
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulation of the biological response to surfaces through the controlled deposition of 3D polymeric surfactants†
Valentina
Cuzzucoli Crucitti
*ab,
Hadi
Hajiali‡
 c,
Adam A.
Dundas
b,
Vineetha
Jayawarna
d,
Dario
Tomolillo
e,
Iolanda
Francolini
f,
Claudia
Vuotto
e,
Manuel
Salmeron-Sanchez
d,
Matthew J.
Dalby
g,
Morgan R.
Alexander
c,
Ricky D.
Wildman
ab,
Felicity R. A. J.
Rose
*c and
Derek J.
Irvine
*ab
c,
Adam A.
Dundas
b,
Vineetha
Jayawarna
d,
Dario
Tomolillo
e,
Iolanda
Francolini
f,
Claudia
Vuotto
e,
Manuel
Salmeron-Sanchez
d,
Matthew J.
Dalby
g,
Morgan R.
Alexander
c,
Ricky D.
Wildman
ab,
Felicity R. A. J.
Rose
*c and
Derek J.
Irvine
*ab
aCentre for Additive Manufacturing, Faculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: Valentina.CuzzucoliCrucitti1@nottingham.ac.uk; Derek.Irvine@nottingham.ac.uk
bDepartment of Chemical and Environmental Engineering, Faculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK
cSchool of Pharmacy, Nottingham Biodiscovery Institute, Faculty of Science, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: Felicity.Rose@nottingham.ac.uk
dCentre for the Cellular Microenvironment, School of Engineering, Advanced Research Centre, University of Glasgow, Glasgow G11 6EW, UK
eNeuromicrobiology Unit, IRCCS Fondazione Santa Lucia, Rome, Italy
fDept of Chemistry, Sapienza University of Rome, Piazzale A. Moro 5, 00185 Rome, Italy
gCentre for the Cellular Microenvironment, School of Molecular Biosciences, College of Medical, Veterinary and Life Sciences, Advanced Research Centre, University of Glasgow, Glasgow G11 6EW, UK
First published on 25th March 2025
Abstract
Biomaterials play a crucial role in modern medicine through their use as medical implants and devices. However, they can support biofilm formation and infection, and lack integration with the surrounding human tissue at the implant site. This work reports the development of novel poly(ethyl acrylate) (PEA) based copolymers that address both issues. These PEA materials were molecularly designed polymeric surfactants (surfmers) synthesised via controlled radical polymerisations to achieve different polymeric architectures, (i.e., statistical and block copolymers). These were both deposited as structured 2D films on glass coverslips and used to manufacture monodisperse 3D micro-particles with functional surfaces (via microfluidics). ToF-SIMS was used to analyse these 2D and 3D surfaces to understand: (a) the surface arrangement of the monomer sequences exhibited by the different polymer structures and (b) how this surface monomer arrangement influenced mammalian fibroblast cell and/or Staphylococcus aureus behaviour at these film/particle surfaces. In addition, the form of the fibronectin (FN) network assembly's importance in promoting growth factor (GF) binding was probed using atomic force microscopy (AFM) on the 2D films. This confirmed that specific surfmer molecular surface organisations were achieved during film/micro-particle fabrication, which presented exterior functionalities that either prevent biofilm attachment or promote the formation of structured FN networks for GF binding.
Introduction
There is a significant body of research focussed on the control of both bacterial and mammalian cell behaviour on materials surfaces to address issues associated with infections related with medical devices1 and as part of regenerative medicine strategies to modulate mammalian cell behaviour and promote tissue repair.2–4Bacterial biofilm formation has been identified as a principal route to device associated infection,5–7 and increased tolerance to antibiotic treatment when compared to planktonic counterparts.8,9 Biomaterial strategies have been developed to prevent the irreversible attachment of biofilms or to kill the surface contacting microbes by the inclusion of biocidal additives, such as silver ions.10–12 Furthermore, this material performance has also been successfully transferred from two dimensional (2D) to three dimensional (3D) device surfaces (i.e., from flat films to surface coatings of 3D structures such as tubes and particles).13,14 These 3D coatings were achieved by synthesising amphiphilic copolymers that included hydrophobic, high-throughput (HT) identified, biofilm resistant monomers. Such copolymer coatings have been demonstrated to avoid bacterial attachment on a commercial urinary catheter.15
Materials research has also focussed on the ability to support tissue regeneration, manipulating the biomaterial surface based on the targeted site which is essential for achieving optimal biological performance.16 As an example, Ranella et al. explored how fibroblast cells adhere and survive on 3D silicon surfaces with varying roughness and wettability. They used femtosecond laser structuring to create surfaces with controlled micro- and nano-scale features and then adjusted surface chemistry without affecting the topography.17 In addition, using different functional groups on the surface of biomaterials can stimulate different biochemical signalling pathways.18–20 Zhang et al. investigated the potential proinflammatory influence of surface functional groups on human pulmonary epithelial cells and macrophages by using quantum dots coated with polymers containing various functional groups (–COOH, –NH2, –OH, –OCH3).18 HT-screening platforms have played a key role in the study of manipulating biomaterial properties to influence stromal cell behaviour.21,22 High-throughput screening experiments identified candidates that drive fibroblasts towards either pro- or antiproliferative functional phenotypes. In this study, poly(tetrahydro furfuryl acrylates) (pTHFuA) was used to support healing in chronic wounds when in particulate form.23 These polymers were used to create bio-instructive surfactant materials termed ‘surfmers’ and then used to produce particles that significantly accelerated wound healing in animal models.
Microparticles, with bio-instructive surfaces that can be easily tailored by simply changing the identity of the surfmer, are of significant interest for cell-based therapies, as they provide a very high surface area for achieving intimate contact with the anatomical treatment area and require minimally invasive surgical procedures. Bone, in the form of bone graft, is the second most transplanted tissue after blood, with bone grafts used to replace tissue that is lost through damage or disease.24 The healing process of a bone is also a multifaceted process that requires mechanical stability and revascularisation along with osteogenesis, osteoinduction and osteoconduction.25 Consequently, several different materials strategies have been developed in recent years to achieve the goal of osteogenic repair, including the use of: (a) allogenic and autologous bone,26–29 (b) decellularized bone matrix,28 and (c) synthetic ceramic.24,30 However, these materials do not actively promote bone healing or face challenges with regards to their supply and use clinically.
One promising route forward in bone repair is the localised delivery of physiologically relevant GF, including bone morphogenetic proteins (BMPs), directly to the implant site to locally regulate mesenchymal stromal cell (MSCs) behaviour and minimise off-target effects associated with supra-physiological doses when administered by injection. They promote differentiation of MSCs to osteoblasts responsible for the deposition of new bone and play a role in the repair of fractures.31,32 The chemistry adopted in these studies included the use of poly(ethyl acrylate) (PEA) coatings, which we have previously shown to trigger the spontaneous assembly of FN molecules into biological networks.33 The key advantage of this strategy resided in the fact that structured FN exposed specific domains shown to be highly effective for GF binding.34 Interaction with these domains promoted simultaneous exposure of the integrin binding (FNIII9–10) and the GF binding (FNIII12–14) regions and so promoted binding and co-localisation of integrins and GF receptors.
An additional complication of bone regeneration, especially after a trauma or surgery, is osteomyelitis; an infection resulting from the formation of a persistent biofilm upon the tissue/implant of the host. As such, if osteomyelitis manifests itself during treatment, it can lead to complicated clinical scenarios often necessitating repeated surgical interventions to clear the infection.35 Thus, the prevention of biofilm formation is a key goal in successfully achieving high quality and long-lasting bone fixation.
In this work, PEA candidate materials were synthesised in the form of surfmers for use in both the generation of structured 2D films and for the stabilisation of oil-in-water emulsions within a droplet-based microfluidics process to produce highly monodisperse 3D MPs in flow. To date, our research has focused on the performance of homo-polymers, statistical copolymers, or blends of homo-polymers in various applications, investigating the behaviour of biological systems on both 2D and 3D surfaces.36 This study represents the first report of block copolymers being utilized in these applications, warranting an investigation into both their 2D and 3D performance. By including the results related to the MPs, we provide a comprehensive evaluation of the block copolymers' potential for innovative biomedical applications. This strategy was driven by the possibility of enabling the derivation of active PEA coatings to provide both non-bacterial killing anti-biofilm-attachment properties and allow the formation of a structured FN network to promote GF binding. Therefore, surface analysis was conducted to study how the surface chemistry and copolymer molecular structure, when applied to both a 2D (glass coverslip substrate) and 3D (MPs), affected mammalian cytotoxicity and adhesion (3T3 fibroblasts), fibronectin network assembly (GF binding promotion), and S. aureus attachment.
Materials and methods
Materials
All the materials were used as received unless stated otherwise. 2-(Dimethylamino)ethyl methacrylate (DMAEMA) (98%), ethyl acrylate (EA) (99%), 2,2′-azobis (2-methylpropionitrile) (AIBN, 98%) and the benzyl mercaptan (BzSH, 99%) were purchased from Sigma Aldrich. The cyclohexanone, acetone and heptane used as solvents for the synthesis and precipitation steps, respectively, were used as received and supplied by Fisher Scientific. In the ATRP, copper(I)bromide (98%) was purchased from Strem Chemicals UK LTD. Copper(I)chloride (99.99%) and copper(II)chloride (97%) were procured from Puratrem and Sigma-Aldrich, respectively. 1,1,4,7,10,10-Hexamethyltriethylenetetramine (HMTETA) (97%), ethyl α-bromoisobutyrate (EBriBru) (99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (99%) were obtained from VWR International LTD. Aluminium oxide (Al2O3) was obtained from Fisher Scientific. Murine 3T3 fibroblast cells (passage number 50–60) were obtained from the European Collection of Cell Cultures (ECACC, UK) and cultured in 75 cm2 flasks (Nunc™, Fisher Scientific, Loughborough, UK) in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Paisley, UK) supplemented with 10% (v/v) foetal calf serum (FCS; Sigma-Aldrich Company Ltd, Dorset, UK), 2 mM L-glutamine solution (Sigma-Aldrich Company Ltd, Dorset, UK) and 1% (v/v) antibiotic/antimycotic solution (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mL penicillin G, 100 mg mL−1 streptomycin sulphate and 25 μg mL−1 amphotericin B; Sigma-Aldrich Company Ltd, Dorset, UK).
000 units per mL penicillin G, 100 mg mL−1 streptomycin sulphate and 25 μg mL−1 amphotericin B; Sigma-Aldrich Company Ltd, Dorset, UK).
Synthetic procedures
 | ||
| Scheme 1 Reaction schemes for the copolymerisations of ethyl acrylate (EA) (red sphere) and 2-(dimethylamino)ethyl methacrylate (DMAEMA) (blue sphere). (a) TFRP of EA-co-DMAEMA producing a random copolymer consisting in random (b) ATRP for EA-b-DMAEMA producing a block copolymer, involving the first step with the synthesis of polyEA (1) and second step with the growing of the second block of DMAEMA (2). | ||
The appropriate quantities of the monomers were introduced into the required volume of cyclohexanone with stirring, such that a 1:3 v/v ratio mixture was achieved. In the case of the copolymers, these quantities also required to reach the targeted molar ratios, 90:10% mol/mol (e.g., EA:DMAEMA 1.71 g:0.29 g). The thiol CTA, benzyl mercaptan (BzSH) was added at the concentration of 1 mol% with respect to the monomers. The initiator, AIBN (0.5% wt with respect to the monomers) was, first, dissolved in cyclohexanone and degassed separately prior to being added to the reaction mixture. Finally, the reaction vessel and the AIBN solution were cooled in ice and then degassed by being purged with argon using a standard Schlenk line for at least 1 h. To commence the reaction, the temperature was raised to 75 °C in an oil bath and the reaction held at this temperature with continual stirring for 18 h. After this, the reaction vessel was cooled to room temperature to cease the reaction and then polymer purification was conducted via precipitation of the cooled reaction mixture into an excess of heptane. The typical non-solvent:reaction media ratio was 5:1 v/v to enhance the precipitation process and, finally, the precipitated materials were collected in a vial and left in a vacuum oven at 25 °C for at least 24 h.
1H-NMR spectroscopic analysis was performed on the crude polymerisation solution to determine polymer conversion and, finally, on the precipitate to establish the actual monomer ratio of the final copolymer composition and to determine that the sample was free of any residual monomer. To evaluate the molar mass of the materials, the purified samples were dissolved in HPLC grade THF for GPC analysis. All the spectra data presented were collected at 400 MHz in CDCl3 and values are quoted as δH ppm.
1H-NMR of EA-co-DMAEMA purified (400 MHz, CDCl3) δ (ppm): 4.12 (4H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OOCH2, m), 2.55 (2H CH2CH2N, s), 2.28 (6H, NCH3CH3, s), 1.26 (3H, OCH2CH3, m).
OOCH2, m), 2.55 (2H CH2CH2N, s), 2.28 (6H, NCH3CH3, s), 1.26 (3H, OCH2CH3, m).
13C-NMR of EA-co-DMAEMA purified (400 MHz, CDCl3) δ (ppm): 174 (CO), 63.09, (COCH2CH2 DMAEMA), 60.25 (COCH2CH3), 56.77 (COCH2CH2 DMAEMA), 45.80 (NCH3CH3), 14.077 (OCH2CH3).
Synthesis of block EA-b-DMAEMA copolymers via atom transfer radical polymerisation
:EBriBru:CuI:PMDETA ratios of 130:1:1:1. After three freeze–pump thaw cycles, the mixture was added to Cu(I)Br (0.25 mmol, 35 mg) under argon atmosphere via canula. The reaction vessel was then purged with argon for 30 min, while stirring in the oil bath with the temperature set at 90 °C. After the reaction was conducted for a further 1 h at 90 °C, it was terminated by opening the flask to air and cooling to room temperature. The reaction mixture was then diluted with 10 mL dicholoromethane (DCM) and passed through a small neutral alumina column to remove the catalyst. The final pure product was obtained after precipitation into an excess of heptane as described in the statistical copolymer method section.
1H-NMR spectroscopic analysis was performed on the crude polymerisation solution to determine polymer conversion and, finally, on the precipitate to establish the actual monomer ratio of the final copolymer composition and to determine that the sample was free of any residual monomer. To evaluate the molar mass and dispersity of the materials, the purified samples were dissolved in HPLC grade THF for GPC analysis. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.23 (m, CH2CH3); 4.14 (m, OCH2CH3).
:2.1 (monomers:acetone). Subsequently, Cu(I)Cl (0.063 mmol, 6.23 mg) and Cu(II)Cl (0.014 mmol, 1.7 mg) were added in the Schlenk flask. Finally, the ligand 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA) (0.062 mmol, 11.42 μL) was introduced immediately before starting the three freeze–pump thaw cycles, to minimise the interaction with the inorganic catalyst during the three cycles. The reaction was conducted for 24 h at a temperature of 50 °C with continuous stirring. The polymerisation was terminated by opening the flask to air and cooling to room temperature. The reaction mixture was then diluted with 10 mL THF and passed through a small neutral alumina column to remove the catalyst. The final pure product was obtained after precipitation into an excess of hexane.
1H-NMR spectroscopic analysis was performed on the crude polymerisation solution to determine polymer conversion and, finally, on the precipitate to establish the actual monomer ratio of the final copolymer composition. To evaluate the molar mass and dispersity of the materials, the purified samples were dissolved in HPLC grade THF for GPC analysis.
1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.23 (m, CH2CH3), 2.28 (s, NCH3CH3) 2.56 (m, CH2CH2N); 4.14 (m, OCH2CH3).
13C-NMR (400 MHz, CDCl3) δ (ppm) = 174 (CO), 63.09, (COCH2CH2 DMAEMA), 60.25 (COCH2CH3), 56.77 (COCH2CH2 DMAEMA), 45.80 (NCH3CH3), 14.077 (OCH2CH3).
Microparticle production method
Microparticle characterisation
Mammalian cytocompatibility testing
000 units per mL penicillin G, 100 mg mL−1 streptomycin sulphate and 25 μg mL−1 amphotericin B). The medium was changed twice a week, with all cultures maintained in a humidified environment at 37 °C, 5% CO2 in air.
000 cells per well in complete 3T3 media (supplemented with 10% (v/v) FCS) and incubated overnight at 37 °C in a controlled, humidified atmosphere (5% CO2 in air). Samples were then investigated for cell adhesion, morphology and viability using Live/Dead™ staining and the Presto Blue assay (as described below).
:9) diluted in sterile PBS and incubated in the dark for 1.5hours at 37 °C. Aliquots (100 μL) aliquots of the reagent were assessed for fluorescence at λexc/λem 560/590 nm using a Tecan Infinite M200 microplate reader (Tecan, UK), and metabolic activity expressed as a percentage of the control (glass coverslip) as a measure of cell viability.
Glass coverslips coated with surfmers of the polymer candidates (EA-co-DMAEMA and EA-b-DMAEMA) were placed on the bottom of the wells of a 24-well plate, and each well filled in with 2 mL of bacterial suspension (OD600 0.1) in tryptic soy broth (TSB) + 1% glucose (w/v). The plate was incubated for 18 h at 37 °C after which time, the bacterial suspension was discarded, and glass coverslips were washed three times with PBS, to remove loosely adherent cells. CFU counts: the glass coverslips were collected into 15 mL-centrifuge tubes with 2 mL of PBS. Cells growing as a biofilm were detached by 10 min-soft sonication and 30 s vortexing. Six 10-fold dilutions were prepared, and 100 μL aliquots of each dilution were plated on Muller Hinton (MH) agar plates. CFUs were counted after overnight incubation at 37 °C, and CFUs per polymer surface unit were determined. FESEM analysis: the polymer coated coverslips with biofilm grown on the top were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer at room temperature for 1 hour and washed twice with 0.1 M cacodylate buffer. Sample dehydration was performed by ethanol/water solutions (30%, 50%, 70%, 85%, 95% and 100% v/v), 10 min each step and two repetitions with 100% (v/v) ethanol, and then by 1:1 ratio ethanol/hexamethyldisilazane (HMDS) solution for 4 min. Lastly, they were treated with 100% HMDS for 5 min, fixed with silver print on aluminium stubs and gold-coated by an automatic sputter coater (Quorum Q150R S). Samples were examined by a field emission scanning electron microscope (Sigma-Zeiss) at an accelerating voltage of 4 kV.
Simultaneously, 100 μL of every sample, at each time point, was serially diluted, and 100 μL of selected dilutions were spotted on MH plates and incubated overnight at 37 °C in duplicate. CFUs grown on MH plates were counted to determine the number of viable bacterial cells present within each culture.
Statistical analysis
In the statistical data analysis (as stated in the text) column represents mean values and error bars represent standard deviation (SD). Microparticle characterisation involved N = 1, n = 3 on a total number of 100 MPs analysed; biological characterisation involved N = 3, n ≥ 3 biologically independent samples.Results and discussion
Thiol-mediated free radical polymerisation (TFRP) was used to synthesise low molar mass (MWt) homo- and statistical co-polymers (i.e., <20000 g mol−1) that contained a target 20 mol% DMAEMA content. Benzyl mercaptan (BzSH) was used as model thiol chain transfer agent (CTA) because its aromatic group allowed for accurate polymer analysis via1H-NMR. Meanwhile, the synthesis of a block copolymer was achieved by sequential addition of DMAEMA to the EA macromonomer in an atom transfer radical polymerisation (ATRP; Scheme 1).
The chemical properties of the final copolymer materials synthesised in this study are reported in Table 1.
:DMAEMA ratios Mn and Đ data for the synthesised homopolymer and statistical (EA-co-DMAEMA) and block (EA-b-DMAEMA) surfactants
| Entry | Polymers | M n (g mol−1) | Đ | Final copolymer ratiod (% mol/mol) |
|---|---|---|---|---|
| a TRFP. b ATRP. c M n and Đ were obtained via permeation chromotography (GPC) using THF as eluent. d Final copolymer ratios were calculated by 1H-NMR. e The Mn of polyDMAEMA was calculated via1H-NMR because the GPC chromatogram was influenced by an interaction between the amino groups and the stationary phase. | ||||
| 1 | EA-co-DMAEMAa | 13100c |
1.3c | 80:20 |
| 2 | PolyEAa | 8900c | 2.2c | — |
| 3 | PolyDMAEMAa | 6000e | n/ae | — |
| 4 | EA-b-DMAEMAb | 15700c |
1.2c | 91:8 |
The TRFP polymers (Table 1, entry 1–4) exhibited relatively low Mn values, between 6000 and 13000 g mol−1, which confirmed that the quantity of CTA employed (1 mol% with respect to the monomers) was sufficient to deliver the required control over the chain lengths. Also, the dispersity values (Đ) were noted to be between 1.3 and 1.8, which are typical for a ‘well-controlled’ TFRP.39 Additionally, in the statistical copolymer (entry 1), the comonomer ratio was found to have reached the desired 20% DMAEMA.
Meanwhile, the synthesis of a block copolymer typically required the use of two step polymerisation. The first step included the synthesis of a macroinitiator (i.e., the reactive first block), from which the second block was subsequently grown. In this study, a poly(EA) macroinitiator was prepared following the synthetic procedure developed by Datta et al.,37 from which the DMAEMA block was grown. The ATRP was conducted in bulk using EBriBru as the initiator and the combination of PMDETA/Cu(I)Br as catalyst. The target theoretical Mn was 13000 g mol−1, to deliver a similar molar mass to those obtained with the random copolymers. The products Mn was recorded as 13190 g mol−1 with a Đ of 1.12 (Fig. 1). The subsequent polymerisation of the DMAEMA second block was performed in acetone using a Cu(I)Cl:Cu(II)Cl:HMTETA complex as catalyst at 50 °C. The 1H-NMR and GPC data (Fig. 1) in Table 1 also demonstrated that, for the EA-b-DMAEMA copolymer, the second block of DMAEMA had successfully grown from the EA block. The chemical shifts attributed to the DMAEMA side chain could be observed in the EA-b-DMAEMA block copolymer 1H-NMR spectra in Fig. 1, confirming the presence of this monomer in the copolymer structure and allowed the monomer (EA/DMAEMA) ratio to be calculated. In addition, the GPC traces (Fig. 1) show a slight shift from the EA macromonomer to the block copolymer, evidencing the growth of the second block. However, the unexpected bimodal nature of this chromatogram was attributed to an interaction between the DMAEMA block and the gel of the column. Similarly, the molar mass of the homopolymer polyDMAEMA was estimated from the 1H-NMR data alone since no traces in the GPC could be observed (Table 1, entry 3).
 | ||
| Fig. 1 Scheme of reaction for EA-b-DMAEMA. Top: 1H-NMR spectra and aligned GPC chromatogram of poly(ethyl acrylate) and bottom: 1H-NMR spectra and the respective GPC chromatograms of the block copolymers EA-b-DMAEMA. | ||
Whilst the relative feed DMAEMA to macroinitiator molar ratios were theoretically required to reach the target 20% mol DMAEMA content, the actual comonomer final product molar ratio achieved was very close to 90:10 mol%:mol% EA:DMAEMA, which was enough to produce stable self-assembling.
Investigation of 3D surfmer surface characteristics via microparticle production with EA-co-DMAEMA and EA-b-DMAEMA copolymers
The same oil-in-water (O/W) droplet flow-focusing chip microfluidic system adopted in previous studies was used with both the block and statistical surfmers to determine their suitability to act as surfactants for the preparation of microparticles (MP).14,40 The dispersed feed stream contained these novel amphiphilic copolymers (2% w/v), a photoinitiator (2% w/v) and 1,6-hexanediol diacrylate (HMDA, 96% w/v) as a core material. The flow rates through the chip of both the dispersed and the continuous phase (water) were optimised to ensure the formation of stable and monodisperse particles. When changing the flow rates of the dispersed phase (Qd) and continuous phase (Qc), different flow regime profiles can be observed within the channels (e.g., dripping, wall wetting, jetting, unstable particle, satellites formation and squeezing), which can affect the performance of the MPs manufacturing process.14 Fig. S1 (ESI†) contains the flow diagrams of the EA-co-DMAEMA and EA-b-DMAEMA surfactants at a chosen concentrations, which were built by systematically varying the Qd and the Qc from 0.1 mL h−1 to 0.5 mL h−1 and from 1 mL h−1 to 9 mL h−1, respectively. Both surfactants displayed similar gradual flow regime changes as the flow rate conditions were altered, with the increasing of Qd and Qc, passing through the main four different flow behaviours: wetting, squeezing, dripping, and jetting. This systematic study demonstrated that both copolymer types formed stable emulsions under the appropriate flow conditions and thus their capability to behave as surfactants. Fig. 2a and b contains the microfluidic particle sizing data and the SEM images of the resulting MPs at 250× and 650× resolution for both surfactants. The conditions under which the block and statistical materials exhibited formation of stable droplets (inside the channels of the microfluidic chip), were found to be similar with both producing a stable emulsion within the flow window of Qc 4–6 mL h−1 and Qd 0.1–0.2/0.3 mL h−1. The data in Fig. 2a demonstrated that the MPs generated were spherical with a diameter (60–70 μm) lower than the orifice width (100 μm), highlighting the efficiency of the surfactant in stabilising the interface between the two phases. The EA-co-DMAEMA seemed to perform less well as a surfactant, exhibiting a higher CV of 9% and a larger size distribution range of ∼60 to ∼70 μm, than the EA-b-DMAEMA (Fig. 2a). This indicated that the molecular surface arrangement of the two surfmers may be different, due to their differing 3D architectures. In the case of the statistical surfmer, this data indicated that the stabilisation given by the charged amine residues (short pendent group length) were not as effective at stabilising the interface as the previously used longer pendant group PEGMA hydrophiles as shown in previous works (%CV in the range of 2.5–3.5).14,23,40 However, such PEG-based hydrophiles could not be utilised in this study as they are known to prevent protein adhesion to surfaces.41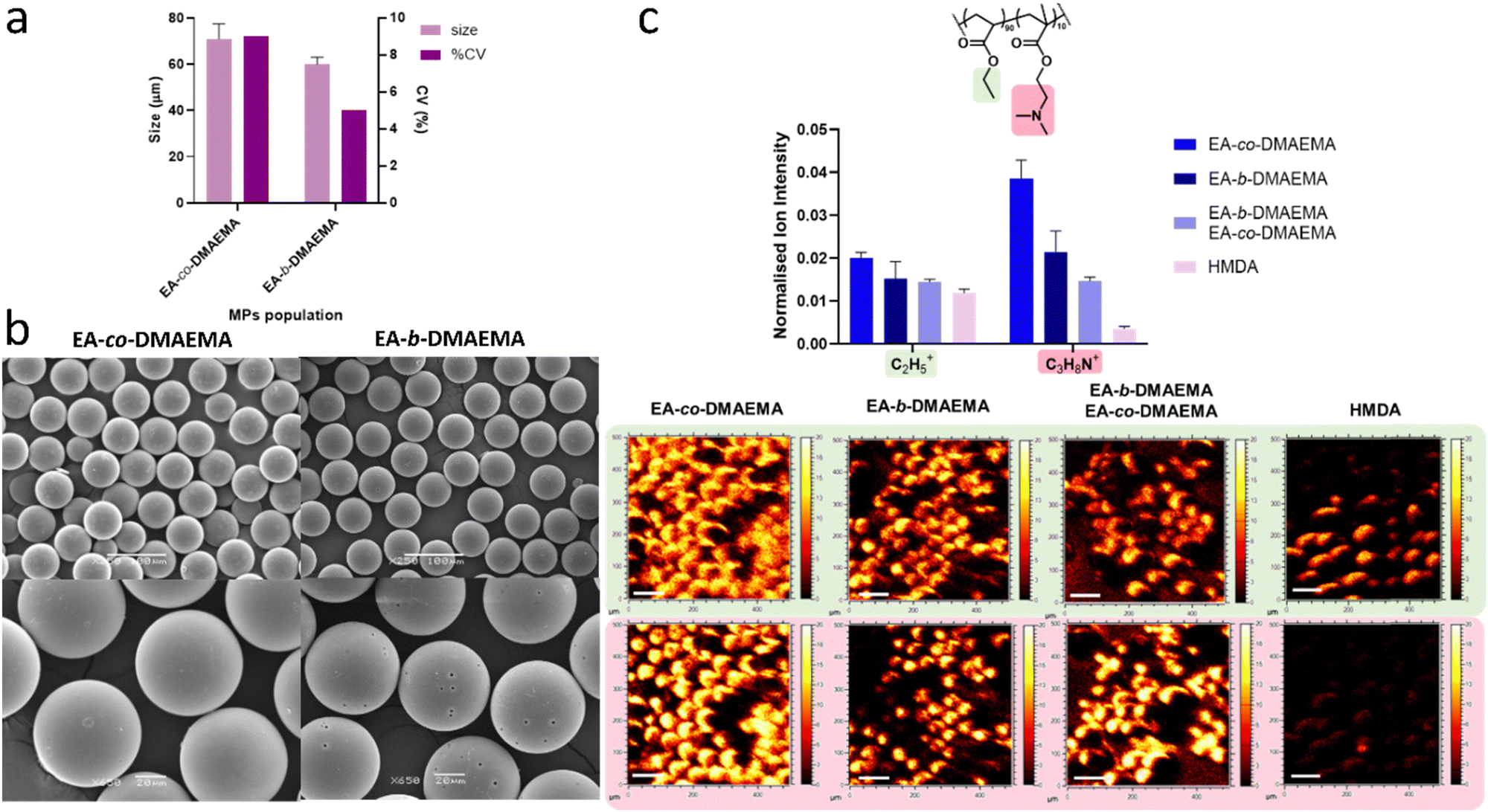 | ||
| Fig. 2 Investigation of 3D surfmer surface characteristics via microparticle production with EA-co-DMAEMA and EA-b-DMAEMA copolymers: (a) plot of particle diameter and the coefficient of variation (%) for the EA-co-DMAEMA and EA-b-DMAEMA surfactants (N = 1, n = 3 total population analysed 100 MPs; error bars represent SD) produced by analysing the representative SEM images (b) SEM images of the polymer MPs (scale bars for upper figures is 100 μm with magnification of 250×; scale bars for the bottom figures is 20 μm with magnification of 650×) (c) ToF-SIMS data showing intensities and the related images of the key ions associated with both EA:DMAEMA copolymers within the surfmer structure at the surface of the MPs compared to the intensity of the same ions present in the core HMDA MPs spectra (ToF-SIMS images of key ions with y and x axes ranging from 0 to 500 μm. The intensity bar spans from 0 to 20 AU, and the scale bar represents 100 μm). | ||
Meanwhile, with the EA-b-DMAEMA microparticles, the SEM analysis also indicated: (a) the appearance of pores on the surface and (b) that they were approximately 10 μm in diameter smaller than the statistical equivalents. Both observations were attributed to a difference in surfmer molecular surface orientation, suggesting that the block copolymer occupied a lower surface area between the particle and water interface. Its partitioned structure allowing it to orientate to be more perpendicular to the interface, with the DMAEMA block towards water and the EA extending towards the core. By comparison, the random structure of the EA-co-DMAEMA's backbone will force it to locate parallel to the interface due to the random distribution of the hydrophilic DMAEMA along its length. As a result, the EA-b-DMAEMA will cover less surface area per molecule, suggesting that an increased concentration of surfmer may be required to completely cover the surface and remove this porosity. However, this proposed change in the orientation of the block surfmer, instead produced MPs that are monodisperse with a CV around 5%. This was attributed to (a) better polymer packing at the interface due to their perpendicular orientation and (b) the block nature of the steric footprint of the DMAEMA in the EA-b-DMAEMA, which resulted in the introduction of steric, as well as charge, stabilisation into the system. However, this steric block hydrophile may also, partially, explain the appearance of the microparticle surface porosity with the increase in hydrophile size concentrated in one area of the molecule leading to increased detachment from the interface prior to polymerisation. To investigate whether pore size might affect cell behaviour and/or fibronectin coating, SEM analysis was conducted on the pores. This estimated the average diameter to be 1.75 ± 0.24 μm. By comparison, the average size of the 3T3 fibroblasts used in this study, when rounded, is in the region of 10 μm, and, when it is spread, this value would be of the order of 100 μm. Thus, the pores are one to two orders of magnitude smaller than both the rounded and spread cells respectively. In addition, Chung et al. reported that fibroblast persistence time was unaffected by the presence of pores, although the cells did exhibit directionality preferences based on pore patterning. So, as our SEM images revealed a random pore distribution, we therefore believe that the porosity of the microparticles produced will not significantly affect cell behaviour.
Finally, similar surface or self-assembly behaviour has been previously reported in other end-use applications such as: micro/nanofabrication of functional materials, the formation of polymersomes and in energy storage/conversion which support the self-assembly conclusions made by the authors.42,43
Microparticle surface characterisation
Time of flight secondary ion mass spectrometry (ToF-SIMS) analysis was used to investigate the bio-instructive surface characteristics of the microparticles, and to investigate how this was dictated by the surfmers used to produce the MPs. The data was collected in both positive and negative ion mode to identify unique ions associated with the hydrophilic and hydrophobic moieties within the copolymers. For these copolymers, the unique identifier found for the DMAEMA molecular structure was the C3H8N+ ion, whilst the C2H5+ was most associated with EA moieties. However, due to the simplicity of the EA structure, there is a small possibility that this ion can also be generated in very small quantities from the DMAEMA. Fig. 2c bar graph represents the total ion counts for the MPs produced using each copolymer alone and a mixed surfmer system containing both surfmers and HMDA core only particles (i.e., MPs prepared without the aid of a surfmer to demonstrate the difference between the unfunctionalized and functionalised MPs).From Fig. 2c, it was possible to confirm the presence of the surfmers on the MPs surfaces thanks to the presence of the detection of the characteristic ion of DMAEMA moieties (C3H8N+) within the structure. The MPs controlled by EA-b-DMAEMA exhibited the highest levels of the unique DMAEMA ion (C3H8N+) of all the samples containing the copolymers. This ion was also found at the surface of both the EA-co-DMAEMA MP's and mixed surfmer samples, but it was more diffusely distributed across the surface when compared to the microparticles prepared using the block copolymer where the individual shape of each MPs could be clearly defined. Again, this was proposed to support the conclusion that the orientation of the block copolymer is different to that of the statistical surfmer. Finally, the intensity for this ion is very low on the HMDA core/no surfactant MP, which was expected as there is no DMAEMA in/on the sample.
Investigation of 2D surfmer surface characteristics via the production of EA-co-DMAEMA statistical and EA-b-DMAEMA block copolymers surface deposited films
As the confirmation of surface structure at the MP surface was complex due to the 3D nature of the MPs, 2D samples were produced to confirm that different surface conformations were exhibited by these two copolymer architectures. This was achieved by dip-coating glass slides with samples of EA-b-DMAEMA, EA-co-DMAEMA, polyDMAEMA and polyEA copolymer and subjecting these to ToF-SIMS analysis. In Fig. S2a and b (ESI†), the C2H5+ ion exhibited high intensities in the polyEA homopolymer film and, as expected, was also found to present in the DMAEMA film in trace amounts. Similarly, again as expected, the C3H8N+ ion is totally absent from the polyEA sample. Meanwhile, in the case of 2D films when contrasting the intensities of the C3H8N+ ion exhibited in films of the block and random copolymers, it was noted that these varied dramatically. The EA-co-DMAEMA coating exhibited intensities from both C2H5+ and C3H8N+ ions, suggesting that the random architecture allowed both chemistries to orientate towards the “outer” surface i.e. the coating: biology surface. This was similar to the behaviour that was observed with the surface of the MPs in Fig. 2c. Meanwhile, the block copolymer data did not show any intensity for the DMAEMA identifier ion. Thus, it was hypothesised that the DMAEMA block was more attracted to the hydrophilic surface of the coverslip, containing OH groups, and so it had arranged itself to face towards the “inner” coating: glass surface only. Thus, the DMAEMA was, in effect, anchoring the block copolymer to the surface. This potentially suggesting an additional benefit of these block structures, namely that the DMAEMA block may, due to its size and high local concentration of amino functionality, provide a better anchor onto the surface of the glass so giving an increased resistance to the coating delaminating. Consequently, the block copolymer will only then present the EA block towards any solution that is it placed into. As a result, the surface will appear to be similar to the polyEA homopolymer. In fact, for films of block copolymers or polymer blends containing chemically distinct components, it is well known that differences in the surface energies of the components can lead to a variety of surface-related phenomena, including surface-induced ordering and orientation.44–47 This observation also supported the conclusions drawn on the trends in surface available functionality observed for the functionalised MPs, which suggests that the orientation of each block is driven by the affinity with the surrounding environment and its surface energy based on its chemical nature (i.e., hydrophobic and/or hydrophilic etc.).48 Therefore, the perpendicular orientation of the block copolymer in an oil in water emulsion will lead to the dominance of the DMAEMA block copolymer on the surface.Effect of surfmer orientation on biological performance
After assessing the difference in surface orientation of the hydrophilic/hydrophobic moieties, the polymer coated coverslips were tested in vitro for the attachment and viability of mammalian and bacterial cells. The ability of 3T3 fibroblast cells to adhere on 2D coverslips coated with polyEA, EA-co-DMAEMA and EA-b-DMAEMA (but without FN adsorption) was conducted using LIVE/DEAD™ staining and their viability assessed using the Presto Blue assay (Fig. 3a and b). Similarly, the biofilm-forming ability of S. aureus (CH 10850) was evaluated on 2D glass coverslips coated with EA-co-DMAEMA and EA-b-DMAEMA. In this experiment, after incubation, bacterial cells adhered onto the glass slides were detached, counted (Fig. 3c) and observed by FESEM (Fig. 3d). Additionally, bacterial cell viability after 3.5 h of incubation with the different surfmer solutions was observed (Fig. 3e). | ||
| Fig. 3 Effect of surfmer orientation on mammalian 3T3 fibroblast cell attachment and viability, bacterial (S. aureus) biofilm formation, and on fibronectin attachment and molecular orientation. (a) Mammalian cell viability of 3T3 fibroblasts following 24-hour incubation on 2D coated coverslips as measured using the Presto Blue assay (n = 3, error bars represent SD; viability expressed as percentage of the glass control). (b) Representative fluorescence images illustrating cell adhesion and viability on the 2D coated coverslips using the Live/Dead assay (Calcein-AM LIVE, GREEN) and propidium iodide (PI – DEAD, RED; scale bar 750 μm); merged fluorescence images shown with the corresponding phase contrast images. Scale bar 750 μm. (c) Biomass quantification after incubation of S. aureus (CH 10850) for 18 h at 37 °C on glass coverslips dip coated with the polymer candidates surfmers (EA-co-DMAEMA and EA-b-DMAEMA). Cell numbers are expressed as a CFU per polymer surface unit (n = 3, N = 1; error bars represent SD) (d) FESEM micrographs of bacterial adhesion on control (glass), EA-co-DMAEMA and EA-b-DMAEMA. Scale bar 10 μm. (e) % of viable cells of S. aureus (CH 10850) following incubation with the polymer surfmers (EA-co-DMAEMA and EA-b-DMAEMA) at two different concentrations (0.2 mg mL−1 and 0.02 mg mL−1). CFUs grown on MH plates were counted to determine the number of viable bacterial cells present within each culture. (f) Correlation between C3H8N+ intensity obtained from Fig. S2a (ESI†) against % cell viability from both S. aureus and 3T3 fibroblast. | ||
Based on the observations from data shown in Fig. 3a and b, the tested materials were not cytotoxic to 3T3 fibroblasts, except for the random copolymer (EA-co-DMAEMA).48 On the EA-b-DMAEMA surface, the FN nanonetwork is visible suggesting that the presence of the EA block has favoured its organisation. This observation further supports our findings from the mammalian cell adhesion experiments. Proposed mechanisms for the cytotoxicity of DMAEMA based copolymers have been reported to be very dependant of both molecular weight and copolymer ratio. For example, Knetsch et al. prepared a series of copolymers containing DMAEMA and MMA in various molar ratios and evaluated the toxicity of both any leachate from the materials and the washed surface.49 They reported that the measured toxicity increased with increasing DMAEMA content. Immuno-staining for phospho-tyrosine or vinculin demonstrated gradual loss of focal adhesions on the increasingly toxic surfaces. The loss of focal adhesions was found to coincide with an increase in paxillin and vinculin protein, indicating cells try compensating for loss of adhesion.
Meanwhile, a high throughput study of DMAEMA copolymers by Weiss et al. synthesized 107 copolymers which varied in charge, hydrophobicity, and molecular weight, and screened them for both cytotoxic behaviour and immunogenic responses.50 They identified the following three compositional regions of interest: (a) highly cationic polymers which disrupted the cellular plasma membrane to induce a toxic phenotype, (b) high molecular weight, hydrophobic polymers which were taken up by the cell via active transport to induce an immunogenic phenotype and (c) tertiary amine- and triethylene glycol-containing polymers that did not invoke immunogenic or toxic responses which they attributed to a combination of their pKa values and high degrees of charge shielding conferred from ethylene glycol moieties. Previous studies have shown that these characteristics could prevent rupture of membranes and facilitate safe, intracellular delivery when complexed with RNA.51,52 Consequently, in this study, we avoided high levels of DMAEMA (molar ratio always less or equal to 20%) and high molecular weight (our copolymer Mn's raged from ∼6000 to 16000 g mol−1) to avoid these potential toxicity triggers. Furthermore, whilst the Knetsch study suggests that lack of surface adhesion may play a role in DMAEMA based toxicity, this work would suggest that this may not be the case with all copolymers, as in this study there was significant FN adhesion, but a toxic response was still induced.49
To further emphasize the influence of surface properties on biological behaviour, glass slides coated with the surfmers bearing cationic functionality were tested against a Gram-positive microorganism, S. aureus, in order to determine their anti-biofilm efficacy with respect to non-coated (control) glass coverslips after 18 h incubation. The FESEM images in Fig. 3d demonstrated that S. aureus adhered to both the control and EA-b-DMAEMA coated surfaces, whereas adhesion was significantly reduced on the EA-co-DMAEMA surface. This finding was supported by the values of adhered biomass (CFU per cm2) shown in Fig. 3c, indicating lower biomass amount on the random copolymer EA-co-DMAEMA compared to the control and the second tested material (EA-b-DMAEMA). Additionally, antimicrobial tests were also performed on bacterial cells in planktonic mode of growth. Incubation with the test polymer EA-co-DMAEMA resulted in killing of bacterial cells during the exponential growth at both concentrations tested (Fig. 3e), similar to the cytotoxicity observed in the experiments carried out on mammalian cells (Fig. 3a). Cationic polymers have been known to possess inherent antimicrobial activity due to their net positive charge, particularly against Gram-positive bacteria.53 The mechanism of action for this class of polymers involves the binding of the agent to the cell wall through electrostatic interactions, thus affecting membrane permeability and finally leading to membrane lysis.53–55
These findings corroborate the observations made during the chemical-surface characterization of the polymers using ToF-SIMS which indicated surface-induced ordering and orientation of the DMAEMA side chain at the surface depending on the surfmers architecture. Interestingly, a correlation between C3H8N+ intensity against % cell viability from both S. aureus and 3T3 fibroblasts (Fig. 3f) showed how the surface is affecting the biological responses in a similar manner. In fact, in the surfaces where the C3H8N+ intensity is low (polyEA and EA-b-DMAEMA), both mammalian and bacterial cells showed low toxicity. On the other hand, when EA-co-DMAEMA was at the surface, the ion uniquely identifying DMAEMA gave the highest intensity corresponding to the killing effect in both experiments.
Based on the authors experience,13–15,23,33 the crucial factor that has been used to predict/design the bio-instructive biological results discussed in this study was related to the molecular pendant group moieties that are prominent on the surface of the coating/nanoparticles. Consequently, in the previous publications, we have reported that statistical copolymers could be used to deliver two types of biological surface influence, e.g. (a) biofilm attachment resistance and/or (b) anti-swarming behaviour.56 This “composite” behaviour has been shown to be as a direct result of the copolymer structures ensuring that both desired bio-instructive moieties are present at the surface.
Meanwhile, in this study, we report a key novelty of adopting a block rather than statistical copolymer structure such that a copolymer coating is derived that presents only one of the copolymer functional pendant moieties at the surface of the coating/nanoparticles. Thus, for the first time, this type of bio-instructive coating has been observed to act as a homo-polymer of only one of the copolymer components. Thus, it was concluded from the corroborating evidence of both the surface chemistry analysis using TOF-SIMS and the subsequent biological assay results, that the block copolymer is surface assembling to present this single functionality. The conclusion is supported by the fact that the DMAEMA-co-EA statistical copolymer presents both copolymer functionalities at the surface showing that a non-surface assembled polymer will not present a homo-EA surface.
Effect of ethyl acrylate chemistry orientation on fibronectin network
Fibronectin fibrillogenesis, a pivotal cell-mediated process driven by integrin activation, instigates conformational alterations in fibronectin molecules and orchestrates the organization of the actin cytoskeleton.32,33,54 It was previously shown that certain chemistries, such as polyethylene amine (PEA), can induce a similar process in the absence of cells. This chemical stimulation augments fibronectin–fibronectin interactions, ultimately giving rise to the formation of a highly potent and biologically active network.32,33,54 Alternatively, in the present study, we investigated the impact of introducing additional moieties to the ethyl acrylate polymer chain on the assembly of fibronectin networks. By doing so, we aimed at expanding the utility of ethyl acrylate, rendering it capable of being manufactured as a surfactant in addition to inducing a biological effect through fibronectin assembly. This strategy could result as a promising avenue for the design and fabrication of enhanced surfactant materials with application in regenerative medicine. We studied the adhesion and cell viability of 3T3 fibroblast mammalian cells on coverslips coated with polyEA, EA-co-DMAEMA and EA-b-DMAEMA after adsorption of FN from human plasma at the concentration of 20 μg mL−1 (Fig. 4) to assess the cytocompatibility of the materials and their ability to support mammalian cell adhesion. | ||
| Fig. 4 Effect of ethyl acrylate chemistry orientation on fibronectin network assembly and on subsequent mammalian 3T3 fibroblast adhesion and viability: (a) mammalian 3T3 fibroblast viability on various coated glass coverslips and following fibronectin adsorption using the Presto Blue viability assay expressed as a percentage of the control (cells on uncoated glass) (data is presented as means ± SD; n = 3). (b) Representative fluorescence (Live/Dead™ stain: Calcein-AM (LIVE, GREEN) and propidium iodide (PI – DEAD, RED) and light microscopy images of 3T3 fibroblasts cultured on glass (control), polyEA, EA-co-DMAEMA and EA-b-DMAEMA coated surfaces (n = 3 N = 1). Scale bar 750 μm. (c) Atomic force microscopy (AFM) images after FN coating on plain glass coverslips and EA-co-DMAEMA and EA-b-DMAEMA coated coverslips. The intensity bar spans from 0 to 20 AU, and the scale bar represents 200 nm. | ||
Our findings from Fig. 4a and b demonstrate that, in general, the tested materials are able to support mammalian cell adhesion and are not cytotoxic, with the exception of the random copolymer, EA-co-DMAEMA. Interestingly, fibronectin adsorption did not improve the viability of the cells when cultured on the EA-co-DMAEMA coated surface. It is important to note that the 70 kDa amino terminal regions of fibronectin are crucial for cell-mediated assembly and conferring FN binding activity. Within this region, FN binding activity determines the ability of FN to bind to surfaces.55 The presence of a positive charge on the surface, in the random copolymer, may have caused a repulsion effect, leading to no adsorption of FN on the surface. Furthermore, as the fibrillogenesis of FN on polyEA has been extensively studied by Llopis-Hernandez et al., demonstrating its significant role in promoting FN organization,32 we investigated the organization of FN at the material interface on the two, novel different chemistries synthesised in this study: EA-co-DMAEMA and EA-b-DMAEMA. AFM images (Fig. 4c) of the plain glass coverslip and polymer coated surfaces, in the absence of FN, do not show any characteristic feature. After the FN adsorption (Fig. 4c), the plain glass and EA-co-DMAEMA surfaces show the typical globular FN conformation, indicating the absence of substrate-induced fibrillogenesis. On the EA-b-DMAEMA surface, the FN nanonetwork is visible suggesting that the presence of the EA block has favoured its organisation. This observation further supports our findings from the mammalian cell adhesion experiments.
Conclusions
This study successfully synthesized two PEG-free amphiphilic copolymers: a statistical copolymer (EA-co-DMAEMA) and a block copolymer (EA-b-DMAEMA) using TFRP and ATRP, respectively. Both possessed the desired low molar mass and exhibited good characteristics for use as microparticle surface modifiers. However, the block copolymer (EA-b-DMAEMA) demonstrated superior performance in several key aspects. It produced monodisperse microparticles with smaller sizes and lower coefficient of variation compared to the statistical copolymer. ToF-SIMS analysis confirmed a preferential orientation of the DMAEMA block at the microparticle interface, suggesting superior surface enrichment compared to the random copolymer. Unlike the cytotoxic statistical copolymer, the block copolymer (EA-b-DMAEMA) exhibited excellent mammalian cell biocompatibility and promoted fibronectin network formation, suggesting great potential for regenerative medicine applications. In addition, the surface induced-ordering orientation of DMAEMA depending on surfmers architecture resulted in killing of bacterial cells with EA-co-DMAEMA, and subsequently, low biofilm formation. Interestingly, a correlation between C3H8N+ intensity against % cell viability from both S. aureus and 3T3 fibroblasts showed how the surface is affecting the biological responses. In fact, in the surfaces where DMAEMA is less exposed to the external environment (polyEA and EA-b-DMAEMA), both mammalian and bacterial cells showed low toxicity. By strategically arranging the monomer sequence (statistical vs. block), we can achieve superior control over surface properties and functionality, promoting advanced materials in drug delivery, tissue engineering, and other biomedical fields and highlighting the importance of copolymer architecture in material design.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant numbers EP/N006615/1 and EP/P001114/1] and the UKRI funded UK Regenerative Medicine Platform 2 – Acellular Hub (MR/R015651/1).References
- N. C. T. Dadi, B. Radochová, J. Vargová and H. Bujdáková, Microorganisms, 2021, 9, 2332 Search PubMed.
- X. Zhao, Q. Li, Z. Guo and Z. Li, Stem Cell Res. Ther., 2021, 12, 1–13 Search PubMed.
- K. Sadtler, A. Singh, M. T. Wolf, X. Wang, D. M. Pardoll and J. H. Elisseeff, Nat. Rev. Mater., 2016, 1, 16040 CAS.
- F. Batool, H. Özçelik, C. Stutz, P. Y. Gegout, N. Benkirane-Jessel, C. Petit and O. Huck, J. Tissue Eng., 2021, 12 DOI:10.1177/20417314211041428.
- L. M. Weiner, A. K. Webb, B. Limbago, M. A. Dudeck, J. Patel, A. J. Kallen, J. R. Edwards and D. M. Sievert, Infect. Control Hosp. Epidemiol., 2016, 37, 1288–1301 CrossRef.
- A. M. C. Maan, A. H. Hofman, W. M. de Vos and M. Kamperman, Adv. Funct. Mater., 2020, 30(32), 2000936 CrossRef CAS.
- T. Tolker-Nielsen, Microbiol. Spectrum, 2015, 3, 1–12 CAS.
- A. Cassini, L. Diaz Högberg, D. Plachouras, A. Quattrocchi, A. Hoxha and G. Skov Simonsen, et al. , Lancet Infect. Dis., 2019, 19, 56–66 CrossRef PubMed.
- A. H. Holmes, L. S. P. Moore, A. Sundsfjord, M. Steinbakk, S. Regmi, A. Karkey, P. J. Guerin and L. J. V. Piddock, Lancet, 2016, 387, 176–187 CrossRef CAS.
- Y. Cheng, G. Feng and C. I. Moraru, Front. Microbiol., 2019, 10, 1–17 CAS.
- T.-F. C. Mah and G. A. O’Toole, Trends Microbiol., 2001, 9, 34–39 CAS.
- C. De la Fuente-Núñez, F. Reffuveille, L. Fernández and R. E. W. Hancock, Curr. Opin. Microbiol., 2013, 16, 580–589 Search PubMed.
- V. Cuzzucoli Crucitti, L. Contreas, V. Taresco, S. C. Howard, A. A. Dundas, M. J. Limo, T. Nisisako, P. M. Williams, P. Williams, M. R. Alexander, R. D. Wildman, B. W. Muir and D. J. Irvine, ACS Appl. Mater. Interfaces, 2021, 13, 43290–43300 CAS.
- A. A. Dundas, V. Cuzzucoli Crucitti, S. Haas, J. Dubern, A. Latif, M. Romero, O. Sanni, A. M. Ghaemmaghami, P. Williams, M. R. Alexander, R. Wildman and D. J. Irvine, Adv. Funct. Mater., 2020, 30, 2001821 CAS.
- A. L. Hook, C.-Y. Chang, J. Yang, J. Luckett, A. Cockayne, S. Atkinson, Y. Mei, R. Bayston, D. J. Irvine, R. Langer, D. G. Anderson, P. Williams, M. C. Davies and M. R. Alexander, Nat. Biotechnol., 2012, 30, 868–875 CAS.
- H. J. Haugen, C. Soc Rev, M. Rahmati, E. A. Silva, J. E. Reseland and C. A. Heyward, Chem. Soc. Rev., 2020, 49, 5178 Search PubMed.
- A. Ranella, M. Barberoglou, S. Bakogianni, C. Fotakis and E. Stratakis, Acta Biomater., 2010, 6, 2711–2720 CrossRef CAS.
- Y. Zhang, H. Pan, P. Zhang, N. Gao, Y. Lin, Z. Luo, P. Li, C. Wang, L. Liu, D. Pang, L. Cai and Y. Ma, Nanoscale, 2013, 5, 5919–5929 RSC.
- Z. Chen, Y. Fan, L. Wang, Z. Bian and L. Hao, RSC Adv., 2021, 57, 36360–36366 RSC.
- L. Wang, M. He, T. Gong, X. Zhang, L. Zhang, T. Liu, W. Ye, C. Pan and C. Zhao, Biomater. Sci., 2017, 5, 2416–2426 RSC.
- A. J. Vegas, O. Veiseh, J. C. Doloff, M. Ma, H. H. Tam, K. Bratlie, J. Li, A. R. Bader, E. Langan, K. Olejnik, P. Fenton, J. W. Kang, J. Hollister-Locke, M. A. Bochenek, A. Chiu, S. Siebert, K. Tang, S. Jhunjhunwala, S. Aresta-Dasilva, N. Dholakia, R. Thakrar, T. Vietti, M. Chen, J. Cohen, K. Siniakowicz, M. Qi, J. McGarrigle, S. Lyle, D. M. Harlan, D. L. Greiner, J. Oberholzer, G. C. Weir, R. Langer and D. G. Anderson, Nat. Biotechnol., 2016, 34, 345 CrossRef CAS PubMed.
- L. Yang, S. Pijuan-Galito, H. S. Rho, A. S. Vasilevich, A. D. Eren, L. Ge, P. Habibović, M. R. Alexander, J. De Boer, A. Carlier, P. Van Rijn and Q. Zhou, Chem. Rev., 2021, 121, 4561–4677 CrossRef CAS PubMed.
- A. Latif, L. E. Fisher, A. A. Dundas, V. Cuzzucoli Crucitti, Z. Imir, K. Lawler, F. Pappalardo, B. W. Muir, R. Wildman, D. J. Irvine, M. R. Alexander and A. M. Ghaemmaghami, Adv. Mater., 2022, 36(43), 2208364 CrossRef PubMed.
- P. V. Giannoudis, E. Jones and T. A. Einhorn, Injury, 2011, 42, 549–550 Search PubMed.
- H.-S. Sohn and J.-K. Oh, Biomater. Res., 2019, 23, 9 CrossRef PubMed.
- Y. Fillingham and J. Jacobs, Bone Joint J., 2016, 98-B, 6–9 CAS.
- V. Campana, G. Milano, E. Pagano, M. Barba, C. Cicione, G. Salonna, W. Lattanzi and G. Logroscino, J. Mater. Sci.: Mater. Med., 2014, 25, 2445–2461 CAS.
- H. S. Sohn and J. K. Oh, Biomater. Res., 2019, 23, 9 Search PubMed.
- G. Kumar and B. Narayan, Classic Papers in Orthopaedics, 2014, pp. 527–530 Search PubMed.
- R. Dimitriou, E. Jones, D. McGonagle and P. V. Giannoudis, BMC Med., 2011, 9, 66 Search PubMed.
- H. Donnelly, M. Salmeron-Sanchez and M. J. Dalby, J. R. Soc. Interface, 2018, 15(145) DOI:10.1098/RSIF.2018.0388.
- V. Llopis-Hernández, M. Cantini, C. González-García, Z. A. Cheng, J. Yang, P. M. Tsimbouri, A. J. García, M. J. Dalby and M. Salmerón-Sánchez, Sci. Adv., 2016, 2(8) DOI:10.1126/sciadv.1600188.
- M. Salmerón-Sánchez, P. Rico, D. Moratal, T. T. Lee, J. E. Schwarzbauer and A. J. García, Biomaterials, 2011, 32, 2099–2105 CrossRef PubMed.
- M. M. Martino and J. A. Hubbell, FASEB J., 2010, 24, 4711–4721 CAS.
- M. Croes, B. C. H. van der Wal and H. C. Vogely, J. Orthop. Res., 2019, 37, 2067–2076 CrossRef PubMed.
- M. Vassey, L. Ma, L. Kämmerling, C. Mbadugha, G. F. Trindade, G. P. Figueredo, F. Pappalardo, J. Hutchinson, R. Markus, S. Rajani, Q. Hu, D. A. Winkler, D. J. Irvine, R. Hague, A. M. Ghaemmaghami, R. Wildman and M. R. Alexander, Matter, 2023, 6, 887–906 CrossRef CAS.
- H. Datta, A. K. Bhowmick and N. K. Singha, Macromolecular Symposia, John Wiley & Sons, Ltd, 2006, vol. 240, pp. 245–251 Search PubMed.
- J. Huang, B. Cusick, J. Pietrasik, L. Wang, T. Kowalewski, Q. Lin and K. Matyjaszewski, Langmuir, 2007, 23, 241–249 CrossRef CAS PubMed.
- J. P. A. Heuts, L. M. Muratore and T. P. Davis, Macromol. Chem. Phys., 2000, 201, 2780–2788 CrossRef CAS.
- C. A. Henshaw, A. A. Dundas, V. C. Crucitti, M. R. Alexander, R. Wildman, F. R. A. J. Rose, D. J. Irvine and P. M. Williams, Molecules, 2021, 26, 1–15 CrossRef.
- E. A. Vogler, Biomaterials, 2012, 33, 1201–1237 CrossRef CAS PubMed.
- S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152–1204 CrossRef CAS.
- C. Pinto-Gómez, F. Pérez-Murano, J. Bausells, L. G. Villanueva and M. Fernández-Regúlez, Polymers, 2020, 12, 2432 CrossRef.
- H. Yokoyama, T. E. Mates and E. J. Kramer, Macromolecules, 2000, 33, 1888–1898 CrossRef CAS.
- M. W. Matsen, Curr. Opin. Colloid Interface Sci., 1998, 3, 40–47 CrossRef CAS.
- J. Chen, X. Wang, S. R. Kline, S. Puri and H. L. Frisch, J. Phys.: Condens. Matter, 1997, 9, 2109–2133 CrossRef.
- X. Y. Lang, Y. F. Zhu and Q. Jiang, Thin Solid Films, 2006, 515, 2765–2770 CrossRef CAS.
- S. H. Kim, M. J. Misner and T. P. Russell, Adv. Mater., 2008, 20, 4851–4856 CrossRef CAS.
- M. L. W. Knetsch, N. Olthof and L. H. Koole, J. Biomed. Mater. Res., Part A, 2007, 82, 947–957 CrossRef PubMed.
- A. M. Weiss, M. A. Lopez, B. W. Rawe, S. Manna, Q. Chen, E. J. Mulder, S. J. Rowan and A. P. Esser-Kahn, Macromolecules, 2023, 56, 7286–7299 CrossRef CAS PubMed.
- F. Hausig, F. H. Sobotta, F. Richter, D. O. Harz, A. Traeger and J. C. Brendel, ACS Appl. Mater. Interfaces, 2021, 13, 35233–35247 CrossRef CAS PubMed.
- L. Du, C. Wang, L. Meng, Q. Cheng, J. Zhou, X. Wang, D. Zhao, J. Zhang, L. Deng, Z. Liang, A. Dong and H. Cao, Biomaterials, 2018, 176, 84–93 CrossRef CAS PubMed.
- L. Parhamifar, A. K. Larsen, A. C. Hunter, T. L. Andresen and S. M. Moghimi, Soft Matter, 2010, 6, 4001–4009 RSC.
- D. Gugutkov, C. González-García, J. C. R. Hernández, G. Altankov and M. Salmerón-Sánchez, Langmuir, 2009, 25, 10893–10900 CrossRef CAS PubMed.
- J. E. Schwarzbauer, J. Cell Biol., 1991, 113, 1463–1473 CrossRef CAS PubMed.
- J. F. Dubern, A. L. Hook, A. M. Carabelli, C. Y. Chang, C. A. Lewis-Lloyd, J. C. Luckett, L. Burroughs, A. A. Dundas, D. J. Humes, D. J. Irvine, M. R. Alexander and P. Williams, Sci. Adv., 2023, 9(4) DOI:10.1126/sciadv.add7474.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01941e |
| ‡ Current address: Healthcare Technologies Institute, Institute of Translational Medicine, School of Chemical Engineering, University of Birmingham, Birmingham, B15 2TT, UK. |
| This journal is © The Royal Society of Chemistry 2025 |