
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRaman imaging investigation of hepatic LX-2 cell reversion under different lipidic treatments†
Gina
Valentino
a,
Assumpta
Widak
 a,
Bernadette
Scopacasa
b,
Luca
Tirinato
c,
Elvira Immacolata
Parrotta
c,
Gerardo
Perozziello
b,
Arturo
Pujia
c,
Giovanni
Cuda
d,
Paola
Luciani
a and
Patrizio
Candeloro
*b
a,
Bernadette
Scopacasa
b,
Luca
Tirinato
c,
Elvira Immacolata
Parrotta
c,
Gerardo
Perozziello
b,
Arturo
Pujia
c,
Giovanni
Cuda
d,
Paola
Luciani
a and
Patrizio
Candeloro
*b
aDepartment of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, 3012 Bern, Switzerland
bBioNEM Lab. and Nanotechnology Research Center, Department of Experimental and Clinical Medicine, University “Magna Graecia” of Catanzaro, 88100 Campus Germaneto, Catanzaro, Italy. E-mail: patrizio.candeloro@unicz.it
cDepartment of Medical and Surgical Sciences, University “Magna Græcia” of Catanzaro, 88100, Catanzaro, Italy
dDepartment of Experimental and Clinical Medicine, University “Magna Graecia” of Catanzaro, 88100 Campus Germaneto, Catanzaro, Italy
First published on 3rd March 2025
Abstract
Liver fibrosis resulting from chronic liver injury is characterized by increased extracellular matrix deposition and inflammation, which leads to excessive scar tissue formation. Targeting activated hepatic stellate cells (HSCs), which are the primary drivers of fibrogenesis, stands out as one of the most compelling therapeutic approaches in this regard. In a healthy liver, HSCs remain quiescent and store vitamin A in cytoplasmic lipid droplets. As a consequence of HSC activation and transdifferentiation to a proliferative myofibroblast-like state upon fibrotic stimuli, the distinctive phenotypic feature of the lipid droplets gets lost. While the reversal of activated HSCs is feasible, understanding the quiescent-like state following injury resolution is crucial for effective fibrosis treatment. This study explores the induced quiescent-like state of naïve immortalized human hepatic stellate (LX-2) cells when treated with soybean phospholipid that contains 75% phosphatidylcholine (S80). The lipid profile of the newly formed lipid droplets was analyzed using Raman imaging, which is a label-free technique well-suited for lipidomics. Results indicate the presence of distinct lipid profiles despite maintaining a quiescent-like state, suggesting that diverse mechanisms govern the active-to-inactive state transition. Additionally, our findings support the fact that each hepatic cell state is composed of heterogeneous subpopulations. This emphasizes the complexity of liver fibrosis and highlights the need for a comprehensive understanding of cellular states to develop targeted therapies.
Introduction
Chronic liver disease (CLD) has emerged as a global health issue owing to the absence of approved treatment options. In CLD, the liver continuously loses its normal parenchyma and ability to regenerate itself.1 Cirrhosis, the end stage of CLD, ranks among the top 10 causes of death, accounting for 0.8 million annual deaths and affecting 160 million people in 2017.2 It often leads to functional liver failure and hepatocellular cancer.3 The pathological sequence of CLD (depicted in Fig. 1) commences with chronic injury caused by various factors, such as metabolic dysfunction-associated steatotic liver disease (MASLD) and viral hepatitis.1,4 This injury induces hepatic steatosis, which is characterized by the accumulation of fat in the liver and can further progress to liver fibrosis. Liver fibrosis is characterized by the increased deposition of extracellular matrix and inflammation of the liver, which precedes the formation of pathological amounts of scar tissue and is a critical intermediate stage in the progression of CLD.1 | ||
| Fig. 1 Pathogenetic sequence of Liver disease progression (Fig. 1 was created with BioRender.). | ||
Given the potential reversibility of fibrogenesis progression, a viable approach to address liver fibrosis involves targeting activated hepatic stellate cells (HSCs), which are the primary fibrogenic cell type in the liver. These cells predominantly reside within the space of Disse in the hepatic lobule. In a healthy liver, HSCs remain quiescent and are characterized by abundant cytoplasmic lipid droplets (LDs) that serve as storage sites for retinoids. However, in a fibrotic liver, these cells undergo transdifferentiation into a proliferative myofibroblast-like state, which is commonly referred to as activation. HSC activation is distinguished by the expression of profibrogenic transcriptional and secretory properties, accompanied by loss of the cytoplasmic LDs.1,4,5 Therapeutic strategies aim to either induce apoptosis in activated HSCs or revert them to their quiescent-like state by restoring the cytoplasmic lipid droplets. The immortalized human cell line LX-2 is commonly used as a culture model to investigate the effects of anti-fibrotic compounds. It exhibits the key features of activated human HSCs, including retinoid metabolism and fibrogenesis, and the naïve LX-2 cells are present in an activated state.1,6,7
Dietary supplements like Essenvita® or Essentiale® are commonly used to manage liver disease, particularly in Eastern European countries. These supplements contain highly purified extracts of essential phospholipids (EPLs) derived from soybeans. Evidences indicate that EPLCs possess antioxidant and anti-inflammatory properties owing to the presence of (>72%) polyenylphosphatidylcholines (PPCs). Clinical reports suggest that EPLs provide hepatoprotective benefits. However, the precise mechanism underlying the potential hepatoprotective effects of EPLs remains to be fully understood.1,6 Our research group investigated the hepatoprotective effects of PPCs in an in vitro study using soybean phospholipid with 75% phosphatidylcholine (S80). The treatment of activated LX-2 cells with S80 liposomes resulted in their reversal to the quiescent-like state.8–12
This study aims to profile the lipid droplets in LX-2 cells using Raman micro-spectroscopy and understand how various treatments alter the composition of the cells and LDs. As documented widely in the literature,13–18 Raman micro-spectroscopy is a well-established technique for the detection and analysis of lipid molecules, even in complex environments, and has also been applied in the study of hepatic cells.19–22
Our results show that the lipid compositions of profibrogenic LX-2 cells and quiescent-like induced cells are very different, as expected. Furthermore, for the first time, we demonstrated two different treatments for the inactivation of LX2-cells, including either the administration of a retinol/palmitic acid mixture23,24 or soybean phospholipid (S80)8 rich in polyenylphosphatidylcholine (>75%). Even though both led to an overexpression of lipids, completely different patterns are observed in terms of saturation and unsaturation levels. This finding suggests that the reversion of LX-2 cells from the active to the inactive state can lead to entirely different cellular phenotypes, supporting the idea of potential hybrid states for quiescent, active and quiescent-like HSCs.25
Experimental details
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1, streptomycin: 10000 μg mL−1), 1% v/v L-glutamine (200 nM), and 2% v/v fetal bovine serum (FBS). LX-2 cells, DMEM, FBS penicillin, streptomycin and L-glutamine were all procured from Merck Millipore, Darmstadt, Germany.
000 U mL−1, streptomycin: 10000 μg mL−1), 1% v/v L-glutamine (200 nM), and 2% v/v fetal bovine serum (FBS). LX-2 cells, DMEM, FBS penicillin, streptomycin and L-glutamine were all procured from Merck Millipore, Darmstadt, Germany.
For cell treatments (see below), LX-2 cells were seeded in 12-well microtiter plates containing 1 mL complete medium/well at a density of 100000 cells per well. A sterilized CaF2 slide (from Crystran Ltd, Dorset, UK) was placed in each well as a substrate for cell culture because of the negligible Raman signal of CaF2. Cells were cultured for 18 h at 37 °C in the presence of 5% CO2 to 70–90% confluency before the treatments. All treatments were performed with 1 mL per well medium in 12-well plates at 37 °C and 5% CO2.
To avoid interference during Raman imaging, the growth medium was replaced with serum-free and phenol-red-free DMEM (from HyClone, Logan, UT, USA) supplemented with 1% v/v penicillin/streptomycin and 1% v/v L-glutamine before Raman experiments.
In the case of cell activation, the serum-free experimental medium was added along with phosphate-buffered saline (PBS, with 0.1% w/v BSA; 10 μg mL−1) containing TGF-β1 up to a final concentration of 10 ng mL−1 (TGF-β1 was supplied by Merck Millipore, Darmstadt, Germany). After discarding the culture medium and rinsing the cells with PBS, LX-2 cells were cultured in this medium for 24 h27,28 to induce a prolonged fibrogenic state.5,29
In the case of cell inactivation, retinol and palmitic acid (ROL + PA, Sigma-Aldrich, Schnelldorf, Germany) or the liposomal formulations described above (S80, DLPC, or DOPC) were mixed with the serum-free experimental medium. The ROL + PA stock solutions were mixed with the experimental serum-free medium up to concentrations of 10 μM ROL and 300 μM PA;24 each liposomal formulation (S80, DLPC, or DOPC) was mixed with the experimental serum-free medium to achieve a final lipid concentration of 5 mM.8 For the inactivation step, after discarding the culture medium and rinsing with PBS, the LX-2 cells were incubated for 24 h in each medium prepared with ROL + PA or one of the liposomal formulations.
Results
Raman micro-spectroscopy and K-means cluster analysis
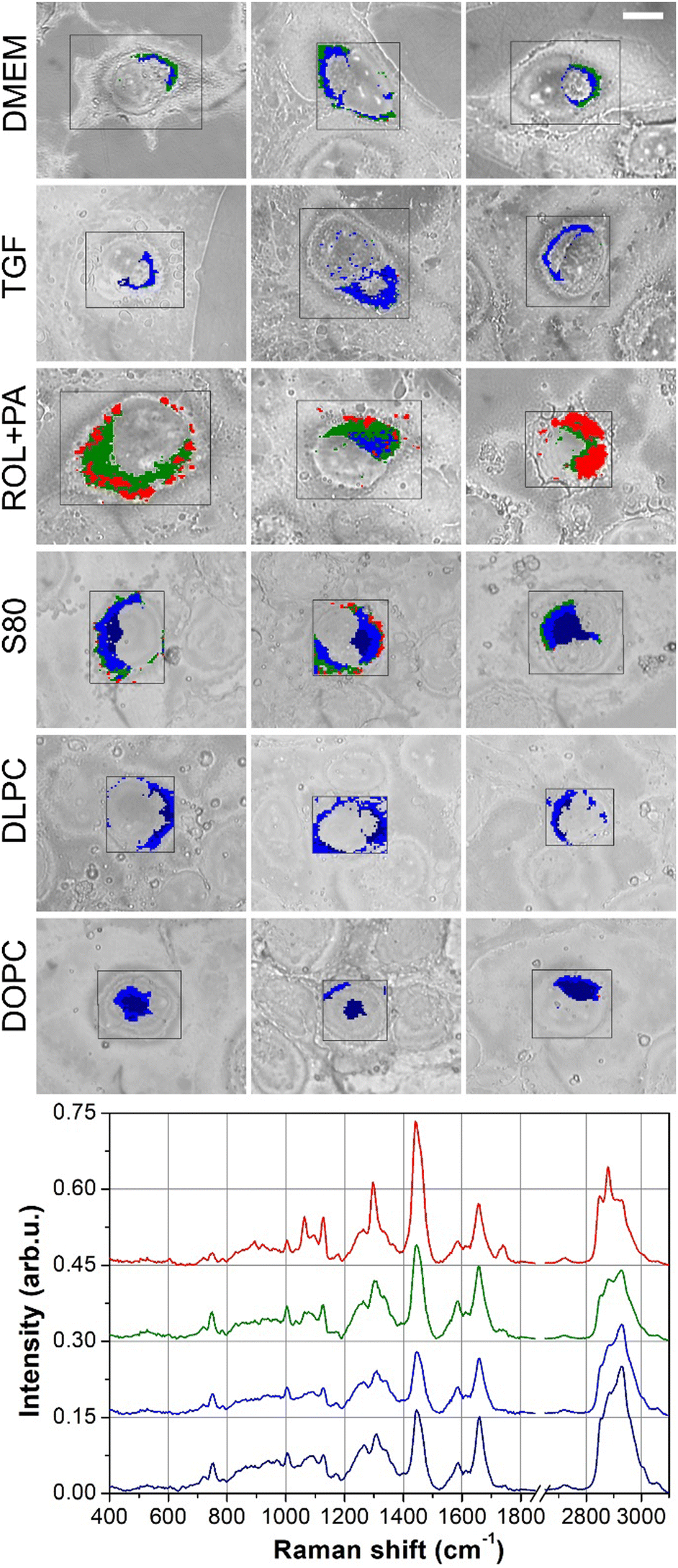
The lipid droplets in LX-2 cells subjected to various treatments were characterized. After incubation and rinsing, the cells were mapped by Raman micro-spectroscopy, and the hyperspectral datasets were analyzed by K-means cluster analysis after proper preprocessing of the spectra. Fig. 2 displays the results of KCA clustering alongside the optical images of the analyzed cells; the red rectangles show the mapped areas. Each treatment group included data from 3 cells. Due to the limited Raman signal recorded from the outer regions of the cells, the scanned areas (red rectangles in the optical images) sometimes covered only the central part of the cell. | ||
| Fig. 2 Optical images and clustering analysis of the different cell treatments based on Raman imaging. Each column of the figure displays 3 cells from each treatment group. DMEM: naïve, profibrogenic LX-2 cells as the control cells; TGF: DMEM cells treated with transforming growth factor to exacerbate the activation of HSCs; ROL + PA: DMEM cells treated with a mixture of retinol and palmitic acid to induce quiescence; S80 and DLPC: DMEM cells treated with soybean phospholipid and 1,2-dilinoleoyl-sn-glycero-3-phosphocholine, respectively, for achieving inactivation; DOPC: DMEM cells treated with 1,2-dioleoyl-sn-glycero-3-phosphocholine as the negative control. Scale bar (top-right optical image) is 10 μm for all images. | ||
In Fig. 3, the average Raman signals of each cluster shown in Fig. 2 are presented; the curve colors correspond to the cluster colors. The average spectra are categorized based on cell treatment, and only the most representative spectra are shown for each treatment. The signal in the high-frequency region in Fig. 3 has been reduced by a factor of 2 to enhance the visibility of the low-frequency region.
 | ||
| Fig. 3 Most representative average Raman spectra of the LX-2 cells. Each graph shows the average spectra computed from the clustered regions marked in Fig. 2, and the spectral colors correspond to the cluster colors. For each cluster map of Fig. 2, only the most representative spectra are shown in the graphs. Across the cell treatments, blue color is ascribed to nuclear regions and reddish colors (dark red, red and magenta) are ascribed to lipids. In all the graphs, the Raman intensities in the high-frequency region from 2650 to 3100 cm−1 are reduced by a factor of 2 for clarity. | ||
For all cell treatments, the blue spectra indicate DNA spectral features; the peaks at 783, 1340, and 1578 cm−1 are assigned to DNA base vibrations,31–33 while the 1090 cm−1 band is typical of the DNA backbone vibration.31,33 Therefore, these clusters are assigned to nuclear regions. The cyan curves display the distinctive peaks of Cytochrome-C (CytC)34–37 at 750, 1125, and 1585 cm−1 among others, suggesting an association with mitochondria.
In contrast to other treatment groups, LX-2 cells treated with S80 exhibited a distinct spectral profile characterized by the presence of both DNA features (blue cluster in Fig. 2) and a combination of DNA and CytC features (dark cyan cluster in Fig. 2). This observation indicates an overlap of the spectral markers associated with nuclear DNA and mitochondrial CytC within the dark cyan cluster. The average curves of the yellow clusters (not shown in Fig. 3) resembled the blue curve very closely but with a significantly smaller intensity and could be attributed to a perinuclear region.
Both the dark and light green clusters exhibited an overall spectral behavior very similar to the average signal of the whole cell but with a decreased general intensity. Notably, the general intensity of the light green signal was smaller than the dark green signal. Both these clusters were assigned to the cytoplasmic and peripheral cellular regions. These observations suggest that the LX-2 cells exhibit strong adhesion to the substrate, and their thin cytoplasmic and outer regions result in a significant reduction in Raman signal intensity.
Finally, the red and magenta clusters (Fig. 2) indicate lipid-rich areas, and the ratio of the peak intensity at 2850 or 2880 cm−1 (CH2 stretching vibrations) to 2930 cm−1 (CH3 vibrations) was used as the lipidic Raman marker (the higher the ratio, the higher the lipidic content).38 In lipidic regions, red and magenta were used to indicate different kinds of lipids based on spectral features observed in the average spectra. To differentiate between the signal intensities, dark red and dark magenta were used for smaller signals, while red and magenta were used for larger signals. The most intense lipidic signals were detected in cells treated with S80 and the positive control ROL + PA, while a lesser intensity was observed in the DLPC-treated cells. This observation aligns with previous results reported using the same treatment,8 indicating that S80 induces LDs and the positive control ROL + PA. These findings suggest the potential restoration of LX-2 cells to a quiescent-like state when treated with S80.
Lipid droplet quantification
The quantification of lipid droplets was performed using a simple geometric ratio of the total count of lipid-expressing pixels to the total number of cell pixels for each treatment. This calculation provided the percentage of lipid area relative to the treated cells, which is presented as a histogram in Fig. 4. The histogram demonstrates that the S80-treated cells produced the highest number of LDs, followed by the ROL + PA-treated cells. | ||
| Fig. 4 Lipid expression (top panel) and the characteristic average Raman spectra recorded over the lipidic regions (bottom panel). Lipid expression is estimated as the ratio of lipid pixels to the total number of pixels for each cell treatment. The bottom panel shows the average spectrum for each cell treatment. In the case of ROL + PA treatment, two different average spectra are displayed owing to the peculiar profile of the red curve. | ||
Additionally, the average spectra of the lipidic pixels are displayed in the bottom panel of Fig. 4. Initially, the spectral profiles of all treated cells appeared similar, except for that of the ROL + PA-treated cells. The Raman spectra of these cells were characterized by unusually high intensities localized to specific narrow regions, with distinctive features markedly different from the other curves, prompting the display of two average spectra for ROL + PA cells in Fig. 4. The black curve represents lipidic signals consistent with the other treated cells, while the red curve corresponds to the recorded average spectrum from the magenta regions of the ROL + PA cells marked in Fig. 2 and the corresponding magenta spectrum in Fig. 3.
All analyses were initially conducted separately for each cell treatment. However, to enable comparison among the treated cells, a unified dataset was created by combining all the lipid Raman data recorded. This integrated dataset was then subjected to KCA for cluster analysis, allowing for quantitative comparisons among lipid clusters across different treatments. The results of this approach are presented in Fig. 5, in which the lipidic pixels are clustered into four distinct groups. These lipidic groups are labeled with different colors, and the clusters are overlaid on the optical images of the cells in the top panel of Fig. 5. In the bottom panel, the average spectrum of each cluster is shown in the corresponding color (Note: the colors used in Fig. 5 are distinct from those in previous figures). It is evident that ROL + PA-treated cells were predominantly linked to the red and green clusters, whereas the other cell lines exhibited significantly higher contents of blue and dark-blue clusters. The average spectra (Fig. 5) provide chemical information about the different lipidic clusters.
 | ||
| Fig. 5 Cluster analysis of the lipid signals as a whole: the lipidic spectra recorded for all cell treatments are pooled into a single dataset and analyzed by KCA-clustering using 4 different classes. Their distribution is shown in the top panel, while the bottom panel reports the average spectra of these lipidic clusters. Red curve, and the corresponding red cluster, exhibit the typical behavior of saturated lipids; green curve is a superposition of a few saturated lipid features with unsaturated lipids; blue and dark blue curves are typical of unsaturated lipids, of which the dark blue curve displays slightly larger spectral indicators of unsaturation. | ||
The sharp and intense peaks in the red spectrum at 2880, 1298, 1128 and 1064 cm−1 are typical markers of saturated fatty acids.15,39 Moreover, the sharpness of these peaks suggests that the lipids in the red regions were in a highly ordered, solid-like phase. The green spectrum exhibits a relatively intense signal (compared to the blue and dark-blue curves) at 2880 cm−1 but with a much broader shape compared with the red curve. The appearance of this distinct peak at 2880 cm−1, rather than a shoulder, further indicates the presence of saturated lipids. However, all indicators of saturated lipids (2880, 1298, and 1128 cm−1) exhibit lower intensities compared with those of the red curve. Moreover, the 1655-to-1440 cm−1 peak ratio is much larger than the other spectra, where the peak at 1655 cm−1 is attributed to the double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of unsaturated fatty acid chains.39 All these features suggest the presence of both saturated and unsaturated lipids in the green region. This aligns with the broader shape of the peak, which is indicative of a more disordered, liquid-like phase typical of unsaturated fatty acids. Finally, in the blue and dark-blue curves, the high-frequency regions are devoid of the characteristic spectral features of saturated lipids and display a concurrent shift from 1298 to 1307 cm−1 along with increasing peak ratios at 1655/1440 cm−1 and 1266/1307 cm−1, all indicating the presence of unsaturated fatty acids.39 These two curves are very similar to each other, and the most remarkable difference is the lower intensity of the blue curve. However, the 1266/1307 cm−1 ratio is slightly increased in the dark-blue spectrum.
C bonds of unsaturated fatty acid chains.39 All these features suggest the presence of both saturated and unsaturated lipids in the green region. This aligns with the broader shape of the peak, which is indicative of a more disordered, liquid-like phase typical of unsaturated fatty acids. Finally, in the blue and dark-blue curves, the high-frequency regions are devoid of the characteristic spectral features of saturated lipids and display a concurrent shift from 1298 to 1307 cm−1 along with increasing peak ratios at 1655/1440 cm−1 and 1266/1307 cm−1, all indicating the presence of unsaturated fatty acids.39 These two curves are very similar to each other, and the most remarkable difference is the lower intensity of the blue curve. However, the 1266/1307 cm−1 ratio is slightly increased in the dark-blue spectrum.
A comparison between the lipidic signals recorded from the cells (red, green, blue and dark blue curves) and measurements from pure fatty acids with different unsaturation degrees is shown in Fig. 6 in order to provide more quantitative evidence for the above comments. The produced calibration chart shows the Raman measurements of the pure fatty acids (black circles), and the data from our lipidic spectra are marked as colored triangles on the same chart.
 | ||
| Fig. 6 Calibration chart based on the Voronoi diagram of the Raman measurements of pure fatty acids. Black full circles indicate the spectral features of pure fatty acids; the colored triangles are obtained from the average lipidic spectra of the cell lines; the colormap intensity increases with the normalized degree of unsaturation (nCC/nCH2) of pure fatty acids. The red triangle is very close to stearic acid and can be assigned to saturated lipids. The green triangle is halfway between stearic and oleic acid and can be assigned to a combination of saturated and mono-unsaturated lipids. The blue and dark blue triangles represent unsaturated lipids, differing solely by spectral intensity (higher intensities correspond to higher molecular concentrations). | ||
In summary, it can be deduced from the spectral analysis that the ROL + PA cells overexpressed large clusters of saturated lipids along with regions of mixed saturated and unsaturated lipids. These high saturation levels are likely a direct consequence of treatment with palmitic acid. In comparison, S80 cells treated with PPC-rich soybean phospholipid also overexpressed lipid regions but they were mostly composed of unsaturated lipids and only a few confined regions with higher saturation levels. Moreover, in this case, the resulting lipid profile could be related to the composition of S80.
The DLPC-treated cells demonstrated relatively higher lipid production compared with untreated cells in DMEM but lower than that observed in the ROL + PA and S80 cells (histogram in Fig. 4). Notably, Raman imaging could only detect unsaturated lipid areas in DLPC-treated cells. As anticipated, cells treated with TGF-β1 and the untreated DMEM control showed the lowest lipid production (Fig. 4). The negative control, DOPC (inactive phospholipid), also exhibited a low level of LD production, which is considered negligible based on previous findings.8 These observations support the fact that, unlike S80, not all unsaturated lipids can induce the accumulation of a significant amount of LDs in cells.
Among the treated cells with the lowest lipid content (DMEM, TGF, and DOPC), the main distinction was the presence of a small region of mixed saturated-unsaturated lipids (green cluster) observed in the DMEM cells, whereas the TGF and DOPC cells only exhibited unsaturated lipid regions (blue and dark blue).
Discussion
In the literature, liver fibrosis resulting from injury and its potential resolution are widely attributed to hepatic stellate cells (HSC).6,25,40 The progression model of liver fibrosis includes quiescent (qHSC), active (aHSCs), and quiescent-like (qlHSCs) states. Quiescent HSCs are characterized by the abundance of cytoplasmic lipid droplets, which store vitamin A in the form of retinyl esters along with other neutralized lipid molecules. Upon liver injury, quiescent HSCs undergo phenotypic transdifferentiation (activation) into a myofibroblast state. This activation process is marked by an increased expression of extracellular matrix (ECM) molecules, such as collagen type 1, and cytoskeletal filaments. The accumulation of ECM components leads to scar tissue formation in the perisinusoidal space, contributing to the loss of endothelial fenestrations.6The transition from quiescent to activated HSCs is associated with a highly fibrogenic state and the concomitant loss of LDs. However, the causal relationship between LD loss and HSC activation remains uncertain.19,41–43 There is an obvious agreement that LD catabolism serves to meet the energetic demands of aHSCs.
As liver damage resolves, the number of aHSCs decreases through two main pathways: apoptosis and cellular reversion to a quiescent-like state. Like quiescent cells, quiescent-like-state HSCs revert to non-fibrogenic cells and form LDs again. However, qlHSC do not recover the same features of quiescent cells, and it has been demonstrated that qlHSC respond more readily to new liver injuries.23,25,44
Based on research evidence that aHSCs can revert to a quiescent-like state, multiple studies are currently focusing on identifying potential therapeutic treatments that target the transition from active to inactive pathways.8,24,45–53 In a previous study,8 the role of lipid droplet pathways in this transition was investigated using soybean phospholipid containing 75% phosphatidylcholine (S80) in an in-vitro model. The results of this study showed that treating naïve LX-2 cells with S80 caused LD formation, leading the cells to a quiescent-like state as compared to the positive control (ROL + PA). Furthermore, DLPC (the main component of S80), which was used as a negative control treatment, did not lead to LD formation in LX-2 cells.
The following discussion mainly focuses on S80 and the positive control ROL + PA. Raman investigation, which is a completely different technique compared to fluorescent studies reported in previous work,8 presented similar results in terms of overall lipidic content. In ROL + PA cells, several lipidic regions were confined to narrow areas (red and magenta clusters of ROL + PA in Fig. 2 and red spots of ROL + PA in Fig. 5) and could be identified as LDs. On the other hand, S80-treated cells exhibited lipidic regions (Fig. 2, red and magenta clusters) that were spread over a wider area, but some narrow spots were also observed (red clusters in Fig. 5), which could be identified as lipid droplets. Since the presence of LDs after S80 treatment has already been proven,8 we hypothesize that cell fixation and the dry conditions prevalent during Raman imaging experiments could have collapsed the small LDs in the broader lipidic regions. In ROL + PA-treated cells, the larger LDs could preserve their morphology and hence are distinguishable in the cluster analysis. Moreover, since Raman imaging has a diffraction-limited spatial resolution, vesicles smaller than or equal to 0.5 μm would be blurred in our optical setup, while vesicles in the 0.5–1.0 μm range could have easily overlapped in the Raman images if they were close to each other. Thus, it can be inferred that S80 treatment induces a quiescent-like state in LX-2 cells with different phenotypes, both in terms of the size and content of LDs. The positive control ROL + PA exhibited mostly large-sized and saturated lipid droplets, while the S80-treated cells produced much smaller droplets with significantly lower levels of saturation.
Former works24,45–48 on HSCs exposed to combinations of FAs and retinol have mostly focused on the modulation of genes, cytokines and proteins involved in retinol metabolism and fibrogenesis. The only lipidomic issue that was properly addressed was the content of lipid droplets since lipid droplet loss is a major marker of HSC activation. However, the lipid profile was poorly investigated. To date, only a few recent works have investigated the origin of lipid droplets and characterized them in HSCs, and only upon activation.19,54,55 Some other older works have dealt with LD cargos under different dietary conditions in rat experiments.56,57
In recent works, different pools of LDs have been proposed to explain lipid dynamics during activation, distinguishing between pre-existing and de-novo-generated LD pools. During activation, LDs were observed to decrease in size and have an increased content of polyunsaturated FAs.19,55 These conclusions on LD pools suggest that different kinds of LDs can occur in HSCs under different conditions, which is in agreement with the difference in LD sizes and saturation levels observed in this work between ROL + PA (positive control) and S80 cells. On the other hand, LD differences have also been reported between quiescent and active cells after activation. In contrast, our results show that the same kind of HSCs, i.e. LX-2 cells reverted to a quiescent–like state, with different lipidic phenotypes depending on the cell treatment. The fact that saturated fatty acids, like palmitic acid, result in higher levels of saturation cannot be a simple coincidence, just as the S80 formulation rich in polyenyl-PPC leads to fewer and smaller saturated droplets with large areas of unsaturated lipids.
The achievement of different quiescent-like states supports the more recent idea that the paradigm of quiescent, active and quiescent-like HSC cells is oversimplified.25,58,59 It does not consider the cellular differences occurring in each state, and that the quiescent, active and quiescent-like HSCs might constitute different subpopulations, thus reflecting the heterogeneity of each state. The conclusion that different treatments lead to different qlHSC subpopulations can be directly extended to the microenvironment surrounding active HSCs in vivo, that is, different local conditions could lead to different final fates for aHSCs.
Conclusion
HSCs play a key role in the immediate response to liver injuries, as well as the progression of liver fibrosis towards cirrhosis, and eventually organ failure. During the last decade, a model consisting of quiescent, active and quiescent-like-HSCs has been proposed to explain the contributions of these cells to the progression or reversion of fibrosis. The fact that active HSCs can be reverted to a quiescent-like state has boosted the quest for treatments for liver fibrosis. Among several pathways, potential treatments have also targeted the inverse relationship between fibrogenic state and formation of lipid droplets. As the search for an effective treatment progressed, the rise in the variety of scenarios encountered and the heterogeneity of HSCs have recently led to the concept of the potential existence of cellular subpopulations within each state of the HSCs.In this context, this study has explored the effect of soybean phospholipid S80 containing 75% PPC on the reversion of active LX-2 hepatic cells, focusing on the lipidic profiles of the newly formed LDs. We have found that formulations based on soybean-derived PPC achieve higher expression of LDs and lipids in the cells, thus leading to a quiescent-like state, which is characterized by low fibrogenesis and higher LD counts. This imitates the quiescent state of LX-cells induced using ROL and PA treatment; however, with ROL + PA treatment, much higher levels of saturation and larger size LDs were observed. This result agrees with the idea of cellular subpopulations in the quiescent-like state, suggesting that cell treatment composition and the local hepatic microenvironment will influence cellular fate upon reversal, leading to several kinds of quiescent-like states in HSCs.
Furthermore, the lipid profile of HSC cells in the quiescent-like state has been poorly investigated so far,60 and in many cases, only the number of LDs has been estimated. Our results indicate that future studies should focus on in-depth lipidomic analyses of the quiescent-like state of HSC cells61 for a better understanding of all the possible mechanisms underlying the transition from an active to a quiescent-like state.
Author contributions
Conceptualization: GV, PL, GP, PC; data curation: GV, PC; formal analysis: PC, BS, LT, GV; funding acquisition: GP, GC, AP, PL; investigation: GV, PC, LT; methodology: GV, PL, LT; project administration: PL, PC; resources: EIP, PC, GC; software: PC, BS; supervision: PC, PL, EIP; visualization: AW, PC, BS; writing – original draft: PC, BS, AW; writing – review & editing: AW, AP, PC, PL.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
No private study sponsors had any involvement in the study design, data collection, or interpretation of data presented in this article. Paola Luciani has consulted for Lipoid GmbH, Sanofi-Aventis Deutschland, and DSM Nutritional Products Ltd and received grants from Lipoid GmbH, Sanofi-Aventis Deutschland, DSM Nutritional Products Ltd and PPM Services SA.Acknowledgements
G. P. and P. C. gratefully acknowledge support by the European Union “NextGenerationEU” through the National Plan of Recovery and Resilience (PNRR), which financed the project PRIN PNRR titled “Automated Microfluidic Platforms for Innovative Cell Screenings” (CUP F53D23008830001; project code P2022NNSFF). L. T. and P. C. gratefully acknowledge support by the European Union “NextGenerationEU” through the National Plan of Recovery and Resilience (PNRR) which financed the project PRIN PNRR titled “Peritumoral Adipose Tissue Lipid Trafficking Investigation through Raman Spectroscopy” (CUP F53D23011350001; project code P2022YAKJY).References
- P. Marcellin and B. K. Kutala, Liver Int., 2018, 38, 2–6 CrossRef PubMed.
- L. Pimpin, H. Cortez-Pinto, F. Negro, E. Corbould, J. V. Lazarus, L. Webber, N. Sheron and EASL HEPAHEALTH Steering Committee, J. Hepatol., 2018, 69, 718–735 CrossRef PubMed.
- S. K. Asrani, H. Devarbhavi, J. Eaton and P. S. Kamath, J. Hepatol., 2019, 70, 151–171 CrossRef PubMed.
- R. Bataller and D. A. Brenner, Semin. Liver Dis., 2001, 21, 437–452 CrossRef CAS PubMed.
- C. R. Gandhi, J. Hepatol., 2017, 67, 1104–1105 CrossRef PubMed.
- T. Tsuchida and S. L. Friedman, Nat. Rev. Gastroenterol. Hepatol., 2017, 14, 397–411 CrossRef CAS PubMed.
- H. She, S. Xiong, S. Hazra and H. Tsukamoto, J. Biol. Chem., 2005, 280, 4959–4967 CrossRef CAS PubMed.
- G. Valentino, C. Zivko, F. Weber, L. Brülisauer and P. Luciani, Pharmaceutics, 2019, 11, 676 CrossRef CAS PubMed.
- M. Carone, R. Gazzi, R. Eugster, R. Gelli, N. Manten, A. A. Ganguin, S. D. Valerio, G. Yadav, P. Castaldo and R. Mezzenga, Adv. Mater. Technol., 2024, 9, 2301930 CrossRef CAS.
- I. Skorup, G. Valentino, S. Aleandri, R. Gelli, A. A. Ganguin, E. Felli, S. E. Selicean, R. A. Marxer, S. Teworte and A. Lucić, Int. J. Pharm., 2023, 646, 123473 CrossRef CAS PubMed.
- C. Zivko, K. Fuhrmann, G. Fuhrmann and P. Luciani, Commun. Biol., 2022, 5, 1155 CrossRef CAS PubMed.
- A. A. Ganguin, I. Skorup, S. Streb, A. Othman and P. Luciani, Adv. Healthcare Mater., 2023, 12, 2300811 CrossRef CAS PubMed.
- C. W. Freudiger, W. Min, B. G. Saar, S. Lu, G. R. Holtom, C. He, J. C. Tsai, J. X. Kang and X. S. Xie, Science, 2008, 322, 1857–1861 CrossRef CAS PubMed.
- J. Cheng and X. S. Xie, Science, 2015, 350, aaa8870 CrossRef PubMed.
- H. Wu, J. V. Volponi, A. E. Oliver, A. N. Parikh, B. A. Simmons and S. Singh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3809–3814 CrossRef CAS PubMed.
- A. Syed and E. A. Smith, Annu. Rev. Anal. Chem., 2017, 10, 271–291 CrossRef CAS PubMed.
- M. Uematsu and T. Shimizu, Commun. Biol., 2021, 4, 1176 CrossRef CAS PubMed.
- C. Zhang, J. Li, L. Lan and J. Cheng, Anal. Chem., 2017, 89, 4502–4507 CrossRef CAS PubMed.
- N. Testerink, M. Ajat, M. Houweling, J. F. Brouwers, V. V. Pully, H. van Manen, C. Otto, J. B. Helms and A. B. Vaandrager, PLoS One, 2012, 7, e34945 CrossRef CAS PubMed.
- K. Galler, R. P. Requardt, U. Glaser, R. Markwart, T. Bocklitz, M. Bauer, J. Popp and U. Neugebauer, Sci. Rep., 2016, 6, 24155 CrossRef CAS PubMed.
- E. Szafraniec, E. Kus, A. Wislocka, B. Kukla, E. Sierka, V. Untereiner, G. D. Sockalingum, S. Chlopicki and M. Baranska, J. Biophotonics, 2019, 12, e201800290 CrossRef PubMed.
- T. Yildirim, C. Matthäus, A. T. Press, S. Schubert, M. Bauer, J. Popp and U. S. Schubert, Macromol. Biosci., 2017, 17, 1700064 CrossRef PubMed.
- L. Xu, A. Y. Hui, E. Albanis, M. J. Arthur, S. M. O’byrne, W. S. Blaner, P. Mukherjee, S. L. Friedman and F. Eng, Gut, 2005, 54, 142–151 CrossRef CAS PubMed.
- T. F. Lee, K. M. Mak, O. Rackovsky, Y. Lin, A. J. Kwong, J. C. Loke and S. L. Friedman, J. Cell. Physiol., 2010, 223, 648–657 CrossRef CAS PubMed.
- P. Trivedi, S. Wang and S. L. Friedman, Cell Metab., 2021, 33, 242–257 CrossRef CAS PubMed.
- L. Rahnfeld, J. Thamm, F. Steiniger, P. van Hoogevest and P. Luciani, Colloids Surf., B, 2018, 168, 10–17 CrossRef CAS PubMed.
- H. Shen, G. Huang and Y. Gong, World J. Gastroenterol., 2003, 9, 784 CrossRef CAS PubMed.
- J. Yu, Y. Hu, Y. Gao, Q. Li, Z. Zeng, Y. Li and H. Chen, Cell Death Discovery, 2018, 4, 93 CrossRef PubMed.
- B. Dewidar, J. Soukupova, I. Fabregat and S. Dooley, Curr. Pathobiol. Rep., 2015, 3, 291–305 CrossRef.
- P. Candeloro, E. Grande, R. Raimondo, D. Di Mascolo, F. Gentile, M. L. Coluccio, G. Perozziello, N. Malara, M. Francardi and E. Di Fabrizio, Analyst, 2013, 138, 7331–7340 RSC.
- I. Notingher, Sensors, 2007, 7, 1343–1358 CrossRef CAS.
- H. van Manen, Y. M. Kraan, D. Roos and C. Otto, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10159–10164 CrossRef CAS PubMed.
- C. Krafft, T. Knetschke, A. Siegner, R. H. Funk and R. Salzer, Vib. Spectrosc., 2003, 32, 75–83 CrossRef CAS.
- D. S. Read, D. J. Woodcock, N. J. Strachan, K. J. Forbes, F. M. Colles, M. C. Maiden, F. Clifton-Hadley, A. Ridley, A. Vidal and J. Rodgers, Appl. Environ. Microbiol., 2013, 79, 965–973 CrossRef CAS PubMed.
- C. Johannessen, P. C. White and S. Abdali, J. Phys. Chem. A, 2007, 111, 7771–7776 CrossRef CAS PubMed.
- F. Sinibaldi, G. Mei, F. Polticelli, M. C. Piro, B. D. Howes, G. Smulevich, R. Santucci, F. Ascoli and L. Fiorucci, Protein Sci., 2005, 14, 1049–1058 CrossRef CAS PubMed.
- E. Parrotta, M. T. De Angelis, S. Scalise, P. Candeloro, G. Santamaria, M. Paonessa, M. L. Coluccio, G. Perozziello, S. De Vitis and A. Sgura, Stem Cell Res. Ther., 2017, 8, 1–12 CrossRef PubMed.
- C. Krafft, T. Knetschke, R. H. Funk and R. Salzer, Vib. Spectrosc., 2005, 38, 85–93 CrossRef CAS.
- K. Czamara, K. Majzner, M. Z. Pacia, K. Kochan, A. Kaczor and M. Baranska, J. Raman Spectrosc., 2015, 46, 4–20 CrossRef CAS.
- S. L. Friedman, Physiol. Rev., 2008, 88, 125–172 CrossRef CAS PubMed.
- L. L. Jophlin, Y. Koutalos, C. Chen, V. Shah and D. C. Rockey, Am. J. Physiol.: Endocrinol. Metab., 2018, 315, G713–G721 CAS.
- W. S. Blaner, S. M. O'Byrne, N. Wongsiriroj, J. Kluwe, D. M. D'Ambrosio, H. Jiang, R. F. Schwabe, E. M. Hillman, R. Piantedosi and J. Libien, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2009, 1791, 467–473 CrossRef CAS PubMed.
- N. Bourebaba and K. Marycz, Pharmacol. Res., 2021, 170, 105739 CrossRef CAS PubMed.
- X. Liu, J. Xu, S. Rosenthal, L. Zhang, R. McCubbin, N. Meshgin, L. Shang, Y. Koyama, H. Ma and S. Sharma, Gastroenterology, 2020, 158(1728–1744), e14 Search PubMed.
- A. El Taghdouini, M. Najimi, P. Sancho-Bru, E. Sokal and L. A. van Grunsven, Fibrog. Tissue Repair, 2015, 8, 1–15 CrossRef PubMed.
- A. M. Hetherington, C. G. Sawyez, E. Zilberman, A. M. Stoianov, D. L. Robson and N. M. Borradaile, Cell. Physiol. Biochem., 2016, 39, 1648–1662 CrossRef CAS PubMed.
- C. Wagner, V. Hois, L. Pajed, L. Pusch, H. Wolinski, M. Trauner, R. Zimmermann, U. Taschler and A. Lass, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2020, 1865, 158730 CrossRef CAS PubMed.
- V. J. Barbero-Becerra, P. J. Giraudi, N. C. Chávez-Tapia, M. Uribe, C. Tiribelli and N. Rosso, Toxicol. In Vitro, 2015, 29, 1753–1758 CrossRef CAS PubMed.
- K. M. Mak and A. C. Shekhar, Anat. Rec., 2024, 307, 2162–2186 CrossRef CAS PubMed.
- Q. Cao, K. M. Mak and C. S. Lieber, J. Lab. Clin. Med., 2002, 139, 202–210 CrossRef CAS PubMed.
- J. E. Adrian, K. Poelstra, G. L. Scherphof, D. K. Meijer, C. Reker-Smit, H. W. Morselt, P. Zwiers and J. A. Kamps, J. Pharmacol. Exp. Ther., 2007, 321, 536–543 Search PubMed.
- Q. Cao, K. M. Mak and C. S. Lieber, Biochem. Biophys. Res. Commun., 2006, 350, 50–55 CrossRef CAS PubMed.
- L. Listenberger, E. Townsend, C. Rickertsen, A. Hains, E. Brown, E. G. Inwards, A. K. Stoeckman, M. P. Matis, R. S. Sampathkumar and N. A. Osna, Cells, 2018, 7, 230 CrossRef CAS PubMed.
- M. Tuohetahuntila, M. R. Molenaar, B. Spee, J. F. Brouwers, R. Wubbolts, M. Houweling, C. Yan, H. Du, B. C. VanderVen and A. B. Vaandrager, J. Biol. Chem., 2017, 292, 12436–12448 CrossRef CAS PubMed.
- I. O. Shmarakov, H. Jiang, J. Liu, E. J. Fernandez and W. S. Blaner, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2019, 1864, 629–642 CrossRef CAS PubMed.
- H. Moriwaki, W. S. Blaner, R. Piantedosi and D. S. Goodman, J. Lipid Res., 1988, 29, 1523–1534 CrossRef CAS.
- M. Yamada, W. S. Blaner, D. R. Soprano, J. L. Dixon, H. M. Kjeldbye and D. S. Goodman, Hepatology, 1987, 7, 1224–1229 CrossRef CAS PubMed.
- D. N. D'Ambrosio, J. L. Walewski, R. D. Clugston, P. D. Berk, R. A. Rippe and W. S. Blaner, PLoS One, 2011, 6, e24993 CrossRef PubMed.
- O. Krenkel, J. Hundertmark, T. P. Ritz, R. Weiskirchen and F. Tacke, Cells, 2019, 8, 503 CrossRef CAS PubMed.
- C. Zivko, F. Witt, A. Koeberle, G. Fuhrmann and P. Luciani, Eur. J. Pharm. Biopharm., 2023, 182, 32–40 CrossRef CAS PubMed.
- S. Maurotti, N. Geirola, M. Frosina, A. Mirarchi, F. Scionti, R. Mare, T. Montalcini, A. Pujia and L. Tirinato, Front. Cell Dev. Biol, 2024, 12, 1404006 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02082k |
| This journal is © The Royal Society of Chemistry 2025 |