
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA carnitine-based BODIPY photosensitizer†
Larissa
Maierhofer‡
 a,
Ruth
Prieto-Montero‡
b,
Tamara
Cubiella
c,
Aitor
Díaz-Andrés
de,
Noelia
Morales-Benítez
a,
David
Casanova
*d,
Virginia
Martínez-Martínez
*b,
María-Dolores
Chiara
*c,
Enrique
Mann
*a and
Jose Luis
Chiara
*a
a,
Ruth
Prieto-Montero‡
b,
Tamara
Cubiella
c,
Aitor
Díaz-Andrés
de,
Noelia
Morales-Benítez
a,
David
Casanova
*d,
Virginia
Martínez-Martínez
*b,
María-Dolores
Chiara
*c,
Enrique
Mann
*a and
Jose Luis
Chiara
*a
aInstituto de Química Orgánica General (IQOG-CSIC), Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: mann@iqog.csic.es; jl.chiara@csic.es
bDepartamento de Química Física, Universidad del País Vasco-EHU, Facultad de Ciencia y Tecnología, Apartado 644, 48080 Bilbao, Spain. E-mail: virginia.martinez@ehu.eus
cInstituto de Investigación Sanitaria del Principado de Asturias, Instituto Universitario de Oncología del Principado de Asturias, Universidad de Oviedo, Hospital Universitario Central de Asturias, 33011 Oviedo, Spain. E-mail: mdchiara.uo@uniovi.es
dDonostia International Physics Center (DIPC), 20018 Donostia, Euskadi, Spain. E-mail: david.casanova@ehu.eus
ePolimero eta Material Aurreratuak: Fisika, Kimika eta Teknologia Saila, Kimika Fakultatea, Euskal Herriko Unibertsitatea (UPV/EHU), PK 1072, 20080 Donostia, Euskadi, Spain
First published on 19th February 2025
Abstract
Organelle-selective photodynamic therapy (PDT) has emerged as a promising approach to enhance the precision and efficacy of cancer treatment by targeting key cellular structures. In this study, we report the design of a novel carnitine-based BODIPY photosensitizer, probe 1, which retains mitochondrial selectivity while acting as both a fluorescent probe and a potent photosensitizer. Building on our previously developed mitochondria-targeting probe (R)-BCT-2, which is transported into the mitochondrial matrix by the inner membrane protein carnitine–acylcarnitine translocase (CAC), probe 1 incorporates two bromine atoms that enhance intersystem crossing, leading to a singlet oxygen quantum yield of ∼80%, while retaining sufficient fluorescence for effective cell staining in fluorescence microscopy. Theoretical calculations indicate that the carnitine moiety distorts chromophore planarity, reducing oscillator strength but enhancing spin–orbit coupling, which, together with the extended triplet lifetime, contributes to increased phototoxicity. Probe 1 co-localizes in both mitochondria and, to a lesser extent, in lysosomes, and this dual targeting may synergistically enhance phototoxic activity by amplifying cellular stress responses. Importantly, probe 1 demonstrated high phototoxicity upon green light irradiation, with IC50 values of 52 nm under normoxia and 117 nm under hypoxia, while remaining non-cytotoxic in the dark. These results suggest that probe 1 is a promising candidate for organelle-targeted PDT, particularly in hypoxic tumor environments where its dual organelle targeting could enhance therapeutic efficacy.
1. Introduction
Photodynamic therapy (PDT) is a non-invasive therapeutic approach that combines light, molecular oxygen, and a photosensitizer (PS) to generate reactive oxygen species (ROS). These ROS are cytotoxic and selectively induce cell death in targeted tissues, such as tumors.1–3 This method offers several advantages, including minimal systemic toxicity, high safety, noninvasiveness, a broad therapeutic range, and low risk of drug resistance, making it a highly attractive option for cancer treatment. Upon photoexcitation, a PS transitions to an excited singlet state (S1) and can subsequently convert to a triplet state through intersystem crossing (ISC). The long lifetime of this triplet allows efficient generation of ROS. ROS production can follow two mechanisms: electron and proton transfer reactions (Type I) produce ROS like hydroxyl radicals (OH˙) and superoxide (O2˙−), while energy transfer reactions (Type II) generate singlet oxygen (1O2), a highly reactive species.4 The latter mechanism is more commonly observed.In contrast to traditional treatment methods, such as surgery or chemotherapy, PDT is far less invasive and presents reduced systemic toxicity thanks to the selective internalization of the PS in the target tissue and the localized irradiation of the affected region.5,6 Despite these advantages, PDT still presents a number of significant shortcomings, some of the most important being the limited penetration depth of visible light, poor selectivity of PS for target cells, and the dependence of most PS on molecular oxygen, which can be problematic due to the common hypoxic environment of solid tumors. To address these challenges, there is a strong interest in improving the efficacy and applicability of PDT through the development of superior photosensitizers. In this context, targeting specific organelles, such as mitochondria and lysosomes,7–10 has emerged as a crucial strategy to enhance PDT efficacy in cancer treatment, given the short lifespan and limited diffusion radius of 1O2 (∼40 ns and ∼20 nm, respectively)11 within cells. Mitochondrial targeting, in particular, improves therapeutic outcomes due to the organelle's central role in apoptosis and its high concentrations of oxygen,12 which promotes ROS production. Some studies have shown that subcellular localization can be even more critical than the photochemical reactivity of the PS in achieving effective cell death.13 To be efficient in PDT, PSs must exhibit low dark toxicity, good aqueous solubility, and strong photophysical properties, including high triplet quantum yields (ΦT), long triplet-state lifetimes (τT), and efficient singlet oxygen generation (ΦΔ).
Herein, we address the challenge of developing the first photosensitizer capable of entering mitochondria in living cells through an active, protein-mediated transport mechanism, rather than the conventional passive and non-specific electrophoretic diffusion process. Our design builds upon previous work by our research group, in which we developed (R)-BCT-2 (Fig. 1), the first mitochondria-specific fluorescent probe actively transported into the mitochondrial matrix via an inner membrane protein, carnitine–acylcarnitine translocase (CAC), a key component of the carnitine shuttle system.14 Notably, this transport mechanism is independent of the mitochondrial transmembrane potential, distinguishing it from most commercially available mitochondria-targeting probes, which rely exclusively on passive electrophoretic uptake driven by the organelle's negative transmembrane potential. These “passive-electrophoretic” probes typically employ delocalized lipophilic cations, such as quaternary ammonium, triphenylphosphonium (TPP), guanidinium, or pyridinium-based groups, as mitochondria-targeting moieties attached to neutral core chromophores, including boron dipyrromethene (BODIPY) derivatives, coumarins, pyrenes, diketopyrrolopyrroles or xanthenes, or by directly using positively charged organic chromophores, such as cyanines or rhodamines.15–17
 | ||
| Fig. 1 General structure of the carnitine-based probe 1 and our previous (R)-BCT-2 bioimaging probe.14 | ||
The new fluorescent probe features the innovative and minimalist design of (R)-BCT-2 incorporating a BODIPY moiety as a highly efficient fluorophore and an L-carnitine molecule as a biotargeting element. These components are directly and orthogonally linked through the boron atom. This streamlined approach, which eliminates the need for polyatomic connectors, was inspired by the promising results of our previous work on synthesizing spiro-O-BODIPYs from F-BODIPYs and di-carboxylic or hydroxy acids.14,18–20 These rigid spiro structures exhibit superior photophysical properties compared to the more commonly studied F-BODIPYs, including enhanced photostability, higher fluorescence quantum yields, and reduced aggregation.19
Building on these promising results, we aimed to engineer a dual-purpose analog of the probe that retains its ability to selectively localize into mitochondria, independent of their membrane potential, while transforming it into an efficient photosensitizer for targeted PDT by incorporating heavy atoms into the BODIPY to increase spin–orbit coupling (SOC) and triplet state generation.
The new L-carnitine-based photosensitizer was photophysically characterized using steady-state and transient spectroscopy, complemented by ground and excited-state theoretical calculations. Its cellular localization and toxicity were assessed under both dark and irradiated conditions in two human squamous cell carcinoma lines, evaluated under normoxic as well as hypoxic conditions.
2. Results and discussion
2.1 Design and synthesis of the new probe
We have designed a new photosensitizer based on our previously developed mitochondria-selective fluorescent probe (R)-BCT-214 by incorporating bromine atoms at the available positions of the BODIPY chromophore unit to give compound 1 (Fig. 1). This small structural modification was expected to retain its mitochondrial selectivity while enhancing photodynamic activity by promoting ISC, and simultaneously preserving some fluorescence emission, thereby yielding a theranostic probe. To this end, two alternative synthetic approaches can be followed, both starting from the corresponding F-BODIPY 2, but differing in the sequence of steps for introducing the carnitine and the bromine atoms. In our first approach, bromine atoms were introduced first at the available positions of F-BODIPY 2 using N-bromosuccinimide under standard conditions yielding dibromo-BODIPY 3.21 This was followed by reaction with L-carnitine hydrochloride in the presence of excess TMSCl in acetonitrile under microwave irradiation, as previously described14 (Scheme 1). While the desired product was successfully obtained, purification proved challenging due to the formation of partially dehalogenated byproducts during the second reaction step. Ultimately, compound 1 was readily obtained in higher purity by directly dibrominating (R)-BCT-2 under the same conditions (Scheme 1). | ||
| Scheme 1 Synthesis of compound 1. | ||
2.2 Photophysical and electrochemical properties
We conducted a comparative study of the photophysical properties of dibromo-BODIPY 3 and its carnitine analog 1 to analyze the effect of introducing the carnitine moiety on the boron atom of the chromophore. UV-vis absorption and fluorescence spectra of both compounds were recorded in four different polar solvents (acetone, acetonitrile, ethanol, and water) (Fig. 2) to obtain relevant photophysical properties, while singlet oxygen production was measured in acetonitrile and methanol. | ||
| Fig. 2 Absorption (solid curves) and fluorescence spectra (dashed curves) of compounds 1 (a) and 3 (b) in different solvents (black: acetone, red; MeCN, green: EtOH, blue: water). | ||
Both compounds displayed the expected absorption and fluorescence profiles of BODIPY dyes in organic solvents, characterized by well-defined, intense, and narrow peaks, with absorption maxima at 525–530 nm and fluorescence maxima at 538–545 nm (Table 1). However, while compound 1 exhibited similar profiles in water due to its good solubility, attributed to the positively charged carnitine moiety, compound 3 showed a broader absorption peak with a shoulder at lower wavelengths, suggesting the formation of H-type aggregates due to its poor solubility in aqueous media.
| Compound | Solvent | λ ab (nm) | ε max (104 M−1 cm−1) | λ fl (nm) | Δν (cm−1) | Φ | τ (ns) | Φ Δ |
|---|---|---|---|---|---|---|---|---|
| λ ab: absorption peak wavelength; ε: molar absorption coefficient at peak; λfl: fluorescence peak wavelength, upon excitation at 490 nm; Δν: Stokes shift; Φ: fluorescence quantum yield using a solution of the commercial BODIPY PM567 in EtOH (Φ = 0.84) as reference; τ: fluorescence lifetime; ΦΔ: singlet oxygen quantum yield using a solution of MeS-BODIPY in MeCN (ΦΔ = 0.95) as ref. 22.a Lifetime with major contribution (>97%).b Same values in methanol-d4. | ||||||||
| 3 | Acetone | 525.0 | 8.10 | 538.0 | 460 | 0.20 | 1.55 | — |
| MeCN | 524.0 | 7.92 | 538.5 | 514 | 0.18 | 1.46 | 0.80b | |
| EtOH | 526.0 | 8.02 | 539.5 | 475 | 0.23 | 1.74 | — | |
| H2O + DMSO | 530.0 | 1.89 | — | — | <0.01 | — | — | |
| 1 | Acetone | 526.0 | 3.36 | 544.5 | 645 | 0.13 | 1.24a | — |
| MeCN | 525.0 | 3.41 | 541.5 | 580 | 0.13 | 1.19a | 0.79b | |
| EtOH | 528.0 | 3.45 | 544.0 | 557 | 0.14 | 1.36a | — | |
| H2O + DMSO | 526.0 | 1.56 | 544.0 | 629 | 0.08 | 1.05a | — | |
Compound 3 exhibited significantly higher molar absorption coefficients than 1 in organic solvents (Table 1). The reduced absorption of 1 can be attributed to the structural constraints introduced by the spiro-junction of the carnitine moiety, which induces a slight distortion of the chromophore's planar geometry, as confirmed by theoretical calculations (see below).
In terms of fluorescence properties, compound 3 showed quantum yields of approximately 0.20 in organic solvents, while 1 was slightly less fluorescent (Φ ≈ 0.10). In water, the fluorescence of 1 was only slightly reduced compared to organic solvents, whereas compound 3 exhibited almost no fluorescence. Compared to their bromine-free analogs (F-BODIPY 2 and (R)-BCT-2), which are highly fluorescent (Φ ≥ 0.85), both 3 and 1 have significantly lower quantum yields due to the heavy atom effect of the bromine atoms, which enhances SOC and ISC, increasing the triplet state population. Consequently, both 1 and 3 showed high singlet oxygen quantum yields, around 80% in polar protic and aprotic solvents (MeOH-d4 and MeCN, respectively).
Since highly populated triplet states are crucial for singlet oxygen generation and thus for the photosensitizing capabilities of a molecule, these states were studied using nanosecond transient absorption spectroscopy (flash photolysis) and the corresponding decay curves under different conditions (samples saturated with nitrogen, air, and oxygen). Compounds 1 and 3 were dissolved in acetonitrile and excited at 525 nm for the experiments. In both cases, we observed a negative signal between 500 and 600 nm, attributed to ground state bleaching associated with the S0 → S1 transition. The transient absorption peaks observed at 410–460 nm and 600–700 nm correspond to triplet-state absorptions T1 → Tn (see ESI,† Fig. S1). One of the key findings from these experiments is the difference in triplet-state lifetimes under deoxygenated conditions between compounds 1 and 3 (Table 2). Given that the triplet-state lifetime of a photosensitizer directly correlates with its phototoxic activity, the observed threefold increase in the triplet lifetime of 1 compared to 3 in the absence of oxygen suggests that the carnitine moiety in 1 contributes not only to mitochondrial targeting but also to enhancing the phototoxic properties of the probe. The lifetimes of both compounds decrease significantly with increasing oxygen concentration (air- and oxygen-saturated solutions), which is typical of triplet states.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 (1:1) for 1 and 3 at 77 K
:CH2Cl2 (1:1) for 1 and 3 at 77 K
| Compound | τ T0 (μs) | τ Tair (μs) | τ Tox (ns) |
|
|
λ ph (nm) | E T (kJ mol−1) | τ ph (ms) |
|---|---|---|---|---|---|---|---|---|
τ
T0: triplet life time under nitrogen; τTair: triplet life time in air; τTox: triplet lifetime in oxygen; 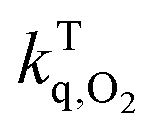 : rate constant for triplet state quenching by O2; : rate constant for triplet state quenching by O2;  : excited triplet fraction, which is deactivated by O2. λph: phosphorescence maxima; ET: triplet energy and τph: lifetimes at 77 K. : excited triplet fraction, which is deactivated by O2. λph: phosphorescence maxima; ET: triplet energy and τph: lifetimes at 77 K. |
||||||||
| 3 | 45 | 0.28 | 57 | 1.96 | 0.99 | 754 | 158.8 | 11 |
| 1 | 135 | 0.39 | 77 | 1.45 | 0.99 | 751 | 159.4 | 7 (74%) |
| 14 (26%) | ||||||||
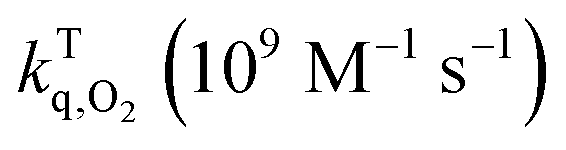

The experimental triplet state lifetimes at different oxygen concentrations were used to determine the triplet quenching constant  , referring to the deactivation of the triplet states by molecular oxygen, as well as the proportion of triplet excited state quenched by O2
, referring to the deactivation of the triplet states by molecular oxygen, as well as the proportion of triplet excited state quenched by O2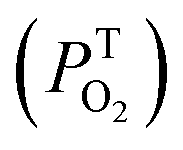 in acetonitrile at room temperature (Table 2). Both
in acetonitrile at room temperature (Table 2). Both  fractions were very close to one, indicating that most of the generated triplets are efficiently quenched by molecular oxygen. By comparing the obtained values to the diffusion coefficient of aromatic hydrocarbons in acetonitrile (kd ≈ 4.0 × 1010 M−1 s−1), we observed that both 1 and 3 adhere to the rule
fractions were very close to one, indicating that most of the generated triplets are efficiently quenched by molecular oxygen. By comparing the obtained values to the diffusion coefficient of aromatic hydrocarbons in acetonitrile (kd ≈ 4.0 × 1010 M−1 s−1), we observed that both 1 and 3 adhere to the rule  suggesting a process controlled by diffusion with a Type II mechanism for singlet oxygen generation in both compounds. This suggests that the main quenching mechanism is energy transfer from the photosensitizer's excited triplet state to molecular oxygen to generate singlet oxygen, rather than the formation of oxidative radical species by an electron transfer process.23
suggesting a process controlled by diffusion with a Type II mechanism for singlet oxygen generation in both compounds. This suggests that the main quenching mechanism is energy transfer from the photosensitizer's excited triplet state to molecular oxygen to generate singlet oxygen, rather than the formation of oxidative radical species by an electron transfer process.23
Phosphorescence experiments were conducted for both compounds. Emission spectra were registered at 77 K in a methanol/dichloromethane (1:1 v/v) solution, obtaining phosphorescence peaks at similar wavelengths, more specifically, 754 nm and 751 nm for 3 and 1, respectively (Fig. S2 and Table 2, ESI†). Phosphorescence lifetimes were measured exciting both samples at 500 nm and recording decay curves at 755 nm for 3 and 745 nm for 1. While 3 exhibits only one lifetime of 11 ms, a bi-exponential behavior with a shorter (7 ms, 74%) and a longer lifetime (14 ms, 26%) was observed for 1 (Table 2 and Fig. S3, ESI†). Explaining the origin of the bi-exponential behavior at 77 K would be outside the scope of this paper, but detailed information can be found in the literature.24
Taking into account the obtained phosphorescence peak wavelengths, the calculated triplet energies (158.8 kJ mol−1 for 3 and 159.4 kJ mol−1 for 1) are higher than the energy gap between the ground (O23Σg) and first excited singlet (O21Δg) states of oxygen, 94.2 kJ mol−1, which confirms that a Type II (energy transfer) process for singlet oxygen generation is feasible.
Despite their high singlet oxygen yields, cyclic voltammetry (Fig. S4, ESI†) reveals that both compounds exhibit reversible reduction waves and quasi-reversible oxidation signals, indicating the formation of a stable radical anion and an unstable radical cation, respectively. Moreover, their relatively low reduction potentials highlight a strong electron-accepting character, potentially enabling the generation of O2˙−via the Type I mechanism.25 In fact, the Type I photosensitization mechanism is enhanced in cellular environments, where biomolecules can participate in electron transfer with the excited-state photosensitizer,26–28 particularly in mitochondria, which have high levels of molecular oxygen and redox activity.29 Furthermore, longer triplet-state lifetimes, as seen in compound 1, would further increase the likelihood of these electron transfer interactions.30,31 Importantly, radical-based Type I mechanism is also effective in hypoxic conditions where the Type II singlet oxygen generation pathway is impaired.28,32,33
2.3 Theoretical calculations
Ground state geometry optimization at the DFT level (see details in ESI†) revealed significant alterations in the molecular geometry of the two BODIPY chromophores, induced by the introduction of the chiral L-carnitine moiety. While probe 3 presents planar conjugated systems aligned along two orthogonal σv symmetry planes (Fig. 3), the addition of L-carnitine displaces the boron atom out of the chromophore's conjugated plane. This displacement disrupts the planarity of the chromophore and alters the symmetry in both planes of probe 1. These structural changes in probe 1, combined with distinct electronic effects, significantly influence its photophysical properties compared to probe 3. In both molecules, the lowest excited singlet (S1) and triplet (T1) states result from the HOMO-to-LUMO electron promotion, with the frontier MOs corresponding to π and π* orbitals localized on the BODIPY unit. Interestingly, the computed oscillator strength associated with the S0 → S1 transition is considerably weaker in 1 (Tables S1 and S2, ESI†), which accounts for the lower molar absorption coefficient observed for different solvents (Table 1). Geometry optimization on the S1 potential energy surface for both probes primarily reveals bond-stretching distortions within the BODIPY unit, driven by the bonding and antibonding π-interactions in the HOMO and LUMO. No significant reorientation is observed between the different molecular units (ESI†). | ||
| Fig. 3 (a) Compound 3 with symmetry planes σ1v and σ2v. (b) Ground state structures optimized at the CAM-B3LYP/cc-pVDZ level of compounds 3 (left) and 1 (right), with perspective views along planes σ1v and σ2v, showing the symmetric and planar structure of probe 3versus the non-planar structure of probe 1. | ||
Following rapid S1 relaxation, molecules 1 and 3 can return to the ground state through fluorescent emission. Consistent with experimental data, the computed S1 vertical deexcitation energies are nearly identical for both compounds, with the S1 state strongly localized on the BODIPY moiety, explaining the weak dependence of emission wavelength on solvent polarity (Tables S3 and S4, ESI†). Alternatively, the S1 state can populate the triplet manifold via ISC. Based on the Fermi Golden Rule, our electronic structure calculations predict efficient ISC to a highly excited triplet state (T3), as the energy gap between these states at the S1 minimum is small, and SOC is relatively large. (Tables S5 and S6, ESI†). Notably, the SOC constant value for the S1 → T3 transition in 1 is twice that of 3, while the energy gap is smaller. This combination of a reduced energy gap and higher SOC in 1 suggests a more efficient ISC process. Molecular orbital analysis further supports these findings. In 3, the ISC transition occurs between a BODIPY-localized S1 and a BODIPY-localized T3 (Fig. 4a). Conversely, in 1, the transition involves a BODIPY-localized S1 state and a T3 state with significant charge transfer between the BODIPY core and the trimethylphenyl (TMP) group (Fig. 4b). Thus, according to El-Sayed's rule, ISC is more favored in 1.34,35 The orthogonal relative orientation between the π orbitals of BODIPY and TMP in 1 enhances the ISC process, emphasizing the impact of the L-carnitine moiety on the photophysical behavior of the chromophore.
 | ||
| Fig. 4 Main MO contribution to T3: (a) HOMO−2 of 3; (b) HOMO−3 of 1. | ||
2.4 Biological assays
Biological assays (live-cell imaging and cell viability assays) were performed to assess the cytotoxicity and phototoxicity of the new photosensitizer 1 using two human squamous cell carcinoma lines, SCC38 and SCC42B. In these experiments, both cell lines were incubated with varying concentrations of probe 1, ranging from 14 μM to 0.07 μM, followed by 20 minutes of green light irradiation to induce singlet oxygen production. Control experiments were conducted for each concentration, where cells were treated with the probe but kept in the dark. Fluorescence and bright-field microscopy conducted 2 hours post-irradiation (Fig. S5–S7, ESI†) revealed the following: (1) probe 1 was successfully internalized by the cells; (2) fluorescence was detectable at concentrations as low as 1.4 μM; and (3) cells kept in the dark remained viable after 2–4 hours, whereas those exposed to green light irradiation exhibited clear signs of apoptosis (rounded shape and increased granularity) at all concentrations. These results indicate that the new probe is phototoxic but not cytotoxic under the conditions tested.After this preliminary visual analysis of probe 1, we performed co-localization assays in SCC38 cells using MitoTracker Red CMXRos and LysoTracker Red DND-99, which selectively stain mitochondria and lysosomes, respectively. As shown in Fig. 5 and 6, probe 1 stained both organelles, with some staining also observed in other regions of the cells. This contrasts with the high mitochondrial selectivity of the non-brominated analog (R)-BCT-2, previously studied by our group.14 Although probe 1 displayed reduced mitochondrial selectivity compared to (R)-BCT-2, its additional localization in lysosomes could influence its phototoxic behavior, as previous studies have suggested potential synergistic effects when photosensitizers target both mitochondria and lysosomes.36–38 However, further investigation is needed to confirm whether this dual localization contributes to the observed phototoxicity.39
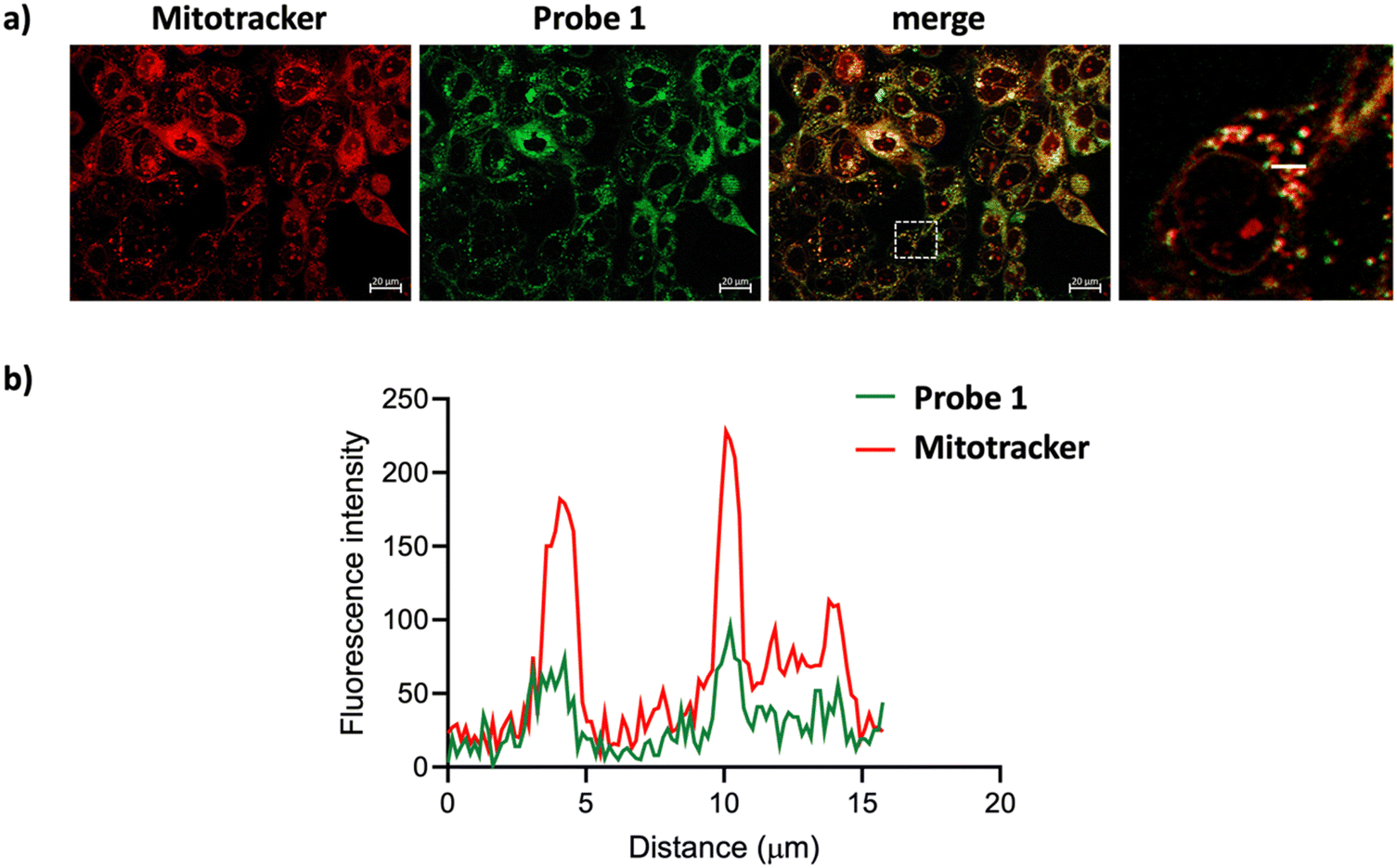 | ||
| Fig. 5 SCC38 cells treated with 14 μM 1 and 10 μM MitoTracker Red CMXRos for 30 min. (a) Fluorescence microscopy images of the sample. (b) Co-localization analysis of 1 and Mitotracker Red CMXRos. | ||
 | ||
| Fig. 6 SCC38 cells treated with 14 μM 1 and 10 nM LysoTracker Red DND-99 for 30 min. (a) Fluorescence microscopy images of the sample. (b) Co-localization analysis of 1 and LysoTracker Red DND-99. | ||
The presence of the two additional bromine substituents in 1 is the sole structural difference from (R)-BCT-2 and likely accounts for its unexpected localization in lysosomes. Bromine is known to increase lipophilicity, with a median ΔlogD7.4 contribution recently described as comparable to that of an ethyl group.40 This increased lipophilicity in probe 1, compared to (R)-BCT-2, likely contributes to its altered distribution in lysosomes, which is attributable to the hydrophobic environment of lysosomes and the enhanced permeability of their membranes to lipophilic molecules.41
Analysis of the images in Fig. 5 and 6 enabled us to calculate the degree of co-localization between probe 1 and MitoTracker or LysoTracker. Pearson's correlation coefficients of 0.826 and 0.865 (p < 0.0001) were observed for probe 1 with MitoTracker (Fig. 5) and LysoTracker (Fig. 6, ROI 1), respectively. These results demonstrate a strong co-localization of probe 1 with both mitochondria and lysosomes. In contrast, ROI 2 in Fig. 6 exhibited a reduced Pearson's coefficient (r = 0.363) for co-localization with LysoTracker. This reduction likely reflects competitive localization dynamics between the two organelles. Cellular regions often display heterogeneity in organelle concentration, activity, or dynamics, which can influence the spatial distribution of both the probe and the co-stain. Probe 1 shows a more uniform co-localization with MitoTracker, reflecting its stronger affinity for this organelle, leading to its preferential localization in mitochondrial regions and reducing its presence in lysosome-rich areas, thereby lowering co-localization metrics. Nonetheless, the possibility of colocalization with other nearby subcellular structures, such as lipid droplets, stress vesicles, or autophagosomes, cannot be excluded.
To evaluate the efficiency of probe 1 as a photosensitizer, we conducted cell viability assays on both cell lines following treatment with the probe and green light irradiation, as previously described. Twenty-four hours post-irradiation, SCC38 cells treated with varying concentrations of 1 were subjected to an MTS assay42 to assess cell proliferation, viability, and cytotoxicity. Consistent with microscopy results, the MTS assay confirmed that 1 was highly phototoxic, killing approximately 80% of the cells exposed to green light, even at submicromolar concentrations. In contrast, control experiments showed that more than 80% of cells kept in the dark remained viable up to 1 μM, the highest concentration tested, confirming the probe's lack of cytotoxicity in the absence of light (Fig. S8, ESI†). Furthermore, microscopy images confirmed that cells remained viable for over two hours following treatment with 14 μM of the probe (Fig. S5 and S6, ESI†).
In light of these promising results, we conducted more extensive cell viability assays using 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H tetrazolium, monosodium salt (WST-8),43 a more sensitive chromogenic indicator for cell viability compared to conventional tetrazolium reagents such as MTT and MTS (Fig. 7). In this experiment, we tested even lower concentrations (140 to 17.5 nM) and compared the potency of probe 1 under oxygenated (normoxic) and hypoxic (1% oxygen) conditions. As expected, control experiments with cells kept in the dark after incubation with 1, under both normoxia and hypoxia, showed high viability. In contrast, cells exposed to light exhibited significantly lower viability. At lower concentrations, probe 1 was more active under normoxic conditions, confirming the role of oxygen in the observed phototoxicity.
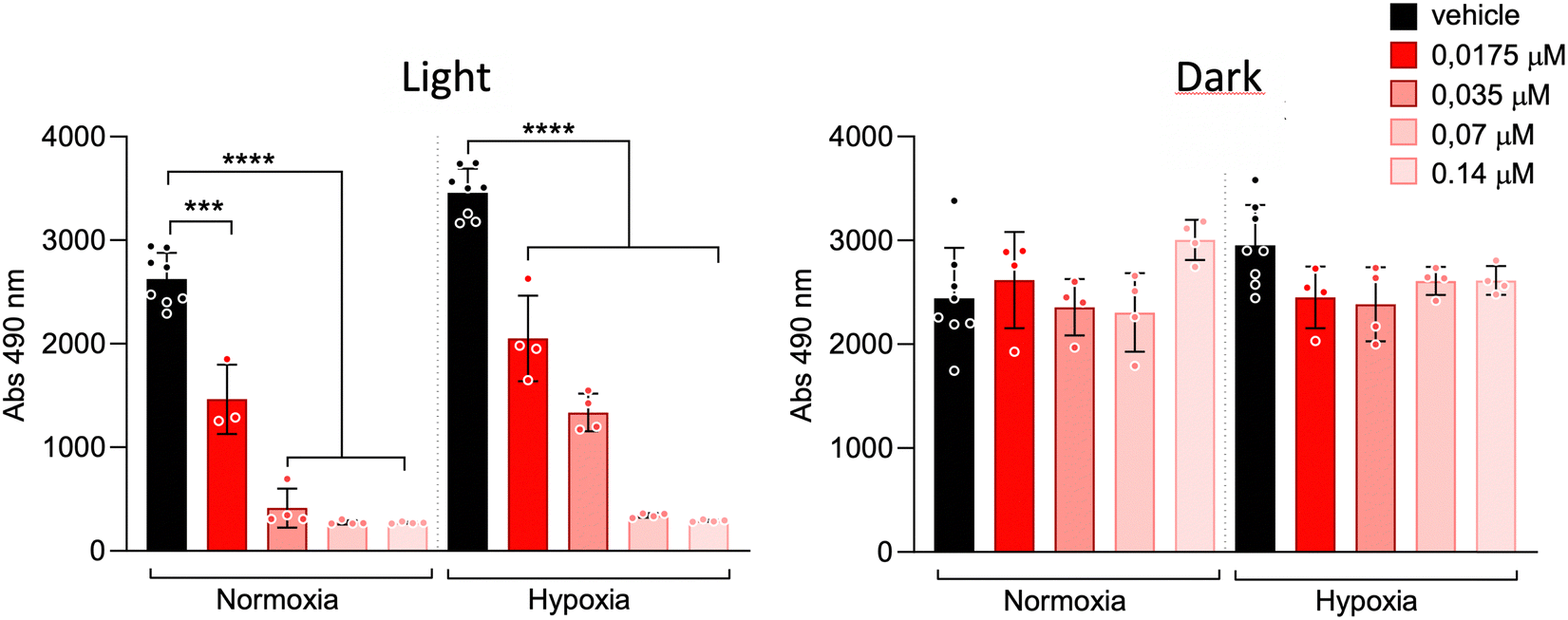 | ||
| Fig. 7 WST-8 cell viability assay of probe 1 at lower concentrations under normoxic and hypoxic conditions in SCC38 cells. | ||
Using the data from this assay, we calculated the IC50 values for probe 1 under normoxic and hypoxic conditions (Fig. S9, ESI†), confirming its high efficiency even at nanomolar concentrations, with 50% cell death at 52 nM in normoxia and 117 nM in hypoxia. The high photoactivity observed under hypoxic conditions may result from the activation of a Type I mechanism within the mitochondria's distinct, oxygen-rich redox microenvironment. This radical-based pathway remains effective even when the Type II singlet oxygen mechanism is impaired due to low oxygen levels.
Flow cytometry was used to assess cell death in SCC38 cells treated with probe 1, both in the dark and under green light irradiation, under normoxic and hypoxic conditions. Cell death generally occurs through two mechanisms: apoptosis, an active, programmed process, and necrosis, a passive form of cell death. A key early marker of apoptosis is the loss of phospholipid asymmetry, indicated by the translocation of phosphatidylserine to the outer plasma membrane. This shift is detected using annexin V, which binds specifically to phosphatidylserine and can be visualized through fluorescent labeling in flow cytometry. Importantly, this externalization occurs while the plasma membrane remains intact, allowing cells to exclude membrane-impermeant dyes, such as 7-AAD, which serves as an indicator of late apoptosis or necrosis. Flow cytometry analysis produced results consistent with those from the MTS and WST-8 assays in SCC38 cells (Fig. 8). In all control experiments (including both, light and dark conditions; in the absence of probe 1; under normoxic or hypoxic conditions), as well as in assays with 0.14 μM of probe 1 kept in the dark, the majority of cells remained viable for over two hours. In contrast, nearly all cells treated with 1 and exposed to green light for 20 minutes were either dead or severely damaged within two hours post-irradiation. Under normoxic conditions, probe 1 induced apoptosis in approximately 50% of the cells, while under hypoxia, the apoptotic rate was still notably high at 30%, demonstrating that probe 1 retains significant photodynamic activity even in low-oxygen conditions.
 | ||
| Fig. 8 Degree of apoptosis under normoxic (Nx) and hypoxic (Hx) conditions, determined by flow cytometry (Annexin V) with SCC38 cells. | ||
The presence of the carnitine moiety in probe 1 plays a critical role in enhancing its photodynamic biological activity compared to structurally related BODIPY photosensitizers reported in the literature. Table 3 presents a set of structurally related BODIPY photosensitizers, featuring iodine atoms at the C-2 and C-6 positions, with fully characterized photophysical, photodynamic, and phototoxicity data in human cell lines. These compounds either lack mitochondria-targeting groups or incorporate conventional targeting moieties such as trimethylammonium or triphenylphosphonium groups. Compounds 4 and 5, which do not contain specific targeting groups, exhibit minimal dark toxicity and moderate phototoxicity. However, their IC50 values are two orders of magnitude higher than those of probe 1, despite the presence of iodine atoms, which are generally associated with greater ISC efficiencies and enhanced singlet oxygen generation. The trimethylammonium-functionalized analog (compound 6) displays high phototoxicity under normoxic conditions but suffers from intrinsic cytotoxicity even in the absence of light and demonstrates low phototoxicity under hypoxic conditions. Conversely, the triphenylphosphonium-functionalized BODIPY derivative (compound 7) displays high phototoxicity but also exhibits significant dark toxicity, which may restrict its therapeutic applicability. In contrast, probe 1 demonstrates strong photodynamic activity upon irradiation, even under hypoxic conditions, while maintaining negligible dark toxicity. These findings highlight the advantageous role of the carnitine moiety, which not only ensures selective mitochondrial accumulation but also enhances phototoxicity even under hypoxia while minimizing adverse effects in the absence of light.
|
|
||||||
|---|---|---|---|---|---|---|
| Compound | Φ Δ (solvent) | Dark toxicitya IC50 (μM) | Light dose (J cm−2) | Phototoxicitya IC50 (μM) | Cell line | Ref. |
| a 24 h Post-treatment. b >10 μM under hypoxic conditions. | ||||||
| 4 | 0.76 (DMSO) | >400 | 1.5 | 27 | HEp2 | 44 |
| 5 | 0.31 (DMSO) | >400 | 1.5 | 28 | HEp2 | 44 |
| 6 | 0.94 (MeOH) | >0.06b | 2.0 | 0.019b | LNCaP | 45 |
| 7 | 0.65 (MeOH) | 0.66 | 6.0 | 0.0015 | HeLa | 46 |

3. Conclusions
Building on the structure of our previously reported mitochondria-selective fluorescent probe (R)-BCT-2,14 which is transported into the mitochondrial matrix by the inner membrane protein carnitine–acylcarnitine translocase (CAC), we designed dually functional probe 1 by incorporating two bromine atoms. This new molecule retains mitochondrial selectivity while acting as both a fluorescent probe and a potent photosensitizer with a singlet oxygen quantum yield of approximately 80%. Although probe 1 exhibits lower molar absorption and fluorescence quantum yield than (R)-BCT-2, it maintains sufficient fluorescence under physiological conditions, enabling effective cell staining in fluorescence microscopy even at concentrations as low as 1.4 μM. Theoretical calculations suggest that the addition of a carnitine moiety at the boron atom distorts the chromophore's planarity, reducing its calculated oscillator strength (and thus its molar absorption coefficient) as compared to the brominated F-BODIPY analog 3, which lacks carnitine, while simultaneously enhancing SOC. The improved SOC, along with the extended triplet lifetimes experimentally observed, contributes to the probe's high phototoxic activity even under hypoxic conditions.The increased lipophilicity, introduced by the bromine substituents, alters the probe's subcellular localization, resulting in co-localization in both mitochondria and lysosomes. The co-localization in these organelles may synergistically enhance phototoxic activity, as both mitochondria and, to a lesser extent, lysosomes are key sites of cellular stress responses, potentially increasing the efficacy of cell damage upon irradiation. Importantly, probe 1 exhibited no significant cytotoxicity in the tested cancer cell lines under dark conditions, but demonstrated pronounced phototoxicity upon green light irradiation, with very low IC50 values of 52 nM under normoxia and 117 nM under hypoxia. Notably, the radical-based Type I mechanism remains effective even under hypoxic conditions, where the Type II singlet oxygen pathway is significantly impaired. These findings suggest that probe 1 holds significant potential as a photosensitizer for PDT, particularly in hypoxic tumor environments, where its dual targeting of mitochondria and lysosomes may enhance therapeutic efficacy.
In conclusion, the carnitine moiety not only enhances solubility, overcoming the poor aqueous solubility limitations of non-carnitine analogs such as compound 3, but also contributes to the unique subcellular localization of probe 1. This active transport mechanism, independent of mitochondrial membrane potential, ensures selective mitochondrial accumulation even in tumor cells with compromised mitochondrial function. Furthermore, the structural impact of the carnitine moiety on the BODIPY chromophore favors intersystem crossing and prolongs the triplet-state lifetime, thereby improving the efficiency of ROS generation. Collectively, these attributes position probe 1 as a superior photosensitizer with a favorable balance between phototoxic potency and safety, highlighting the advantage of carnitine conjugation in the design of next-generation organelle-targeted PDT agents.
Author contributions
Larissa Maierhofer: investigation, methodology, data curation, visualization, writing – original draft, writing – review & editing; Ruth Prieto-Montero: investigation, methodology, data curation, visualization, writing – original draft, writing – review & editing; Tamara Cubiella: investigation, methodology, visualization, writing – review & editing; Aitor Diaz-Andres: investigation, methodology, data curation, visualization, writing – review & editing; Noelia Morales: investigation, methodology; David Casanova: conceptualization, methodology, investigation, data curation, visualization, validation, supervision, funding acquisition, writing – original draft, writing – review & editing; Virginia Martínez-Martínez: conceptualization, methodology, investigation, data curation, visualization, validation, supervision, funding acquisition, writing – original draft, writing – review & editing; María D. Chiara: conceptualization, methodology, investigation, data curation, visualization, validation, supervision, funding acquisition, writing – original draft, writing – review & editing; Enrique A. Mann: conceptualization, methodology, investigation, data curation, validation, supervision, funding acquisition, writing – original draft, writing – review & editing; Jose Luis Chiara: conceptualization, methodology, investigation, data curation, validation, supervision, funding acquisition, writing – original draft, writing – review & editing.Data availability
DFT computational details, photophysical characterization data, live-cell microscopy images, and copies of 1H and 13C NMR spectra of compounds (PDF) can be found in the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge financial support from MCIN/AEI/10.13039/501100011033 (projects PID2020-114347RB-C31, PID2020-114347RB-C32, PID2022-136231NB-I00, PID2023-151388OB-I00; and TED2021-132122B-C22, which is co-funded by the European Union “NextGenerationEU/PRTR”), Gobierno Vasco-Eusko Jaurlaritza (project IT1639-22), and Instituto de Salud Carlos III/co-funded by the European Union ERDF/ESF, “A way to make Europe“/”Investing in your future” (project 20/01754). L. M., A. D.-A and T. C. thank MCIN (PRE2018-086180 and PRE2020-092036), and Consejería de Ciencia, Innovación y Universidad (Principado de Asturias) (BP20-033), for predoctoral contracts, respectively.References
- U. Chilakamarthi and L. Giribabu, Chem. Rec., 2017, 17, 775–802 CrossRef CAS PubMed.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- H. V. Tappeiner and A. Jodlbauer, Die sensibilisierende Wirkung fluorescierender Substanzen: Gesammelte Untersuchungen über die photodynamische Erscheinung: aus dem pharmakologischen Institute der K. Universität München, Verlag von F.C.W. Vogel, Leipzig, 1907 Search PubMed.
- M. Bregnhøj, F. Thorning and P. R. Ogilby, Chem. Rev., 2024, 124, 9949–10051 CrossRef PubMed.
- Á. Juarranz, P. Jaén, F. Sanz-Rodríguez, J. Cuevas and S. González, Clin. Transl. Oncol., 2008, 10, 148–154 CrossRef PubMed.
- A. F. Dos Santos, D. R. Q. De Almeida, L. F. Terra, M. S. Baptista and L. Labriola, J. Cancer Metastasis Treat., 2019, 5, 25 Search PubMed.
- S. M. Mahalingam, J. D. Ordaz and P. S. Low, ACS Omega, 2018, 3, 6066–6074 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- X. Guo, N. Yang, W. Ji, H. Zhang, X. Dong, Z. Zhou, L. Li, H. Shen, S. Q. Yao and W. Huang, Adv. Mater., 2021, 33, 2007778 CrossRef CAS PubMed.
- S. Wang, L. Gai, Y. Chen, X. Ji, H. Lu and Z. Guo, Chem. Soc. Rev., 2024, 53, 3976–4019 RSC.
- J. Moan and K. Berg, Photochem. Photobiol., 1991, 53, 549–553 CrossRef CAS PubMed.
- H. Kurokawa, H. Ito, M. Inoue, K. Tabata, Y. Sato, K. Yamagata, S. Kizaka-Kondoh, T. Kadonosono, S. Yano, M. Inoue and T. Kamachi, Sci. Rep., 2015, 5, 10657 CrossRef CAS PubMed.
- C. S. Oliveira, R. Turchiello, A. J. Kowaltowski, G. L. Indig and M. S. Baptista, Free Radical Biol. Med., 2011, 51, 824–833 CrossRef CAS PubMed.
- A. Blázquez-Moraleja, I. Sáenz-De-Santa María, M. D. Chiara, D. Álvarez-Fernández, I. García-Moreno, R. Prieto-Montero, V. Martínez-Martínez, I. López Arbeloa and J. L. Chiara, Chem. Sci., 2020, 11, 1052–1065 RSC.
- H. Crawford, M. Dimitriadi, J. Bassin, M. T. Cook, T. F. Abelha and J. Calvo-Castro, Chem. – Eur. J., 2022, 28, e202202366 CrossRef CAS PubMed.
- Y. Huang, J. Liang and Z. Fan, Talanta, 2023, 259, 124529 CrossRef CAS PubMed.
- M. K. Goshisht, N. Tripathi, G. K. Patra and M. Chaskar, Chem. Sci., 2023, 14, 5842–5871 RSC.
- A. Blázquez-Moraleja, D. Álvarez-Fernández, R. Prieto Montero, I. García-Moreno, V. Martínez-Martínez, J. Bañuelos, I. Sáenz-de-Santa-María, M. D. Chiara and J. L. Chiara, Dyes Pigm., 2019, 170, 107545 CrossRef.
- H. Manzano, I. Esnal, T. Marqués-Matesanz, J. Bañuelos, I. López-Arbeloa, M. J. Ortiz, L. Cerdán, A. Costela, I. García-Moreno and J. L. Chiara, Adv. Funct. Mater., 2016, 26, 2756–2769 CrossRef CAS.
- A. Blázquez-Moraleja, L. Cerdán, I. García-Moreno, E. Avellanal-Zaballa, J. Bañuelos, M. L. Jimeno, I. López-Arbeloa and J. L. Chiara, Chem. – Eur. J., 2018, 24, 3802–3815 CrossRef PubMed.
- A. Stafford, D. Ahn, E. K. Raulerson, K.-Y. Chung, K. Sun, D. M. Cadena, E. M. Forrister, S. R. Yost, S. T. Roberts and Z. A. Page, J. Am. Chem. Soc., 2020, 142, 14733–14742 CrossRef CAS PubMed.
- R. Prieto-Montero, R. Sola-Llano, R. Montero, A. Longarte, T. Arbeloa, I. López-Arbeloa, V. Martínez-Martínez and S. Lacombe, Phys. Chem. Chem. Phys., 2019, 21, 20403–20414 RSC.
- F. Wilkinson, D. J. McGarvey and A. F. Olea, J. Phys. Chem., 1994, 98, 3762–3769 CrossRef CAS.
- Z. Wang, A. Toffoletti, Y. Hou, J. Zhao, A. Barbon and B. Dick, Chem. Sci., 2021, 12, 2829–2840 RSC.
- J. An, S. Tang, G. Hong, W. Chen, M. Chen, J. Song, Z. Li, X. Peng, F. Song and W.-H. Zheng, Nat. Commun., 2022, 13, 2225 CrossRef CAS PubMed.
- H. Görner, J. Photochem. Photobiol., B, 2007, 87, 73–80 CrossRef PubMed.
- Z. Duan, L. Li, Q. Zhan, J. Chen, Q. Li, R. Liu and Y. Tu, Int. J. Nanomed., 2025, 20, 125–139 CrossRef PubMed.
- J. Zhuang, B. Wang, H. Chen, K. Zhang, N. Li, N. Zhao and B. Z. Tang, ACS Nano, 2023, 17, 9110–9125 CrossRef CAS PubMed.
- G. Gunaydin, M. E. Gedik and S. Ayan, Front. Chem., 2021, 9, 691697 CrossRef CAS PubMed.
- M. S. Baptista, J. Cadet, A. Greer and A. H. Thomas, Photochem. Photobiol., 2023, 99, 313–334 CrossRef CAS PubMed.
- D. Kim and T. S. Teets, Chem. Phys. Rev., 2022, 3, 21302 CrossRef CAS.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CrossRef CAS.
- D. Kessel, Photochem. Photobiol., 2017, 93, 609–612 CrossRef CAS PubMed.
- D. Kessel and J. J. Reiners, Autophagy, 2020, 16, 2098–2101 CrossRef CAS PubMed.
- J. J. Reiners Jr, J. A. Caruso, P. Mathieu, B. Chelladurai, X.-M. Yin and D. Kessel, Cell Death Differ., 2002, 9, 934–944 CrossRef CAS PubMed.
- I. J. MacDonald, J. Morgan, D. A. Bellnier, G. M. Paszkiewicz, J. E. Whitaker, D. J. Litchfield and T. J. Dougherty, Photochem. Photobiol., 1999, 70, 789–797 CAS.
- M. L. Landry and J. J. Crawford, ACS Med. Chem. Lett., 2020, 11, 72–76 CrossRef CAS PubMed.
- N. Mukherjee, R. Gaur, S. Shahabuddin and P. Chandra, Mater. Today Proc., 2022, 62, 7082–7087 CrossRef CAS.
- J. A. Barltrop, T. C. Owen, A. H. Cory and J. G. Cory, Bioorg. Med. Chem. Lett., 1991, 1, 611–614 CrossRef CAS.
- H. Tominaga, M. Ishiyama, F. Ohseto, K. Sasamoto, T. Hamamoto, K. Suzuki and M. Watanabe, Anal. Commun., 1999, 36, 47–50 RSC.
- J. H. Gibbs, L. T. Robins, Z. Zhou, P. Bobadova-Parvanova, M. Cottam, G. T. McCandless, F. R. Fronczek and M. G. H. Vicente, Bioorg. Med. Chem., 2013, 21, 5770–5781 CrossRef CAS PubMed.
- J. Piskorz, W. Porolnik, M. Kucinska, J. Dlugaszewska, M. Murias and J. Mielcarek, ChemMedChem, 2021, 16, 399–411 CrossRef CAS PubMed.
- E. R. H. Walter, P. K.-K. Leung, L. C.-C. Lee, K. K.-W. Lo and N. J. Long, J. Mater. Chem. B, 2024, 12, 10409–10415 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary tables and figures, computational details, additional live-cell microscopy images, copies of 1H and 13C NMR spectra of new compounds. See DOI: https://doi.org/10.1039/d4tb02782e |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |