mg of NaIO4 was added and kept under constant stirring for 1 h. The obtained PVA solution was vigorously stirred by IKA Ultra-Turrax T-25 disperser with Teflon coated tip at 8000 rpm for 3 h at room temperature. Fine foam generated at the top of the solution (air–water interface) suggests successful PVA-MBs synthesis. Afterward, suspension was transferred to the separating funnel where MBs were separated and washed 5 times.2.3. In situ growth of Au nanocluster on PVA-MBs (Au/MB)
Au nanocluster was grown on the surface of MBs by direct reduction of Au ions in the PVA-MBs solution. 800 μL of MBs solution was mixed with 150 μL of Au precursor HAuCl4 (10 mM) and 150 μL of AA (0.1 mM) was introduced, acting as a reducing agent. The solution colour rapidly changes to reddish as the Au reduction occurs. Then the solution obtained was left undistributed for 3 h until the resultant Au/MB is floating on the water surface and followed by washing three times.2.4. Au/MB modification with 4-MBA and metal ions
4-MBA (1 mM, 2 μL) was added into 1 mL Au/MBs under vigorous shaking for 24 h. 4-MBA will attach to the Au nanocluster by forming Au–S bond. Then the 4-MBA decorated Au/MB is left undisturbed to allow the Au/MB separation from bulk solution and it is washed for 4 times with deionized water. To decorate metal ions on 4-MBA molecule, 1 mM of CuAc2, PbAc2 and FeAc3 were dissolved in deionized water, then 5 uL of the dissolved ion was added into 1 mL Au/MB solution under vigorous shaking for 2 h. Finally, Au/MBs was washed three times to remove the excessive metal ions.2.5. Aβ1–40 monomer and oligomer preparation
To obtain the Aβ1–40 monomer, 200 μl of hexafluoro-2-propanol (HFIP) was added to dissolve the Aβ1–40 monomer into concentration of 1 mM and it is left undisturbed for 1.5 h.5 Then the HFIP is removed under the content flow of nitrogen gas, and a transparent film is obtained at the bottom of the tube. To resuspend the Aβ1–40, 2 μL of DMSO was added followed by adding Tris-buffered saline (TBS) buffer (20 μM, pH 7.4, 150 μM NaCl) and dilute the Aβ protein into 25 μM. To obtained Aβ1–40 oligomer, the monomer in TBS was incubated in 37 °C for 24 h and 48 h. Then, the size of Aβ1–40 was characterized by dyamics light scattering system (Zetasizer Nano ZS instrument; Malvern Instruments, Worcestershire, UK). Prior to the measurement, 5 uM of Aβ1–40 was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 min at 4 °C. The DLS measurements were conducted at a constant temperature at 25 °C. All signals were recorded for 20 runs.
000g for 20 min at 4 °C. The DLS measurements were conducted at a constant temperature at 25 °C. All signals were recorded for 20 runs.
2.6. Circular dichroism (CD) measurement
The CD spectra of Aβ1–40 were measured from JASCO J-810 Spectrometer (JASCO Co., Tokyo, Japan) with a standard quartz cuvette in 1 mm optical length. Briefly, the pre-treated Aβ1–40 in different time pointed were diluted to 15 μM with Ultrapure water and the CD spectra were obtained for at least for three times under the N2 gas purging. To analyse the Aβ1–40 conformational we consider the change of CD peak in 195 and 215 nm, respectively. Using the built-in “Smooth” function in the software, the CD curve was applied with a 11-point Savitzky–Golay smooth to the baseline.2.7. Atomic force microscope (AFM) analysis
5 μL of each Aβ1–40 sample (25 μM) was deposited on clean silicon wafer pre-treated with Piranha solution. Until the sample dries, a few drops of water were applied to remove the large aggregate on top. AFM images were obtained under tapping mode with a Nano-FTIR (neaspec). The Image J software was used for AFM image analysis. The background image value was first obtained, and the standard derivation is used for determining the image threshold.2.8. SERS measurement of Cu2+/4-MBA
Aβ1–40 and other proteins were incubated with 50 μL of Cu2+/4-MBA decorated Au/MB in PBS buffer (pH 7.5) for 30 min. The mixture was then transferred to a flat silicon substrate and waited for 20 min to allow the Au/MB floats to settle on the water surface. To obtain the spectrum, the Raman system was operated in LASER measurement power at 0.5 mW. The exposure time was set to 5 s. All obtained SERS spectra were normalized to the 4-MBA at 1078 cm−1. Angle-resolved SERS measurements were performed to investigate the orientation-dependent vibrational behavior of 4-MBA molecules on the Au/MB substrate under different molecular interaction states. Raman spectra were acquired using a confocal Raman microscope equipped with a 633 nm laser. The samples were mounted on a rotational stage that allowed precise adjustment of the incident laser angle relative to the substrate surface. Two sample groups were prepared: (i) the Cu2+/4-MBA-modified Au/MB substrate on silicon surface, and (ii) the same substrate after incubation with Aβ1–40 (1 μM) for 30 min. Raman spectra were collected at incident angles of 0°, 45°, and 70°. The intensity of the ν(CCC) vibrational mode (∼718 cm−1) was extracted from the spectra after baseline correction and normalization. The angle-dependent signal ratios ( and
and 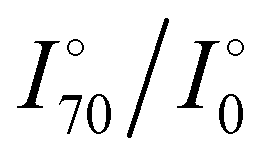 ) were calculated to quantitatively assess the changes in molecular orientation.
) were calculated to quantitatively assess the changes in molecular orientation.
2.9. Molecular dynamics simulations
The initial structure of Aβ1–40 protein was predicted using HelixFold 3 and preprocessed with OpenBabel. Molecular dynamics simulations were performed using GROMACS 2021 with the AMBER99SB force field for proteins and GAFF2 for small molecules. The system was solvated in a TIP3P water box and neutralized with Na+/Cl− ions. After energy minimization and 100 ps equilibration, a 100 ns production run was conducted using the leap-frog algorithm with a 2 fs time step. Temperature and pressure were maintained at 298.15 K and 1.0 bar using V-rescale and Parrinello–Rahman coupling, respectively. PME and dispersion correction were applied for long-range electrostatic and van der Waals interactions. Snapshots were extracted every 20 ns to capture the precise state of the simulation system at specific time points. These snapshots enabled detailed analysis of the dynamic behavior of key species, including Cu2+, 4-MBA, and Aβ1–40, such as intermolecular interactions, molecular trajectories, and structural evolution.2.10. Statistical analysis
The statistical results in this study were presented as the mean value with error bars indicating the standard deviation. GraphPad Prism 8.0 was utilized for statistical analysis and the creation of graphs. Statistical testing was conducted using a one-way ANOVA test, with subsequent post hoc analyses employed for multiple comparisons. Statistical significance was a probability value (p) less than 0.05.3. Results and discussion
3.1. Preparation and characterization of buoyant plasmonic Au/MB substrate
The fabrication process of the Au/MB substrate is illustrated in Fig. 2(a). Initially, air-filled microbubbles (MBs) were synthesized by the polymerization of water-soluble polyvinyl alcohol (PVA) using a high-speed homogenizer. The obtained PVA-MBs exhibited an average hydrodynamic diameter of 4.78 μm and demonstrated good stability in aqueous solution (Fig. S1, ESI†). Subsequently, chloroauric acid (HAuCl4) was reduced by ascorbic acid, resulting in the in situ growth of gold (Au) nanoclusters on the surface of the PVA-MBs. Notably, no surfactants were required during the fabrication process, as the intrinsic buoyancy of the MBs effectively prevented aggregation at the nanoscale. Consequently, the Au/MB substrate preserved monodispersity in aqueous solutions. A series of analytical techniques was employed to characterize the fabricated Au/MB substrate. First, elemental mapping demonstrated the colocalization of gold (Au), oxygen (O), and carbon (C) elements, providing additional evidence that the Au nanoclusters were reduced on the MB surface (Fig. 2(b)). Furthermore, zeta potential measurements revealed that pure MBs exhibited a surface potential of approximately −10 mV, which shifted to a more negative value of −16 mV following the in situ growth of Au nanoclusters (Fig. 2(c)). This change in surface potential confirmed the successful deposition of Au on the MB surface. The UV-vis measurement of the pure MB has no significant absorption over the entire visible region. For Au/MB fabricated in low Au+ concentration (0.1 mM), a peak arises at around 523 nm (Fig. 2(d)), suggesting that the reduced Au nanocluster has a dipolar plasmonic resonance mode as observed from spherical Au nanoparticles.27 As Au+ concentration increases, the fabricated Au/MB exhibits an extended absorption in NIR region at around 650 nm (Fig. 2(d)), indicating that higher Au+ precursor results in the increased dimension and anisotropy of Au nanocluster.28 Scanning electron microscopy (SEM) and corresponding dark-field imaging were then used to investigate the morphology of the synthesized Au/MB substrate (Fig. 2(e)). The results confirmed the spherical morphology of the MBs and demonstrated that the Au nanostructures were uniformly attached to their surfaces. At low Au+ precursor concentrations (0.1 mM), isolated Au nanoclusters were observed on the MB surface, consistent with the dipolar resonance mode identified in the UV-vis spectra. In contrast, at higher Au+ concentrations (0.15 mM and 0.25 mM), the dimensions and density of the Au nanoclusters increased significantly (Fig. 2(e)), corroborating the enhanced absorption observed in the UV-vis spectra. Additionally, dark-field images revealed pronounced light-scattering behavior for Au/MB substrates synthesized at higher Au+ concentrations (Fig. 2(e)), further confirming the optical properties of the Au nanostructures. Furthermore, the SERS performance of the Au/MB substrates synthesized with different Au+ precursor concentrations was evaluated by measuring the Raman intensity of the 4-MBA reporter on the substrates. The results show that the SERS intensity of the characteristic peak at ∼1590 cm−1 increased with the Au+ concentration and reached its maximum at 0.25 mM (Fig. S2a–c, ESI†). | ||
| Fig. 2 Preparation and characterization of Au/MB substrate. (a) Schematic illustration of the fabrication of air-filled PVA MBs and preparation of Au/MB substrate. (b) Element mapping of synthesized Au/MB. Oxygen (O), carbon (C) and gold (Au). The Overlay figure shows the co-localization of the mapped element on Au/MB. (c) Zeta potential measurement pure PVA MB and Au/MB. (d) Normalized UV-vis absorption spectra of the Au/MB synthesized with varying final concentrations of Au+ precursor (0.1 mM, 0.15 mM and 0.25 Mm). (e) Scanning electron microscope (SEM) micrograph and the corresponding dark-field image of Au/MB synthesized in different final Au+ concentration at 0.1 mM, 0.15 mM and 0.25 mM. The scale bars in the SEM images (left) represent 3 μm, while the scale bars in the dark-field images (right) represent 30 μm. (f) The Photograph of Au/MB synthesized in different final Au+ concentrations. The lower panel shows the same batch of Au/MB rested undisturbed for 25 min. (g) The stability of Au/MB in H2O and PBS. Au/MB was left undistributed for 2 months. | ||
After the reduction process, the Au/MB composites remained well-dispersed in solutions. Upon standing for 25 min, the Au/MB substrates spontaneously separated from the bulk solution, forming a stable layer at the liquid surface while leaving a transparent solution underneath (Fig. 2(f)). This self-separation behavior further confirmed the buoyant property of the Au/MB substrate and suggested that the Au nanoclusters were successfully loaded onto the MBs. However, it was observed that excessively high Au+ precursor concentrations (e.g., 0.4 mM) led to overloading of the Au nanoclusters, resulting in the formation of the skinned Au/MB structures with reduced buoyancy and lower yield (Fig. S2c and d, ESI†). Furthermore, the stability of the Au/MB substrate was evaluated in biological buffers. The results demonstrated that the buoyancy of the Au/MB composites was maintained for more than two months in both deionized water and phosphate-buffered saline (PBS) with no observable detachment of Au nanoclusters from the MBs (Fig. 2(g)). Together, we successfully fabricated a composite Au/MB heterostructure with two distinct features: (1) the buoyant property conferred by the air-filled MBs and (2) the plasmonic properties provided by the attached Au nanoclusters. The plasmonic resonance of the Au nanoclusters significantly enhances Raman signals through the plasmonic enhancement mechanism. Additionally, the buoyant property not only enables the self-separation of the Au/MB substrate from the sensing buffer but also allows the formation of a stable SERS detection layer at the liquid surface. This minimizes Brownian motion, thereby improving the signal-to-noise ratio and generating a robust SERS signal suitable for sensitive detection. These features collectively make the Au/MB substrate an excellent candidate for SERS-based biosensing applications.
3.2. Fabrication of SERS-based Au/MB sensing platform
To evaluate the Raman signal generated by the Au/MB platform, the Au/MBs were functionalized with 4-mercaptobenzoic acid (4-MBA) and incubated for SERS analysis. The Raman signals were collected by focusing the laser on the surface of the buoyant Au/MB layer. In this study, Au/MB substrates synthesized using 0.25 mM Au+ precursor were selected, as the UV-vis spectra shown in Fig. 2(d) indicated enhanced absorbance across the visible region, which is advantageous for SERS performance.29 The collected SERS spectra of 4-MBA revealed that the Au/MB platform exhibited excellent SERS intensity with a remarkably low signal-to-noise ratio (Fig. 3(a)). Two prominent SERS peaks were observed at approximately 1078 cm−1 and 1590 cm−1, corresponding to the ν(C–S) stretching vibration and the ν8a aromatic ring vibration, respectively. These peaks are consistent with the typical vibrational modes of 4-MBA as reported in previous studies.30,31 By monitoring the ν8a aromatic ring vibration at ∼1590 cm−1, the Au/MB buoyant SERS substrate demonstrated the ability to detect 4-MBA concentrations as low as 10−13 M (Fig. 3(b)), signifying its exceptional sensitivity. To further investigate the performance of the substrate, the SERS intensity was compared across different substrates and laser excitation wavelengths (Fig. 3(c)). When the Au/MB platform was excited with a 633 nm laser, the SERS intensity was approximately 6 times higher than that of the Au NPs suspension and 5 times higher than the signal collected under 532 nm laser excitation (Fig. 3(c)). This enhanced SERS response of the Au/MB substrate can be attributed to the synergistic effects of the plasmonic and buoyant properties. Specifically, the extended plasmonic absorption resulting from the overgrowth of Au nanoclusters at a 0.25 mM Au+ precursor enabled efficient LSPR excitation under 633 nm laser irradiation. Additionally, the buoyancy of the Au/MB substrate ensured the formation of a stable detection layer at the liquid–air interface, minimizing Brownian motion and improving signal stability. Together, these factors contributed to the observed high SERS signal intensity and low noise interference.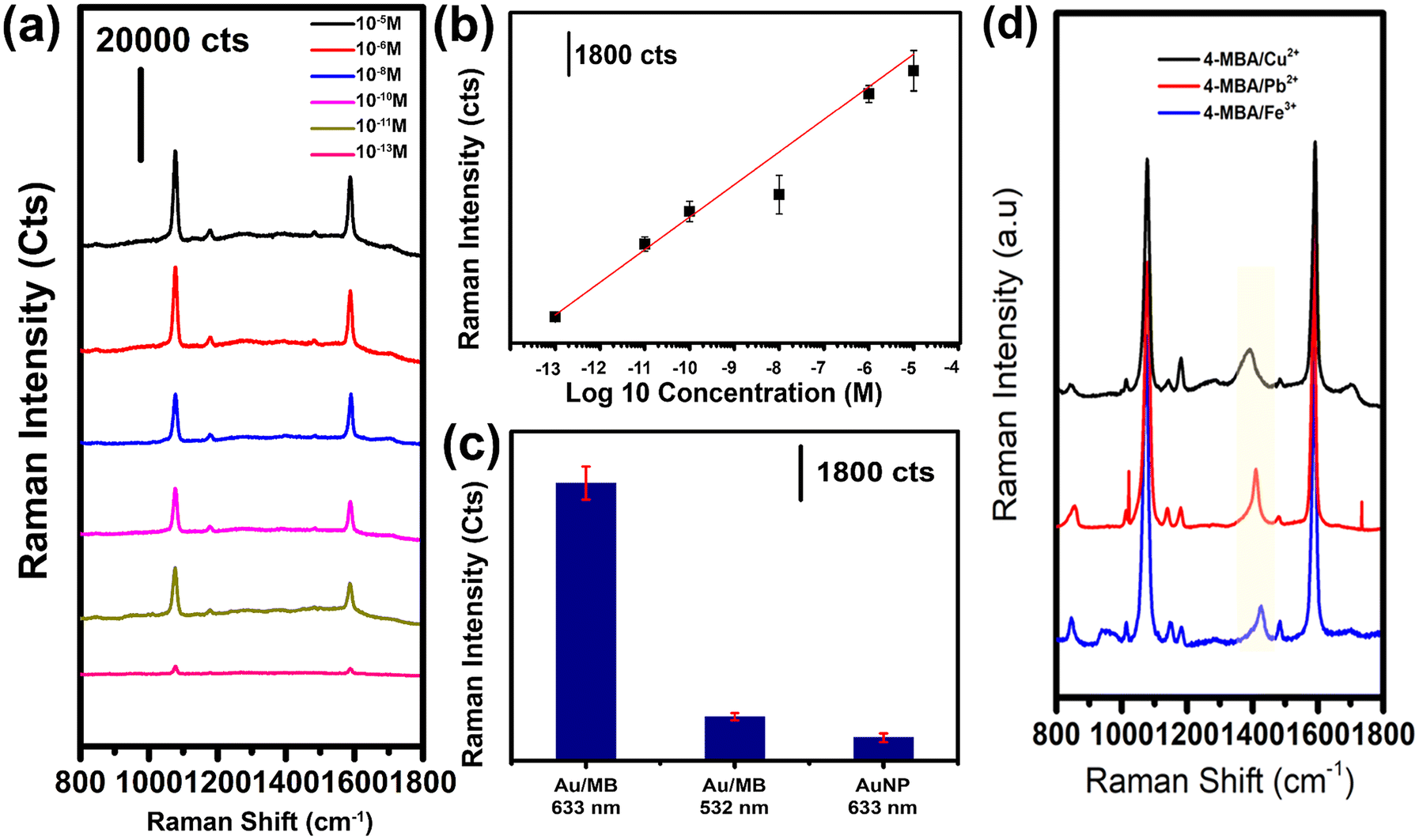 | ||
| Fig. 3 The collection of Raman signals of 4-MBA with Au/MB SERS substrate. (a) The SERS signal of 4-MBA molecule collected from 10−13 M to 10−3 M. (b) The log10 concentration of 4-MBA molecule plotted against the SERS signal intensity. (c) The 4-MBA SERS signal intensity collected under different experiment conditions. With Au/MB excited by 633 nm laser (left), with Au/MB excited by 532 nm laser (middle), with suspension Au nanoparticle excited by 633 nm laser (right). (d) The 4-MBA signal after the coordination with Cu2, Pb2+, and Fe3+. | ||
To explore the functional versatility of the 4-MBA-modified Au/MB platform, the free carboxyl groups of 4-MBA were utilized for metal ion coordination. Previous studies have demonstrated that carboxyl groups can coordinate with divalent and trivalent metal ions through ligand interactions, forming stable metal ion–ligand complexes.32 By incubating the Au/MB substrate with different ions (Cu2+, Pb2+, and Fe3+), distinct spectral changes were observed in the SERS signals (Fig. 3(d)). Specifically, the intensities of the protonated carboxyl vibrational bands at ∼1280 cm−1 and ∼1700 cm−1 decreased, while the deformation band δ(COO−) at ∼850 cm−1 and the symmetric stretching mode νs(COO−) at ∼1400 cm−1 (associated with deprotonated carboxylate groups) exhibited notable frequency shifts and intensity changes (Fig. 3(d)). Among these, the νs(COO−) band proved to be the most sensitive to metal ion coordination, with peak frequencies observed at 1390 cm−1 (Cu2+), 1402 cm−1 (Pb2+), and 1430 cm−1 (Fe3+), respectively (Fig. 3(d)). These results confirm the successful coordination of metal ions to the carboxyl groups of the 4-MBA molecules.33,34 The ability of the Au/MB platform to support both SERS enhancement and functionalization with metal ions demonstrates its versatility as a sensing platform for probing biomolecular interactions involving metal ions.
3.3. Detection of Aβ1–40 protein by Au/MB SERS sensing platform
The 4-MBA on the surface of Au/MB sensing platform were further functionalized with Cu2+ ions to enable selective detection of Aβ proteins, as Aβ contains several amino acids that act as coordination sites for Cu2+.23,24 Here, we exploited Au/MB SERS platform for Aβ1–40 protein detection. The Cu2+/4-MBA-functionalized Au/MB substrate was first exposed to freshly prepared Aβ1–40 monomer solution. Interestingly, Aβ1–40 monomer induced only a reduction in the intensity of the νs(COO−–Cu2+) band, while no significant changes were observed in other characteristic SERS peaks (Fig. S3, ESI†). As the detection mechanism of this sensing system relies on conformational changes or deformations of the SERS reporter molecules induced by the target analyte, this limited spectroscopic response may be explained by the low protein mass of the freshly prepared Aβ1–40, which primarily exists in its monomeric form with an average molecular weight of ∼4 kDa.35 To address this, we pretreated Aβ1–40 in TBS buffer at 37 °C for 48 h to promote oligomerization. The Cu2+/4-MBA-functionalized Au/MB was then incubated with the obtained Aβ1–40 oligomer. As expected, the oligomerized Aβ1–40 protein induced significant and distinct changes in the 4-MBA SERS spectra (Fig. 4(a)). Specifically, both frequency shifts and reductions in relative peak intensity were observed for the ν8a aromatic ring vibration (∼1592 cm−1) and the νs(COO−–Cu2+) symmetric stretching mode (∼1388 cm−1). The ν8a peak and νs(COO−–Cu2+) peak exhibited shifts to lower and higher wavenumbers, respectively (Fig. 4(b)). The fitted frequency of the ν8a vibration shifted from ∼1592 cm−1 to ∼1588 cm−1, while the νs(COO−–Cu2+) peak shifted from ∼1388 cm−1 to ∼1394 cm−1. Additionally, the relative intensities of both peaks decreased following incubation with Aβ1–40 oligomer. Specifically, the intensity of the ν8a vibration decreased from ∼1.05 to ∼0.8, while that of the νs(COO−–Cu2+) band decreased from ∼0.26 to ∼0.19 (Fig. 4(c)). | ||
| Fig. 4 The detection of Aβ1–40 fibrils protein by Cu2+/4-MBA/Au/MB SERS sensing platform. (a) The SERS signal of Cu2+/4-MBA system before and after the exposure to Aβ1–40 protein at a concentration of 10−6 M. (b) The peak intensity of ν8a vibration (1592 cm−1) and νs(COO–Cu2+) vibration (1388 cm−1) of the SERS signal in (a). (c) The summarized peak intensity of ν8a vibration (1592 cm−1) and νs(COO–Cu2+) vibration (1388−1). All peak intensity is normalized to breathing vibration mode at 1078 cm−1. | ||
To rule out the possibility that the intensity changes were due to the detachment of Cu2+ from the 4-MBA molecules, we examined the protonated carboxyl-associated peaks at ∼1280 cm−1 (ν(C–OH)) and ∼1700 cm−1 (ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)). Without coordination of ions, these peaks would increase in intensity. However, no significant changes were observed for these peaks upon Aβ incubation, indicating that Cu2+ ions remained bound to the 4-MBA molecules (Fig. S4, ESI†). Furthermore, the potential influence of the protein dilution buffer (PBS without Aβ protein) was excluded, as the blank buffer did not induce any significant spectral changes over time (Fig. S5, ESI†). These results confirm that the observed spectral changes in the Cu2+/4-MBA system were specifically caused by interactions with the oligomerized Aβ1–40 protein. The interaction between Aβ1–40 oligomers and Cu2+ ions induced nanoscale stress on the SERS reporter molecules, leading to depolarization of the carboxyl groups and deformation of the phenyl rings of 4-MBA, which were reflected in the spectroscopic changes.36
O)). Without coordination of ions, these peaks would increase in intensity. However, no significant changes were observed for these peaks upon Aβ incubation, indicating that Cu2+ ions remained bound to the 4-MBA molecules (Fig. S4, ESI†). Furthermore, the potential influence of the protein dilution buffer (PBS without Aβ protein) was excluded, as the blank buffer did not induce any significant spectral changes over time (Fig. S5, ESI†). These results confirm that the observed spectral changes in the Cu2+/4-MBA system were specifically caused by interactions with the oligomerized Aβ1–40 protein. The interaction between Aβ1–40 oligomers and Cu2+ ions induced nanoscale stress on the SERS reporter molecules, leading to depolarization of the carboxyl groups and deformation of the phenyl rings of 4-MBA, which were reflected in the spectroscopic changes.36
Subsequently, we investigated the effects of Aβ1–40 oligomerization on the SERS signals by incubating the Cu2+/4-MBA-functionalized substrate with Aβ1–40 proteins incubated in TBS buffer for various time (Fig. 5(a)). The observed peak shifts in the ν8a vibration were hypothesized to result from the progressive aggregation of Aβ proteins, which exerted increasing nanoscale stress on the 4-MBA molecules. To verify this hypothesis, freshly prepared monomeric Aβ1–40 solutions were incubated at 37 °C for varying durations (24 and 48 h) to simulate different stages of Aβ aggregation. Dynamic light scattering (DLS) measurements confirmed a significant increase in the hydrodynamic size of Aβ proteins, from approximately 4 nm for freshly prepared monomers to ∼15 nm and ∼30 nm for samples incubated for 24 and 48 h, respectively (Fig. 5(b)). Consistently, atomic force microscopy (AFM) revealed a structural transition in Aβ1–40, progressing from isolated monomers at 0 hours to larger oligomers and fibrils at 24 and 48 hours (Fig. 5(c)). Furthermore, circular dichroism (CD) spectroscopy demonstrated a clear transition from a prevalently unordered conformation (characterized by a weak negative band at ∼205 nm) to a β-strand structure (characterized by a stronger negative band centered at 220 nm) (Fig. 5(d)).37–39 These changes in the spatial structure and protein size of Aβ were strongly correlated with progressive shifts in the ν8a peak position, from ∼1592 cm−1 at 0 hours to ∼1590 cm−1 at 24 h and ∼1587 cm−1 at 48 h (Fig. 5(e)). The summarized peak positions confirm that the pre-incubation time of Aβ protein induced a gradual shift in the ν8a resonance mode. Furthermore, the relative intensity of the ν8a peak decreased after incubation with oligomerized Aβ protein, dropping from ∼1.07 to ∼0.89 (Fig. 5(f)). Together, these results confirm that the frequency shifts and intensity reductions in the ν8a vibration are primarily caused by the increased size of the Aβ aggregates. The larger protein size induces greater deformation of the 4-MBA phenyl rings, resulting in the observed spectroscopic changes. The accumulation and oligomerization of Aβ protein are widely recognized as primary contributors to neurotoxicity in AD and are strongly correlated with disease severity.4,5 Therefore, the ability to monitor Aβ protein oligomerization using the Cu2+/4-MBA-functionalized Au/MB substrate highlights its potential as a sensitive and effective tool for tracking AD progression.
 | ||
| Fig. 5 Cu2+/4-MBA/Au/MB SERS sensing platform for detecting Aβ1–40 proteins with varying degrees of oligomerization. (a) Schematic diagram of the detection assay and sensing mechanism. (b) DLS measurement of Aβ1–40 protein after incubation in TBS buffer at 37 °C for 0 h, 24 h and 48 h. (c) Atomic force microscopy (AFM) images of Aβ1–40 protein after incubation for various durations. (d) Circular dichroism (CD) spectra of Aβ1–40 protein after incubation for various durations. (e) Peak position of ν8a vibration mode after incubation with Aβ1–40 protein. (f) The relative intensity of the ν8a peak after incubation with Aβ1–40 protein. The SERS signal of (g) ν8a vibration and (h) νs(COO–Cu2+) vibration of Cu2+/4-MBA-functionalized Au/MB platform after incubation with different concentration of Aβ1–40 oligomers (pre-treated 48 h). The linear relationship of the peak intensity of band (i) ν8a and (j) νs(COO–Cu2+) with the logarithmic concentration (log10) of Aβ1–40 (pre-treated 48 h). | ||
Thereafter, to evaluate the sensitivity of the Cu2+/4-MBA-functionalized Au/MB platform, we incubated it with varying concentrations of Aβ1–40 protein, which was incubated in TBS buffer at 37 °C for 48 h to promote oligomerization. As shown in Fig. 5(g) and (h), frequency shifts and intensity changes were observed in the ν8a and νs(COO−–Cu2+) bands, with a limit of detection (LOD) of 10−9 M for Aβ1–40, which offers comparable sensitivity to existing Au nanostructure-based SERS systems (Table S1, ESI†).40–47 Specifically, the ν8a vibration at ∼1592 cm−1 exhibited a concentration-dependent frequency shift due to deformation of the 4-MBA phenyl ring (Fig. 5(g)). Similarly, a frequency shift was also noted for the νs(COO−–Cu2+) band at ∼1389 cm−1 (Fig. 5(h)). Most notably, the intensity of both peaks showed a good linear relationship with the concentration of Aβ1–40 from to 10−9 M to 10−6 M (Fig. 5(i) and (j)). Based on linear fitting of the ν8a and νs(COO−–Cu2+) peak intensities, the limit of detection (LOD) for Aβ1–40 was estimated to be 10−8 M and 10−9 M, respectively (Fig. 5(i) and (j)). To further assess the interference resistance of the sensing platform under complex biological conditions, we performed additional experiments by spiking Aβ1–40 into various biological buffer systems, including PBS, Tris–HCl, and artificial cerebrospinal fluid (aCSF). The sensing platform could detect Aβ1–40 in all tested matrices, exhibiting observable frequency shifts and intensity changes in both the ν8a and νs(COO−–Cu2+) vibrational bands. Specifically, compared to the reference spectrum obtained in H2O, the ν8a vibration and the νs(COO−–Cu2+) band demonstrated similar frequency shifts and intensity attenuations in the other biological media (Fig. S6, ESI†), confirming the reliable anti-interference capability of our sensing platform. The observed Raman intensity reduction can be attributed to the deformation of the 4-MBA phenyl ring and the depolarization of the carboxylate–Cu2+ complex, induced by the interaction with the large oligomerized Aβ1–40 protein. These interactions further confirm the sensitivity of the sensing platform to Aβ1–40 protein and its ability to detect changes in its aggregation state.
In addition to the spectroscopic changes observed in the ν8a and νs(COO−–Cu2+) bands, we identified a significant SERS peak at 718 cm−1, attributed to the out-of-plane ν(CCC) vibration of the 4-MBA phenyl ring, which reflects the molecular orientation of 4-MBA.36 The intensity of the ν(CCC) peak increased upon coordination of Cu2+ ions (Fig. S7, ESI†). However, when the Cu2+/4-MBA system was exposed to oligomerized Aβ1–40 (48-hour TBS treatment), the ν(CCC) intensity decreased again, showing a concentration-dependent relationship with Aβ1–40 (Fig. 6(a)). This change in ν(CCC) intensity reveals molecular orientation changes in 4-MBA molecules during the detection process. Initially, before Cu2+ coordination, 4-MBA molecules predominantly adopt an upright orientation (Fig. 6(b)). Upon coordination with Cu2+, the 4-MBA molecules are tilted due to repulsive forces induced by the ion–ligand interactions.48 In contrast, when Aβ oligomers bind to the Cu2+/4-MBA system, the interaction induces intermolecular attractive forces, partially restoring the upright orientation of the 4-MBA molecules. The sensing platform (Cu2+/4-MBA-Au/MB) was fixed on silicon surface, and Raman spectra were collected at incident angles of 0°, 45°, and 70° with respect to the substrate. The results show distinct angle-dependent variations in the intensity of the ν(CCC) band (∼718 cm−1). Cu2+/4-MBA-Au/MB exhibited weaker angle dependence of the ν(CCC) mode at ∼718 cm−1 (Fig. S8a and c, ESI†), which is consistent with a tilted orientation of the 4-MBA aromatic ring induced by Cu2+ coordination. In contrast, the Raman spectra showed a more pronounced increase in ν(CCC) intensity at higher angles after incubation with Aβ1–40 (Fig. S8b and c, ESI†), indicating a partial reorientation toward an upright configuration upon Aβ1–40 binding. These angle-dependent variations in Raman intensity provide strong spectroscopic evidence supporting our conclusion regarding molecular orientation changes during the sensing process. To investigate the molecular-level mechanism, we performed molecular dynamics (MD) simulation to explore the binding interactions among Cu2+, 4-MBA, and Aβ1–40. The interaction analysis of MD simulations exhibited multiple types of non-covalent interactions among Cu2+, 4-MBA, and Aβ1–40 (Fig. 6(c)). Cu2+ exhibited strong coordination with the carboxyl group of 4-MBA (Fig. 6(c)). These interactions remained stable throughout the simulation, with a final LJ-SR energy of −4319.43 kJ mol−1 and a Coulomb-SR energy of 226.88 kJ mol−1 (Fig. S9a, ESI†), indicating strong and persistent Cu2+–COO− binding. After Aβ1–40 was introduced into the simulation system, Cu2+ formed significant non-covalent interactions with amino acid residues of Aβ protein via both electrostatic and hydrophobic interactions (Fig. 6(c)). The final LJ-SR energy for Cu2+-Aβ was −3362.78 kJ mol−1, and the Coulomb-SR energy was 158.74 kJ mol−1 (Fig. S9b, ESI†), confirming the affinity of Cu2+ for binding pockets on the Aβ protein. In addition, the phenyl ring of 4-MBA was found to interact with Aβ through hydrogen bonding, hydrophobic interactions, and electrostatic forces, involving residues such as histidine (HIS), phenylalanine (PHE), valine (VAL), and isoleucine (ILE) (Fig. 6(c) and Fig. S9c, ESI†). The results support the proposed mechanism in which Cu2+ acts as a coordination bridge between 4-MBA and Aβ, inducing rearrangement of the phenyl ring of 4-MBA after binding to Aβ (Fig. 6(b)). These molecular orientation changes, in conjunction with the deformation of the phenyl ring, contribute to the observed intensity variations in the ν(CCC), ν8a, and νs(COO−–Cu2+) bands, providing additional insights into the detection mechanism at the molecular level.
 | ||
| Fig. 6 The signal intensity of out-of-plane vibration of ν(CCC) vibration at 718 cm−1 of the Au/MB sensing platform. (a) The signal intensity of out-of-plane vibration of ν(CCC) after incubation with different concentrations of oligomerized Aβ1–40 protein changes from 10−6 M to 10−9 M. (b) The schematic diagram illustrating the 4-MBA orientation upon incubating with Cu2+ ions and oligomerized Aβ protein. (c) Molecular interactions among Cu2+, 4-MBA, and Aβ at the final time frame of the molecular dynamics (MD) simulations. | ||
3.4. Specificity of Au/MB SERS sensing platform
Aβ1–40 protein demonstrated a strong binding affinity toward Cu2+ ions compared to other divalent metal ions.49,50 To validate this, we evaluated a Pb2+/4-MBA-functionalized Au/MB platform under identical conditions. After incubation with oligomerized Aβ1–40, no significant intensity changes were observed for either the νs(COO−–Pb2+) band (∼1404 cm−1) or the ν8a vibration (∼1590 cm−1), and the peak frequencies remained unchanged (Fig. 7(a) and (b)). These results suggest that oligomerized Aβ1–40 exhibits minimal interaction with the Pb2+/4-MBA complex, further highlighting the strong affinity between Aβ1–40 and the Cu2+/4-MBA system. | ||
| Fig. 7 The specificity of Au/MB SERS sensing platform. The SERS signal of (a) νs(COO–Cu2+) vibration and (b) ν8a vibration of Pb2+/4-MBA after incubation with oligomerized Aβ1–40 protein with different concentrations. The SERS signals of (c) ν8a vibration and (d) νs(COO–Cu2+) vibration of Cu2+/4-MBA-Au/MB after incubation with different proteins, including RGV, Aβ1–40 oligomers, BSA, and HSA. Summarized peak position of (e) band ν8a and (f) band νs(COO–Cu2+) of the SERS spectra. | ||
To evaluate the specificity of the Cu2+/4-MBA-functionalized Au/MB sensing platform for the detection of Aβ1–40, we selected human serum albumin (HSA), bovine serum albumin (BSA), and rabies virus glycoprotein (RVG) protein as control analytes. The Cu2+/4-MBA substrate was incubated separately with Aβ1–40 oligomer, HSA, BSA, and RVG protein to compare their effects on the SERS spectra. Notably, the RVG protein did not induce any significant changes in the ν8a or νs(COO−–Cu2+) vibration modes (Fig. 7(c) and (d)). The results suggest that the RVG protein does not interact with the coordinated Cu2+ ions, thereby causing no observable perturbation in the SERS signals. In contrast, distinct changes in the ν8a and νs(COO−–Cu2+) bands were observed when the platform was exposed to Aβ1–40, HSA, or BSA (Fig. 7(c) and (d)). Previous studies have shown that both HSA and BSA can interact with Cu2+ ions, which likely explains their ability to perturb the Cu2+/4-MBA system and cause significant spectroscopic changes.51,52 However, upon closer examination of the SERS spectra, each of these proteins induced unique frequency shifts in both the ν8a and νs(COO−–Cu2+) bands, which reflect differences in their interactions with the Cu2+/4-MBA complex. For the ν8a band, the peak shifted from 1592 cm−1 (baseline) to 1584 cm−1 for Aβ1–40, 1585 cm−1 for HSA, and 1588 cm−1 for BSA (Fig. 7(e) and (f)). For the νs(COO−–Cu2+) band, the peak shifted from 1388 cm−1 (baseline) to 1394 cm−1 for Aβ1–40, and 1399 cm−1 for both HSA and BSA (Fig. 7(e) and (f)). These distinct frequency shifts indicate that the conformation of the protein–Cu2+/4-MBA complex varies depending on the interacting protein. The unique frequency shifts observed in both the ν8a and νs(COO−–Cu2+) bands demonstrate the ability of the Au/MB SERS sensing platform to distinguish between different proteins based on their interactions with the Cu2+ ions. This specificity highlights the versatility of the Cu2+/4-MBA-functionalized Au/MB platform for the selective detection of oligomerized Aβ1–40 protein in complex biological environments. With its ability to distinguish the aggregation states of Aβ1–40, this Au/MB SERS sensing platform shows great potential for early diagnosis and monitoring of AD progression.
4. Conclusion
In this study, we developed a buoyant SERS substrate based on air-filled PVA microbubbles MBs with in situ growth of Au NPs on their surface, resulting in the Au/MB composite structure. Benefiting from its enhanced NIR plasmonic resonance properties and inherent buoyancy, the Au/MB platform demonstrated excellent potential for biomedical SERS-based applications. The platform was further functionalized with SERE reporter 4-MBA molecules and Cu2+ ions to enable the detection of AD-related Aβ protein. Our results revealed that freshly prepared Aβ1–40 monomers did not induce observable spectroscopic changes in the 4-MBA SERS spectra. However, oligomerized Aβ1–40 proteins caused significant molecular deformation of 4-MBA, leading to frequency shifts and intensity changes in the ν8a and νs(COO−–Cu2+) vibrational modes. By correlating these spectral changes with the protein concentration, the platform achieved a detection sensitivity as low as 10−9 M. Moreover, the SERS signals provided valuable insights into the dynamic molecular orientation of 4-MBA molecules upon interaction with Aβ protein. Specifically, the coordination between Cu2+ ions and Aβ oligomers induced nanoscale stress, resulting in molecular reorientation of the reporter molecules. In addition to detecting oligomerized Aβ protein, our platform demonstrated the ability to distinguish spectroscopic features induced by other proteins, including HSA and BSA. While these proteins also perturbed the Cu2+/4-MBA system, the unique conformational changes they induced resulted in distinguishable frequency shifts. In conclusion, this work establishes a microbubble-based SERS sensing platform capable of detecting Aβ protein in different aggregation states with high sensitivity and specificity. Beyond its role as a diagnostic tool for AD-related biomarker detection, this platform also provides fundamental insights into the design of microbubble-based SERS sensing systems for the quantitative detection of disease-associated protein biomarkers. Its promising applications in early-stage diagnosis of AD pave the way for further advancements in biomolecular sensing.Author contributions
W. K. H. H. designed and performed experiments. Q. Z. analyzed the data and wrote the manuscript. F. Z. and Y. T. G. conducted the AFM test. M. Y. and Q. Z. conceived the idea, edited the paper. M. Y. supervised the whole study and supported funding. All authors reviewed and approved the paper.Conflicts of interest
All authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Data availability
All data needed to evaluate the conclusions are present in the article and its ESI.†Acknowledgements
This work was supported by the Shenzhen Science and Technology Program-Basic Research Scheme (JCYJ20220531090808020), the Research Grants Council (RGC) of Hong Kong Collaborative Research Grant (C5005-23W and C5078-21E), the Research Grants Council (RGC) of Hong Kong General Research Grant (PolyU 15217621 and PolyU 15216622), the Guangdong-Hong Kong Technology Cooperation Funding Scheme (GHP/032/20SZ and SGDX20201103095404018), the Hong Kong Polytechnic University Shenzhen Institute Bai Cheng Bai Yuan Fund (I2022A002), PolyU Internal Fund (1-YWB4, 1-WZ4E, 1-CD8M, 1-WZ4E, 1-CEB1, 1-YWDU, 1-CE2J, 1-W02C, W40F, and WZ5Z).References
- B. Dubois, H. Hampel, H. H. Feldman, P. Scheltens, P. Aisen, S. Andrieu, H. Bakardjian, H. Benali, L. Bertram, K. Blennow, K. Broich, E. Cavedo, S. Crutch, J. F. Dartigues, C. Duyckaerts, S. Epelbaum, G. B. Frisoni, S. Gauthier, R. Genthon, A. A. Gouw, M. O. Habert, D. M. Holtzman, M. Kivipelto, S. Lista, J. L. Molinuevo, S. E. O’Bryant, G. D. Rabinovici, C. Rowe, S. Salloway, L. S. Schneider, R. Sperling, M. Teichmann, M. C. Carrillo, J. Cummings, C. R. Jack, Jr. and G. Proceedings of the Meeting of the International Working, A. D. the American Alzheimer's Association on The Preclinical State of, July and U. S. A. Washington Dc, Alzheimer's Dementia, 2016, 12, 292–323 CrossRef PubMed.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS PubMed.
- Q. Zhang, C. Li, B. Yin, J. Yan, Y. Gu, Y. Huang, J. Chen, X. Lao, J. Hao, C. Yi, Y. Zhou, J. C. W. Cheung, S. H. D. Wong and M. Yang, Bioact. Mater., 2024, 42, 165–177 CAS.
- J. J. Palop and L. Mucke, Nat. Neurosci., 2010, 13, 812–818 CrossRef CAS PubMed.
- J. Wang, Y. Fan, Y. Tan, X. Zhao, Y. Zhang, C. Cheng and M. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 36615–36621 CrossRef CAS PubMed.
- J. Wang, Y. Gu, X. Liu, Y. Fan, Y. Zhang, C. Yi, C. Cheng and M. Yang, Int. J. Mol. Sci., 2022, 23, 9890 CrossRef PubMed.
- Y. Gu, Q. Zhang, H. Huang, K. H. W. Ho, Y. Zhang, C. Yi, Y. Zheng, R. C. C. Chang, E. S. Wang and M. Yang, Theranostics, 2024, 14, 6218–6235 CrossRef CAS PubMed.
- Q. Zhang, B. Yin, Y. Huang, Y. Gu, J. Yan, J. Chen, C. Li, Y. Zhang, S. H. D. Wong and M. Yang, Biosens. Bioelectron., 2023, 230, 115270 CrossRef CAS PubMed.
- Y. A. R. Mahaman, K. S. Embaye, F. Huang, L. Li, F. Zhu, J. Z. Wang, R. Liu, J. Feng and X. Wang, Ageing Res. Rev., 2022, 74, 101544 CrossRef CAS PubMed.
- H. P. Karki, Y. Jang, J. Jung and J. Oh, J. Nanobiotechnol., 2021, 19, 72 CrossRef CAS PubMed.
- Q. Li, H. Huo, Y. Wu, L. Chen, L. Su, X. Zhang, J. Song and H. Yang, Adv. Sci., 2023, 10, e2202051 CrossRef PubMed.
- B. Yin, Q. Zhang, X. Xia, C. Li, W. K. H. Ho, J. Yan, Y. Huang, W. P. Wang, J. Hao, J. Wang, H. Chen, S. H. D. Wong and M. Yang, Theranostics, 2022, 12, 5914–5930 CrossRef CAS PubMed.
- H. Lai, R. Zhang, H. Chen, W. Yin, X. Yan, S. Xiao, C. Y. K. Lam, Y. Gu, J. Yan, K. Hu, J. Shi and M. Yang, ACS Sens., 2024, 9, 4860–4869 CrossRef PubMed.
- B. P. Nanda, P. Rani, P. Paul, S. S. Ganti and R. Bhatia, J. Pharm. Anal., 2024, 14, 100959 CrossRef PubMed.
- B. Yin, W. K. H. Ho, X. Xia, C. K. W. Chan, Q. Zhang, Y. M. Ng, C. Y. K. Lam, J. C. W. Cheung, J. Wang, M. Yang and S. H. D. Wong, Small, 2023, 19, e2206762 CrossRef PubMed.
- K. Kant, R. Beeram, Y. Cao, P. S. S. Dos Santos, L. Gonzalez-Cabaleiro, D. Garcia-Lojo, H. Guo, Y. Joung, S. Kothadiya, M. Lafuente, Y. X. Leong, Y. Liu, Y. Liu, S. S. B. Moram, S. Mahasivam, S. Maniappan, D. Quesada-Gonzalez, D. Raj, P. Weerathunge, X. Xia, Q. Yu, S. Abalde-Cela, R. A. Alvarez-Puebla, R. Bardhan, V. Bansal, J. Choo, L. C. C. Coelho, J. de Almeida, S. Gomez-Grana, M. Grzelczak, P. Herves, J. Kumar, T. Lohmueller, A. Merkoci, J. L. Montano-Priede, X. Y. Ling, R. Mallada, J. Perez-Juste, M. P. Pina, S. Singamaneni, V. R. Soma, M. Sun, L. Tian, J. Wang, L. Polavarapu and I. P. Santos, Nanoscale, 2024, 9, 2085–2166 RSC.
- H. Liu, X. Gao, C. Xu and D. Liu, Theranostics, 2022, 12, 1870–1903 CrossRef CAS PubMed.
- Y. Chen, Y. Bai, X. Wang, H. Zhang, H. Zheng and N. Gu, Biosens. Bioelectron., 2023, 219, 114744 CrossRef CAS PubMed.
- R. Chen, S. Li, S. Ren, D. Han, K. Qin, X. Jia, H. Zhou and Z. Gao, Adv. Colloid Interface Sci., 2024, 331, 103235 CrossRef CAS PubMed.
- S. Ghosh, A. D. Ranjan, S. Das, R. Sen, B. Roy, S. Roy and A. Banerjee, Nano Lett., 2021, 21, 10–25 CrossRef CAS PubMed.
- Y. Zhang, X. Mi, X. Tan and R. Xiang, Theranostics, 2019, 9, 491–525 CrossRef CAS PubMed.
- N. Feliu, M. Hassan, E. Garcia Rico, D. Cui, W. Parak and R. Alvarez-Puebla, Langmuir, 2017, 33, 9711–9730 CrossRef CAS PubMed.
- M. Saini and K. K. Sadhu, Sens. Actuators, B, 2020, 303, 127086 CrossRef CAS.
- S. K. Singh, V. Balendra, A. A. Obaid, J. Esposto, M. A. Tikhonova, N. K. Gautam and B. Poeggeler, Metallomics, 2022, 14, 1–14 CrossRef CAS PubMed.
- V. Da Ros, L. Oddo, Y. Toumia, E. Guida, S. Minosse, L. Strigari, S. Strolin, G. Paolani, F. Di Giuliano, R. Floris, F. Garaci, S. Dolci, G. Paradossi and F. Domenici, Pharmaceutics, 2023, 15, 217 CrossRef CAS PubMed.
- A. D’Andrea, L. Severini, F. Domenici, S. Dabagov, V. Guglielmotti, D. Hampai, L. Micheli, E. Placidi, M. Titubante, C. Mazzuca, G. Paradossi and A. Palleschi, ACS Appl. Mater. Interfaces, 2021, 13, 24207–24217 CrossRef PubMed.
- J. Olesiak-Banska, M. Waszkielewicz, P. Obstarczyk and M. Samoc, Chem. Soc. Rev., 2019, 48, 4087–4117 RSC.
- S. M. van de Looij, E. R. Hebels, M. Viola, M. Hembury, S. Oliveira and T. Vermonden, Bioconjugate Chem., 2022, 33, 4–23 CrossRef CAS PubMed.
- M. Hembury, N. Beztsinna, H. Asadi, J. B. van den Dikkenberg, J. D. Meeldijk, W. E. Hennink and T. Vermonden, Biomacromolecules, 2018, 19, 2841–2848 CrossRef CAS PubMed.
- E. A. Carter, Spectrochim. Acta, Part A, 2011, 80, 1 CrossRef CAS PubMed.
- B. Nieto-Ortega and T. Burgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS PubMed.
- J. Shen, Y. Dai, F. Xia and X. Zhang, Prog. Polym. Sci., 2022, 135, 101622 CrossRef CAS.
- L. Guerrini and R. A. Alvarez-Puebla, ACS Omega, 2021, 6, 1054–1063 CrossRef CAS PubMed.
- S. Chatterjee, X.-Y. Lou, F. Liang and Y.-W. Yang, Coord. Chem. Rev., 2022, 459, 214461 CrossRef CAS.
- G. Festa, F. Mallamace, G. M. Sancesario, C. Corsaro, D. Mallamace, E. Fazio, L. Arcidiacono, V. Garcia Sakai, R. Senesi, E. Preziosi, G. Sancesario and C. Andreani, Int. J. Mol. Sci., 2019, 20, 4126 CrossRef CAS PubMed.
- H. Ma, S. Liu, N. Zheng, Y. Liu, X. X. Han, C. He, H. Lu and B. Zhao, Anal. Chem., 2019, 91, 9376–9381 CrossRef CAS PubMed.
- H. Amijee, C. Bate, A. Williams, J. Virdee, R. Jeggo, D. Spanswick, D. I. Scopes, J. M. Treherne, S. Mazzitelli, R. Chawner, C. E. Eyers and A. J. Doig, Biochemistry, 2012, 51, 8338–8352 CrossRef CAS PubMed.
- E. Y. Hayden, G. Yamin, S. Beroukhim, B. Chen, M. Kibalchenko, L. Jiang, L. Ho, J. Wang, G. M. Pasinetti and D. B. Teplow, J. Neurochem., 2015, 135, 416–430 CrossRef CAS PubMed.
- A. De Simone, M. Naldi, D. Tedesco, A. Milelli, M. Bartolini, L. Davani, D. Widera, M. L. Dallas and V. Andrisano, ACS Omega, 2019, 4, 12308–12318 CrossRef CAS PubMed.
- M. Li, D. Lu, R. You, H. Shen, L. Zhu, Q. Lin and Y. Lu, J. Phys. Chem. C, 2022, 126, 12651–12659 CrossRef CAS.
- H. Li, Y. Tian, S. Yan, L. Ren, R. Ma, W. Zhao, H. Zhang and S. Dou, Coatings, 2024, 14, 530 CrossRef CAS.
- C. Dallari, E. Lenci, A. Trabocchi, V. Bessi, S. Bagnoli, B. Nacmias, C. Credi and F. S. Pavone, ACS Sens., 2023, 8, 3693–3700 CrossRef CAS PubMed.
- L. Guerrini, R. Arenal, B. Mannini, F. Chiti, R. Pini, P. Matteini and R. A. Alvarez-Puebla, ACS Appl. Mater. Interfaces, 2015, 7, 9420–9428 CrossRef CAS PubMed.
- M. Shorie, V. Kumar, H. Kaur, K. Singh, V. K. Tomer and P. Sabherwal, Mikrochim. Acta, 2018, 185, 158 CrossRef PubMed.
- S. He, Y. M. E. Kyaw, E. K. M. Tan, L. Bekale, M. W. C. Kang, S. S. Kim, I. Tan, K. P. Lam and J. C. Y. Kah, Anal. Chem., 2018, 90, 6071–6080 CrossRef CAS PubMed.
- J. Wu, Y. Zhang, J. Wang, Z. Ling, X. Yan, X. Lyu, J. Fang, M. Cheng, M. Zhao, T. Ban, Y. Liu and Y. Li, Anal. Chem., 2024, 96, 15735–15745 CrossRef CAS PubMed.
- A. Foti, C. D’Andrea, V. Villari, N. Micali, M. G. Donato, B. Fazio, O. M. Marago, R. Gillibert, M. Lamy de la Chapelle and P. G. Gucciardi, Materials, 2018, 11, 440 CrossRef PubMed.
- W. K. H. Ho, Z. Y. Bao, X. Gan, K. Y. Wong, J. Dai and D. Lei, J. Phys. Chem. Lett., 2019, 10, 4692–4698 CrossRef CAS PubMed.
- T. Marino, N. Russo, M. Toscano and M. Pavelka, Interdiscip. Sci., 2010, 2, 57–69 CrossRef CAS PubMed.
- G. Drochioiu, M. Manea, M. Dragusanu, M. Murariu, E. S. Dragan, B. A. Petre, G. Mezo and M. Przybylski, Biophys. Chem., 2009, 144, 9–20 CrossRef CAS PubMed.
- T. Kirsipuu, A. Zadoroznaja, J. Smirnova, M. Friedemann, T. Plitz, V. Tougu and P. Palumaa, Sci. Rep., 2020, 10, 5686 CrossRef CAS PubMed.
- M. Sendzik, M. J. Pushie, E. Stefaniak and K. L. Haas, Inorg. Chem., 2017, 56, 15057–15065 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb00632e |
| ‡ Willis Kwun Hei Ho and Qin Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |