
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Topical siRNA therapy of diabetic-like wound healing†
Eva
Neuhoferova‡
ab,
Marek
Kindermann‡
 cd,
Matej
Buzgo
ef,
Karolina
Vocetkova
e,
Dalibor
Panek
g,
Petr
Cigler
*c and
Veronika
Benson
*ah
cd,
Matej
Buzgo
ef,
Karolina
Vocetkova
e,
Dalibor
Panek
g,
Petr
Cigler
*c and
Veronika
Benson
*ah
aInstitute of Microbiology of the Czech Academy of Sciences, Videnska 1083, 142 20, Prague 4, Czechia. E-mail: veronika.benson@tul.cz
bFaculty of Science, Charles University, Hlavova 2030, Prague 2, 128 40, Czechia
cInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo namesti 2, 166 10, Prague 6, Czechia. E-mail: petr.cigler@uochb.cas.cz
dDepartment of Physical Chemistry, University of Chemistry and Technology Prague, Technicka 5, 166 28 Prague 6, Czechia
eInstitute of Experimental Medicine of the Czech Academy of Sciences, Videnska 1083, 142 20, Prague, Czechia
fInoCure s.r.o., Politickych veznu 13, 100 00, Prague, Czechia
gFaculty of Biomedical Engineering, Czech Technical University in Prague, Namesti Sitna 3105, Kladno 2, 272 01, Czechia
hTechnical University of Liberec, Faculty of Health Studies, Trebizskeho 1402, 46001, Liberec, Czechia
First published on 22nd November 2024
Abstract
Non-healing wounds are a serious complication in diabetic patients. One of the detrimental factors contributing to limited wound healing is the accumulation of metalloproteinase-9 (MMP-9) in the wound. Selective inhibition of MMP-9 is one of the established therapeutic targets for diabetic wound healing. Here, a functional and biocompatible wound dressing is developed to enable a controlled release of a traceable vector loaded with the antisense siRNA against MMP-9 in the wound. The dressing consists of degradable polymer nanofibers embedded with a vector nanosystem – polymer-coated fluorescent nanodiamonds optimized for the binding of siRNA and colloidal stability of nanodiamond–siRNA complexes in a physiological environment. The developed dressing is tested on murine fibroblasts and also applied to wounds in a diabetic murine model to evaluate its suitability in terms of in vivo toxicity, biological efficacy, and handling. The treatment results in significant local inhibition of MMP-9 and a shortening of the wound healing time. The scar formation in treated diabetic-like mice becomes comparable with that in non-treated diabetes-free mice. Our results suggest that the application of our biocompatible dressing loaded with a non-toxic vector nanosystem is an effective and promising approach to gene therapy of non-healing wounds.
Introduction
Cutaneous wound healing is a physiological process involving a complex overlap of cascade events resulting in the reconstruction of the injured skin.1,2 Once a human wound does not follow the normal healing pattern (haemostasis, inflammation, proliferation and remodelling phase) within a period of roughly 6–8 weeks, it is typically considered chronic.3 Because of the high complexity of the healing process, chronic wounds are quite common.4 Among the numerous causes of impaired wound healing,5 diabetes mellitus attracts attention as a growing international health concern with no end in sight.3,6 The chronicity (wound healing delay) observed in diabetes mellitus can be triggered via different mechanisms, including prolonged inflammation, poor blood (oxygen) delivery, sustained infection, degradation of growth factors, and increased levels of matrix metalloproteinases (MMPs).3,7 Therefore, targeting these pathological triggers can serve as an efficient tool for the faster treatment of chronic diabetic wounds, which represent a heavy burden on public health and the healthcare system.8Despite the mentioned findings elucidating wound repair mechanisms, the most common treatment for non-healing diabetic ulcers is based on the removal of non-viable tissue from the wound followed by conventional dressing – gauzes, semipermeable films, hydrocolloids, alginates, etc.4,9 This kind of dressing typically has only a limited ability to bring chronicity-causing triggers back to their normal levels and thus facilitating faster wound healing.4,10 On the other hand, a lot of effort has been made to develop bioactive and biodegradable dressings containing antimicrobial agents,11,12 materials for gene therapy,13–16 or even living cells.4,17,18 These active compounds or cells can be effectively released from the biodegradable scaffold due to hydrolytic enzymes in wound exudates,19 the presence of bacteria in the infected wound,20 or other, rather mechanical, effects.3 Although these dressings could significantly reduce hospitalization time, their preparation is costly and they are yet to be put to widespread use.4
Recently, electrospinning technology applied in the fabrication of biodegradable/biomimetic nanofiber dressing has gained exceptional interest due to its simplicity, robustness and the possibility to construct even portable electrospinning machines21 offering an attractive way for tailored wound care.2,9 The nanofiber character of the dressing and its high porosity enables the embedding of bioactive moieties, absorption of the wound exudate, wound hydration, and permeation of oxygen.22 Furthermore, in contrast to conventional dressing, the nanofiber arrangement mimics the structure of the extracellular matrix (ECM) and thus promotes the attachment, proliferation and migration of cells in the wound.23
One crucial prerequisite for successful wound reconstruction is appropriate ECM remodelling (i.e. the final phase of wound healing) and the formation of a primary scar.1 It is therefore obvious that a balanced synthesis–degradation process of ECM is essential. The degradation of damaged ECM during wound healing is driven by zinc-dependent MMPs. During the normal healing process (acute wound without comorbidities), MMPs are transiently expressed and extracellularly activated to enable their proteolytic activity.1,24 However, persistently elevated MMP levels and abnormally low levels of tissue inhibitors of metalloproteinases have been found in chronic wounds.24,25 This pathological state leads to an undesirable imbalance of the ECM synthesis–degradation process and ECM loss.
In this article, we present a biodegradable nanofiber dressing which mediates the downregulation of MMP-9 (gelatinase B) levels in wounds via an RNA interference mechanism. Our approach combines the advantages of the synthetic nanofiber mesh (NF) embedded with a non-toxic nanosystem delivering siRNA targeted against MMP-9 mRNA in cells. As delivery particles we used a recently developed hybrid nanosystem based on nanodiamonds coated with a cationic statistical copolymer poly{(2-dimethylaminoethyl methacrylate)-co-[N-(2-hydroxypropyl) methacrylamide]} (Cop+-FND; Fig. 1).26,27 The complex of Cop+-FND with siRNA, Cop+-FND:siRNA, eliminates the enzymatic cleavage of siRNA, is non-toxic and is effective as an in vivo delivery platform.28 The nanofiber mesh composite NF{Cop+-FND:siRNA} is fabricated by electrospinning and serves as a scaffold for the controlled release of Cop+-FND:siRNA complexes into the wound. In addition, the nanodiamonds contain nitrogen-vacancy defects in the crystal lattice which produce near-infrared fluorescence upon excitation by a green laser and have a relatively long emission lifetime (≈20–40 ns).29 We utilized these specific features to easily distinguish the nanodiamonds from the polymer scaffold autofluorescence.28 Fluorescent lifetime imaging provided us with in-depth information about the spatial distribution of Cop+-FND:siRNA complexes inside nanofibers. Finally, we tested the performance of NF{Cop+-FND:siRNA} system on murine fibroblasts and in a diabetic murine wound model to evaluate its suitability in terms of in vitro and in vivo toxicity, biological efficacy and handling. We evaluate and compare the inhibition of MMP-9, wound healing time and scar formation in treated diabetic-like and diabetes-free mice.
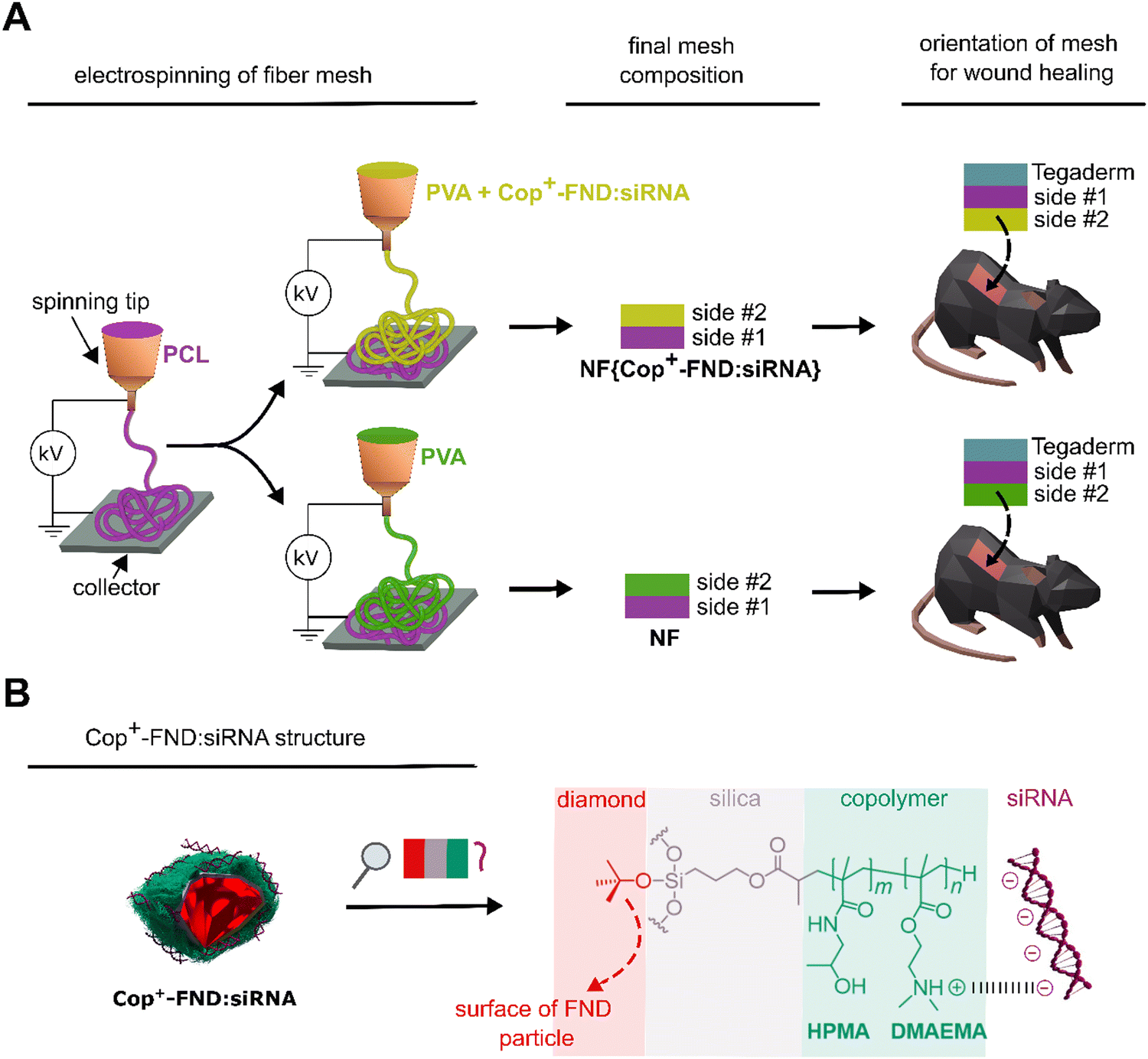 | ||
| Fig. 1 (A) Preparation procedure of the hybrid PVA/PCL fibrous mesh containing Cop+-FND:siRNA complexes, NF{Cop+-FND:siRNA} (upper part), and the control mesh without nanodiamond complexes, NF (bottom part). Side #2 faces the wound during the treatment. (B) Schematic structure of the cationic copolymer coating on the surface of a fluorescent nanodiamond (Cop+-FND), which binds electrostatically the siRNA molecule. | ||
Results and discussion
The NF{Cop+-FND:siRNA} composite – preparation and characterization
In our recent studies, we focused on the colloidal robustness,26 intracellular fate,27 and biodistribution of Cop+-FND:siRNA complexes (Fig. 1).28 We have observed that the localized intratumoral application of the colloidally stable Cop+-FND:siRNA leads to a high inhibition efficacy in mice bearing xenografted Ewing sarcoma tumours.Here, we utilized the potential of this delivery system for localized in vivo therapy and took advantage of its combination with a hybrid biodegradable PVA/PCL nanofiber mesh designed for topical wound healing. Unmodified nanodiamonds have been successfully incorporated in nanofibers of various compositions.30–32 We have designed and fabricated by electrospinning the nanofiber mesh composite NF{Cop+-FND:siRNA}, which serves as a scaffold for the controlled release of Cop+-FND:siRNA complexes into the wound.
Our previous studies26–28 demonstrate an optimal mass ratio of Cop+-FNDs to siRNA exceeding 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for efficient delivery. With an average of approximately 1360 siRNA duplexes per nanoparticle at this ratio,27 Cop+-FNDs offer a high loading capacity. This optimal ratio ensures: (i) minimal free siRNA after complexation with Cop+-FNDs (∼2%, ref. 28), (ii) a positive apparent ζ-potential of the complexes (ref. 26 and Table S1, ESI†), (iii) a reduced quantity of Cop+-FNDs in downstream applications, and (iv) the maintenance of robust colloidal stability of the system.26 The ratio is usually influenced by factors such as the specific batch of Cop+-FNDs and the siRNA manufacturer.28 In line with this, we used ∼30:1 ratio for our experiments in this study.
:1 for efficient delivery. With an average of approximately 1360 siRNA duplexes per nanoparticle at this ratio,27 Cop+-FNDs offer a high loading capacity. This optimal ratio ensures: (i) minimal free siRNA after complexation with Cop+-FNDs (∼2%, ref. 28), (ii) a positive apparent ζ-potential of the complexes (ref. 26 and Table S1, ESI†), (iii) a reduced quantity of Cop+-FNDs in downstream applications, and (iv) the maintenance of robust colloidal stability of the system.26 The ratio is usually influenced by factors such as the specific batch of Cop+-FNDs and the siRNA manufacturer.28 In line with this, we used ∼30:1 ratio for our experiments in this study.
The coating process results in stable, unaggregated nanoparticles, as evidenced by TEM imaging (Fig. 2(A)). The electrosteric repulsion provided by the copolymer coating effectively prevents particle aggregation, even in high ionic strength environments like 10× PBS (see Fig. 2(B)). In such conditions, the linear polymer chains may collapse due to electrostatic screening, leading to a decrease in particle size. Alternatively, a small fraction of particles may aggregate rapidly prior the measurement, contributing to decrease in the average particle size. Furthermore, we tested the stability in physiologically relevant conditions, such as 90% fetal calf serum at 37 °C (Fig. 2(C)). While slow aggregation was observed after 20 minutes in static conditions, the nanoparticles demonstrated good resistance to rapid aggregation in this challenging environment.
 | ||
| Fig. 2 Characterization of nanoparticle stability. (A) TEM image of raw FND nanoparticles (inset) and the particles after polymerization (Cop+-FND). (B) DLS measurement of hydrodynamic diameter of Cop+-FND in water and 10× PBS at 25 °C. (C) DLS measurement of Cop+-FND:siRNA complexes with mass ratio of 30:1 (Cop+-FND to siRNA) in 90% fetal calf serum at 37 °C. | ||
These complexes also allow for direct complexation of Cop+-FND with siRNA, even at high Cop+-FND concentrations (75 mg mL−1), without aggregation. This stability is maintained in the presence of polyvinyl alcohol used for electrospinning, as demonstrated in Fig. 3(A). Thus, highly concentrated/low-volume samples can easily be produced, which is a typical requirement for in vivo applications. The apparent Z-average diameter of the Cop+-FND:siRNA complexes (Cop+-FND to siRNA mass ratio ∼30:1) in the presence of the main dispersing component of the nanofibers, PVA, was 91.3 ± 1.5 nm (measured by DLS, see Fig. 3(A)). The magnitude of the cumulant-based polydispersity index (PDI) of 0.12 ± 0.02 represents a lower limit of mid-range PDI values and the sample can be considered nearly monodisperse. The relative measure of one part of the double-layer charge, which also contributes to the interparticle interaction, is apparent ζ-potential. Measuring it by ELS, we found a value 43.3 ± 2.2 mV (measured by ELS). All values represent means ± the standard deviation; more detailed information can be found in Table S1 in the ESI.†
 | ||
| Fig. 3 (A) DLS measurement of Cop+-FND:siRNA in 5% PVA (each sample was further diluted with Milli-Q water as described in Section 2.4). The intensity size distribution was inferred from the intensity autocorrelation function g(2)(τ) − 1. The vertical black dashed lines coupled with the histogram represent particle diameters Di0.1, Di0.5 and Di0.9 at the 10th, 50th, and 90th percentiles of the size distribution; DZ represents the Z-average diameter. The error bars represent standard deviations over three single measurements from separately prepared samples. (B) Fiber diameter quantification (box plot and all measured data points organized in histogram bins) obtained from image analysis of FESEM micrographs (for all images analysed, see Fig. S1, ESI†). The box plots characterize the fiber diameter distribution using the 25th, 50th, and 75th percentiles; whiskers: 90th and 10th percentiles; the horizontal line in the box: median of the distribution; square: mean of the distribution. The number of fibers analysed per sample was 2000. (C) Comparison of NF{Cop+-FND:siRNA} nanofibers with a control nanofiber sheet without particles (NF). Only side #2, facing the wound, is shown for both samples – NF{Cop+-FND:siRNA} (top images) and NF (bottom images). Left column: FESEM micrographs – for all images see Fig. S1 (ESI†); middle column: FLIM images with green arrows indicating locations of single-point measurements. (D) Values of fluorescence decay acquired for 10 s from a single point from both sides of NF{Cop+-FND:siRNA} and NF, as indicated in the legend. The inset presents intensity traces of the measurements from side #2. (E) Phasor plot of all recorded fluorescence decays; each point corresponds to a FLIM image or point measurement. | ||
Next, we incorporated the Cop+-FND:siRNA complex in the hybrid PVA/PCL nanofiber mesh by blend electrospinning, providing NF{Cop+-FND:siRNA}. We utilized a combination of a highly hydrophilic polyvinyl alcohol homopolymer and a slowly biodegradable poly-ε-caprolactone homopolyester,33 which provides a balanced nanofiber for wound healing. As shown in,34 PCL and PVA are highly suitable for the formation of nanofibrous wound dressings. In the current setting, the PCL fibers were used as the integrative part of a wound dressing stimulating the adhesion and proliferation of skin cells. The PCL fibers were spun as the first layer subsequently covered by a PVA mesh containing the bioactive nanoparticles. PVA was used as a hydrophilic polymer, electrospun from aqueous solutions, that enables the dispersion of Cop+-FND:siRNA nanoparticles. The controlled release of PVA was regulated by the crosslinking of PVA chains, which alternates nanofiber degradation kinetics.35 We determined the morphology of the electrospun scaffold containing the NF{Cop+-FND:siRNA} using field emission scanning electron microscopy (FESEM). A PVA/PCL mesh without particles served as a control. The resulting FESEM micrographs revealed the fibrous morphology of both samples (Fig. 3, left column; Fig. S1, ESI†). However, side #2 of the NF{Cop+-FND:siRNA} sample showed a higher content of microfibrous fraction in comparison with side #2 of the nanoparticle-free control (see Fig. 3(B), (C), and Fig. S1, ESI†). The different morphology could be explained by the rheological behaviour of highly concentrated nanoparticle dispersion stabilized by a linear polyelectrolyte (Cop+-FND). Similar colloidal systems are typically influenced by electroviscous effects (e.g. swelling of linear chains), which are caused by overlapping of the electrical double layers in the first approximation. Consequently, an increase in solution viscosity and surface tension can be observed. These parameters are among the most important and affect the diameter of fibers as well as the resulting mesh morphology.
To assess the distribution of Cop+-FND:siRNA in the NF{Cop+-FND:siRNA} composite, we investigated the particles using FLIM. The long emission lifetime of fluorescent nitrogen-vacancy centres in FND (approximately 25 ns)36 enabled us to distinguish the Cop+-FND:siRNA complexes from the omnipresent short-living autofluorescence background of the nanofibers (Fig. 3(C), FLIM). The presence of Cop+-FND:siRNA complexes on side #2 of NF{Cop+-FND:siRNA} was unambiguously confirmed by both FLIM images recorded from several areas and single-point measurements from both sides of NF{Cop+-FND:siRNA} and NF. Exemplary fluorescence decays are presented in Fig. 3(D). Furthermore, photobleaching was observed during image acquisition from side #2 of NF whereas no decrease of signal was observed either during imaging or single-point measurements from NF{Cop+-FND:siRNA}, as can be expected for photo-stable luminescence of nitrogen-vacancy centres. Intensity traces recorded during 10-s single-point measurements are presented in the inset of Fig. 3(D). To represent the rather complex fluorescence decay profiles, we employed the graphical phasor approach. Fig. 3(E) is a phasor plot of all time-resolved data recorded from NF{Cop+-FND:siRNA} and NF. Each point in the plot corresponds to one measurement, namely either a FLIM image, or a single-point decay recording. Clustering of the points in the phasor plot demonstrates the consistency of the time-resolved measurements and that FLIM can reliably reveal the presence of long-lived NF{Cop+-FND:siRNA}. Further, as the FLIM images were recorded from several distinct locations and for various scanned area sizes (ranging from 20 × 20 to 80 × 80 μm), we can conclude that the Cop+-FND:siRNA complexes can be found in abundance and are distributed homogeneously in the NF{Cop+-FND:siRNA} composite.
In summary, we confirmed the intended structural arrangement of the sample and found a high loading of Cop+-FND:siRNA on side #2 of NF{Cop+-FND:siRNA} (facing the wound). By contrast, we did not detect the Cop+-FND:siRNA complex either on side #1 of NF{Cop+-FND:siRNA} or on any side of the NF control sample.
Cell culture – NF{Cop+-FND:siRNA} release, inhibition efficacy, and cytotoxicity
For effective topical treatment, the controlled release of Cop+-FND:siRNA complexes from the hybrid NF{Cop+-FND:siRNA} mesh into the wound is essential. In a simple release experiment in PBS (Fig. 4(A)) we found a gradual liberation of approximately 25% of all embedded complexes within 144 h. Interestingly, this hybrid structure, produced by blend electrospinning, did not exhibit any undesirable initial burst release, which is typical for scaffolds manufactured this way. | ||
Fig. 4 (A) Time-dependent release of Cop+-FND:siRNA from the nanofiber mesh under physiological-like conditions (PBS buffer, pH = 7.4, 37 °C). Control samples: NF – nanofiber mesh without nanoparticles, 1× PBS without additives. (B) Posterior distribution of the average effect size of NF{Cop+-FND:siRNA} and NF nanofiber mesh (2.0 cm2 mL−1) on MMP-9 mRNA inhibition in NIH/3T3 cells 144 hours post-treatment; the effect size (![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) ![[C with combining macron]](https://www.rsc.org/images/entities/i_char_0043_0304.gif) ![[t with combining macron]](https://www.rsc.org/images/entities/i_char_0074_0304.gif) ) reflects a difference between experimental group and no treatment control; assuming 100% release efficiency: ∼50 μg siRNA mL−1; data collected from three independent experiments. (C) Cell cytotoxicity: LDH assay assessing the release of lactate dehydrogenase from damaged cells 48 h post-treatment. A nanofiber mesh with an area of 0.3 cm2/200 μL was used; assuming 100% release efficiency: ∼30 μg siRNA mL−1. Data for analysis—(A) and (C), respectively—were obtained from a single experiment where the samples were analysed in triplicates; the error bars represent standard deviations over these replicates. ) reflects a difference between experimental group and no treatment control; assuming 100% release efficiency: ∼50 μg siRNA mL−1; data collected from three independent experiments. (C) Cell cytotoxicity: LDH assay assessing the release of lactate dehydrogenase from damaged cells 48 h post-treatment. A nanofiber mesh with an area of 0.3 cm2/200 μL was used; assuming 100% release efficiency: ∼30 μg siRNA mL−1. Data for analysis—(A) and (C), respectively—were obtained from a single experiment where the samples were analysed in triplicates; the error bars represent standard deviations over these replicates. | ||
While the elevated MMP-9 levels are associated with chronic wounds, fibroblasts, such as NIH/3T3 cells, exhibit basal levels of MMP-9 expression. This provides a suitable model system to evaluate the efficacy of our siRNA in modulating MMP-9 expression and its potential impact on wound healing processes. After 144 h of incubation with the NF{Cop+-FND:siRNA} complex, MMP-9 mRNA was extracted and its fraction was measured compared to a reference by RT-qPCR. Fig. 4(B) illustrates that the NF{Cop+-FND:siRNA} treatment reduced the expression of MMP-9 mRNA (PM (): −3.28, 89% CI [−5.95, −0.53]) compared to the no treatment control. In other words, the estimated fold change in MMP-9 mRNA expression for NF{Cop+-FND:siRNA}was markedly downregulated, with a fold change of approximately 0.1 (PM (2): 0.10, 89% CI [0.02, 0.69]). To assess the potential cytotoxicity of the tested scaffolds, we monitored the release of lactate dehydrogenase from damaged cells (Fig. 4(C)). We did not observe any adverse effects after 48 h. The interval 48 hours ensures good quality of tested cells (cell overgrowth avoided) and appropriate assessment of solely the direct toxic effect of the composite. We note that cellular uptake and cytotoxicity of nanoparticles without the nanofiber mesh were studied in our previous works.26,27
Diabetic-like animal model – in vivo NF{Cop+-FND:siRNA} application
To evaluate the in vivo efficacy of our delivery system, we used a diabetic-like mouse model of chronic wound healing (Scheme 1). This model system involves surface application enabling the direct visual observation of changes triggered by the NF{Cop+-FND:siRNA} composite used. Importantly, the improved wound healing described here relies on the re-establishment of a disease-impaired balance and can be achieved by targeting one particular metalloproteinase (such as MMP-9), thus constituting an easy model for gene therapy.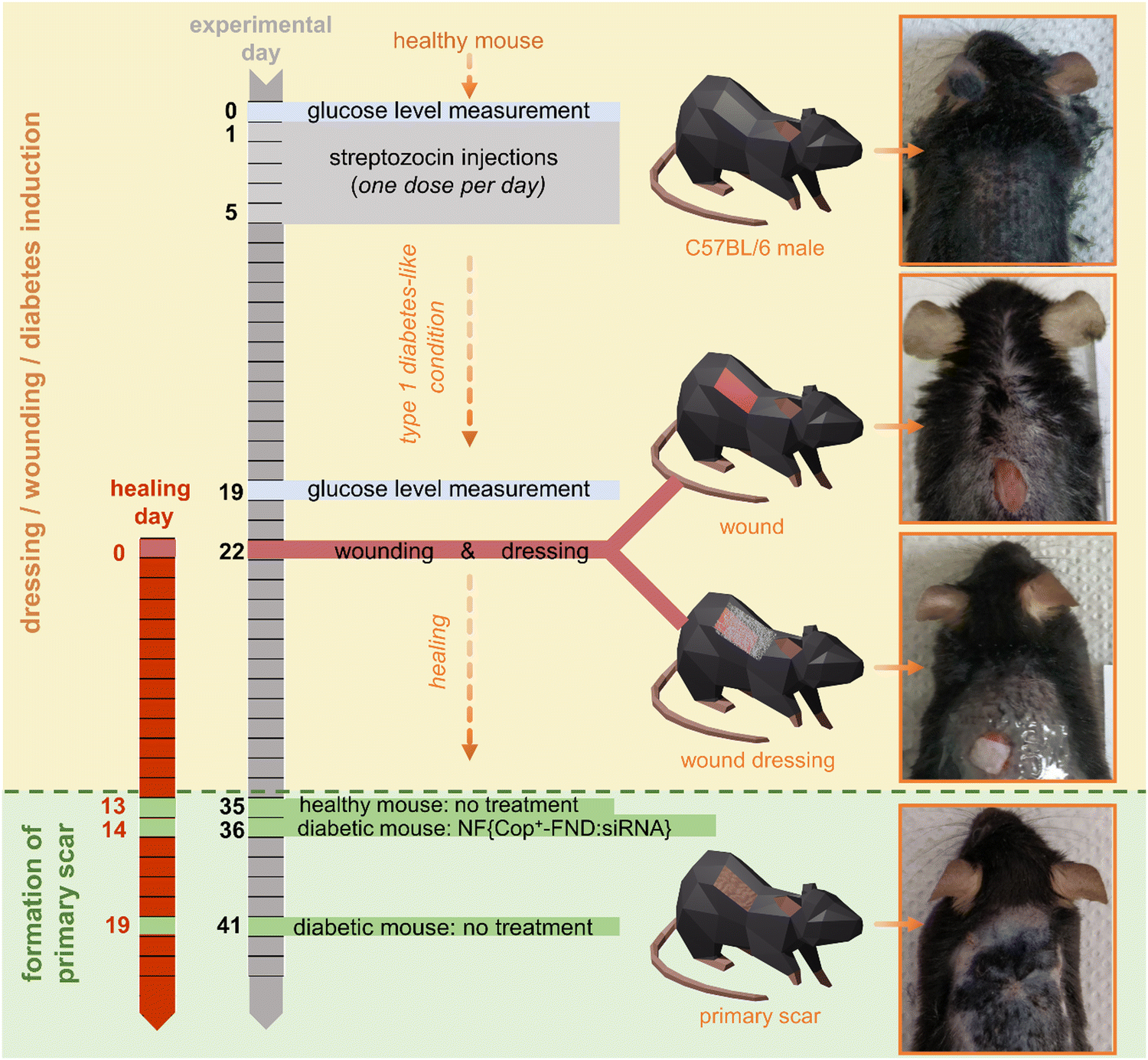 | ||
| Scheme 1 Timeline of wound healing model preparation. Orange top area: induction of diabetes-like conditions in healthy C57BL/6 mouse models (days 0–22) followed by wound creation and the application of primary (nanofiber-based) and secondary (Tegaderm) dressing (day 22). Green bottom area: the wounds were considered healed when primary scars were formed. The experiment was performed independently three times. | ||
The diabetic-like animals developed high glucose levels two weeks (on experimental day 19) after being injected with streptozotocin (STZ) (Fig. 5(C)), in agreement with the previously published protocol.37 To maximize the homogeneity of the diabetes-like conditions among the animals, they were grouped (group #1, #2, and #3) based on the reached glucose level (see Fig. 5(C) and Fig. S5, ESI†). Dorsal wounds were then created (experimental day 22/healing day 0) and subjected to a wound healing treatment with the NF{Cop+-FND:siRNA} composite and controls. To distinguish the healing efficacy after the application of our NF{Cop+-FND:siRNA} composite, we observed the wound until a primary scar was formed (Fig. S2–S4, ESI†).
 | ||
Fig. 5 (A) Healing kinetics of healthy and diabetic-like control wounds. (B) Comparison of healing kinetics between diabetic-like animals treated with nanofiber-based composites. Step functions represent measured data (n = 3/group), while dashed curves SW(t) show Weibull survival function estimates with 89% credible intervals. T50 indicates time to 50% wound closure (see Section 3.3). (C) Posterior distribution of the difference in blood glucose levels post- and pre-streptozotocin treatment, calculated from all individual data points. Healthy animals did not undergo the streptozotocin treatment and were therefore not included in this analysis. Data points for individual animals are jittered for better visualization and do not represent exact values on the y-axis. Grouping of data points shows assignment of individuals for wound healing experiment; original glucose levels are shown in Fig. S5 (ESI†). (D) Posterior distribution of the difference in 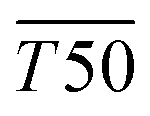 between experimental groups and diabetic-like control. Shaded areas represent 89% credible intervals and vertical dashed lines indicate means of posterior distributions. Study design: three experimental groups (group #1, group #2, group #3; n = 5/group; see Fig. 5(C)); were established based on glucose levels: diabetic-like (4 animals) and healthy control (1 animal; not shown in Fig. 5(C)); each group received all treatments. Results from these groups are shown in Fig. 5(A), (B) and (D). between experimental groups and diabetic-like control. Shaded areas represent 89% credible intervals and vertical dashed lines indicate means of posterior distributions. Study design: three experimental groups (group #1, group #2, group #3; n = 5/group; see Fig. 5(C)); were established based on glucose levels: diabetic-like (4 animals) and healthy control (1 animal; not shown in Fig. 5(C)); each group received all treatments. Results from these groups are shown in Fig. 5(A), (B) and (D). | ||
To quantify the macroscopic effect, we assessed a mean T50 time ( ) until 50% of mice experienced wound closure under a given treatment (SW(t) = 0.5) – see estimated survival functions SW(t) in Fig. 5(A) and (B). Bayesian analysis inherently provides a posterior distribution of possible
) until 50% of mice experienced wound closure under a given treatment (SW(t) = 0.5) – see estimated survival functions SW(t) in Fig. 5(A) and (B). Bayesian analysis inherently provides a posterior distribution of possible  times for each treatment that are compatible with data and applied statistical model. Based on data shown in Fig. 5(A) and (B), we estimated the effect of the applied treatment on the wound delay, which is expressed as a difference in
times for each treatment that are compatible with data and applied statistical model. Based on data shown in Fig. 5(A) and (B), we estimated the effect of the applied treatment on the wound delay, which is expressed as a difference in  between experimental groups and diabetic-like control (see Fig. 5(D)). We found that the application of our NF{Cop+-FND:siRNA} composite promoted healing by ∼6 days (PM: −5.7, 89% CI [−6.4, −5.0]) in comparison with diabetic-like controls without treatment. This shows that the treated wounds in diabetic-like animals were healed markedly sooner than in diabetic-like animals without the nanofibrous dressing. The healing efficacy of NF{Cop+-FND:siRNA}-treated diabetic-like animals was then similar to healthy animals without diabetes-like conditions (PM: –6.7, 89% CI [–7.3, –6.0]). Taking into account the overall position of the posterior distributions in Fig. 5(D) for NF and NF{Cop+-FND:ctrlRNA}, the results are most compatible with no important effect. By contrast, posteriors for healthy control and NF{Cop+-FND:siRNA}-treated mice are considerably separated from the control samples.
between experimental groups and diabetic-like control (see Fig. 5(D)). We found that the application of our NF{Cop+-FND:siRNA} composite promoted healing by ∼6 days (PM: −5.7, 89% CI [−6.4, −5.0]) in comparison with diabetic-like controls without treatment. This shows that the treated wounds in diabetic-like animals were healed markedly sooner than in diabetic-like animals without the nanofibrous dressing. The healing efficacy of NF{Cop+-FND:siRNA}-treated diabetic-like animals was then similar to healthy animals without diabetes-like conditions (PM: –6.7, 89% CI [–7.3, –6.0]). Taking into account the overall position of the posterior distributions in Fig. 5(D) for NF and NF{Cop+-FND:ctrlRNA}, the results are most compatible with no important effect. By contrast, posteriors for healthy control and NF{Cop+-FND:siRNA}-treated mice are considerably separated from the control samples.
To assess the effectiveness of the siRNA treatment, we evaluated MMP-9 expression and activity in wound tissue. Levels of the mRNA were quantified using RT-qPCR (Fig. 6(A)), protein abundance was determined by western blot (Fig. 6(B)), and enzyme activity was analysed by zymography (Fig. S6, ESI†).
 | ||
| Fig. 6 (A) Expression of MMP-9 mRNA in tissues before treatment (experimental day 22) and in scar tissues (experimental days 35–42) excised from healthy and diabetic-like animals. (B) Level of the MMP-9 protein in excised tissues after 7 days of treatment (experimental day 29, samples denoted as “wound”) and after scar formation (denoted as “scar”) in healthy and diabetic-like tissues analysed densitometrically from a western blot (C); uncropped blot image available in Fig. S7–S9 (ESI†). | ||
Relative MMP-9 mRNA expression (Fig. 6(A)) decreased by 54 ± 10% when comparing scar tissue exposed to the NF{Cop+-FND:siRNA} (experimental days 35–42) and tissue before treatment (experimental day 22). Further comparison of relative MMP-9 mRNA downregulation values between NF{Cop+-FND:siRNA} and NF{Cop+-FND:ctrlRNA} reveals a 2.5 ± 0.4-fold decrease (54 ± 10%: 22 ± 1%) of MMP-9 expression (Fig. 6(A)). A similar result was achieved for the diabetic control. These results confirm the successful delivery of siRNA molecules into the cytoplasm of wound cells and the degradation of MMP-9 mRNA transcripts. Contrary to our initial expectation of observing elevated MMP-9 mRNA levels in diabetic mice compared to healthy controls, our investigation revealed similar expression in both groups. In diabetic wounds, there are several mechanisms regulating the MMP-9 gene expression38 such as chronic inflammation promoted by pro-inflammatory cytokines like IL-1β and TNF-α,39 alongside potential dysregulation of intracellular signalling,40,41 or elevated levels of reactive oxygen species.42 Therefore, the MMP-9 mRNA level reflects dynamic changes during different stages of healing.43 The similar level of MMP-9 mRNA in both, healthy and diabetic-like, groups respond to the need for MMP-9 protein during healing and may be also affected by small sample size in each group.
In contrast to similar mRNA levels, the western blot analysis revealed a distinction in protein abundance between diabetic-like and healthy control animals (Fig. 6(B)). Diabetic animals displayed a higher accumulation of MMP-9 protein within scar tissue compared to healthy control. This observation suggests a potential dysregulation in protein translation or increased protein stability specific to the diabetic wound environment.44 Comparing MMP-9 protein levels in diabetic mice at the beginning of scar formation (healing day 7) and the day of scar formation (Fig. 6(B)), revelaed 38% increase in the untreated control group and 45% in the treated group. Importantly, treatment with NF{Cop+-FND:siRNA} significantly reduced MMP-9 protein levels in diabetic mice. We observed a decrease of 31% and 22% in protein abundance during healing and scar formation, respectively, compared to the non-treated diabetic-like control group. By reducing MMP-9 protein production, the treatment has the potential to promote a more balanced remodeling process within diabetic wounds.41
Complementing our findings on MMP-9 mRNA and protein, zymography analysis (Fig. S6, ESI†) revealed specific activity of MMP-9 in wound and scar tissues. Notably, the activity of MMP-9 was reduced in animals treated with the NF{Cop+-FND:siRNA} composite compared to the control group. Its activity is not solely dependent on mRNA levels but also influenced by activators (like MMP-3), tissue inhibitors of metalloproteinases, and the wound's redox state.42 Therefore, the observed reduction in MMP-9 activity in the NF{Cop+-FND:siRNA} group likely reflects a combined effect of these factors.
To evaluate the in vivo biocompatibility of NF{Cop+-FND:siRNA}, we investigated its potential to induce cutaneous allergic responses or organ toxicity. Except a minor skin damage due to the redressing process and presence of secondary dressing (Tegaderm), no signs of skin irritation were visually observed in any treatment group (see Fig. S2–S4, ESI†). Mice tolerated the application of NF{Cop+-FND:siRNA} dressings and controls well, with no signs of erythema, rash formation, or development of papules in the peri-wound area. These observations suggest good skin tolerance to the applied dressings. Our previous study28 employed systemic/regional administration of Cop+-FND:siRNA nanoparticles. This study revealed the liver as one of the primary sites where Cop+-FND:siRNA accumulated/aggregated.28,45 Given this finding, we evaluated serum ALT levels in this study. ALT is a well-established marker of liver damage, allowing us to assess potential toxicity from NF{Cop+-FND:siRNA}. As expected with topical application, Fig. S10 (ESI†) shows no significant differences in ALT levels between treated groups and the negative control (untreated control animal). This finding aligns with the minimized systemic exposure achieved by delivering Cop+-FND:siRNA via a nanofiber mesh.
These results, showcasing both the effectiveness of our NF{Cop+-FND:siRNA} treatment in preclinical studies and the benefits of MMP-9 restoration via gene therapy in damaged tissue with minimal impact on resident cells, provide preclinical evidence for its therapeutic potential. From a critical standpoint, streptozotocin-treated mice represent a basic model for investigating diabetic wound healing because it cannot fully mimic the pathological wound healing in humans. Their ability to replicate key features of type 1 diabetes, such as hyperglycemia, insulin deficiency, and impaired glucose tolerance, makes them a relevant tool for studying the mechanisms underlying delayed wound healing in this condition. However, it is essential to acknowledge the limitations of this model. The rapid onset of diabetes induced by streptozotocin may not fully capture the gradual progression often observed in human diabetes. Additionally, while the model can replicate hyperglycemia and insulin deficiency, it may not accurately represent the full spectrum of complications associated with human diabetes, such as neuropathy and retinopathy. A typical disadvantage of this model is also significant wound contraction observed in the normal process of mouse wound healing whereas bottom-up epithelialization and granulation represent normal wound healing in humans. More advanced models, such as the Zucker diabetic fatty rat, may offer greater physiological relevance for the follow-up studies. Despite these limitations, streptozotocin-treated mice still provide valuable insights into the pathophysiology of diabetic wounds and contribute to the development of potential therapeutic interventions.
However, application of nanofibers, and to lesser extent also the Tegaderm dressing, improves the quality of scar tissue.46 Due to moisture retention, the nanofiber-based dressings promote re-epithelialization and reduce epidermal thickness, thus resulting in good imitation of human wound healing.47 It is reasonable to expect that MMP-9 imbalance also affects wound contraction.48
On the other hand, the close similarity between murine and human MMP-9 and the possibility to incorporate fully degradable nanoparticles (instead of Cop+-FND) inside a fibrous mesh49 would facilitate the translation of this preclinical model into clinical research. Notably, the ongoing research on multifunctional scaffolds that combine siRNA delivery with other wound healing functionalities, such as promoting angiogenesis or delivering growth factors, shows promising results.50
Further, the size and covalent crystalline character of FNDs makes these nanoparticles essentially non-biodegradable and their size (>10 nm) prevents thus their excretion via the renal pathway. For this reason, FNDs are probably not suitable for routine systemic administration. Nevertheless, topical and locoregional51 treatments have different requirements for excretion and FNDs can still be considered a promising delivery vehicle for these applications.
Finally, exploring the use of siRNA pools targeting multiple mRNA sites within MMP-9 could further reduce potential off-target effects.52 Combining such siRNA strategies with those targeting other relevant diabetic wound healing factors could create a powerful tool for personalized medicine. It's important to acknowledge that, while existing approved single-target siRNA treatments53 represent successful examples, multitargeting siRNA pool-based therapy for wound healing remains an area with significant regulatory hurdles to overcome.
Conclusion
In summary, we have designed a hybrid biodegradable PVA/PCL fibrous mesh embedded with cationic copolymer-grafted fluorescent nanodiamonds carrying siRNA targeted against MMP-9 mRNA (Cop+-FND:siRNA). We assembled the hybrid nanomaterial using blend electrospinning and characterized its structure by fluorescence lifetime imaging microscopy and scanning electron microscopy. The fibrous mesh enabled the controlled release of the Cop+-FND:siRNA complexes without any undesirable initial burst release. The dressing facilitated the conservation and sustained release of the siRNA in murine diabetic-like wounds. We have demonstrated delayed wound healing in diabetic-like animals compared to animals without diabetic conditions caused by MMP-9 deregulation. The biofunctionality of the fibrous mesh containing Cop+-FND:siRNA complexes was demonstrated by a significant reduction in healing time: scar formation in treated diabetic-like animals was comparable with that in non-treated diabetes-free mice. Our healing system emphasizes the importance of selective gene therapy which may supersede current, commonly applied non-specific treatments of topical chronic wounds such as non-healing diabetic ulcers. The restoration of MMP-9 balance in damaged tissue seems to be a promising direction, which may ease the translation of topical gene therapy into clinical practice.Experimental methods
Materials
NIH/3T3 CRL-1658™ cells (ATCC®) were obtained from the Institute of Experimental Medicine of the Czech Academy of Sciences. TaqMan Universal Master Mix, MMP-9 specific qPCR assay (Mm00442991_m1), High-Capacity cDNA Reverse Transcription Kit, Power SYBR™ Green PCR Master Mix, LDH Cytotoxicity Assay Kit, RNAlater™ Stabilization Solution, M-PER™ Mammalian Protein Extraction Reagent, Pierce™ Coomassie Plus™ (Bradford) Assay Kit, Nunc™ MicroWell™ 96-Well Microplates, Novex™ Tris-Glycine SDS Sample Buffer (2X), Novex™ Sharp Pre-stained Protein Standard, SimplyBlue™ SafeStain, NuPAGE™ LDS Sample Buffer (4X), NuPAGE™ Sample Reducing Agent (10X), NuPAGE™ 4–12% Bis-Tris Mini gel, SuperSignal™ West Pico PLUS Chemiluminescent Substrate, and XCell SureLock Mini-Cell Electrophoresis System were purchased from Thermo Fisher Scientific. Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), streptozotocin and duplexed siRNA against MMP-9 mRNA (MW = 14,604) sense strand: 5′-r(GCC UAU UUC UGC CAU GGC AAA) d(AA)-3′ and antisense strand: 5′-r(UUU GCC AUG GCA GAA AUA GGC) d(TT)-3′ were purchased from Sigma–Aldrich. Fluorescently labelled siRNA duplex against GAPDH mRNA used for in vitro release studies was purchased from Generi BioTech, sense strand: 5′-AlexaFluor488-r(GAA GGU CGG UGU GAA CGG AU) d(TT)-3′ and antisense strand: 5′-r(AUC CGU UCA CAC CGA CCU UC) d(TT)-3′. High Pure RNA Isolation Kit and Cell Proliferation Reagent WST-1 were purchased from Roche. QIAshredder and RNeasy Fibrous Tissue Mini Kit were purchased from Qiagen. Gene-specific TBP primers: forward: 5′-ACC CTT CAC CAA TGA CTC CTA TG-3′ and reverse: 5′-TGA CTG CAG CAA ATC GCT TGG-3′ and control RNA (ctrlRNA) forward: 5′-r(CCU GCA CCA CCA ACU GCU UAG) d(AA)-3′ and reverse: 5′-r(CUA AGC AGU UGG UGG UGC AGG) d(TT)-3′were obtained from Integrated DNA Technologies IDT. TPP Petri dishes and TPP 24-well plates from BioTech. Gentamicin from Sandoz, Novartis Company. MMP-9 Monoclonal Antibody (5G3) and GAPDH Loading Control Monoclonal Antibody (GA1R) were purchased from Invitrogen. 3M™ Tegaderm™ Transparent Film Dressing was obtained from I.T.A. Interact, Tissue Adhesive was obtained from Surgibond, Glucometer FreeStyle Freedom Lite with cartridges were purchased from Abbott, and Disposable Biopsy Punch was obtained from Stiefel. Poly(vinyl alcohol) (PVA 40–88 and PVA 5–88) was purchased from Merck and poly(ε-caprolactone) from Sigma Aldrich. Chemicals (synthesis, purification and origin) for the preparation of copolymer-functionalized nanodiamonds in Section 2.2 were described in detail in ref. 26.Synthesis of cationic copolymer-functionalized fluorescent nanodiamonds (Cop+-FND)
Commercially available high pressure high temperature diamond nanocrystals (Microdiamond MSY 0–0.05 μm, type Ib with nitrogen impurity ∼200 ppm) were oxidized by air in a furnace at 510 °C for 4 h and treated with a mixture of HF and HNO3 (2:1) at 160 °C for 2 days. The nanocrystals were washed with water, 1 M NaOH, 1 M HCl and water (for detailed centrifugal parameters, see ref. 54). To create NV centres in the diamond lattice, we used a previously published protocol (see ESI† in ref. 55). Briefly, the powder was irradiated with a 16.6 MeV electron beam followed by annealing (900 °C, 1 h) and air oxidation (510 °C, 4 h). The powder was again treated with a mixture of HF and HNO3, washed with NaOH, HCl, water and then freeze-dried. The resulting powder was redispersed in Milli-Q water (60 mL, 2 mg mL−1) by probe sonication (for sonication parameters, see ref. 26). The transparent colloid containing fluorescent nanodiamonds (FNDs) was incubated for 30 min at room temperature and filtered using a 0.2-μm PVDF filter. The particles were coated with a methacrylate-terminated silica layer using a modified Störber procedure;56–58 the amount of all components needed for the silication procedure was linearly recalculated (according to ref. 26) to the amount of FND powder (120 mg). The terminal methacrylate groups on the FND surface allow to proceed with a radical polymerization, resulting in a dense layer of statistical cationic copolymer poly{(2-dimethyl-aminoethyl methacrylate)-co-[N-(2-hydroxypropyl)methacrylamide]} (poly(DMAEMA-co-HPMA)); the detailed preparation (including the purification of the chemicals) and a characterization of FND@silica@poly[DMAEMA-co-HPMA] complexes were presented in ref. 26 (specifically, sample denoted as “80+/200” in ref. 26 was used in this study). Briefly, HPMA and AIBN were freshly recrystallized prior to use. Both monomers, DMAEMA (1.968 mg) and HPMA (656 mg), were dissolved in DMSO (7.5 mL); AIBN (752 mg) was added to the mixture and filtered using a 0.2-μm PTFE filter. Methacrylate-terminated FNDs in DMSO (120 mg, 752 μL) were added and the stirred mixture was secured in argon. The polymerization proceeded for 3 days under an argon atmosphere at 55 °C. The reaction was terminated by MeOH addition, and the resulting Cop+-FND sample was purified by centrifugation using nuclease-free water (the sample was handled in a laminar flow box). The final concentration after purification was 74.6 mg mL−1 and the sample was stored at 4 °C. Cop+-FND was stored long-term in MeOH at −20 °C.
Electrospinning of hybrid PVA/PCL nanofiber mesh containing Cop+-FND:siRNA/siRNA-A488/ctrlRNA
PVA 40–88 powder was dissolved in nuclease-free water (nfH2O) at a concentration of 15% (w/v) and heated for 30 min at 90 °C under stirring. After cooling to RT, the PVA solution was sonicated for 1 min (Bandelin Sonorex™ RK 31, 30/240 W, 35 kHz) and autoclaved (121 °C, 15 min). Desalted siRNA lyophilizate was dissolved in nfH2O (500 μm, 7.3 mg mL−1); 32.2 μL of the siRNA stock solution was mixed with 66.8 μL of autoclaved PVA solution (15% (w/v)). To create a Cop+-FND:siRNA complex with the desired Cop+-FND:siRNA mass ratio of 32:1, an siRNA–PVA mixture was placed in a sonication bath filled with DEPC-treated water and 101.0 μL of Cop+-FND (74.6 mg mL−1) dissolved in nfH2O were slowly pipetted (not dropwise) directly into the sonicated mixture of siRNA–PVA. The resulting dispersion was sonicated for 30 s after mixing (the sample was placed in the sonication hotspot). The result was a stable, deep brown colloidal dispersion (200 μL) of Cop+-FND:siRNA in a 5% (w/v) PVA solution. A typical picture of this dispersion was presented in ref. 28 (see ESI,† Fig. S4e of ref. 28). This procedure was repeated ten times to obtain 2 mL of the dispersion. All samples (including the sonication bath) were handled in a laminar flow box. The same procedure was used to complex Alexa Fluor 488-labelled siRNA (siRNA-A488) and non-active siRNA (ctrlRNA).
Nanofibers were prepared using an InoSPIN MINI electrospinning unit (InoCure) with a needle electrode (G10) and a rotating drum collector (100 mm diameter) covered with aluminium foil. The voltage in the system was set at potential difference of 45 kV at a temperature of 30 °C. Composites were prepared by spinning 20 mL of PCL nanofibers from an acetic: formic acid (7:3) solution with a concentration of 20% (w/v). At the top of the solution, nanofibers from a PVA solution containing either active or non-active siRNA were deposited. The solution of PVA with Cop+-FND:siRNA (2 mL) was mixed with 2.5 mL 30% PVA 5–88, 3.75 mL 20% PVA 40–88, and 6.75 mL dH2O with total 10 (w/v)% PVA concentration. In addition, 40000 ppm of glyoxal (wglyox/wPVA) and 30000 ppm of phosphoric acid (wglyox/wphosphoric acid) were added for further crosslinking of the mesh. After spinning, the mesh was stored in vacuum foil and crosslinked in an oven at 60 °C for 24 hours. The resulting nanofiber mesh composite, NF{Cop+-FND:siRNA}, was approximately estimated as 10 × 10 cm in size and contained 20 μg siRNA per cm2 of NF. The NF{Cop+-FND:siRNA} was cut into round/square pieces with an area of 0.3 and 2.0 cm2, respectively; 0.3 cm2 round piece contains roughly 7 μg siRNA and 200 μg Cop+-FND; 2.0 cm2 round piece contains roughly 50 μg siRNA and 1500 μg Cop+-FND. Additionally, a PVA/PCL nanofiber mesh without Cop+-FND:siRNA complexes was used as a control and was prepared with parameters similar to those used for NF{Cop+-FND:siRNA}. All samples for in vitro/in vivo testing were sterilized in UV light for 15 minutes prior to use.
Cop+-FND:siRNA colloidal characterization evaluated by DLS and ELS
To evaluate the colloidal stability of Cop+-FND:siRNA in 5% (w/v) PVA, dynamic (DLS) and electrophoretic (ELS) light scattering analysis was performed. Z-Average diameter and apparent ζ-potential were measured using a Zetasizer Nano ZSP system equipped with a He–Ne laser emitting a power of 10 mW at a 633 nm wavelength. Size results were inferred from the second-order time intensity autocorrelation function g(2)(τ) − 1 considering 3rd order cumulant analysis in Zetasizer Software 7.13; each sample was measured three times with an automatic duration at a backscatter angle of 173°; the dynamic viscosity and refractive index of the solvent at 25 °C was set at 0.887 cP and 1.33, respectively. Apparent ζ-potential was measured using phase analysis light scattering at an angle of 13°. Each sample was measured two times with a dip cell, considering Monomodal analysis. To calculate the apparent ζ-potential value from Henry's equation, we used the Smoluchowski approximation for spherical non-coated particles without inspecting the value of the ratio of particle size to Debye length. For both analyses, Cop+-FND:siRNA in 5% (w/v) PVA was diluted with pure Milli-Q water to give a final volume of 0.6 mL and a Cop+-FND concentration of approximately 0.1 mg mL−1; measurements were performed in disposable cuvettes and the dilution of the Milli-Q water by the testing sample was lower than 1%. The values reported represent means of these measurements.Fluorescence lifetime imaging microscopy (FLIM) of nanofiber mesh composites
The spatial distribution of Cop+-FND:siRNA complexes embedded in the nanofiber mesh was characterized using fluorescence lifetime imaging microscopy (FLIM). Images were recorded using an inverted time-resolved MicroTime 200 (PicoQuant GmbH) confocal fluorescence microscope. FLIM and point measurements were performed using 531-nm sub-nanosecond pulsed laser excitation with a repetition rate of 20 MHz and an average power of approximately 35 μW. Nanofiber samples were placed between two round glass coverslips which were gently pushed together and imaged using an Olympus 60× UPlanSApo water-immersion objective with a numerical aperture of 1.2. A single-photon avalanche photodiode was used to detect fluorescence in the 641 ± 75 nm spectral region corresponding to FND luminescence. The final image resolution was 256 × 256 pixels and the pixel dwell time was 2 μs. Fluorescence decay and lifetime images were reconstructed and analysed using custom-written TTTR Data Analysis software.Electron microscopy
Each sample was fixed to a holder with double-sided sticky tape and coated with a thin layer of Au/Pd. The morphology of samples was observed using a Hitachi S-4700 field emission scanning electron microscope (FESEM) at 15kV. The image analysis was performed in ImageJ 1.53k. All micrographs from Fig. S1 (ESI†) were displayed separately in ImageJ at 200% magnification. Each image was then virtually dived into five different areas (each of the image corners and the middle part) and 250 manual measurements of fiber diameter were taken from these selected areas (50 measurements per area; 250 measurements per image; 8 × 250 measurement/sample). Micrographs (8 images per sample) were recorded from distinct locations.
Release of Cop+-FND:siRNA from the nanofiber mesh under physiological-like conditions
The release of the complexes from the nanofiber mesh was assessed spectrophotometrically using Alexa Fluor 488-labelled siRNA (siRNA-A488). The round NF{Cop+-FND:siRNA-A488} composite with an area of 0.3 cm2 was incubated for 14 days in 1 mL of PBS in a humid atmosphere consisting of 5% CO2 at 37 °C. Controls (PBS buffer and NF without nanoparticles in PBS buffer) were incubated in the same manner. At given incubation times (at 24, 72, 144, and 216 hours of incubation), the fluorescence intensity was directly measured in the supernatant using an Infinite M200 Pro plate reader (Tecan), Ex/Em 488 nm/525 nm. The maximum releasable amount of Cop+-FND:siRNA-A488 from the composite was determined after the total dissolution of the NF{Cop+-FND:siRNA-A488} composite in CHCl3 without further purification.Inhibition of MMP-9 metalloproteinase expression
000 cells per well in 1 mL of DMEM supplemented with 10% FBS in plastic 24-well plates. Cells were left for 4 hours to adjust and adhere to the bottom. Round NF{Cop+-FND:siRNA} composite with an area of 2.0 cm2 was added to the media on top of the cells and the cells were incubated for a further 144 hours at 37 °C, 5% CO2. Cells without the addition of NF{Cop+-FND:siRNA} were used as a control. Cell lysis and total RNA extraction were performed using the High Pure RNA Isolation Kit.
Cytocompatibility of the NF{Cop+-FND:siRNA} composite
The NIH/3T3 cell line was seeded in 96-well plastic plates, 20000 cells per well in 200 μL of DMEM containing 1% FBS. The NF{Cop+-FND:siRNA} composite (with a round area of 0.3 cm2) was placed on top of the cells and incubated for 48 hours at 37 °C in an atmosphere containing 5% of CO2.
Induction of diabetes-like conditions in animal models
In this study, fifteen animals were assigned to three independent experimental groups of five animals each (group #1, group #2, group #3 – see Fig. 5(C) and Fig. S5, ESI†). Animals were divided in these groups based on the reached glucose level. This grouping was performed to maximize the homogeneity of the diabetes-like conditions among the animals within one experimental group. Each group included four diabetic-like animals and one healthy control. Every group received all of the tested treatments. Diabetic-like conditions were established by the administration of a low-dose STZ injection to 8–10 weeks old C57BL/6 mice (Mus musculus, male) following the method described in ref. 37. All procedures involving mice followed the Czech law regarding animal protection, and the Ethical Committee of the Czech Academy of Sciences approved the experimental plan under the reference numbers 36/2015 and 80/2017. Glucose levels were measured with an Abbott glucometer after 6 hours of starving on day 0 of the experiment. STZ was dissolved in sodium citrate (pH 4.5) immediately before use and was administered intraperitoneally for five consecutive days. The daily dose comprised 1 mg of STZ per 25 g of animal weight. The mice were starved for 4 hours before each STZ injection. On day 19, the glucose level was measured to assess how it differed from that on day 1. As a result, 90% of animals had elevated glucose levels of >8.3 mmol L−1 after 6 hours of starving. These mice were considered to mimic diabetic-like conditions. Prior to wounding (day 22), the animals were anaesthetized using a ketamine–xylazine cocktail (100 μL per 20 g of animal weight, containing 0.16 mg of xylazine and 1 mg of ketamine) and shaved. Wounds were created using the Stiefel Disposable Biopsy Punch 0.6 cm in diameter (0.3 cm2). The tissue sampled during the wounding was kept in an RNAlater™ stabilization solution for further analysis. For treated groups, each wound was covered with a pre-sized (0.3 cm2) square of the nanofiber mesh. These meshes were then moistened with 50 μL of PBS to ensure proper adhesion and hydration. Wounds in the healthy control and diabetic-like control groups received only 50 μL of PBS, without the application of the nanofiber mesh. Transparent film dressing (Tegaderm™) was then used to protect the wound site and was affixed with tissue adhesive (Surgibond) placed on the non-wounded part of the skin. Wounds were visually inspected and redressed every two days, e.g. all the animals in particular group obtained a new dressing. Samples for wound analysis were taken on the day of wounding (day 22), on day 29, and on days when primary scars were formed. Wounds were considered healed when primary scars were formed. At this point, blood was collected first, followed by euthanasia of the animals. Subsequently, scar tissue was excised and stored in RNAlater™ for further analysis. Blood was collected from the heart of anaesthetized animals. ALT levels were measured using a diagnostic kit for determining the catalytic ALT/GPT concentration in plasma samples (Erba Lachema). To each 10 μL plasma sample, 80 μL of reagent 1 from the kit was added, and the mixture was incubated for 5 minutes at 37 °C. Following this, 200 μL of reagent 2 was added to each sample, and the mixture was incubated for an additional 1 minute at 37 °C. Absorbance was measured at 340 nm using an Infinite M200 Pro multimode plate reader (Tecan).Activity and amount of MMP-9 protein in healing and scar tissues
Excised tissues were lysed with the M-PER™ Mammalian Protein Extraction Reagent. Total protein concentrations in the lysates were measured using the Pierce™ Coomassie Plus™ (Bradford) Assay Kit. A bovine serum albumin standard of a known concentration (from 0 to 10 μg μL−1) was used to define a standard curve. Seven μL of the standard or the sample were mixed with 193 μL of the Bradford reagent and pipetted onto a transparent 96-well plate. The plate was incubated for 10 min and absorbance at 595 nm was measured using an Infinite M200 Pro plate reader (Tecan). The protein concentrations in samples were determined from the standard curve.MMP-9 enzymatic activity was assessed using zymography. Each protein-containing sample (5 μg of protein per 5 μL) was mixed with 5 μL of Novex Tris-Glycine SDS Sample Buffer. Electrophoresis was executed in an XCell SureLock™ Mini-Cell using Novex Running Buffer and the following settings: running time 90 min, constant voltage 125 V, and starting current 40 mA. The Novex Sharp Pre-Stained Protein Standard served for band identification. Renaturing Buffer was used to wash the gel after electrophoresis for 30 min at room temperature. The gel was then incubated in Developing Buffer for 90 min. Bands on the gel were visualized by incubation with SimplyBlue SafeStain for 1 hour, followed by 1-hour incubation in dH2O.
The MMP-9 protein level in the wound was assessed by Western blotting and compared to a GAPDH protein load control. A total of 160 μg of the protein sample (adjusted up to 16.3 μL with dH2O) was mixed with 2.5 μL of Reducing Agent and 6.3 μL of LDS Sample. The sample mixture (25 μL) was loaded onto a 10-well NuPAGETM 4–12% Bis-Tris Mini gel. The Novex Sharp Pre-Stained Protein Standard was loaded to determine the molecular weight and transfer efficiency. Electrophoresis ran for 35 minutes at a constant 200 V using the XCell SureLockTM Mini-Cell system and NuPAGETM MES 1× Running Buffer. Samples were then transferred onto the PVDF membrane using a semi-dry blotter and the following settings: 90 minutes, constant voltage of 7 V, and starting current of 30 mA. The membrane was washed in 0.1% Tween-20/PBS (T-PBS), blocked in 5% non-fat milk for 1 hour, and incubated with 1 mL of the primary antibody MMP-9 Monoclonal Antibody (5G3) overnight (1:1000 antibody in T-PBS). Next day, the membrane was washed in 0.1% T-PBS and incubated in 10 mL of the secondary antibody for 60 minutes (1:200 antibody in T-PBS; Goat Anti-Mouse IgG Peroxidase Conjugated). After washing in 0.1% T-PBS, the membrane was incubated with the SuperSignal™ West Pico PLUS Chemiluminescent Substrate. A G: Box Chemi XRQ and GeneSys software (both Syngene) were used for band detection and analyses. The GAPDH control was detected accordingly using the primary antibody GAPDH Loading Control Monoclonal Antibody (GA1R).
Statistical analysis
Traditional frequentist methods can provide a point estimate (e.g. mean value Ȳ) and a confidence interval for a measured variable Y. In contrast, Bayesian inference provides a distribution of plausible values for Ȳ (given the observed data and the model assumptions). This distribution, known as the posterior distribution, is summarized by credible intervals (CIs) and the mean of the posterior distribution (PM). In this study, the posterior estimates are presented in the abbreviated form: (PM: value, 89% CI: [lower limit, upper limit]). An 89% credible interval (Bayesian) indicates that there is an 89% probability that the true value of Ȳ lies within that interval. In comparison, an 89% confidence interval (frequentist) indicates that if we were to repeat the experiment many times, approximately 89% of the calculated intervals would contain the true value of Ȳ. Data analysis was performed using a regression models in a Bayesian framework utilizing software packages in R.59–66 The Bayesian models were fitted with the ‘brms’ package64 that employs Stan software for probabilistic sampling. The source data used for this publication including complete statistical procedure and R code are available online.67 The graphical output was modified for clarity using the Inkscape graphics editor.68Data availability
Data for this article, including the data used for construction of graphs in Fig. 1–5 are available at E. Neuhoferova, M. Kindermann, M. Buzgo, K. Vocetkova, D. Panek, P. Cigler and V. Benson, 2024, Zenodo, https://zenodo.org/records/12588170, DOI: 10.5281/zenodo.12588170.Conflicts of interest
All authors declare no financial/commercial conflict of interests.Acknowledgements
The authors are grateful to Dr David Chvatil (Institute of Nuclear Physics of the Czech Academy of Sciences) for the irradiation of nanodiamonds by electrons, to Dr Zuzana Zlamalova Cilova (University of Chemistry and Technology, Prague) for her assistance with FESEM measurements and to Dr Filip Tichanek (Institute of Clinical and Experimental Medicine) for his guidance with the statistical analysis. This work was supported by the Czech Academy of Sciences – Strategy AV21 (VP29), Czech Science Foundation project no. 23-04876S, European Union project C-QuENS (grant no. 101135359), Horizon Europe MSCA-SE project FLORIN (grant agreement ID: 101086142), Technology Agency of the Czech Republic, project TH90010001, EXTRASENS (ERA-NET/QuantERA Cofund Project), and by the Ministry of Education, Youth, and Sports of the Czech Republic (project No. CZ.02.01.01/00/22_008/0004558, co-funded by the European Union). The irradiation of HPHT nanodiamond by electrons was supported through Czech Academy of Sciences project no. RVO61389005.References
- A. Atala, D. J. Irvine, M. Moses and S. Shaunak, MRS Bull., 2010, 35, 597–606 CrossRef CAS PubMed.
- A. Memic, T. Abudula, H. S. Mohammed, K. Joshi Navare, T. Colombani and S. A. Bencherif, ACS Appl. Bio Mater., 2019, 2, 952–969 CrossRef CAS PubMed.
- N. K. Rajendran, S. S. D. Kumar, N. N. Houreld and H. Abrahamse, J. Drug Delivery Sci. Technol., 2018, 44, 421–430 CrossRef CAS.
- G. Han and R. Ceilley, Adv. Ther., 2017, 34, 599–610 CrossRef PubMed.
- S. F. Spampinato, G. I. Caruso, R. De Pasquale, M. A. Sortino and S. Merlo, Pharmaceuticals, 2020, 13, 60 CrossRef CAS PubMed.
- U. A. Okonkwo and L. A. DiPietro, Int. J. Mol. Sci., 2017, 18, 1419 CrossRef PubMed.
- M. Gao, T. T. Nguyen, M. A. Suckow, W. R. Wolter, M. Gooyit, S. Mobashery and M. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15226–15231 CrossRef CAS PubMed.
- E. Gianino, C. Miller and J. Gilmore, Bioengineering, 2018, 5, 51 CrossRef CAS PubMed.
- E. Mele, J. Mater. Chem. B, 2016, 4, 4801–4812 RSC.
- V. Jones, J. E. Grey and K. G. Harding, Br. Med. J., 2006, 332, 777–780 CrossRef PubMed.
- I. Negut, V. Grumezescu and A. M. Grumezescu, Molecules, 2018, 23, E2392 CrossRef PubMed.
- A. GhavamiNejad, A. Rajan Unnithan, A. Ramachandra Kurup Sasikala, M. Samarikhalaj, R. G. Thomas, Y. Y. Jeong, S. Nasseri, P. Murugesan, D. Wu, C. Hee Park and C. S. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 12176–12183 CrossRef CAS PubMed.
- C. Pang, K. S. Fan, L. Wei and M. K. Kolar, Wound Repair Regen., 2021, 29, 225–239 CrossRef PubMed.
- S. A. Castleberry, B. D. Almquist, W. Li, T. Reis, J. Chow, S. Mayner and P. T. Hammond, Adv. Mater., 2016, 28, 1809–1817 CrossRef CAS PubMed.
- C. Pinese, J. Lin, U. Milbreta, M. Li, Y. Wang, K. W. Leong and S. Y. Chew, Acta Biomater., 2018, 76, 164–177 CrossRef CAS PubMed.
- L. Wu, Y. Chen, G. Zeng, N. Mao, N. Li, L. Li, X. Xu and L. Yan, Chem. Eng. J., 2023, 457, 141244 CrossRef CAS.
- J. S. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- N. Kosaric, H. Kiwanuka and G. C. Gurtner, Expert Opin. Biol. Ther., 2019, 19, 575–585 CrossRef CAS PubMed.
- J. W. DuBose, C. Cutshall and A. T. Metters, J. Biomed. Mater. Res., Part A, 2005, 74, 104–116 CrossRef PubMed.
- Y. Suzuki, M. Tanihara, Y. Nishimura, K. Suzuki, Y. Kakimaru and Y. Shimizu, J. Biomed. Mater. Res., 1998, 42, 112–116 CrossRef CAS PubMed.
- S.-C. Xu, C.-C. Qin, M. Yu, R.-H. Dong, X. Yan, H. Zhao, W.-P. Han, H.-D. Zhang and Y.-Z. Long, Nanoscale, 2015, 7, 12351–12355 RSC.
- A. Venault, K.-H. Lin, S.-H. Tang, G. V. Dizon, C.-H. Hsu, I. V. B. Maggay and Y. Chang, J. Membr. Sci., 2020, 598, 117648 CrossRef CAS.
- B. P. Antunes, A. F. Moreira, V. M. Gaspar and I. J. Correia, Carbohydr. Polym., 2015, 130, 104–112 CrossRef CAS PubMed.
- S. Wall and G. Murphy, Int. J. Exp. Pathol., 2000, 81, A27–A28 Search PubMed.
- N. J. Trengove, M. C. Stacey, S. MacAuley, N. Bennett, J. Gibson, F. Burslem, G. Murphy and G. Schultz, Wound Repair Regen., 1999, 7, 442–452 CrossRef CAS PubMed.
- M. Kindermann, J. Neburkova, E. Neuhoferova, J. Majer, M. Stejfova, V. Benson and P. Cigler, Adv. Funct. Mater., 2024, 2314088 CrossRef CAS.
- J. Majer, M. Kindermann, D. Pinkas, D. Chvatil, P. Cigler and L. Libusova, Nanoscale, 2024, 16, 2490–2503 RSC.
- S. Claveau, M. Kindermann, A. Papine, Z. V. Díaz-Riascos, X. Délen, P. Georges, R. López-Alemany, Ò. M. Tirado, J.-R. Bertrand, I. Abasolo, P. Cigler and F. Treussart, Nanoscale, 2021, 13, 9280–9292 RSC.
- J.-J. Greffet, J.-P. Hugonin, M. Besbes, N. D. Lai, F. Treussart and J.-F. Roch, arXiv, 2011, preprint DOI:10.48550/arXiv.1107.0502.
- V. Guarino, I. Cruz-Maya, P. Reineck, H. Abe, T. Ohshima, K. Fox, A. D. Greentree, B. C. Gibson and L. Ambrosio, ACS Appl. Nano Mater., 2020, 3, 10814–10822 CrossRef CAS.
- A. Khalid, D. Bai, A. N. Abraham, A. Jadhav, D. Linklater, A. Matusica, D. Nguyen, B. J. Murdoch, N. Zakhartchouk, C. Dekiwadia, P. Reineck, D. Simpson, A. K. Vidanapathirana, S. Houshyar, C. A. Bursill, E. P. Ivanova and B. C. Gibson, ACS Appl. Mater. Interfaces, 2020, 12, 48408–48419 CrossRef CAS PubMed.
- J. C. Price, S. J. Levett, V. Radu, D. A. Simpson, A. M. Barcons, C. F. Adams and M. L. Mather, Small, 2019, 15, 1900455 CrossRef PubMed.
- L. N. Woodard and M. A. Grunlan, ACS Macro Lett., 2018, 7, 976–982 CrossRef CAS PubMed.
- M. Buzgo, M. Plencner, M. Rampichova, A. Litvinec, E. Prosecka, A. Staffa, M. Kralovic, E. Filova, M. Doupnik, V. Lukasova, K. Vocetkova, J. Anderova, T. Kubikova, R. Zajicek, F. Lopot, K. Jelen, Z. Tonar, E. Amler, R. Divin and F. Fiori, Regener. Med., 2019, 14, 423–445 CrossRef CAS PubMed.
- M. Buzgo, J. Greplová, M. Soural, D. Bezděková, A. Míčková, O. Kofroňová, O. Benada, J. Hlaváč and E. Amler, Polymer, 2015, 77, 387–398 CrossRef CAS.
- A. Beveratos, R. Brouri, T. Gacoin, J.-P. Poizat and P. Grangier, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 64, 061802(R) CrossRef.
- B. L. Furman, Curr. Protoc. Pharmacol., 2015, 70, 5.47.1 Search PubMed.
- M. Chang, Pharmacol. Res., 2016, 107, 243–248 CrossRef CAS PubMed.
- S. Nirenjen, J. Narayanan, T. Tamilanban, V. Subramaniyan, V. Chitra, N. K. Fuloria, L. S. Wong, G. Ramachawolran, M. Sekar, G. Gupta, S. Fuloria, S. V. Chinni and S. Selvaraj, Front. Immunol., 2023, 14, 1216321 CrossRef CAS PubMed.
- F. Al-Mulla, S. J. Leibovich, I. M. Francis and M. S. Bitar, Mol. BioSyst., 2011, 7, 3006 RSC.
- J. Chen, S. Qin, S. Liu, K. Zhong, Y. Jing, X. Wu, F. Peng, D. Li and C. Peng, Front. Immunol., 2023, 14, 1089001 CrossRef CAS PubMed.
- C. K. Sen, Wound Repair Regen., 2009, 17, 1–18 CrossRef PubMed.
- C. Yang, P. Zhu, L. Yan, L. Chen, R. Meng and G. Lao, J. Am. Podiatr. Med. Assoc., 2009, 99, 489–496 CrossRef PubMed.
- M. Hariono, S. H. Yuliani, E. P. Istyastono, F. D. O. Riswanto and C. F. Adhipandito, Wound Med., 2018, 22, 1–13 CrossRef.
- S. Claveau, É. Nehlig, S. Garcia-Argote, S. Feuillastre, G. Pieters, H. A. Girard, J.-C. Arnault, F. Treussart and J.-R. Bertrand, Nanomaterials, 2020, 10, 553 CrossRef CAS PubMed.
- B. S. Atiyeh, C. A. Amm and K. A. El Musa, Aesthetic Plast. Surg., 2003, 27, 411–417 CrossRef PubMed.
- S. Ahn, C. O. Chantre, A. R. Gannon, J. U. Lind, P. H. Campbell, T. Grevesse, B. B. O’Connor and K. K. Parker, Adv. Healthcare Mater., 2018, 7, 1701175 CrossRef PubMed.
- M. Reiss, Y. Han, E. Garcia, M. Goldberg, Y. Hong and W. Garner, Surgery, 2010, 147, 295 CrossRef PubMed.
- A. Mickova, M. Buzgo, O. Benada, M. Rampichova, Z. Fisar, E. Filova, M. Tesarova, D. Lukas and E. Amler, Biomacromolecules, 2012, 13, 952–962 CrossRef CAS PubMed.
- P. K. Mandapalli, S. Labala, A. Jose, S. Bhatnagar, R. Janupally, D. Sriram and V. V. K. Venuganti, AAPS PharmSciTech, 2017, 18, 809–820 CrossRef CAS PubMed.
- K. Kvakova, M. Ondra, J. Schimer, M. Petrik, Z. Novy, H. Raabova, M. Hajduch and P. Cigler, Adv. Funct. Mater., 2022, 32, 2109960 CrossRef CAS.
- J. Neumeier and G. Meister, Front. Plant Sci., 2021, 11, 526455 CrossRef PubMed.
- I. Ahn, C. S. Kang and J. Han, Exp. Mol. Med., 2023, 55, 1283–1292 CrossRef CAS PubMed.
- I. Rehor and P. Cigler, Diamond Relat. Mater., 2014, 46, 21–24 CrossRef CAS.
- T. Rendler, J. Neburkova, O. Zemek, J. Kotek, A. Zappe, Z. Chu, P. Cigler and J. Wrachtrup, Nat. Commun., 2017, 8, 14701 CrossRef CAS PubMed.
- I. Rehor, H. Mackova, S. K. Filippov, J. Kucka, V. Proks, J. Slegerova, S. Turner, G. Van Tendeloo, M. Ledvina, M. Hruby and P. Cigler, ChemPlusChem, 2014, 79, 21–24 CrossRef CAS PubMed.
- J. Neburkova, F. Sedlak, J. Zackova Suchanova, L. Kostka, P. Sacha, V. Subr, T. Etrych, P. Simon, J. Barinkova, R. Krystufek, H. Spanielova, J. Forstova, J. Konvalinka and P. Cigler, Mol. Pharmaceutics, 2018, 15, 2932–2945 CrossRef CAS PubMed.
- J. Slegerova, M. Hajek, I. Rehor, F. Sedlak, J. Stursa, M. Hruby and P. Cigler, Nanoscale, 2015, 7, 415–420 RSC.
- H. Wickham, R. François, L. Henry and K. Müller, dplyr: A Grammar of Data Manipulation. R package version 1.1.4, 2023, https://github.com/tidyverse/dplyr, https://dplyr.tidyverse.org Search PubMed.
- A. Vehtari, J. Gabry, M. Magnusson, Y. Yao, P.-C. Bürkner, T. Paananen and A. Gelman, loo: Efficient leave-one-out cross-validation and WAIC for Bayesian models. R package version 2.6.0, 2023, https://mc-stan.org/loo/ Search PubMed.
- J. Gabry and T. Mahr, bayesplot: Plotting for Bayesian Models. R package version 1.10.0, 2022, https://mc-stan.org/bayesplot/ Search PubMed.
- H. Wickham, ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag, New York, 2016 Search PubMed.
- R Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2021.
- P.-C. Bürkner, J. Stat. Softw., 2017, 80, 1–28 Search PubMed.
- A.-N. Spiess, propagate: Propagation of Uncertainty, https://CRAN.R-project.org/package=propagate, 2018.
- H. Wickham, D. Vaughan and M. Girlich, tidyr: Tidy Messy Data, https://CRAN.R-project.org/package=tidyr, 2023.
- E. Neuhoferova, M. Kindermann, M. Buzgo, K. Vocetkova, D. Panek, P. Cigler and V. Benson, Topical siRNA Therapy of Diabetic-Like Wound Healing: repository https://github.com/KindermannMarek/Topical-siRNA-therapy-1.
- Inkscape Project, Inkscape (version 1.2) https://inkscape.org 2023.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01547a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |