
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating the anti-cancer photodynamic efficiency of molybdenum-iodide nanoclusters via ligand design†
Tomáš
Přibyl
 *ab,
Karel
Škoch
b,
Dmytro
Bavol
b,
Ivana
Křížová
c,
Jaroslav
Zelenka
a,
Tomáš
Ruml
a,
Kamil
Lang
b and
Kaplan
Kirakci
*b
*ab,
Karel
Škoch
b,
Dmytro
Bavol
b,
Ivana
Křížová
c,
Jaroslav
Zelenka
a,
Tomáš
Ruml
a,
Kamil
Lang
b and
Kaplan
Kirakci
*b
aDepartment of Biochemistry and Microbiology, University of Chemistry and Technology Prague, Technická 5, 166 28 Praha 6, Czech Republic
bInstitute of Inorganic Chemistry of the Czech Academy of Sciences, Řež 1001, 250 68 Husinec-Řež, Czech Republic. E-mail: kaplan@iic.cas.cz
cDepartment of Biotechnology, University of Chemistry and Technology Prague, Technická 5, 166 28 Praha 6, Czech Republic
First published on 29th April 2025
Abstract
Ligand-stabilized metallic nanoclusters are emerging as promising agents for photodynamic therapy (PDT). This study explores how precisely tailored ligands can optimize the anti-cancer PDT efficiency of molybdenum-iodide nanoclusters. Utilizing click chemistry, we synthesized a series of triazolate-capped nanoclusters by reacting Na2[Mo6I8(N3)6] with dibenzo[a,e]cyclooctyne (DBCO) derivatives. Dynamic light scattering and luminescence measurements confirmed that the bulky DBCO moieties effectively stabilized the nanoclusters in aqueous media, while the appended side chains dictated their colloidal behavior and photosensitizing capabilities. In vitro experiments with HeLa cancer cells revealed that the side chain's nature critically influences cellular uptake and phototoxicity. Positively charged nanoclusters exhibited enhanced cell membrane interactions and potent phototoxic effects, whereas negatively charged counterparts displayed reduced internalization and diminished PDT efficacy. Notably, the nanoclusters maintained consistent phototoxicity even after prolonged exposure to aqueous media, demonstrating the robust stability conferred by the DBCO ligands. These results higlight the potential for fine-tuning molybdenum-iodide nanocluster properties to optimize PDT applications, achieving a delicate balance between water stability, cellular interaction, and phototoxicity, thereby isolating key parameters that govern PDT efficiency.
Introduction
Photodynamic therapy (PDT) is an emerging non-invasive modality that utilizes light-activated photosensitizers to generate reactive oxygen species (ROS) for selective cancer cell destruction. Recently, ligand-stabilized nanoclusters have gained significant attention in PDT due to their tunable optical properties, enhanced biocompatibility, and potential for targeted delivery.1–3 The ligand design plays a crucial role in determining the photodynamic efficiency of nanoclusters by influencing their stability, cellular uptake, and ROS generation capability.4–6 Understanding the interplay between ligand chemistry and photodynamic performance is essential for advancing nanocluster-based PDT strategies.The octahedral molybdenum cluster (Mo6) complexes are attractive ligand-stabilized nanocluster biological applications such as photodynamic therapy,3,7–13 radiodynamic therapy14–16 photoinactivation of bacteria3,17–20 or optical sensing in solution.21,22 These complexes, denoted [Mo6Li8La6]n, are constituted of an octahedron of molybdenum atoms surrounded by eight strongly bonded inner ligands (Li) and six labile inorganic/organic apical ligands (La). Upon UV-visible light or X-ray excitation, they display red phosphorescence and produce singlet oxygen O2(1Δg) in high quantum yields.23–27 As opposed to organic dyes/photosensitizers such as porphyrin, they are less prompt to photobleaching due to their inorganic nature and do not experience self-quenching of their excited states at high concentrations or in the solid state.16,19 Being made of relatively abundant elements, they represent a more sustainable alternative to phosphors or photosensitizers based on noble metals- or rare-earth.28
Chemical stability of the photosensitizer in biological medium is paramount to understanding the ligand's role in the photodynamic efficiency. Unfortunately, Mo6 complexes generally undergo a displacement of apical ligands by water molecules. This feature constitutes an obstacle to their use in biological media, as it generally leads to severe aggregation and deterioration of their photosensitizing and cellular uptake properties.3,8,9 In a few cases, hydrolysis could be mitigated by using bulky hydrophobic apical ligands such as diphenylphosphinate or trifluoroacetate.7,14 Recently, we demonstrated that the click-chemistry reaction between Na2[Mo6I8(N3)6] and dibenzo[a,e]cyclooctyne (DBCO)-functionalized biocompatible polymers led to water-stable nanoparticles where the photophysical properties of the attached Mo6 complexes remained unchanged for a long time period when in phosphate-buffered saline (PBS).29,30 We surmise that the bulky DBCO moieties were the reason for the enhanced water stability. More generally, nanoparticle stability in biological environments depends on the ligand chemistry and the overall nanostructure design and shape. Steric hindrance and surface hydrophilicity are key factors in preventing aggregation, limiting hydrolysis, and maintaining colloidal stability in phosphate-buffered saline (PBS) and serum-containing media.31
Herein, we prepared by click chemistry a series of Mo6 complexes with triazolate apical ligands bearing the DBCO moieties (Fig. 1). The organic function of the side-chain of the DBCO moieties (carboxylic acid, amine, or PEG) were chosen to vary the charge of the complexes and to evaluate the corresponding effect on the properties of the complexes in aqueous media and on their activity towards cancer cells. The complexes were characterized by 1H and 13C NMR, elemental analysis and high-resolution mass spectroscopy. Dynamic light scattering in deionized water and PBS was used to study their colloidal stability and zeta potentials. Their luminescence properties were measured in the solid state and PBS dispersions as well as their photosensitizing ability. Biological activity against cancerous HeLa cells was tested in order to understand how the apical ligand influences interactions with cells. This knowledge is crucial in identifying potential routes for further research.
 | ||
| Fig. 1 Structure of complexes 1, 2, and 3 with indicated apical ligand (L). | ||
Material and methods
Reagents and general procedures
Na2[Mo6I8(N3)6] was prepared using the previously published procedure.32 Molybdenum, iodine, sodium azide, 11,12-didehydro-oxodibenz[b,f]azocine-5(6H)-hexanoic acid (C21H19NO3, DBCO–COOH), 3-amino-1-(11,12-didehydrodibenz[b,f]azocin-5(6H)-yl)-1-propanone (C18H16N2O, DBCO–NH2), m-dPEG®12-DBCO (C45H68N2O14, DBCO–PEG), phosphate-buffered saline (10× concentrate, BioPerformance Certified) (PBS), and 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) were obtained from Sigma-Aldrich and used as received. Solvents for the syntheses were purchased from Penta (Czech Republic).Preparation of Na2[Mo6I8(N3–DBCO–COOH)6] (1)
A mixture containing 48 mg (25 μmol) of Na2[Mo6I8(N3)6] and 60 mg (180 μmol) of DBCO–COOH (C21H19NO3) in 1 mL of dimethyl sulfoxide (DMSO) was stirred at room temperature for 48 h. Subsequently, 40 mL of diethyl ether was added, resulting in the formation of a red precipitate. The precipitate was then washed once with 5 mL of acetone and twice with 40 mL of diethyl ether to yield 81 mg (84%) of orange powder after drying under reduced pressure. HRMS (ESI−) calcd for [Mo6I8(C21H19N4O3)6]2−m/z 1926.77, found: 1926.8; elemental analysis calcd (%) for C126H114N24Na2O18I8Mo6: C, 38.91; H, 2.95; N, 8.64; found: C, 38.62; H, 3.15; N 8.92; 1H NMR (d6-DMSO, 600 MHz, 293 K): 11.95 (br, 1H, COOH), 7.77–7.10 (m, 8H, CHAr), 5.82 (m, 1H, NCH2), 4.73 (m, 1H, NCH2), 1.91 (m, 1H, CH2D), 1.83 (m, 1H, CH2D), 1.74 (m, 1H, CH2A), 1.56 (m, 1H, CH2A), 1.32–1.09 (m, 4H, CH2B and CH2C); 13C{1H} NMR (d6-DMSO, 151 MHz, 293 K): 174.8 (COOH), 171.5 (CO-amide), [143.9, 141.8](C-triazole), [141.0, 134.6, 133.8, 133.4, 130.9, 130.3, 129.4, 128.9, 128.8, 128.0, 127.1, 126.8](CAr and CHAr), 53.2 (NCH2DBCO), 33.9 (CH2D), 33.1 (CH2A), 24.6 (CH2B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) orC), 24.3 (CH2BorC).
orC), 24.3 (CH2BorC).
Preparation of Na2[Mo6I8(N3–DBCO–NH2)6] (2)
A mixture containing 98 mg (52 μmol) of Na2[Mo6I8(N3)6] and 100 mg (362 μmol) of DBCO–NH2 (C18H16N2O) in 1 mL of DMSO was stirred at room temperature for 48 h. Subsequently, 40 mL of diethyl ether was added, resulting in the formation of a red precipitate. The precipitate was then washed once with 5 mL of acetone and twice with 40 mL of diethyl ether to yield 175 mg (95%) of orange powder after drying under reduced pressure. HRMS (ESI−) calcd for [Mo6I8(C18H16N5O)6(H2O)4]2−m/z 1791.75, found: 1791.75; elemental analysis calcd (%) for C108H96N30Na2O6I8Mo6: C, 36.57; H, 2.73; N, 11.85; found: C, 36.95; H, 2.79; N 12.14; 1H NMR (d6-DMSO, 600 MHz, 293 K) selected resonances: [7.67, 7.56, 7.39, 7.15](all br, CHAr), [5.82, 4.38](br s, CH2NDBCO); 13C{1H} NMR (d6-DMSO, 151 MHz, 293 K): [171.0, 166.8](CO-amide), 53.1 [CH2NDBCO].(NB: NMR analysis showed two organic fragments in approximately 2:1 ratio. The 1H and 13C{1H} NMR shifts were similar in the aromatic region, with a notable difference mainly for the amine-substituted pendant arm. This suggests that 2 contains two protonated amine groups and exhibits zwitterionic character in solution. As a result, we report the chemical shifts only for the characteristic moieties. Details are available in the ESI†)
Preparation of Na2[Mo6I8(N3–DBCO–PEG)6] (3)
A mixture containing 32 mg (17 μmol) of Na2[Mo6I8(N3)6] and 100 mg (116 μmol) of m-dPEG®12-DBCO (C45H68N2O14) in 1 mL of DMSO was stirred at room temperature for 48 h. Subsequently, 40 mL of diethyl ether was added, resulting in the formation of a red precipitate. The precipitate was then washed once with 5 mL of acetone and twice with 40 mL of diethyl ether to yield 103 mg (87%) of orange powder after drying under reduced pressure. HRMS (ESI−) calcd for [Mo6I8(C45H68N5O14)4(OH)2(CH3OH)(H2O)]2−m/z 2648.31, found: 2648.31; elemental analysis calcd (%) for C270H408N30Na2O84I8Mo6: C, 45.96; H, 5.83; N, 5.96; found: C, 46.08; H, 6.07; N 5.82; 1H NMR (d6-DMSO, 600 MHz, 293 K) selected resonances: 7.67 (br, 1H, NH), 7.72–7.49 (m, 2H, CHAr), 7.45–7.32 (m, 3H, CHAr), 7.21–7.11 (m, 3H, CHAr), 5.82 (br, 1H, NCH2DBCO), 4.37 (br, 1H, NCH2DBCO), 3.53–3.33 (m, PEG-11), 3.22 (s, 3H OMe), 3.02 (br, 2H, CH2α), [1.89, 1.80, 1.71, 1.55, 1.54, 1.30](each br, 1H, CH2ABC); 13C{1H} NMR (d6-DMSO, 151 MHz, 293 K): [171.1, 171.0](CO), [143.4 (m), 141.3 (m)](C-triazole), [140.5, 134.0, 133.4, 133.0, 130.1, 129.8, 129.0, 128.5, 128.2, 127.5, 126.6, 126.3](CAr and CHAr), 71.3 (CH2β), 69.8 (PEG-11), 58.1 (OMe), 52.7 (NCH2DBCO), 38.3 (CH2α), 34.4 (CH2A/C), 32.2 (CH2A/C), 20.6 (CH2B).Instrumental techniques
NMR spectra were recorded on a JEOL Delta 600 spectrometer at 20 °C. 1H and 13C NMR chemical shifts (δ in ppm) are given relative to TMS and referenced to the residue solvent signal (d6-DMSO: δH = 2.50, δC = 39.5).33 Assignment of the peaks was supported by 2D COSY, HSQC and HMBC experiments. C, H, N elemental analyses were performed using a Vario EL Cube analyser (Elementar GmbH). Mass spectrometry measurements were performed on an Orbitrap Exploris 120 spectrometer equipped with heated electrospray ionisation (HESI) in negative mode using nitrogen (5.0 Messer) as a collision gas. Samples were dissolved in acetonitrile. The dispersions in deionized water and PBS were prepared by adding 100 μL of DMSO solution of 1, 2, or 3 (1 mg mL−1) to 10 mL of aqueous medium. The size distribution and zeta potentials in PBS were determined by dynamic light scattering on a particle size analyser, Zetasizer Nano ZS (Malvern, UK). UV-vis absorption spectra of solutions were measured on a PerkinElmer Lambda 35 spectrometer. Phosphorescence properties were analysed on an FLS1000 spectrometer (Edinburgh Instruments, UK) using a cooled PMT-980 photon detection module (Edinburgh Instruments, UK). X-ray excitation was performed using a XS-1 X-ray sample chamber (Edinburgh Instruments, UK). The signal of singlet oxygen, O2(1Δg), was measured on a FLS1000 spectrometer (Edinburgh Instruments, UK) using a Hamamatsu H10330-75 photomultiplier (Hamamatsu, Japan). The FLS1000 spectrometer was also used for time-resolved phosphorescence measurements (λex = 405 nm, VPLED Series), and the recorded decay curves were fitted to exponential functions by the Fluoracle software (v. 2.13.2, Edinburgh Instruments, UK). Phosphorescence quantum yields were recorded using a Quantaurus QY C11347-1 spectrometer (Hamamatsu, Japan). Dispersions were saturated with air or argon to assure different oxygen concentrations for phosphorescence analyses. Evaluation of the photoinduced oxidation of DCFH-DA was performed by illuminating water dispersions of 1, 2, or 3 (1.5–25 μM) and DCFH-DA (10 μM) with a 12 × 10 W LED source (Cameo) (460 nm, 18 mW cm−2) for 5 min, and measuring the fluorescence at 488/525 nm.Cell cultivation and viability measurement
Human cervical adenocarcinoma cells (HeLa) were cultured in Eagle's Medium Essential Medium (EMEM) supplemented with 5% fetal bovine serum (FBS) and 0.5 mM L-glutamine in a humidified chamber tempered to 37 °C and a 5% CO2 atmosphere. The cell tests were conducted after seeding. On the next day, the medium was replaced with fresh phenol red-free medium, with (full medium) or without FBS supplementation, depending on the experimental condition. The Mo6 complexes were dissolved in DMSO, and the fixed volumes with different concentrations of the compounds were mixed with cells. The final concentration of DMSO was less than 1% v/v DMSO. After 2 h of incubation with the Mo6 complexes, the cells were illuminated with blue light (460 nm, 15 min, 18 mW cm−2) or kept in the dark. The next day, the cells were tested for viability using the resazurin method, and the fluorescence was measured at an excitation/emission wavelength of 560/590 nm. Control experiments were performed under the same conditions in the absence of the Mo6 complexes.Confocal microscopy
Spinning disc confocal microscopy (Andor XD Revolution) was used for the intracellular localization of Mo6 complexes. The cells were seeded to a 96-well plate with a glass bottom (Cellvis) and treated in the same way as used for the viability measurement in the full medium. After a 2-h incubation, living cell microscopy was performed. Lysosomes were stained with LysoTracker Green for localization.Uptake
HeLa cells were seeded into 12-well plates in the full medium. After 24 h, cells were treated with indicated concentrations of 1, 2, and 3 for indicated times, then washed with PBS, trypsinized, and analysed by flow cytometry (BD FACSaria III). The excitation wavelength was set to 405 nm, and emission was recorded within 655–685 nm.ROS measurement
HeLa cells were seeded into a 96-well plate and treated as for the viability measurement. After irradiation (460 nm, 15 min, 18 mW cm−2) or keeping in the dark, cells were treated with DCFH-DA (10 μM) and incubated for 30 min. The medium was then aspirated, and fluorescence was measured using an excitation wavelength of 488 nm and an emission wavelength of 525 nm. Confocal microscopy was utilised to visualise intracellular ROS.Cell death
The cells were seeded into a 6-well plate in the same way as described for the viability testing. Cells were exposed to light (460 nm, 15 min, 18 mW cm−2), collected, and processed according to the manufacturer's protocol using the Dead Cell Apoptosis Kit with Annexin V Alexa Fluor™ 488 & Propidium Iodide (PI) (Thermo Fisher Scientific).Results and discussion
Synthesis and characterization
1, 2, and 3, with the general formula Na2[Mo6I8L6] (Fig. 1), were prepared by the Michael-Huisgen 1,3-dipolar cycloaddition of dibenzo[a,e]cyclooctyne (DBCO) derivates to azido apical ligands of Na2[Mo6I8(N3)6] in DMSO, as previously described.9 The formation of 1, 2, and 3 was evidenced by NMR measurements (Fig. S1–S12, ESI†). Initially, the DBCO-based ligands were characterized using 1H, 13C{1H} NMR, and the individual signals were assigned based on 2D NMR experiments. For individual peak assignment see ESI.† Subsequently, complexes were measured after the reaction with Na2[Mo6I8(N3)6]. A significant difference was the broadening of signals typical for Mo6 cluster complexes with organic ligands;9 nevertheless, characteristic changes in the NMR spectra were still observable (for full description, see ESI†). Firstly, there was a significant downfield shift of the methylene linker signals by approximately 0.5 ppm in 1H NMR after the cycloaddition. However, the most important change was the absence of signals corresponding to the alkyne bond in 13C NMR. Signals of acetylene moiety (100–115 ppm) were replaced by a new set of signals corresponding to the triazolate fragment (140–145 ppm). This region is typical for Mo-coordinated triazolate complexes, and the observed values are consistent with related complexes previously described in the literature.9,34,35 In the case of the DBCO complex containing an amine pendant (2), we observed the aforementioned characteristic features indicating complete reaction of the alkyne, resulting in the formation of the triazolate complex. However, NMR analysis revealed the presence of two organic fragments in an approximate 2:1 ratio. The sets of 1H and 13C NMR shifts were highly similar in the aromatic region, with a significant difference primarily observed for the amine-substituted pendant arm. We, therefore, hypothesize that the resulting complex contains two protonated amine groups, suggesting that 2 exhibits a zwitterionic character in the solution. HRMS-ESI and CHN analysis showed high purity of prepared compounds (Fig. S13, ESI†).
Colloidal characteristics
As the complexes were not readily soluble in water, they were dissolved in DMSO and added to deionized water (pH ∼ 5.5, low ionic strength) or phosphate-buffered saline (PBS) (pH 7.4, high ionic strength), at a concentration of 0.01 mg mL−1 (= 1% v/v). The size distributions of the resulting dispersions were studied by dynamic light scattering, revealing the presence of nanoaggregates (Table 1 and Fig. 2A, B and Fig. S14, ESI†).| Complex | Solvent | Number mean d/nm | Z-average d/nm | PDI | Zeta potential /mV |
|---|---|---|---|---|---|
| a Zeta potential distribution. b Average from three independent measurements. | |||||
| 1 | H2O | 64 ± 7 | 189 | 0.33 | −40 ± 8a |
| PBS | 25 ± 8 | 97 | 0.43 | −13 ± 1b | |
| PBS, 8 days | 28 ± 8 | 165 | 0.25 | −15 ± 2b | |
| 2 | H2O | 73 ± 34 | 165 | 0.38 | 40 ± 5a |
| PBS | 127 ± 53 | 180 | 0.21 | 14 ± 1b | |
| PBS, 8 days | 250 ± 113 | 311 | 0.17 | 10 ± 1b | |
| 3 | H2O | 222 ± 61 | 307 | 0.31 | −20 ± 10a |
| PBS | 50 ± 26 | 179 | 0.28 | −13 ± 1b | |
| PBS, 8 days | 72 ± 36 | 177 | 0.28 | −14 ± 1b | |
 | ||
| Fig. 2 Size distributions by number of fresh dispersions of 1, 2, and 3 in deionized water as obtained by dynamic light scattering (A). Size distributions by number of fresh (B) and 8-days old (C) PBS dispersions of 1, 2, and 3 as obtained by dynamic light scattering. Zeta potentials distributions of fresh dispersions of 1, 2, and 3 in deionized water (D). | ||
The mean size by number of freshly prepared dispersions of 1 was 64 ± 7 nm in deionized water and 25 ± 8 nm in PBS. The smaller size obtained in PBS when compared to deionized water could be attributed to the deprotonation of the carboxylic acid side chain at high pH. The mean size by number of freshly prepared dispersions of 2 was 73 ± 34 nm in deionized water and 127 ± 53 nm in PBS. In this case, the smaller size obtained in deionized water when compared to PBS could arise from the protonation of the amine group of the apical ligand's side chain at low pH. The mean size by number of freshly prepared dispersions of 3 was 222 ± 61 nm in deionized water and 50 ± 26 nm in PBS. There, the smaller size of aggregates could be due to the stabilizing effect of the sodium cations interacting with the PEG side-chain.15
After 8 days in PBS, the size distribution of dispersions of 1 and 3 did not significantly change, while dispersion of 2 showed a moderate increase in its mean size, contrasting with previously reported bicyclo[6.1.0]non-4-yne (BCN) derivatives which showed considerable aggregation after 8 days in PBS (Fig. 2C).9 The zeta potential values in deionized water were −40 ± 8, 40 ± 5, and −20 ± 10 mV, for 1, 2, and 3 respectively, reflecting the apparent charge of the functional groups of the ligands (Fig. 2D). Study of the pH dependence of the zeta potentials from pH ∼ 4 to pH ∼ 8 confirmed these trends. A decrease in the zeta potential of 1 was observed when increasing pH due to deprotonation of the carboxylic acid, while the zeta potential of 2 decreased due to deprotonation of the amine group. The zeta potential of 3 decreased when increasing pH despite the absence of ionizable functional group, possibly due to interactions between the PEG functional group and ionic species in solution (Fig. S15, ESI†). In PBS, a mitigation of the surface charge of the nanoaggregates was observed, with zeta potentials of −13 ± 1 mV, 14 ± 1 mV, and −13 ± 1 mV, for 1, 2, and 3 respectively. The relative stability over time suggests that the bulky DBCO derivates represent more water-protective moieties than BCN derivatives, resulting in the prevention of the hydrolysis of the complex, which can lead to severe aggregation.9
Photophysical properties
The phosphorescence properties of the complexes were first examined in the solid state in an air atmosphere. Upon excitation at 400 nm, red phosphorescence, typical for Mo6 complexes, was observed with maxima around 690 nm, high emission quantum yields of 0.58, 0.49, and 0.25 and long emission lifetimes of 124, 97, and 41 μs for 1, 2 and 3, respectively (Fig. S16 (ESI†) and Table 2). As Mo6 complexes have previously been reported to be efficient scintillators,14,15,26 their radioluminescence upon irradiation with an X-ray source (60 KeV, 200 mA) was also recorded. It showed comparable spectral features with its phosphorescence counterpart, although a small red shift was observed for 2 (Fig. S16, ESI†). While in PBS dispersions prepared in the same way as for the DLS experiments, the Mo6 complexes maintained their phosphorescence properties with similar emission spectra and high emission quantum yields of 0.22, 0.23, and 0.34 and lifetimes of 79, 78, and 105 μs for 1, 2 and 3, respectively (Fig. 3A). Photostability of 1, 2 and 3 was tested by illuminating air-saturated PBS dispersions under the same conditions as those used for the phototoxicity experiments, revealing no significant changes in the intensity and position of emission spectra (Fig. S17, ESI†). The 8 days old PBS dispersions showed comparable phosphorescence spectra, quantum yields and lifetimes relative to fresh PBS dispersions, which confirmed the long-term stability of the Mo6 complexes in this medium (Fig. S18 and S19, ESI†).| Complex | λ L/nm | ϕ L | ϕ air | τ L/μs | τ air/μs |
|
|---|---|---|---|---|---|---|
a
λ
L – phosphorescence maximum (λexc = 400 nm); τL and τair – amplitude average lifetimes in Ar- and air-saturated PBS, recorded at the maximum of emission (λexc = 405 nm); ΦL and Φair phosphorescence quantum yield in Ar- and air-saturated PBS (λexc = 320–400 nm);  is the fraction of the formed triplet states quenched by oxygen in air atmosphere is the fraction of the formed triplet states quenched by oxygen in air atmosphere  . .
|
||||||
| 1 solid | 689 | n.a. | 0.58 ± 0.03 | n.a. | 124 | n.a. |
| 1 PBS, fresh | 685 | 0.22 ± 0.02 | 0.05 ± 0.01 | 79 | 19 | 0.76 |
| 1 PBS,8 days | 692 | 0.22 ± 0.02 | 0.06 ± 0.01 | 79 | 20 | 0.76 |
| 2 solid | 685 | n.a. | 0.49 ± 0.03 | n.a. | 97 | n.a. |
| 2 PBS, fresh | 688 | 0.23 ± 0.02 | 0.11 ± 0.01 | 78 | 43 | 0.45 |
| 2 PBS,8 days | 688 | 0.22 ± 0.02 | 0.11 ± 0.01 | 72 | 40 | 0.44 |
| 3 solid | 684 | n.a. | 0.25 ± 0.02 | n.a. | 41 | n.a. |
| 3 PBS, fresh | 694 | 0.34 ± 0.03 | 0.07 ± 0.01 | 105 | 22 | 0.79 |
| 3 PBS,8 days | 695 | 0.34 ± 0.03 | 0.07 ± 0.01 | 104 | 20 | 0.79 |

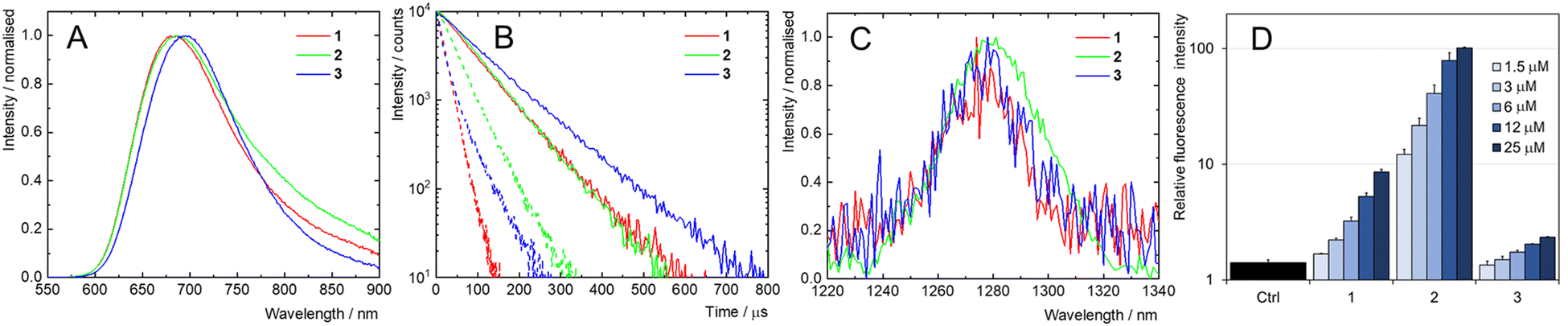 | ||
| Fig. 3 (A) Phosphorescence spectra of argon-saturated PBS dispersions of 1, 2 or 3, λex = 400 nm. (B) Phosphorescence decay kinetics of dispersions of 1, 2 or 3 in oxygen-free (full line) or air-saturated (dotted line) PBS, λex = 405 nm, λem = 700 nm. (C) Normalized phosphorescence spectrum of O2(1Δg) in oxygen-saturated PBS dispersions of 1, 2 or 3, λex = 400 nm. (D) Photoinduced (460 nm, 18 mW cm−2) formation of ROS stimulated by 1, 2, and 3 (1.5–25 μM) in PBS, determined using DCFH-DA (10 μM). Increase in relative fluorescence intensity indicates oxidation of DCFH-DA to highly green fluorescent 2′,7′-dichlorofluorescein. The results were normalized to the fluorescence intensity of DCFH-DA without illumination. Ctrl represents the intensity of DCFH-DA when irradiated alone. | ||
In air-saturated PBS dispersions, the phosphorescence quantum yields and lifetimes were considerably reduced, indicating the quenching of the emissive triplet states by molecular oxygen (Fig. 3B). The fraction of the formed triplet states quenched by oxygen in air atmosphere, 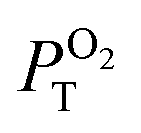 , was 0.76, 0.45 and 0.79 for 1, 2, and 3 respectively suggesting the formation of O2(1Δg), whose production was further confirmed by measuring its phosphorescence signal at 1270 nm (Fig. 3C). The potential of Mo6 complexes to oxidize substrates in PBS was further evaluated using the oxidation sensitive probe DCFH-DA which converts to green fluorescent 2′,7′-dichlorofluorescein upon oxidative stress (Fig. 3D). In this set of experiment, the ranking of the oxidation rate was 2 ≫ 1 > 3, contrasting with the oxygen quenching trends gained from the photophysical measurements (3 > 1 > 2). The observed differences seem related to the surface charge of the nanoaggregates of the Mo6 complexes, i.e., negatively charged DCFH-DA is attracted to positively charged 2, resulting in a higher oxidation ability, whereas negatively charged 1 and 3 produce lower oxidation yield of DCFH-DA due to electrostatic repulsion forces. The nature of the side-chain could also play a role by scavenging O2(1Δg), as PEG derivates were reported to display antioxidant properties,36 which would explain why 3 shows the lowest oxidation rate of DCFH-DA despite having the highest
, was 0.76, 0.45 and 0.79 for 1, 2, and 3 respectively suggesting the formation of O2(1Δg), whose production was further confirmed by measuring its phosphorescence signal at 1270 nm (Fig. 3C). The potential of Mo6 complexes to oxidize substrates in PBS was further evaluated using the oxidation sensitive probe DCFH-DA which converts to green fluorescent 2′,7′-dichlorofluorescein upon oxidative stress (Fig. 3D). In this set of experiment, the ranking of the oxidation rate was 2 ≫ 1 > 3, contrasting with the oxygen quenching trends gained from the photophysical measurements (3 > 1 > 2). The observed differences seem related to the surface charge of the nanoaggregates of the Mo6 complexes, i.e., negatively charged DCFH-DA is attracted to positively charged 2, resulting in a higher oxidation ability, whereas negatively charged 1 and 3 produce lower oxidation yield of DCFH-DA due to electrostatic repulsion forces. The nature of the side-chain could also play a role by scavenging O2(1Δg), as PEG derivates were reported to display antioxidant properties,36 which would explain why 3 shows the lowest oxidation rate of DCFH-DA despite having the highest 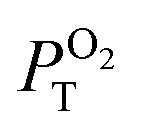 .
.
Photodynamic therapy of cancer cells
The biological properties of 1, 2, and 3 were investigated on human cervical cancer cells (HeLa) in the context of photodynamic therapy. HeLa cells are one of the most common models of cancer cells and were employed in previous studies investigating the phototoxicity of Mo6 complexes, which allows us a broader comparison.3,7–9,14 Because of their red phosphorescence, the uptake and intracellular localization of the Mo6 complexes could be investigated by flow cytometry and fluorescence microscopy (Fig. 4). Flow cytometry evidenced a more efficient uptake for 2 compared to 1 and 3. This feature is probably related to the positive charge of 2 in PBS, which allows strong interaction with negatively charged cell surfaces. Confocal microscopy showed that 2 is localized at the cell membrane, similar to the previously reported Mo6 complex with aminated ligands.9 On the other hand, 1 was localized in vesicles, while 3 showed colocalization with lysosomes, which are the storage organelle for some of the previously reported Mo6 complexes.7,8,29 | ||
| Fig. 4 Uptake of 1, 2, and 3 in HeLa cells measured by flow cytometry, (A) in the concentration range of 2.5–10 μM, 120 min incubation, (B) at a concentration of 5 μM, incubation time range 30–120 min. The results were normalized to the highest response. (C) Confocal microscopy images of 1 (5 μM), 2 (1 μM), and 3 (20 μM), (in red), LysoTracker Green (in green), and merged channels. | ||
No dark toxicity was observed for the studied Mo6 complexes at concentrations up to 40 μM. We attribute the effect of slightly increased viability to subtoxic oxidative stress (Fig. 5A and Fig. S20B, ESI†). The elevation of metabolic activity in response to mild stress was previously reported.37 After being illuminated with blue light, 1 and 2 caused a concentration-dependent reduction in viability, with IC50 values of 9.3 ± 0.5 and 0.42 ± 0.01 μM, respectively (Fig. 5, Table 3). In contrast, 3 did not exhibit a phototoxic effect despite being located in lysosomes, probably because of limited uptake and low photosensitizing ability (Fig. 5 and Table 3). Overall, 2 displays strong phototoxic effects towards HeLa cells relative to other reported Mo6 complexes, probably due to localization at the cell membrane as previously reported for the BCN derivative with amino group.9
 | ||
| Fig. 5 Effect of complex 1, 2, and 3 after 2-hour incubation on HeLa cells in the full medium (A) kept in the dark and (B) illuminated with blue light (460 nm, 15 min, 18 mW cm−2). | ||
| Complex | Phototoxicity (IC50/μM) | Dark toxicity (IC50/μM) | ||
|---|---|---|---|---|
| w/ FBS | w/o FBS | w/ FBS | w/o FBS | |
| 1 | 9.26 ± 0.54 | 0.67 ± 0.02 | >40 | >40 |
| 8-days aged | 18.2 ± 0.6 | 1.23 ± 0.02 | >40 | >40 |
| 2 | 0.42 ± 0.01 | 0.22 ± 0.01 | >40 | >40 |
| 8-days aged | 0.18 ± 0.01 | 0.23 ± 0.01 | >40 | >40 |
| 3 | >40 | >40 | >40 | >40 |
| 8-days aged | >40 | >40 | >40 | >40 |
When nanoparticles are present in biological fluids, they become coated by a protein corona, which can impact their cellular uptake.38 Therefore, we studied the influence of FBS during incubation of the Mo6 complexes with cells prior to illumination. The phototoxic effect of 2 was only marginally influenced when incubation was performed in the medium without serum proteins, (IC50 = 0.22 ± 0.01 μM) (Fig. S20 (ESI†) and Table 3). In contrast, the phototoxicity of 1 was dramatically increased when using FBS-free serum (IC50 = 0.67 ± 0.02), probably owing to its negative charge which leads to stronger interaction with serum proteins, a feature that was already observed for several Mo6 complexes (Fig. S20 (ESI†) and Table 3).8,9
In the case of 3, no increase in phototoxicity was observed after incubation without FBS, suggesting that the cause of its low efficiency is not related to interaction with serum proteins. Hydrolytic stability under physiological conditions and related robustness of the biological activity is a critical issue in Mo6 cluster development. Thus, we have also examined how an 8-days ageing treatment in water/DMSO (v/v = 9/1) affected the efficiency of the Mo6 complexes. Thereafter, the efficiency of 1 and 2 remained relatively unchanged when compared to previously reported BCN derivates which experienced a dramatic decrease of their phototoxicity upon ageing (Fig. S21 (ESI†) and Table 3).9
Assessment of the oxidative stress in cells after administration of 1, 2, and 3 was performed by using DCFH-DA added after illumination. In line with the trends gained from phototoxicity experiments, increased ROS levels were found in cells treated with micromolar concentrations of 2 and one order of magnitude higher concentrations of 1, while 3 caused negligible oxidative stress (Fig. 6A, B and D). Finally, the cell death mode was determined using Annexin-V staining. A strong population of Annexin-V-positive cells, which suggests apoptosis, was found in cells treated with 80 μM of 1 and 2.5 μM of 2, 4 h after illumination, while no effects were observed for 3 at a concentration of 80 μM. (Fig. 6C). Overall, there is a clear correlation between cellular uptake, phototoxicity, oxidative stress, and apoptotic cell death induction.
 | ||
| Fig. 6 The formation of secondary ROS in HeLa cells as a result of primary generation of ROS during illumination (A) and kept in the dark (B), determined by DCFH-DA (10 μM). (C) The cell death mode after illumination (1 – 80 μM, 2 – 2.5 μM, 3 – 80 μM). (D) Representative confocal images of formed secondary ROS in the illuminated HeLa cells, using DCFH-DA, excitation wavelength 488 nm, emission wavelength 525 nm. | ||
Conclusion
This study highlights the potential of water-stable triazolate complexes of octahedral molybdenum clusters for fine-tuning of the photodynamic efficiency, focusing on how the structure of apical ligands influences the colloidal and photosensitizing properties as well as the biological activity. 1, 2, and 3, featuring bulky DBCO derivatives, exhibited greater stability in the aqueous environment than the previously studied BCN derivatives. Dynamic light scattering showed that the Mo6 complexes form nanoaggregates in aqueous medium and suggested that the steric bulk of DBCO derivatives effectively protects the molybdenum cluster core from hydrolysis, resulting in stable dispersions in PBS over time. Zeta potential measurements highlighted the influence of the side chain on the surface charge of the aqueous dispersion of the Mo6 complexes.The photophysical characterization confirmed the stability of all Mo6 complexes, which maintained high luminescence quantum yields and O2(1Δg) generation in PBS, even after extended storage, underscoring their robustness. The nature of the apical ligands affected the differential behaviour during photodynamic therapy. The positively charged 2, featuring an amino group at the terminal position, exhibited the highest phototoxicity due to enhanced electrostatic interaction with the negatively charged cellular membranes and effective O2(1Δg) production. In contrast, negatively charged 1, bearing the carboxylate, showed moderate phototoxicity that improved significantly in the absence of serum proteins, likely due to strong interaction with serum proteins. Finally, 3 carrying PEG-functionalized ligands displayed lysosomal localization, but showed no phototoxic effects due to poor uptake, emphasizing the crucial role of ligand design in determining biological activity.
These findings highlight the versatility of Mo6 complexes as tunable photodynamic agents. The insights gained from this work provide a foundation for the rational design of water-stable molybdenum-based photosensitisers tailored for specific biological applications. Future research will focus on optimising ligand functionalisation to further enhance selectivity and efficacy for photobiological applications.
Author contributions
Conceptualization (K. K.), supervision (K. L, and T. R.), investigation (K. K, K. S., D. B, T. P., and I. K.), writing – original draft (T. P., K. K.), writing – review and editing (J. Z., and K. L.), project administration (T. R., K. L.), and funding acquisition (K. K.).Data availability
All relevant data are included in the manuscript and its ESI.† And detailed data are available from the corresponding authors upon reasonable request. Data are stored and archived at the digital repository of the Institute of Inorganic Chemistry operated by the Library of the Czech Academy of Sciences. Data are accessible on https://asep.lib.cas.cz/arl-cav/en/search/, and alternatively on request at data_repository@iic.cas.cz.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was supported by the EIG Concert Japan (No. 8I24001).Notes and references
- S. M. van de Looij, E. R. Hebels, M. Viola, M. Hembury, S. Oliveira and T. Vermonden, Bioconjugate Chem., 2021, 33, 4–23 CrossRef.
- H. C. D. Medeiros, C. Yang, C. K. Herrera, D. Broadwater, E. Ensink, M. Bates, R. R. Lunt and S. Y. Lunt, Chem. – Eur. J., 2023, 29, e202202881 CrossRef CAS PubMed.
- K. Kirakci, J. Zelenka, M. Rumlová, J. Cvačka, T. Ruml and K. Lang, Biomater. Sci., 2019, 7, 1386–1392 RSC.
- H. Fakhouri, M. P. Bakulić, I. Zhang, H. Yuan, D. Bain, F. Rondepierre, P.-F. Brevet, Ž. S. Maršić, R. Antoine, V. Bonačić-Koutecký and D. Maysinger, Commun. Chem., 2023, 6, 97 CrossRef CAS.
- B. A. Thomas-Moore, S. Dedola, D. A. Russell, R. A. Field and M. J. Marín, Nanoscale Adv., 2023, 5, 6501–6513 RSC.
- C. Fan, S. Zhai, W. Hu, S. Chi, D. Song and Z. Liu, RSC Adv., 2021, 11, 21384–21389 RSC.
- K. Kirakci, J. Demel, J. Hynek, J. Zelenka, M. Rumlová, T. Ruml and K. Lang, Inorg. Chem., 2019, 58, 16546–16552 CrossRef CAS PubMed.
- K. Kirakci, J. Zelenka, I. Křížová, T. Ruml and K. Lang, Inorg. Chem., 2020, 59, 9287–9293 CrossRef CAS PubMed.
- K. Kirakci, M. Kubáňová, T. Přibyl, M. Rumlová, J. Zelenka, T. Ruml and K. Lang, Inorg. Chem., 2022, 61, 5076–5083 CrossRef CAS PubMed.
- N. Brandhonneur, T. Hatahet, M. Amela-Cortes, Y. Molard, S. Cordier and G. Dollo, Eur. J. Pharm. Biopharm., 2018, 125, 95–105 CrossRef CAS PubMed.
- N. Brandhonneur, Y. Boucaud, A. Verger, N. Dumait, Y. Molard, S. Cordier and G. Dollo, Int. J. Pharm., 2021, 592, 120079 CrossRef CAS.
- A. Verger, G. Dollo, S. Martinais, Y. Molard, S. Cordier, M. Amela-Cortes and N. Brandhonneur, J. Pharm. Sci., 2022, 111, 3377–3383 CrossRef CAS PubMed.
- A. O. Solovieva, Y. A. Vorotnikov, K. E. Trifonova, O. A. Efremova, A. A. Krasilnikova, K. A. Brylev, E. V. Vorontsova, P. A. Avrorov, L. V. Shestopalova and A. F. Poveshchenko, J. Mater. Chem. B, 2016, 4, 4839–4846 RSC.
- K. Kirakci, J. Zelenka, M. Rumlová, J. Martinčík, M. Nikl, T. Ruml and K. Lang, J. Mater. Chem. B, 2018, 6, 4301–4307 RSC.
- K. Kirakci, T. N. Pozmogova, A. Y. Protasevich, G. D. Vavilov, D. V. Stass, M. A. Shestopalov and K. Lang, Biomater. Sci., 2021, 9, 2893–2902 RSC.
- M. Koncošová, M. Rumlová, R. Mikyšková, M. Reiniš, J. Zelenka, T. Ruml, K. Kirakci and K. Lang, J. Mater. Chem. B, 2022, 10, 3303–3310 RSC.
- R. Guégan, X. Cheng, X. Huang, Z. Nemeckova, M. Kubáňová, J. Zelenka, T. Ruml, F. Grasset, Y. Sugahara and K. Lang, Inorg. Chem., 2023, 62, 14243–14251 CrossRef.
- E. D. Novikova, E. V. Pronina, Y. A. Vorotnikov, L. S. Adamenko, A. Y. Alekseev, A. M. Shestopalov, A. R. Tsygankova, T. Y. Gusel’nikova, P. Kubát and K. Kirakci, J. Environ. Chem. Eng., 2023, 11, 110796 CrossRef CAS.
- K. Kirakci, T. K. N. Nguyen, F. Grasset, T. Uchikoshi, J. Zelenka, P. Kubát, T. Ruml and K. Lang, ACS Appl. Mater. Interfaces, 2020, 12, 52492–52499 CrossRef CAS.
- A. Beltrán, M. Mikhailov, M. N. Sokolov, V. Pérez-Laguna, A. Rezusta, M. J. Revillo and F. Galindo, J. Mater. Chem. B, 2016, 4, 5975–5979 RSC.
- J. Elistratova, M. Mikhailov, V. Burilov, V. Babaev, I. Rizvanov, A. Mustafina, P. Abramov, M. Sokolov, A. Konovalov and V. Fedin, RSC Adv., 2014, 4, 27922–27930 RSC.
- S. Muñoz, L. Alvarado-Soto, J. Gaete, C. Morales-Verdejo and R. Ramírez-Tagle, ACS Omega, 2022, 7, 19152–19157 CrossRef PubMed.
- A. W. Maverick, J. S. Najdzionek, D. MacKenzie, D. G. Nocera and H. B. Gray, J. Am. Chem. Soc., 1983, 105, 1878–1882 CrossRef CAS.
- K. Kirakci, P. Kubát, M. Dušek, K. Fejfarová, V. Šícha, J. Mosinger and K. Lang, Eur. J. Inorg. Chem., 2012, 3107–3111 CrossRef CAS.
- M. N. Sokolov, M. A. Mihailov, E. V. Peresypkina, K. A. Brylev, N. Kitamura and V. P. Fedin, Dalton Trans., 2011, 40, 6375–6377 RSC.
- K. Kirakci, P. Kubát, K. Fejfarová, J. Martinčík, M. Nikl and K. Lang, Inorg. Chem., 2016, 55, 803–809 CrossRef CAS PubMed.
- D. V. Evtushok, A. R. Melnikov, N. A. Vorotnikova, Y. A. Vorotnikov, A. A. Ryadun, N. V. Kuratieva, K. V. Kozyr, N. R. Obedinskaya, E. I. Kretov and I. N. Novozhilov, Dalton Trans., 2017, 46, 11738–11747 RSC.
- L. Smith, T. Ibn-Mohammed, I. M. Reaney and S. L. Koh, Resour., Conserv. Recycl., 2021, 166, 105317 CrossRef CAS.
- K. Kirakci, R. Pola, M. R. Tavares, M. Pechar, T. Přibyl, I. Křížová, J. Zelenka, T. Ruml, T. Etrych and K. Lang, Mater. Adv., 2023, 4, 6389–6395 RSC.
- T. Přibyl, M. Rumlová, R. Mikyšková, M. Reiniš, A. Kaňa, K. Škoch, J. Zelenka, K. Kirakci, T. Ruml and K. Lang, Inorg. Chem., 2024, 63, 4419–4428 CrossRef.
- H. Li, H. Liu, T. Nie, Y. Chen, Z. Wang, H. Huang, L. Liu and Y. Chen, Biomaterials, 2018, 178, 620–629 CrossRef CAS.
- K. Kirakci, P. Kubát, M. Kučeráková, V. Šícha, H. Gbelcová, P. Lovecká, P. Grznárová, T. Ruml and K. Lang, Inorg. Chim. Acta, 2016, 441, 42–49 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- F.-C. Liu, J.-E. Liang, J.-Y. Jin, Y.-L. Lin, Y.-J. Chu, P.-S. Yang, G.-H. Lee and S.-M. Peng, J. Organomet. Chem., 2013, 735, 1–9 CrossRef CAS.
- A. D. Mironova, M. A. Mikhailov, K. A. Brylev, A. L. Gushchin, T. S. Sukhikh and M. N. Sokolov, New J. Chem., 2020, 44, 20620–20625 RSC.
- K. Juarez-Moreno, M. Ayala and R. Vazquez-Duhalt, Appl. Biochem. Biotechnol., 2015, 177, 1364–1373 CrossRef CAS.
- B. P. Dranka, G. A. Benavides, A. R. Diers, S. Giordano, B. R. Zelickson, C. Reily, L. Zou, J. C. Chatham, B. G. Hill, J. Zhang, A. Landar and V. M. Darley-Usmar, Free Radicals Biol. Med., 2011, 51, 1621–1635 CrossRef CAS PubMed.
- T. Kopac, Int. J. Biol. Macromol., 2021, 169, 290–301 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR characterizations, HR-MS spectra, additional luminescence data, and additional in vitro data. See DOI: https://doi.org/10.1039/d5tb00401b |
| This journal is © The Royal Society of Chemistry 2025 |