
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single-ion-conducting polymer and high-entropy Li-garnet composite electrolyte with simultaneous enhancement in ion transport and mechanical properties†‡
Ji-young
Ock
 *a,
Michelle
Lehmann
a,
Chang
Li
b,
Yangyang
Wang
c,
Harry M.
Meyer III
a,
Alexei P.
Sokolov
a,
Zhezhen
Fu
*b and
Xi Chelsea
Chen
*a
*a,
Michelle
Lehmann
a,
Chang
Li
b,
Yangyang
Wang
c,
Harry M.
Meyer III
a,
Alexei P.
Sokolov
a,
Zhezhen
Fu
*b and
Xi Chelsea
Chen
*a
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, USA. E-mail: ockj@ornl.gov; chenx@ornl.gov
bMechanical Engineering, School of Science, Engineering and Technology, Pennsylvania State University-Harrisburg, Middletown, PA 17057, USA. E-mail: zwf5065@psu.edu
cCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, USA
First published on 18th June 2025
Abstract
Enabling the lithium metal anode has been the holy grail for improving the energy density for the next generation advanced batteries. Developing electrolytes that will suppress Li dendrite growth and provide sufficient ionic conductivity remains a major challenge in this field. In this study, we develop a polymer–ceramic composite electrolyte for lithium metal batteries. The polymer matrix is a vinyl ethylene carbonate (VEC) based single-ion-conducting polymer electrolyte. The ceramic filler is a Li7La3Zr0.5Nb0.5Ta0.5Hf0.5O12 high-entropy Li-garnet (HE Li-garnet) ceramic, which is less prone to surface Li2CO3 formation compared to Al-doped Li garnets. The addition of HE Li-garnet leads to a 7-fold increase in the ionic conductivity (8.6 × 10−5 S cm−1 at 30 °C) compared to the pure polymer, while maintaining a high Li+ transference of 0.73. Proton nuclear magnetic resonance and thermogravimetric analysis results suggest that the addition of HE Li-garnet results in a lower degree of polymerization of VEC, leaving more unpolymerized VEC monomers in the matrix, serving as the governing mechanism for conductivity enhancement. The favorable interactions between HE Li-garnet particles and the polymer matrix lead to a stable and well-mixed composite with 2-fold enhancement of storage modulus at 40 °C. The simultaneous ion transport and mechanical property enhancement significantly improves the composite electrolyte's dendrite resistance and cycle life in Li symmetric cells. This work highlights the positive role HE Li-garnet can play in improving polymer electrolytes to enable lithium metal anodes.
Introduction
Single-ion-conducting polymers (SICs) with anions covalently attached to the polymer chains have emerged as promising electrolyte candidates.1–3 Unlike traditional polymer electrolytes with dissolved salts, SICs exhibit lithium-ion (Li+) transference numbers approaching unity, meaning they predominantly facilitate the transport of Li+ while effectively suppressing anion mobility.1,3–9 Accordingly, they greatly decrease concentration polarization during electrochemical cycling and thereby enhancing maximum current density as well as promoting homogeneous Li plating and stripping.10–13 Nevertheless, SICs struggle to reach room temperature ionic conductivity comparable to that of liquid electrolytes (>10−3 S cm−1), mainly due to low Li+ mobility.3The ionic conductivity of SICs may be enhanced through the incorporation of lithium-ion conductive ceramic fillers. This approach is based on the synergistic combination of ceramics and polymers, utilizing the high Li+ conductivity of ceramics and the mechanical and adhesive properties of polymers, to produce polymer-ceramic composite electrolytes with enhanced ionic conductivity, improved electrochemical stability, and adhesion to electrodes. Considerable research has focused on employing various ceramic fillers, such as LISICON-like (lithium superionic conductors), NASICON-like (sodium superionic conductors), argyrodites, garnets, and perovskites in these composites.14–28 Among the different types of ionically conductive inorganic fillers, the Li+ conducting garnet (e.g., Li7La3Zr2O12, LLZO) is a promising ceramic electrolyte, with high ionic conductivity, wide electrochemical window, and relatively good stability against Li metal.29–34 Many studies focus on the crystal chemistry of LLZO-based inorganics, studying the effect of iso- or aliovalent substitution in the crystal to achieve higher conductivity. Particularly, high-entropy (HE) Li garnets, which involve multiple equal or close molar ratio cations on the same site, can fundamentally reduce the Li ion migration energy barrier, alter the crystal structures (e.g., Li ion occupancy in Li-garnet), and consequently boost the ionic conductivity.35–38
In a polymer-ceramic composite electrolyte, the ion conducting mechanism is highly dependent on the interactions between the polymer and the ceramic.21,39 Active ceramic fillers may enhance the ionic conductivity by the following proposed mechanisms: (1) fast Li-ion dissociation along the polymer-ceramic interface and (2) through the highly ionically conductive ceramics themselves. Zhang et al. utilized 6Li/7Li magic angle spinning nuclear magnetic resonance (MAS-NMR) spectroscopy to investigate the local environments of Li ions and their contributions to forming Li-ion transport pathways in poly(ethylene oxide) (PEO)-LiClO4-LLZO composites containing tetraethylene glycol dimethyl ether (TEGDME) plasticizer.40 They observed that at 5 wt% TEGDME, Li-ion transport predominantly occurs through the LLZO phase, similar to the one without TEGDME. However, with 20 wt% TEGDME, Li ions mainly conduct through TEGDME associated phases (decomposed LLZO and LiClO4 in PEO/TEGDME complex), not the LLZO phase. In a previous study of our own,41,42 we developed a composite by incorporating Li perovskite nanorods into an SIC polymer. Our results showed that the enhancement in the composite's ionic conductivity was due to the formation of a percolated interfacial polymer layer around the perovskite nanorods with enhanced Li+ mobility and estimated the thickness of the interfacial layer to be ∼5 nm.
In this study, we developed a polymer-ceramic composite electrolyte for lithium metal batteries. The polymer host is a vinyl ethylene carbonate (VEC)-based SIC polymer electrolyte. The ceramic filler is a four-element-substituted high-entropy Li-garnet (HE Li-garnet) with a chemical formula of Li7La3Zr0.5Nb0.5Ta0.5Hf0.5O12. This HE Li-garnet is less prone to surface passivation and Li2CO3 formation compared to the traditional Al doped LLZO. Surface Li2CO3 is known to block the interactions between the polymer and ceramics, thereby limiting ion transport enhancement both along and across the polymer-ceramic interface.43 The formation of Li2CO3 on LLZO surface is dopant-dependent, as the dopants influence the decomposition energy of the Li-garnet structure, shown in literature by density functional theory simulation.44 In the HE Li-garnet, the dopants substituting the Zr site were selected due to their stable valence states and favorable defect formation energy, which lead to enhanced air stability, as demonstrated in our previous work.36 The composite electrolyte was prepared by in situ polymerization using a mixture of the HE Li-garnet fillers and the precursors of the SIC polymer. The composite electrolyte with 30 wt% HE Li-garnet showed a 7-fold enhancement in ionic conductivity (8.6 × 10−5 S cm−1 at 30 °C) and a two-fold enhancement in mechanical modulus, compared to the neat SIC polymer. The simultaneous enhancement in ion transport and mechanical properties leads to greatly prolonged cycle life in a Li symmetric cell at 40 °C. The origin of the ion transport enhancement as well as the mechanical moduli enhancement is elucidated by a combination of thermogravimetric analysis (TGA), proton nuclear magnetic resonance (1H NMR), and rheological measurements.
Experimental section
Synthesis of high-entropy Li-garnet
To synthesize HE Li-garnet (Li7La3Zr0.5Nb0.5Ta0.5Hf0.5O12), a solid-state synthesis method was used. Stoichiometric amounts of LiOH·H2O with 10% excess (Li is easy to evaporate during synthesis and sintering therefore excess amount were added, 98% minimum purity, Thermo Scientific), La2O3 (99.9% purity, Tokyo Chemical Industry America, dried at 950 °C), ZrO2 (99.9% purity, Inframat Advanced Materials), Nb2O5 (99.9% purity, Alfa Aesar), Ta2O5 (99.99% purity, Inframat Advanced Materials), and HfO2 (99%, Thermo Scientific) were ball milled for 1 hour using zirconia milling tank and balls and isopropyl alcohol (IPA, >99.5%, VWR) as the liquid media on a shaker mill (SPEX 8000, SPEX SamplePrep). The mixture was dried at 105 °C, ground, and pressed into pellets. Calcination occurred in an alumina crucible at 900 °C for 12 hours in air. Subsequent 2 hours of ball milling using zirconia milling media in IPA was performed on the shaker mill to reduce the particle size.36Synthesis of the single-ion-conducting polymer and composite electrolytes
The SIC polymer electrolyte was prepared by the copolymerization of vinyl ethylene carbonate (VEC, Sigma-Aldrich), azobisisobutyronitrile (AIBN, Sigma-Aldrich) and lithium sulfonyl(trifluoromethane sulfonyl)imide methacrylate (LiMTFSI, SPECIFIC POLYMERS). The details of the precursor storage and polymer synthesis are reported elsewhere.41 VEC and LiMTFSI had a molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The polymer had a gel-like consistency.
:1. The polymer had a gel-like consistency.
The composite electrolytes were prepared by combining VEC and LiMTFSI in a molar ratio of 10:1 in a polypropylene vial, followed by the addition of LLZO powder with predetermined weight fraction and AIBN with a molar ratio of 1:100 of AIBN:VEC. The weight ratios of SIC to LLZO varied from 90:10, 70:30, to 50:50. The weighing took place in an Ar glovebox. After weighing all the components, yttria-stabilized zirconia milling balls were added to the vial and the vial was tightly closed and sealed with parafilm. This polymer precursor–ceramic mixture was then moved outside the glovebox and vigorously mixed using a Turbula mixer (GlenMills INC.) for 10 min. The vial containing the mixture was then moved back into the glovebox and sealed in an airtight mason jar. Finally, in situ polymerization was performed by heating the samples for 12 hours in a thermostat chamber (Heratherm OGH60, Thermo Scientific) at 80 °C. The resulting composite had a sticky clay-like consistency.
Ionic conductivity measurements
The ionic conductivity of the electrolytes was examined using both broadband dielectric spectroscopy (BDS) and impedance spectroscopy (IS). The BDS was measured using an Alpha-A spectrometer (Novocontrol) in the frequency range between 106 Hz to 10−2 Hz. The polymer and composite electrolytes were loaded into a dielectric cell in an Ar glovebox. The dielectric cell had a design with two parallel electrode disks with a diameter of 10.2 mm and separation of 0.3 mm. The cell was sealed with an O-ring to ensure air-tightness. Measurements were conducted from low to high temperatures, and the sample was equilibrated to achieve thermal stabilization within 0.2 K prior to each measurement. For IS measurement (Biologic, SP-300), the same dielectric cell was used. One heating/cooling cycle ranging from −20 °C to 80 °C was applied using a temperature-controlled oven (ESPEC, SU-222) in the frequency range from 7 MHz to 0.1 Hz with a voltage amplitude of 6 mV. Before each measurement, the cell was allowed to equilibrate at a set temperature for one hour. The IS data obtained during the heating process was used in the Results and discussion section.Electrochemical and battery tests
For all the electrochemical and battery tests, we assembled separator-free 2032-type coin cells inside an Ar-filled glovebox. Two Mylar washers (outer diameter: 5/8′′, inner diameter: 7/16′′) were stacked and glued together, to contain the electrolyte and provide a defined thickness of 198 μm. All the battery tests were performed at 40 °C.The oxidative stability of SIC and SIC-30 wt% HE Li-garnet were examined using linear sweep voltammetry (LSV). The LSV testing cell assembly was the following: Mo foil as the working electrode, followed by the electrolyte confined in the Mylar washer; then Li anode (600 μm thick, MTI Corporation) was placed on top of the electrolyte, serving as both the reference and counter electrode. LSV measurements were carried out at a scan rate of 0.05 mV s−1 from the open circuit voltage to 6 V (vs. Li/Li+).
The Li transference number (t+) was determined using the Bruce-Vincent method (electrochemical impedance and chronoamperometry measurements) in a Li symmetric cell. To assemble the cell, the mylar washer was placed on a piece of Li metal anode and filled with polymer or composite electrolytes. Another piece of Li was placed on top and the stack was assembled in a coin cell. The t+ value was calculated based on the equation proposed by Bruce et al.45

 and
and  are the initial and steady-state interfacial resistances, and ΔV (10 mV in this work) is the applied potential to the Li symmetric cell. The initial current (IΩ) was calculated using IΩ = ΔV/RΩ (where RΩ is the total resistance of the cell, proposed by Balsara et al.46)
are the initial and steady-state interfacial resistances, and ΔV (10 mV in this work) is the applied potential to the Li symmetric cell. The initial current (IΩ) was calculated using IΩ = ΔV/RΩ (where RΩ is the total resistance of the cell, proposed by Balsara et al.46)
Li stripping/plating test was performed on the same Li symmetric cell. Electrochemical impedance spectroscopy (EIS) was measured before cycling in a frequency range of 7 MHz to 100 mHz with a voltage amplitude of 6 mV. Then, Li stripping/plating tests were carried out using a Maccor system at a constant current density of 0.1 mA cm−2 with an areal capacity of 0.5 mA h cm−2 at 40 °C.
Galvanostatic charge/discharge measurements were performed on a Li metal|SIC or SIC-30 wt% HE Li-garnet|LiFePO4 (LFP) cathode cell. The LFP cathode was composed of LFP (89 wt%, Hydro-Quebec), carbon black (5 wt%, Timcal) and polyvinylidene fluoride (PVDF) (6 wt%, Kynar-flex). The cathode was punched into a diameter of 7/16′′ discs and mass loading of LFP was 9.7 mg cm−2. Al foil was the cathode current collector. The Li/LFP cells were cycled within a voltage range of 2.9–3.6 V vs. Li/Li+. IS was conducted prior to cycling, followed by galvanostatic charge/discharge testing with the fixed current density of 0.1 mA cm−2 at 40 °C.
Thermogravimetric (TGA) measurements
The thermal properties of the precursors and their composites were evaluated by TGA using a TG/DTA instrument (TGA Q500) from room temperature to 600 °C at a heating rate of 10 °C min−1 under an air atmosphere.Scanning electron microscopy (SEM) and transmission electron microscopy (TEM)
The HE Li garnet powders were observed by a scanning electron microscope (SEM) spectroscopy using a Hitachi TM3030Plus table top instrument with a 15 kV acceleration voltage as well as a Talos F200X TEM (Thermo Scientific) equipped with energy-dispersive X-ray spectroscopy (EDS).Neutron powder diffraction
HE Li-garnet powders were characterized by neutron powder diffraction performed on VULCAN in Oak Ridge National Laboratory and the obtained patterns were refined through the GSAS software package to obtain crystallographic structural parameters.X-ray photoelectron spectroscopy (XPS)
The HE Li garnet powders and Al doped LLZO powders were loaded into a vacuum-transfer holder in an Ar glovebox for transport to the XPS instrument. Once in the load-lock of the XPS instrument, the vacuum transfer holder was unsealed, and the sample was inserted into the analysis chamber under vacuum. For each sample a wide energy range survey spectrum was acquired to determine all elements present. Next, a set of narrow energy range core level spectra were acquired for each identified element.Rheology measurements
The linear viscoelastic properties of the SIC and SIC-30 wt% HE-LLZO were characterized using an HR2 rheometer (TA Instruments) equipped with an environmental test chamber. Small-amplitude oscillatory shear was performed in the frequency range of 1–100 rad s−1 and temperature range of 25–80 °C under the protection of dry nitrogen gas.Nuclear magnetic resonance (NMR)
1H-NMR analysis was performed using a Bruker Avance III 400 Hz spectrometer and were collected in dimethylsulfoxide (DMSO-d6). DMSO-d6 was added to the composite and left for 24 hours to dissolve the polymer, then the ceramic was removed from the polymer solution by centrifugation.Results and discussion
The ceramic fillers used in this work are Li7La3Zr0.5Nb0.5Ta0.5Hf0.5O12 HE Li-garnet.36 The size and structure of milled HE Li-garnet powders are characterized by TEM and neutron diffraction. As shown in Fig. 1a, the HE Li-garnet powders have a fine particle size, which ensure the dispersion in the composite as an effective filler. The associated energy dispersive spectroscopy (EDS) element mapping indicates uniform element distribution at the nanometer scale. A high-resolution TEM micrograph in Fig. 1b clearly shows the lattice structure and the interplanar spacing is determined to be 0.34 nm, which is associated with the (321) crystallographic planes in cubic Li-garnet. Neutron powder diffraction (Fig. 1c) of the HE Li-garnet demonstrates a single cubic Li-garnet phase (space group of Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d, No. 230), which is consistent with the EDS mapping results. Rietveld refinement (Rwp 3.58%, χ2 3.370, see full Rietveld refinement results in ESI Table S1‡) shows a relatively high occupancy on the high mobility Li2(48 g/96 h), which ensures high conductivity of the materials.47 As indicated in Fig. S1,‡ the bulk conductivity of HE Li-garnet is approximately ∼5 × 10−4 S cm−1 at room temperature.
d, No. 230), which is consistent with the EDS mapping results. Rietveld refinement (Rwp 3.58%, χ2 3.370, see full Rietveld refinement results in ESI Table S1‡) shows a relatively high occupancy on the high mobility Li2(48 g/96 h), which ensures high conductivity of the materials.47 As indicated in Fig. S1,‡ the bulk conductivity of HE Li-garnet is approximately ∼5 × 10−4 S cm−1 at room temperature.
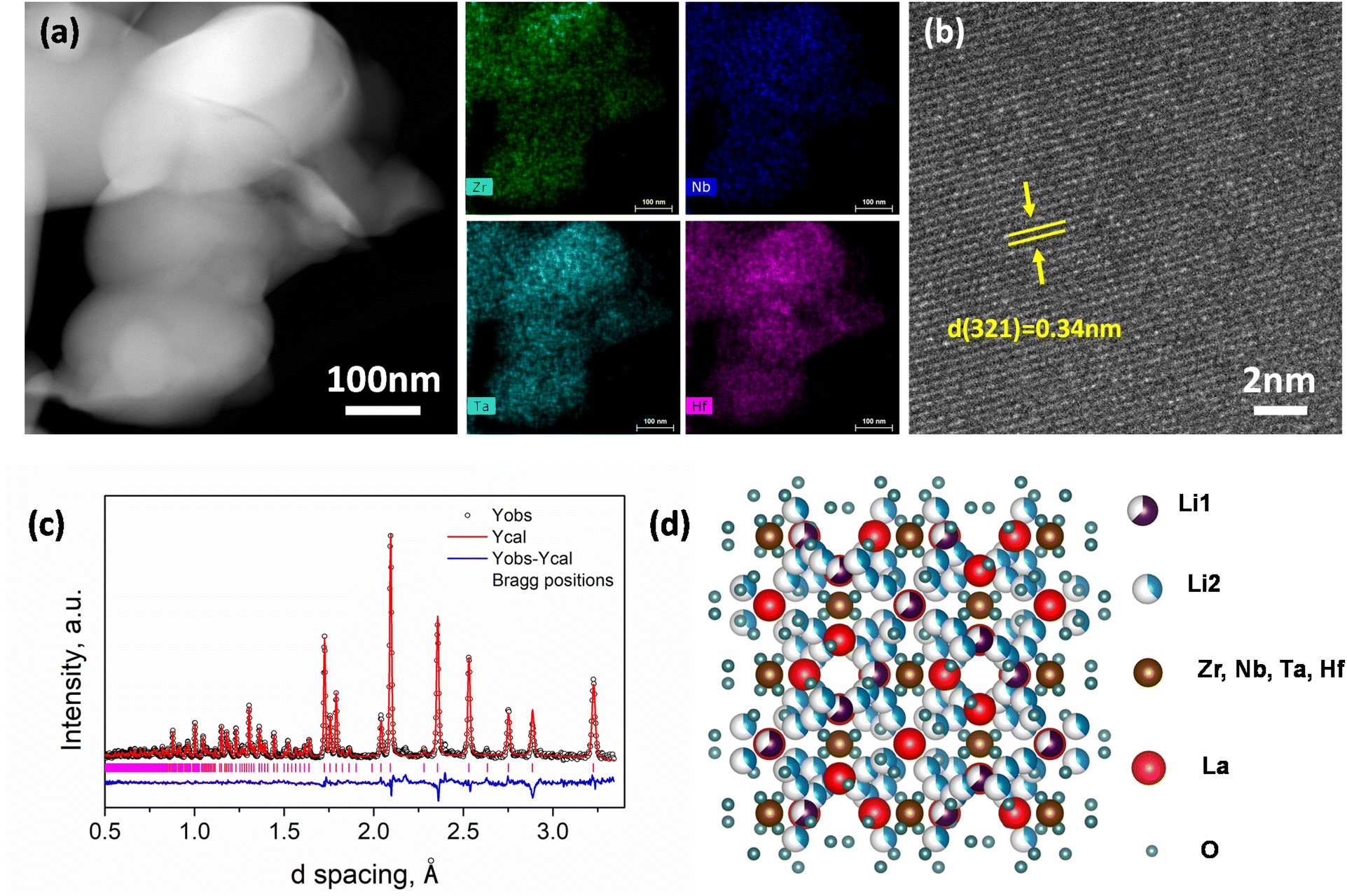 | ||
| Fig. 1 Characterizations of the HE Li-garnet. (a) TEM micrograph along with EDS mapping, (b) a high-resolution TEM micrograph to show the lattice structure, (c) neutron powder diffraction and Rietveld refinement to show the (d), refined crystal structures. | ||
The polymer electrolyte used in this work is a vinyl ethylene carbonate (VEC) based single-ion conducting polymer (SIC), as shown in Fig. 2a.41 The composite electrolyte was prepared by in situ polymerization using a mixture of precursors of the polymer electrolytes and the ceramic fillers, as shown by Fig. 2b. In our previous work, we found that VEC monomer is a slow-reacting component and does not completely polymerize. The unpolymerized VEC monomer plasticizes the polymer, leading to the formation of SIC gel polymer electrolyte.41 Therefore, we anticipate that the composite also contains unpolymerized VEC. This will be discussed in detail later.
 | ||
| Fig. 2 Reaction scheme and schematic of (a), SIC pure polymer and (b), SIC-HE Li-garnet composite electrolyte. | ||
Fig. 3a shows the ionic conductivities of the composite electrolytes as a function of the weight fraction of the HE Li-garnet at 30 °C. The conductivity increased as HE Li-garnet's loading increased from 10 wt% to 30 wt%. At 30 wt%, the ionic conductivity of composite was 8.6 × 10−5 S cm−1, an 8-fold increase from that of neat SIC (1.1 × 10−5 S cm−1). When the loading further increased to 50 wt%, the conductivity decreased to almost half of that of the 30 wt% composite (4.5 × 10−5 S cm−1vs. 8.6 × 10−5 S cm−1). Conductivity as a function of frequency (Bode spectra) of SIC and composite is shown in Fig. S2.‡ Three distinct regimes appeared as a function of frequency, (i) the decay at low frequencies due to ion-blocking electrode polarization effect, (ii) frequency independent plateau region, which is identified as the DC conductivity, and (iii) the increase at high frequency representing the AC conductivity. A single DC plateau is observed in the Bode spectra of both the SIC polymer and the composite.
 | ||
| Fig. 3 Ionic conductivity of the composite electrolytes. (a) Conductivity at 30 °C as a function of HE Li-garnet loading. (b) Arrhenius plot of the conductivity. | ||
Fig. 3b shows the Arrhenius plot of conductivity in SIC polymer and composites. Both show Vogel–Fulcher–Tammann (VFT) temperature dependence, typical for polymer electrolytes above their glass transition temperatures.48 To assess the time-dependent stability of the composites, we compared the conductivity of 50 wt% composite electrolyte prepared fresh and aged for 2 weeks in the glovebox (Fig. 3a and b). The conductivity of the composite was largely stable over 2 weeks, showing approximately 10% decrease from the fresh sample. This minor decrease can be explained by several factors: continued polymerization of the sample, partial evaporation of unpolymerized VEC especially under elevated temperatures (the BDS cell is airtight for the conductivity measurements but may not stay airtight for 2 weeks), and possible variations in sample's contact with cell.
It is worth noting that we also incorporated 50 wt% Al doped LLZO powder into the SIC polymer, using the same synthesis procedure as the HE Li-garnet composite (Fig. S3‡). The ionic conductivity of 50 wt% Al doped LLZO composite was 1.6 × 10−6 S cm−1 at 30 °C, which is almost an order of magnitude lower than that of the pure polymer matrix (1.1× 10−5 S cm−1). This result indicates that regular Al doped LLZO could not achieve similar ionic transport enhancement in the SIC polymer matrix. XPS on the two powders revealed significantly more carbonate species on the surface of Al doped LLZO (Fig. S4 and Table S2‡) than on the surface of HE Li garnet. The formation of Li2CO3 on the surface will affect the interface between SIC and Li-garnet and the associated Li-ion migration pathways.43,49 Since the surface of HE Li garnet is significantly less prone to carbonate formation, a more favorable interface between the SIC and HE Li garnet can be realized.
To reveal the mechanism of the conductivity enhancement in the HE Li-garnet composites, we first quantified the amount of unpolymerized VEC in SIC and in the HE Li-garnet composites, using two methods – TGA and 1H-NMR. Fig. 4a shows TGA profiles of VEC monomer, SIC, and SIC-30 wt% HE Li-garnet composite. VEC monomer showed rapid weight loss at around 155 °C due to evaporation. For SIC, we observed 3% weight loss occurring at 100 °C. This weight loss probably comes from residual water from the precursors as well as the polymerization process. The weight loss of unpolymerized VEC is represented by the red arrow in Fig. 4a, which is 44%. The weight loss after the evaporation of unpolymerized VEC is from the decomposition of polymerized VEC and LiMTFSI. Since the total weight percentage of the VEC (polymerized + unpolymerized) is 77%, we calculated that unpolymerized VEC in SIC was 57%. Similarly, the weight loss coming from unpolymerized VEC in the SIC-30 wt% HE Li-garnet composite was marked by a purple arrow. Note that the weight loss was normalized by the weight polymer components only, excluding the ceramics' weight, as they didn't experience weight loss in this temperature range. The unpolymerized VEC in the SIC-30 wt% HE Li-garnet composite calculated from the TGA curve was 78%, significantly higher than neat SIC (57%) (Table 1). The detailed calculation of the unpolymerized VEC amount was explained in Note S1 in the ESI.‡
 | ||
| Fig. 4 (a) Thermogravimetric analysis (TGA) curves of VEC, SIC, and the composite electrolyte. The data of the composite excluded the weight of the ceramic. (b) 1H-NMR spectra of SIC and the composite electrolyte. | ||
| TGA | 1H-NMR | ||||
|---|---|---|---|---|---|
| Mass loss of VEC unpolymerized component (%) | Unpolymerized VEC (%) | Peak c (LiMTFSI) | Peak a (unpolymerized VEC) | Unpolymerized VEC (%) | |
| SIC using stirring | 44 | 57 | 2.00 | 5.78 | 57.8 |
| SIC-30 wt% HE Li-garnet (w.r.t. polymer components) | 60 | 78 | 2.00 | 7.96 | 79.6 |
| SIC-50 wt% HE Li-garnet | — | — | 2.00 | 8.07 | 80.7 |
We further evaluated the degree of polymerization in the pure polymer and the composites using 1H-NMR (Fig. 4b). To quantitatively identify the degree of polymerization, we calculated the molar ratio between VEC monomers and LiMTFSI, in the SIC and SIC-30 wt% HE Li-garnet, by comparing the integrals of the VEC C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C signal at 6 ppm with that of LiMTFSI at 3 ppm (detailed peak assignments have been previously reported).41 In Fig. 4b, peak a is 1 proton from the vinyl group in VEC. Peak b is 1 proton corresponding to the vinyl group of LiMTFSI, and peak c is the 2 protons on the carbon next to the ester group of LiMTFSI. Before polymerization, the precursor mixtures showed the molar ratio of VEC:LiMTFSI is 10:1, as indicated by the ratio between the integrals of peaks a and b. After polymerization, the integral of peak a decreased relative to peak c, while peak b completely disappeared, indicating partial polymerization of VEC and complete polymerization of LiMTFSI. The complete polymerization of LiMTFSI means that the MTFSI anions are covalently bonded to the polymer chains and are immobilized.41 The ratio of the integrals of peaks a and c can be used to calculate the amount of unpolymerized VEC. The unpolymerized VEC content in the SIC polymer was 57.8% and that in SIC-30 wt% HE Li-garnet composite was 79.6%. These numbers are consistent with TGA results. Both TGA and 1H-NMR indicate that there is significantly more unpolymerized VEC present in the HE Li-garnet composites compared to the pure polymer. The increased amount of unpolymerized VEC might be related to the surface composition of the HE Li-garnet, which slows the already sluggish reaction kinetics of the VEC monomer, resulting in more unpolymerized VEC in the HE Li-garnet composites.
C signal at 6 ppm with that of LiMTFSI at 3 ppm (detailed peak assignments have been previously reported).41 In Fig. 4b, peak a is 1 proton from the vinyl group in VEC. Peak b is 1 proton corresponding to the vinyl group of LiMTFSI, and peak c is the 2 protons on the carbon next to the ester group of LiMTFSI. Before polymerization, the precursor mixtures showed the molar ratio of VEC:LiMTFSI is 10:1, as indicated by the ratio between the integrals of peaks a and b. After polymerization, the integral of peak a decreased relative to peak c, while peak b completely disappeared, indicating partial polymerization of VEC and complete polymerization of LiMTFSI. The complete polymerization of LiMTFSI means that the MTFSI anions are covalently bonded to the polymer chains and are immobilized.41 The ratio of the integrals of peaks a and c can be used to calculate the amount of unpolymerized VEC. The unpolymerized VEC content in the SIC polymer was 57.8% and that in SIC-30 wt% HE Li-garnet composite was 79.6%. These numbers are consistent with TGA results. Both TGA and 1H-NMR indicate that there is significantly more unpolymerized VEC present in the HE Li-garnet composites compared to the pure polymer. The increased amount of unpolymerized VEC might be related to the surface composition of the HE Li-garnet, which slows the already sluggish reaction kinetics of the VEC monomer, resulting in more unpolymerized VEC in the HE Li-garnet composites.
To gauge the effect of unpolymerized VEC on the ionic conductivity of SIC, we mixed the SIC precursors and let the mixture polymerize at 80 °C for different time spans (1 h, 2 h, 3 h, 4 h and overnight (12 h)) and quickly quenched the reaction by transferring the mixture to a freezer. We also prepared the SIC using two different ways – by mechanical stirring and vigorous shaking (using a Turbula, a procedure used to prepare the composites). The amount of unpolymerized VEC in these samples and the corresponding conductivity at 30 °C were quantified by 1H-NMR and IS and listed in Table 2. It is worth mentioning that in all these samples, essentially complete polymerization (>98%) of LiMTFSI was observed. In general, a higher amount of unpolymerized VEC led to higher conductivity, indicating that unpolymerized VEC monomer is an effective plasticizer. The 1 h polymerized SIC exhibited the highest conductivity of 2.3 × 10−4 S cm−1, with a corresponding 98.7% unpolymerized VEC. As the degree of polymerization increased, the ionic conductivity of the polymer electrolyte decreased, reaching as low as 1.1 × 10−5 S cm−1 with 50.5% unpolymerized VEC after a 12 h polymerization period (Turbula), while the ionic conductivity of SIC after 12 hours using the mechanical stirring method was 1.4 × 10−5 S cm−1 with 57.8% unpolymerized VEC.41 Notably, 2 h polymerized SIC and (overnight polymerized) SIC-30 wt% HE Li-garnet composite exhibit very similar levels of unpolymerized VEC, 78.3% and 79.6%, respectively. These samples also had similar ionic conductivities of 9.4 × 10−5 S cm−1 and 8.6 × 10−5 S cm−1, respectively. This suggests that the conductivity enhancement in these composites mainly arises from an increase in the amount of unpolymerized VEC which serves as a plasticizer. It is well known that plasticizers facilitate segmental motion of polymer chains, reduce the glass transition temperature, and solvate ions, thereby enhancing ion mobility and overall ionic conductivity.
| Conductivity at 30 °C (S cm−1) | % of unpolymerized VEC | |
|---|---|---|
| SIC-1 h polymerized | 2.3 × 10−4 | 98.7 |
| SIC-2 h polymerized | 9.4 × 10−5 | 78.3 |
| SIC-3 h polymerized | 4.5 × 10−5 | 80.5 |
| SIC-4 h polymerized | 1.9 × 10−5 | 61.5 |
| SIC-30 wt% HE Li-garnet | 8.6 × 10−5 | 79.6 |
| SIC-50 wt% HE Li-garnet | 4.5 × 10−5 | 80.7 |
| SIC using stirring | 1.4 × 10−5 | 57.8 |
| SIC using Turbula | 1.1 × 10−5 | 50.5 |
The mechanical properties of SIC and SIC-30 wt% HE Li-garnet composite were evaluated using a rheometer. A master curve was constructed by applying appropriate temperature-dependent shift factor aT to the angular frequency ω and shown in Fig. 5a. The rheology measurements at each individual temperature are provided in Fig. S5.‡ The storage modulus G′ and loss modulus G′′ of the SIC are approximately equal over the entire measured frequency range and exhibit a power-law behavior of G′ ≈ G′′ ∼ ω1/2, which is indicative of Rouse-like dynamics. In contrast, a different scaling behavior is observed for G′ and G′′ of the SIC-30 wt% HE Li-garnet composite: G′,G′′ ∼ ω1/4, corresponding to some gelation regime with G′ ∼2 times higher than G′′. At low frequencies, the dynamic moduli (G′ and G′′) of the composite are higher than those of the SIC. The distinctly different frequency dependence of G′ and G′′between the SIC and composite suggests that the observed mechanical reinforcement in the composite arises from interactions between HE Li-garnet and the polymer matrix. HE Li-garnet may serve as physical crosslinking points in the polymer matrix. We also observed that the HE Li-garnet particles formed very stable suspension in the polymer precursors, and no precipitation of the particles occurred during in situ polymerization. At lower temperatures and higher frequencies, the composite appeared to have similar G′ and G′′to neat SIC, indicating that the composite had shorter chains than the SIC polymer, consistent with 1H-NMR analysis. Recall from Table 2 that the composite contained more unpolymerized VEC monomers, which is a small-molecule plasticizer. It should be noted that plasticizer incorporation often compromises the mechanical modulus of the polymer matrix as a result of the plasticization effect, a trade-off commonly observed in gel polymer electrolytes.50–52 Herein, by adding HE Li-garnet, the positive effect of ceramics on the mechanical properties offset the detrimental effect from the unpolymerized VEC monomers. HE Li-garnet-SIC composite electrolyte combined the high modulus from the ceramic–polymer interactions and good surface adhesion from the polymer phase, which are desirable mechanical properties for battery performance.
 | ||
| Fig. 5 (a) Storage modulus (G′) and loss modulus (G′′) of SIC and SIC-30 wt% HE Li-garnet as a function of angular frequency (ω) times the shift factor, aT. Inset: temperature dependence of the shift factor aT. (b)Ionic conductivity and G′ of SIC and SIC-30 wt% HE Li-garnet measured at various temperatures. G′ was data at 10 Hz. The green arrows highlight simultaneous enhancement in ionic conductivity and mechanical modulus in 30 wt% HE Li-garnet composite at 40 °C. | ||
In Fig. 5b, we compare the temperature-dependent conductivity and mechanical properties (measured at 10 Hz) of the SIC polymer and SIC-30 wt% HE Li-garnet composite. It can be seen that the conductivity and the mechanical properties have the opposite temperature dependencies, as an increase in temperature led to increased conductivity and decreased mechanical moduli. The comparison clearly indicates that HE Li-garnet incorporation simultaneously improved both conductivity and storage modulus over an extended temperature range.
The Li+ transference number and Li+ conductivity at 40 °C are summarized in Fig. 6a. The Li+ transference number of the SIC polymer is ∼0.9, while a slightly lower transference number of 0.73 is observed for SIC-30 wt% HE Li-garnet composite (Fig. S6‡). The high Li+ transference number is intrinsic to SIC polymer electrolytes due to anions immobilization.41 The Li+ conductivity, obtained by multiplying the Li+ transference number and ionic conductivity, is a critical factor to achieve high performance of Li batteries. The Li+ conductivity of SIC-30 wt% HE Li-garnet is 9.8 × 10−5 S cm−1 at 40 °C, representing an almost 6-fold increase compared to that of the SIC polymer (1.7 × 10−5 S cm−1), indicates faster Li-ion transport promoted in the bulk electrolyte with HE Li-garnet owing to the higher amounts of VEC monomers in the composite than in the neat polymer. Fig. 6b shows the electrochemical stability window of SIC polymer and SIC-30 wt% HE Li-garnet measured by linear sweep voltammetry (LSV). The oxidative current for SIC polymer started to increase at 3.6 V vs. Li/Li+ (V), which is consistent with our previous report.42 The oxidative current of SIC-30 wt% HE Li-garnet started to rise at around 4.1 V vs. Li/Li+ (V), indicating better oxidative stability in composite.
 | ||
| Fig. 6 (a) Li+ transference number and Li+ conductivity of SIC and SIC-30 wt% HE Li-garnet composite. (b) Linear sweep voltammogram of SIC and SIC-30 wt% HE Li-garnet composite at a scan rate of 0.05 mV s−1. (c) Nyquist plots of the Li symmetric cells made with SIC and SIC-30 wt% HE Li-garnet composite at open circuit voltage, before cycling. (d) Lithium stripping/plating profiles of the SIC and SIC-30 wt% HE Li-garnet composite at 0.1 mA cm−2 (a–d) Measurements were carried out at 40 °C. To evaluate cell performance, we constructed separator-free 2032-type coin cells within an Ar-filled glovebox, as illustrated schematically in the inset of panel d. | ||
Fig. 6c shows the Nyquist plots of the SIC polymer and SIC-30 wt% HE Li-garnet composite in Li symmetric cell before cycling. The impedance spectra of both cells showed two depressed semicircles, representing ion transport through the bulk electrolyte and across the Li metal/electrolyte interfaces. A lower total resistance in the SIC-30 wt% HE Li-garnet composite compared to the SIC polymer is consistent with its higher Li+ conductivity due larger concentration of VEC monomers. Fig. 6d shows Li symmetric cell cycling at a constant current of 0.1 mA cm−2 at 40 °C. At an early stage of the cycling, the cell made with SIC polymer showed larger overpotential compared to that of the SIC-30 wt% HE Li-garnet composite. Specifically, the SIC polymer initially exhibited an overpotential of 0.11 V with flat voltage profiles (Fig. S7a–e‡). The voltage profiles for the 3rd, 5th, 10th, and 15th cycles exhibited a gradual shift from a flat to a rounded shape, with voltage fluctuations appearing at the 10th cycle. This observation may be due to Li dendrite growth in the polymer, eventually leading to a large and abrupt potential drop at 175 hours. In contrast, the voltage profile of SIC-30 wt% HE Li-garnet was much more stable showing a smaller initial overpotential of 0.04 V and much slower overpotential evolution (Fig. S8a–e‡). At the end of 700 hours of cycling, the overpotential only increased to 0.07 V. The cell incorporating HE Li-garnet exhibited no signs of short-circuiting, in contrast to the pure SIC polymer. These results demonstrate that the incorporation of HE Li-garnet into the SIC polymer reinforces mechanical properties and boosts ionic conductivity, both promoting outstanding Li plating/stripping performance with prolonged cycling stability.
We further evaluated full cell performance using Li|LiFePO4 (LFP). As shown in Fig. S9a,‡ the internal cell resistance of the SIC-30 wt% HE Li-garnet composite is significantly lower, around 300 Ohm, compared to approximately 900 Ohm for the neat polymer. This trend is consistent with our transport property measurements. Fig. S9 b and c‡ shows the cycling performance of the Li/LFP cell. Overall, the cell made with the composite showed better cycle stability than that of the pure polymer. The Li|SIC|LFP cell exhibited a first cycle charge capacity of 85 mA h g−1, and subsequently delivered a first cycle discharge capacity of 82 mA h g−1. The discharge capacity decreased to 68 mA h g−1 by the 5th cycle and couldn't charge to 3.6 V from 7th cycle. Meanwhile, the Li|SIC-30 wt% HE Li-garnet|LFP cell delivered a first cycle charge capacity of 167 mA h g−1, and subsequently delivered a first cycle discharge capacity of 144 mA h g−1. The discharge capacity dropped from 142 mA h g−1 in the 2nd cycle to 122 mA h g−1 in the 10th cycle, but didn't show any charging issues like the one made with SIC. We believe the primary reason for the observed capacity decay is likely due to unoptimized interface between the LFP cathode and the electrolyte (e.g., loss of contact or undesired reactions). Further optimization of the cathode-electrolyte interface using HE Li-garnet is ongoing and will be addressed in future work.
Conclusions
In this study, we incorporated a Li7La3Zr0.5Nb0.5Ta0.5Hf0.5O12 high entropy Li garnet (HE Li-garnet) ceramic in a vinyl ethylene carbonate (VEC) based single-ion conducting (SIC) polymer. The addition of HE Li-garnet led to a 7-fold increase in the ionic conductivity (8.6 × 10−5 S cm−1 at 30 °C) compared to the neat polymer, while maintaining a high Li+ transference of 0.73. 1H-NMR and TGA results suggest that the addition of HE Li-garnet resulted in lower degrees of polymerization, leaving more unpolymerized VEC monomer in the matrix, serving as the governing mechanism for conductivity enhancement. This conductivity enhancement mechanism is a less well-known mechanism, as most active fillers enhance conductivity by promoting Li ion dissociation at the polymer-ceramic interface or by forming percolated ion-conducting paths themselves. HE Li-garnet particles also serve as physical crosslinks in the polymer matrix, leading to a stable and well-mixed composite with 2-fold enhanced storage modulus (G′) at 40 °C, offsetting the plasticization effect (which is detrimental to mechanical modulus) from increased amount of unpolymerized VEC. The simultaneous ion transport and mechanical property enhancement significantly improved the dendrite resistance and cycle life against the Li metal anode. This work reveals an uncommon conductivity enhancement mechanism in the polymer-ceramic composite electrolytes and highlights the positive role ceramic fillers can play in improving polymer electrolytes for advanced battery technologies.Data availability
All data supporting the findings of this study are included in the article or available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported as part of the Fast and Cooperative Ion Transport in Polymer-Based Materials (FaCT), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences at Oak Ridge National Laboratory under contract DE-AC05-00OR22725. CL and ZF would like to acknowledge the support of the National Science Foundation under Grant No. CMMI-2347492. The rheology experiments were conducted at the Center for Nanophase Materials Sciences, a US Department of Energy Office of Science User Facility operated at Oak Ridge National Laboratory.References
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- R. Meziane, J.-P. Bonnet, M. Courty, K. Djellab and M. Armand, Electrochim. Acta, 2011, 57, 14–19 CrossRef CAS.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- Z. Song, F. Chen, M. Martinez-Ibanez, W. Feng, M. Forsyth, Z. Zhou, M. Armand and H. Zhang, Nat. Commun., 2023, 14, 4884 CrossRef CAS PubMed.
- V. Bocharova and A. P. Sokolov, Macromolecules, 2020, 53, 4141–4157 CrossRef CAS.
- Q. Ma, H. Zhang, C. Zhou, L. Zheng, P. Cheng, J. Nie, W. Feng, Y. S. Hu, H. Li, X. Huang, L. Chen, M. Armand and Z. Zhou, Angew Chem. Int. Ed. Engl., 2016, 55, 2521–2525 CrossRef CAS PubMed.
- K. Deng, Q. Zeng, D. Wang, Z. Liu, Z. Qiu, Y. Zhang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2020, 8, 1557–1577 RSC.
- Y. Lu, M. Tikekar, R. Mohanty, K. Hendrickson, L. Ma and L. A. Archer, Adv. Energy Mater., 2015, 5, 1402073 CrossRef.
- M. Doyle, T. F. Fuller and J. Newman, Electrochim. Acta, 1994, 39, 2073–2081 CrossRef CAS.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- L. C. Merrill, X. C. Chen, Y. Zhang, H. O. Ford, K. Lou, Y. Zhang, G. Yang, Y. Wang, Y. Wang, J. L. Schaefer and N. J. Dudney, ACS Appl. Energy Mater., 2020, 3, 8871–8881 CrossRef CAS.
- R. Sahore, K. D. Owensby, B. L. Armstrong, J. Ock, M. L. Lehmann, A. M. Ullman, S. Kalnaus and X. C. Chen, ACS Appl. Energy Mater., 2024, 7, 11714–11723 CrossRef CAS.
- H. Mazor, D. Golodnitsky, E. Peled, W. Wieczorek and B. Scrosati, J. Power Sources, 2008, 178, 736–743 CrossRef CAS.
- L. Porcarelli, P. Sutton, V. Bocharova, R. H. Aguirresarobe, H. Zhu, N. Goujon, J. R. Leiza, A. Sokolov, M. Forsyth and D. Mecerreyes, ACS Appl. Mater. Interfaces, 2021, 13, 54354–54362 CrossRef CAS PubMed.
- S. Yu, Q. Xu, X. Lu, Z. Liu, A. Windmuller, C. L. Tsai, A. Buchheit, H. Tempel, H. Kungl, H. D. Wiemhofer and R. A. Eichel, ACS Appl. Mater. Interfaces, 2021, 13, 61067–61077 CrossRef CAS PubMed.
- H. Kwon, H. J. Choi, J. K. Jang, J. Lee, J. Jung, W. Lee, Y. Roh, J. Baek, D. J. Shin, J. H. Lee, N. S. Choi, Y. S. Meng and H. T. Kim, Nat. Commun., 2023, 14, 4047 CrossRef CAS PubMed.
- T. Zhang, W. He, W. Zhang, T. Wang, P. Li, Z. Sun and X. Yu, Chem. Sci., 2020, 11, 8686–8707 RSC.
- G. M. Overhoff, M. Y. Ali, J. P. Brinkmann, P. Lennartz, H. Orthner, M. Hammad, H. Wiggers, M. Winter and G. Brunklaus, ACS Appl. Mater. Interfaces, 2022, 14, 53636–53647 CrossRef CAS PubMed.
- L.-Z. Fan, H. He and C.-W. Nan, Nat. Rev. Mater., 2021, 6, 1003–1019 CrossRef CAS.
- S. C. Sand, J. L. M. Rupp and B. Yildiz, Chem. Soc. Rev., 2025, 54, 178–200 RSC.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- A. S. Pandian, X. C. Chen, J. Chen, B. S. Lokitz, R. E. Ruther, G. Yang, K. Lou, J. Nanda, F. M. Delnick and N. J. Dudney, J. Power Sources, 2018, 390, 153–164 CrossRef CAS.
- X. C. Chen, X. M. Liu, A. S. Pandian, K. Lou, F. M. Delnick and N. J. Dudney, ACS Energy Lett., 2019, 4, 1080–1085 CrossRef CAS.
- X. C. Chen, R. L. Sacci, N. C. Osti, M. Tyagi, Y. Wang, M. J. Palmer and N. J. Dudney, Mol. Syst. Des. Eng., 2019, 4, 379–385 RSC.
- J. Peng, Y. Xiao, D. A. Clarkson, S. G. Greenbaum, T. A. Zawodzinski and X. C. Chen, ACS Appl. Polym. Mater., 2020, 2, 1180–1189 CrossRef CAS.
- X. C. Chen, R. L. Sacci, N. C. Osti, M. Tyagi, Y. Wang, J. K. Keum and N. J. Dudney, Front. Chem., 2021, 8, 592604 CrossRef PubMed.
- X. C. Chen, Y. M. Zhang, L. C. Merrill, C. Soulen, M. L. Lehmann, J. L. Schaefer, Z. J. Du, T. Saito and N. J. Dudney, J. Mater. Chem. A, 2021, 9, 6555–6566 RSC.
- R. Murugan, V. Thangadurai and W. Weppner, Angew Chem. Int. Ed. Engl., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- J. C. Bachman, S. Muy, A. Grimaud, H. H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- A. J. Samson, K. Hofstetter, S. Bag and V. Thangadurai, Energy Environ. Sci., 2019, 12, 2957–2975 RSC.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 120, 4257–4300 CrossRef CAS PubMed.
- T. Krauskopf, H. Hartmann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2019, 11, 14463–14477 CrossRef CAS PubMed.
- Y. Zeng, B. Ouyang, J. Liu, Y.-W. Byeon, Z. Cai, L. J. Miara, Y. Wang and G. Ceder, Science, 2022, 378, 1320–1324 CrossRef CAS PubMed.
- Z. Fu and J. Ferguson, J. Am. Ceram. Soc., 2022, 105, 6175–6183 CrossRef CAS.
- C.-H. Kuo, A.-Y. Wang, H.-Y. Liu, S.-C. Huang, X.-R. Chen, C.-C. Chi, Y.-C. Chang, M.-Y. Lu and H.-Y. Chen, APL Mater., 2022, 10, 121104 CrossRef CAS.
- R.-Z. Zhang and M. J. Reece, J. Mater. Chem. A, 2019, 7, 22148–22162 RSC.
- M. Keller, A. Varzi and S. Passerini, J. Power Sources, 2018, 392, 206–225 CrossRef CAS.
- J. Zheng, H. Dang, X. Feng, P.-H. Chien and Y.-Y. Hu, J. Mater. Chem. A, 2017, 5, 18457–18463 RSC.
- J.-y. Ock, A. Bhattacharya, T. Wang, C. Gainaru, Y. Wang, K. L. Browning, M. Lehmann, M. A. Rahman, M. Chi, F. Wang, J. K. Keum, L. Kearney, T. Saito, S. Dai, R. J. Clément, A. P. Sokolov and X. C. Chen, Macromol., 2024, 57, 7489–7498 CrossRef CAS.
- T. Wang, J. Ock, X. C. Chen, F. Wang, M. Li, M. S. Chambers, G. M. Veith, L. B. Shepard, S. B. Sinnott, A. Borisevich, M. Chi, A. Bhattacharya, R. J. Clément, A. P. Sokolov and S. Dai, Adv. Sci., 2025, 12, 2408805 CrossRef CAS PubMed.
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS.
- L. Huang, J. Gao, Z. Bi, N. Zhao, J. Wu, Q. Fang, X. Wang, Y. Wan and X. Guo, Energies, 2022, 15, 3206 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polym., 1987, 28, 2324–2328 CrossRef CAS.
- M. D. Galluzzo, J. A. Maslyn, D. B. Shah and N. P. Balsara, J. Chem. Phys., 2019, 151, 020901 CrossRef PubMed.
- T. Thompson, A. Sharafi, M. D. Johannes, A. Huq, J. L. Allen, J. Wolfenstine and J. Sakamoto, Adv. Energy Mater., 2015, 5, 1500096 CrossRef.
- C. A. Angell and D. L. Smith, J. Phys. Chem., 1982, 86, 3845–3852 CrossRef CAS.
- H. Huo, J. Luo, V. Thangadurai, X. Guo, C.-W. Nan and X. Sun, ACS Energy Lett., 2019, 5, 252–262 CrossRef.
- Y. M. Kim and H. C. Moon, Adv. Funct. Mater., 2019, 30, 1907290 CrossRef.
- S. Xu, Z. Sun, C. Sun, F. Li, K. Chen, Z. Zhang, G. Hou, H. M. Cheng and F. Li, Adv. Funct. Mater., 2020, 30, 2007172 CrossRef CAS.
- G. Lu, Y. Zhang, J. Zhang, X. Du, Z. Lv, J. Du, Z. Zhao, Y. Tang, J. Zhao and G. Cui, Carbon Energy, 2022, 5, e287 CrossRef.
Footnotes |
| † This manuscript has been authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). |
| ‡ Electronic supplementary information (ESI) available: Neutron powder diffraction; BDS spectra; XPS spectra; rheology data; voltage profiles. See DOI: https://doi.org/10.1039/d5ta02665b |
| This journal is © The Royal Society of Chemistry 2025 |