
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liquid–liquid–solid interfacial polymerization approach to rapid fabrication of large-sized, self-standing structured COF membranes†
Chenhui
Ding
a,
Yingying
Du
a,
Tamara
Fischer
b,
Jana
Timm
c,
Roland
Marschall
c,
Jürgen
Senker
 b and
Seema
Agarwal
*a
b and
Seema
Agarwal
*a
aMacromolecular Chemistry and Bavarian Polymer Institute, University of Bayreuth, Universitätsstrasse 30, 95440 Bayreuth, Germany. E-mail: agarwal@uni-bayreuth.de
bDepartment of Chemistry, Inorganic Chemistry III, and Northern Bavarian NMR Centre, University of Bayreuth, Universitätsstrasse 30, 95440 Bayreuth, Germany
cDepartment of Chemistry, Physical Chemistry III, University of Bayreuth, Universitätsstrasse 30, 95440 Bayreuth, Germany
First published on 20th May 2025
Abstract
Covalent organic framework (COF) membranes, with their customizable chemical structures and regular nanochannels, are widely used for separation. However, challenges remain in the rapid and high-yield fabrication of robust, self-standing, flexible, large-sized, highly crystalline COF membranes, especially as self-standing or supported membranes. Here, we introduce a liquid–liquid–solid interfacial polymerization technique at high temperature for the rapid production (in a few hours) of large-sized, robust, flexible, and thickness-adjustable highly crystalline self-standing all COF membranes with hierarchical structure in which a nano COF membrane is integrated on COF hollow fiber networks of the same material, achieving impressive mechanical stability (tensile strength ∼23 MPa), which is maintained even after 500 bending cycles and 80% compression. Furthermore, we demonstrate the universality of this technique by preparing various imine-linked COFs on different porous substrates. Laboratory filtration experiments produced excellent results, highlighting the considerable potential of COF membranes for use in industrial wastewater purification applications going forward.
Introduction
Covalent organic frameworks (COFs) feature distinctive topologies, adjustable functionalities, highly ordered channels over long distances, high levels of porosity, and exceptional thermochemical stability.1–4 They are widely used in membrane technology, including molecular/ion separation,5,6 proton/ion conduction,7,8 energy storage,9–12 and carrier migration.13,14 Typically, the formation of COFs relies on thermodynamically controlled chemical reactions, synthesizing high-quality but insoluble and difficult-to-process COF powders under solvothermal conditions. Further processing of these powders into large-sized flexible membranes remains a significant challenge, hindering their broader application in membrane technology.15–17Recent studies have successfully produced self-standing or substrate-supported COF membranes through methods such as interfacial polymerization (IP),18–21in situ solvothermal synthesis,22–24 and layer-by-layer stacking.25–28 Liquid–liquid IP method is the most widely employed technique for fabricating self-standing COF membranes, owing to its straightforward process and mild reaction conditions, typically carried out at ambient temperature and pressure.29–32 In this method, two reactive monomers react at the interface (organic–water), first forming an amorphous polymer membrane mostly by stacking of COF nanosheets, which then transforms into a crystalline COF membrane. However, the mild reaction conditions (low temperature) result in a significant mismatch between the timing of polymerization and crystallization, leading to long reaction times and COF membranes with low crystallinity.33–36 At the same time, the fragile nature of these COF membranes poses challenges for handling and transferring to a substrate. The weak interaction between COF membranes and substrates in substrate-supported COFs restricts their practical applications in real-world scenarios.37,38 For substrate-supported COF membranes, in situ solvothermal synthesis is usually employed. In this method, high temperatures accelerate the reaction rate and promote the transformation of amorphous polymers into crystalline COFs, thereby forming high crystallinity COF membranes in situ on the substrate surface.39–42 The substrate must have a continuous or nanoporous surface to ensure the integrity of the COF membranes formed on it, which greatly limits the choice of substrates.43,44
Therefore, this work combines the advantages of liquid–liquid IP and in situ solvothermal synthesis, proposing a novel liquid–liquid–solid IP technique, where a covalent organic framework (COF) is synthesized both on the surface and within the bulk of a porous template substrate via modified liquid–liquid interfacial polymerization at 80 °C. In this technique, the porous solid substrate absorbs and retains the aqueous phase containing one of the COF reactants within the pores. When the organic phase, with the second COF component, is introduced, the reaction between the two COF components takes place at the interface of the two liquids. This process creates multiple liquid–liquid interfaces within the substrate pores in addition to a large liquid–liquid interface at the surface. The high reaction temperature accelerates the reaction rate, akin to a solvothermal process, and facilitates the formation of COF particles and a complete COF film at the interface through symbiosis between COF crystals, unlike COF nanosheet stacking in conventional liquid–liquid–IP. This approach enables the rapid and scalable production of robust, flexible, and highly crystalline COF membranes, which can be either seamlessly directly integrated into a porous polymer substrate or elegantly grown and self-standing on a network of COF hollow fibers made from the same COF material by removal of the substrate. By controlling the reaction time, temperature monomer concentration, and monomer type, COF membranes of varying thicknesses and types can be constructed. Furthermore, to evaluate the practical potential of the prepared COF membranes, they have been successfully applied in dye removal from wastewater in model experiments. These membranes exhibit remarkable water permeance, high dye rejection efficiency, and stability during the filtration process.
Experimental
Materials
Polyacrylonitrile (PAN, Mw = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Carl Roth). 2,5-diaminobenzenesulfonic acid (Pa-SO3H), p-phenylenediamine (Pa), 2,5-dimethyl-1,4-phenylenediamine (Pa-(CH3)2), benzidine (BD), [2,2′-bipyridine]-5,5′-diamine (Bpy), 1,3,5-triformylphloroglucinol (Tp), methyl blue (MeBe, MW = 799.8), Congo red (CR, MW = 696.5), rhodamine B (RhB, MW = 479), and methylene blue (MB, MW = 319.8) were bought from Sigma-Aldrich, whereas N,N-dimethylformamide (DMF, 99.9%), dichloromethane (DCM, 99.9%), and acetic acid (AcOH, 99.7%) were supplied by Fisher Chemical.
000, Carl Roth). 2,5-diaminobenzenesulfonic acid (Pa-SO3H), p-phenylenediamine (Pa), 2,5-dimethyl-1,4-phenylenediamine (Pa-(CH3)2), benzidine (BD), [2,2′-bipyridine]-5,5′-diamine (Bpy), 1,3,5-triformylphloroglucinol (Tp), methyl blue (MeBe, MW = 799.8), Congo red (CR, MW = 696.5), rhodamine B (RhB, MW = 479), and methylene blue (MB, MW = 319.8) were bought from Sigma-Aldrich, whereas N,N-dimethylformamide (DMF, 99.9%), dichloromethane (DCM, 99.9%), and acetic acid (AcOH, 99.7%) were supplied by Fisher Chemical.
PAN electrospun fiber membrane
A PAN solution was prepared by dissolving 1.5 g in DMF (8.5 g) and stirring for 3 h. The electrospinning parameters were set as follows: voltage 15 kV voltage; the distance between the tip and the collector 20 cm, and the flow rate 0.8 mL h−1.Growth of COF membranes on PAN fiber membrane (TpPa/PAN)
First, a p-phenylenediamine (Pa) aqueous solution was prepared (20 mg mL−1, designated as solution A). Next, a circular PAN electrospun fiber membrane with a diameter of 4 cm is placed flat at the bottom of the glass pressure reaction vessel, and an ample amount of solution A is added to thoroughly wet the membrane. The excess solution is removed. By measuring the weight of the reaction vessel before and after the process, it was determined that the PAN fiber membrane absorbed 0.325 g of solution A. This means the PAN electrospun fiber membrane has absorbed 6.5 mg (0.06 mmol) of Pa. Next, based on the stoichiometric ratio of Pa to 1,3,5-triformylphloroglucinol (Tp) (3:2), 8.4 mg (0.04 mmol) of Tp is dissolved in 5 mL of a mixed solution of DCM and AcOH (v/v = 49/1), referred to as solution B. Solution B is then slowly added to the reaction vessel, creating a water–organic interface on the surface of the PAN fiber membrane, forming liquid–liquid–solid interface. Finally, the reaction bottle was sealed and reacted at 80 °C for 3 h, and a TpPa membrane was successfully grown on the PAN fiber membrane, which was called TpPa/PAN membrane. Additionally, the reaction was swiftly halted by placing the reaction vessel in a refrigerator at −20 °C, allowing for precise control of the reaction time at intervals of 2 min, 5 min, 15 min, 30 min, 1 h, 2 h, and 3 h, respectively. Several other experiments were carried out in the similar way but changing the concentration of Pa aqueous solution (solution A, 2, 5, 10, 20, 30, and 40 mg mL−1, respectively).
Furthermore, the same method was also used prepare TpPa/PAN at room temperature (about 23 °C) with a Pa aqueous solution (solution A) of 20 mg mL−1.
The preparation method was also applied to other four imine-linked COFs by taking different diamines (diaminobenzenesulfonic acid (Pa-SO3H), 2,5-dimethyl-1,4-phenylenediamine (Pa-(CH3)2), benzidine (BD), [2,2′-bipyridine]-5,5′-diamine (Bpy)), keeping the Tp same. The concentration of each diamine monomer in water was fixed at 10 mg mL−1 (solution A), the reaction time was 6 h at 80 °C. The resulting membranes were called TpPa-SO3H/PAN, TpPa-(CH3)2/PAN, TpBD/PAN, and TpBpy/PAN, respectively.
Preparation of self-standing TpPa-COF membrane
For making a self-standing TpPa-COF membrane, in the next PAN was removed by selective dissolution in DMF. For this, the TpPa/PAN membrane was immersed in a DMF solution at 80 °C for 12 h, during which fresh DMF solution was replaced several times.Preparation of TpPa/cellulose membrane
The same method is used to form a TpPa membrane on cellulose paper, and it is called TpPa/Cellulose membrane. During experiment, the concentration of Pa in water was 20 mg mL−1, the reaction temperature was 80 °C, the reaction time was 3 h, cellulose paper with a diameter of 4 cm was used as the carrier, and the rest of the steps remained unchanged. In the first step of wetting the cellulose with Pa solution, it was determined that the cellulose membrane absorbed 0.384 mg (7.68 mg of Pa) of solution A. For the next step, 9.94 mg of Tp in a mixed solution of 5 mL DCM and AcOH (v/v = 49/1) (solution B) was slowly to the reaction vessel.Synthesis of TpPa powder at room temperature (about 23 °C)
6.5 mg (0.06 mmol) Pa and 8.4 mg (0.04 mmol) Tp were added to 5 mL of DCM and AcOH (v/v = 49/1) and reacted at room temperature for 24 h to obtain TpPa powder with a stoichiometric ratio of Pa to Tp of 3:2. 6.5 mg (0.06 mmol) Pa and 16.8 mg (0.08 mmol) Tp were added to 5 mL of DCM and AcOH (v/v = 49/1) and reacted at room temperature for 24 h to obtain TpPa powder with a stoichiometric ratio of Pa to Tp of 3:4.
Characterization and instruments
The morphology was checked by scanning electron microscope (FEI Quanta FEG 250) and transmission electron microscope (JEOL JEM-2200FS, accelerating voltage 200 kV). The FT-IR spectra were taken between 600–4000 cm−1 (Digilab Excalibur FTS-3000). 13C and 15N solid-state NMR spectra were acquired on a Bruker Avance III HD spectrometer operating at a B0 field of 9.4 T (ν0(13C) = 100.6 MHz and ν0(15N) = 40.6 MHz). The samples were spun at 15.0 kHz (13C) and 10 kHz (15N) in a 3.2 mm MAS triple resonance probe. 13C MAS spectra were obtained with ramped cross-polarization (CP) experiments where the 13C nutation was set to 50 kHz and the 1H nutation frequency νnut was varied linearly from 50–100% to match the Hartmann–Hahn conditions. The contact time was set to 3 ms. The 15N CP MAS NMR spectra were acquired with a proton ramp (70–100%) with a contact time of 5 ms where the 15N nutation was set to 21 kHz. Proton broadband decoupling with spinal-64 and νnut = 70 kHz was applied during acquisition for both the 13C and 15N MAS NMR spectra. The spectra are referenced with respect to tetramethylsilane (13C) and CH3NO2 (15N). The powder X-ray diffraction (PXRD) patterns were carried out on a Bragg–Brentano type diffractometer (XPERT-PRO, PANalytical B.V.) using Cu Kα radiation (λ = 1.540598 Å). The measuring range of each sample is 2θ = 2° to 40° at a rate of 1° min−1. N2 adsorption/desorption isotherms were measured on ASiQ-MP-MP-AG setup (Anton Paar QuantaTec, Boynton Beach, USA) and kept at 77 K by a liquid nitrogen bath. The specific surface area was calculated with Brunauer–Emmet–Teller (BET) model considering the Rouquerol correction for microporous materials. For the pore size analysis a quasistationary density functional theory (QSDFT) kernel for carbon-based materials was used. The samples were degassed under vacuum (10−2 kPa) at 120 °C for 16 h before starting the physisorption experiment. Use a tensile testing machine (ZwickiLine Z0.5; BT1-FR0.5 TN.D14; Zwick/Roell, Germany) for tensile testing (sample length 30 mm, width 5 mm and initial effective tensile length 20 mm), and cyclic bending test were performed under the same measurement conditions. The water contact angle was measured by an automatic goniometer/tensiometer 7 (KRUSS, DSA100, Germany). A water droplet (3 μL) was placed on the membrane surface and allowed to stabilize for 5 s to determine the contact angle. The average pore size of PAN fiber membrane and cellulose paper was measured by PSM 165 Pore Size Meter (Topas GmbH, Germany). Optical microscope images of all samples were provided by ZEISS Smartzoom 5. Ultraviolet-visible (UV-vis) spectra were recorded on a Jasco Spectrometer V-670 with a measurement range of 200–750 nm (scan speed 200 nm min−1).Nanofiltration test
The nanofiltration performance of TpPa/PAN membranes was evaluated using a laboratory-scale filtration device. The device had a maximum operating pressure of 5.3 bar and the effective filtration area of 4.9 cm2. Before testing the nanofiltration performance, each COF membrane was treated with deionized water at 3 bar for 30 min to have a stable state for testing the nanofiltration performance. The tested dye solution concentration was 100 mg L−1, and a transmembrane pressure of 3 bar was used for filtration.The permeation flux (J, L m−2 h−1) and permeance (P, L m−2 h−1 bar−1) of each membrane were calculated by the following equations:
 | (1) |
 | (2) |
The rejection (R) of dyes was calculated as the following equation:
 | (3) |
Results and discussion
The most commonly used imine-linked COF (TpPa, Fig. S1† and 1) was selected as the research model, and an electrospun polyacrylonitrile (PAN) fiber membrane was used as a solid sacrificial porous template. | ||
| Fig. 1 Diagrammatic representation of the preparation process of TpPa/PAN and self-standing TpPa-COF membranes. | ||
First, a suitably sized PAN fiber membrane (average fiber diameter 332 ± 56 nm, average pore size 1.8 μm, and average thickness 31.4 ± 2.7 μm) prepared by electrospinning (Fig. S2a†) was positioned at the base of a glass pressure resistant Schott bottle, and an appropriate amount of p-phenylenediamine (Pa) aqueous solution was added to fully wet it, without dripping of the solvent on tilting (Fig. S2b†). Subsequently, a dichloromethane (DCM) solution of 1,3,5-triformylphloroglucinol (Tp) was slowly added onto the wet PAN membrane surface, forming an aqueous-organic solvent interface on the solid PAN fibers (liquid–liquid–solid interface). In the final step, the reaction system was sealed, and heated at 80 °C for 3 h, resulting in the growth of TpPa (red color) on the PAN fiber membrane. Optically, only one side of the template PAN fiber membrane showed a red color; the other side was colorless (Fig. S2c–h†). The resulting TpPa composite membrane is designated as the TpPa/PAN membrane.
Scanning electron microscopy (SEM) was employed to capture the progressive morphological changes of TpPa over the course of the reaction, by examining both the surface and cross-sectional structures (Fig. 2 and S3†). At the initial stage of the reaction (2 min, Fig. 2b, b′ and S3b, b′†), numerous TpPa nanocrystals formed and covered PAN fibers. This was due to the high temperature accelerating both the polymerization and crystallization rates, leading to the rapid formation of TpPa nanocrystals, similar to solvothermal synthesis.45,46 Effectively balancing the polymerization rate and crystallization rate is crucial yet challenging for forming high crystallinity COF membranes. Notably, the TpPa crystal growth occurs not only on the PAN fibers in the top layer of the PAN template membrane but also on the inner bulk PAN fibers. As the reaction proceeded (5 min, Fig. 2c, c′ and S3c, c′†), more TpPa nanocrystals formed at the liquid–liquid–solid interface, exhibiting intergrowth, which provided the foundation for the growth of the TpPa film in between the PAN fibers in addition to the wrapping of PAN fibers. At 15 min of reaction time, TpPa was also seen as a separate film (thickness ∼140 nm, Fig. 2d, d′ and S3d, d′†) composed of TpPa nanocrystals on top of the PAN-wrapped TpPa fibers. As the reaction time was increased to 3 h, the top TpPa film exhibited a more distinct nanocrystal morphology, and the film thickness increased progressively to ∼530 nm (Fig. 2h, h′ and S3h, h′†). By comparing the thickness of the top TpPa film at various reaction times (Table S1†), it was observed that the thickness stabilized between 2 and 3 h, thus establishing 3 h as the optimal reaction time. The majority of TpPa grew on PAN support, with an overall yield exceeding 85%. The complete TpPa/PAN membrane exhibited a fascinating hierarchical three-layer morphology: a continuous top TpPa film (top layer) (∼530 nm) grown on a middle layer (∼10 μm) of PAN-wrapped TpPa fibers randomly laid onto each other, followed by a bottom layer of pure PAN fibers (∼20 μm) without COF (Fig. 2i, j). In addition to this special hierarchical morphology, the complete process is fast in comparison to solvothermal process of growing COF on PAN elctrospun fibers.46 The procedure is reproducible as confirmed by repeating the same experiment and by carrying out the similar experiments by changing the Pa concentration, template material and fiber diameter, and the type of the diamine used as described later.
 | ||
| Fig. 2 Surface and cross-sectional SEM pictures of TpPa/PAN membranes at varying reaction times at 80 °C (a, a′ 0 min), (b, b′ 2 min) (c, c′ 5 min), (d, d′ 15 min), (e, e′ 30 min), (f, f′ 1 h) (g, g′ 2 h), and (h, h′, i, and j 3 h). And the corresponding FT-IR spectra (k) and PXRD patterns (l). | ||
The chemical and crystal structural changes over reaction time were analyzed using Fourier-transform infrared (FT-IR) spectra and powder X-ray diffraction (PXRD) patterns (Fig. 2k and l). First, all samples showed the signature peaks (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N at 2243 cm−1) and reflexes (2θ = 17.2°) of PAN in the electrospun PAN fiber membrane.47 Secondly, in a very short reaction time (2 min), the signature peaks (C
N at 2243 cm−1) and reflexes (2θ = 17.2°) of PAN in the electrospun PAN fiber membrane.47 Secondly, in a very short reaction time (2 min), the signature peaks (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 1577 cm−1, C–N 1240 cm−1) and reflexes (2θ = 4.8° and 27°) belonging to TpPa could be clearly observed, indicating that polymerization and crystallization have begun to occur.48,49 Combined with the SEM image, it can be found that high temperature (80 °C) accelerates the reaction polymerization rate and crystallization rate, directly forming TpPa nanocrystals instead of amorphous polymer films. Finally, with the increase of reaction time, these characteristic peaks and reflexes gradually become clearer. Notably, at 3 h, the reflex at 2θ = 4.8° exhibits a rather sharp shape, indicating the formation of TpPa with higher crystallinity.
C at 1577 cm−1, C–N 1240 cm−1) and reflexes (2θ = 4.8° and 27°) belonging to TpPa could be clearly observed, indicating that polymerization and crystallization have begun to occur.48,49 Combined with the SEM image, it can be found that high temperature (80 °C) accelerates the reaction polymerization rate and crystallization rate, directly forming TpPa nanocrystals instead of amorphous polymer films. Finally, with the increase of reaction time, these characteristic peaks and reflexes gradually become clearer. Notably, at 3 h, the reflex at 2θ = 4.8° exhibits a rather sharp shape, indicating the formation of TpPa with higher crystallinity.
Additionally, the effect of reaction temperature on TpPa formation was examined by conducting the reaction at room temperature (approximately 23 °C). SEM images (Fig. S4 and S5†) revealed that at 2 min of reaction time, the PAN fibers were already covered with TpPa, and a small amount of TpPa was grown as a film between the fibers. As the reaction progressed, the TpPa gradually expanded between the fibers, eventually forming a complete film on top of the PAN-wrapped TpPa fibers (15 min). The thickness of the top TpPa film continued to increase until it stabilized after 1 h (∼100 nm, Table S2†), though it remained much thinner than the membrane formed at 80 °C. Concurrently, rod-like particles and nanowires began to appear on the top TpPa film's surface, progressively growing over time. The rod-like particles gradually extended to the micron scale, while the nanowires slowly covered the top TpPa film. After 24 h, a dense layer of nanowires had formed, resulting in an overall hierarchical three-layer TpPa/PAN membrane structure similar to that observed at 80 °C. Using FT-IR spectra and PXRD patterns, the chemical and crystalline structure evolution of the TpPa was analyzed (Fig. S4m and n†). At 2 min, prominent signature peaks at 1577 cm−1 (CC) and 1240 cm−1 (C–N) indicated that polymerization had occurred. By 30 min, a weak reflex at 2θ = 4.8° marked the onset of crystallization. This reflex sharpened over time, suggesting increased crystallinity, though significantly lower than that of TpPa formed at 80 °C.
To investigate the formation of rod-like TpPa particles and nanowires, Pa and Tp were mixed in different stoichiometric ratios (3:2 and 3:4 molar ratio) in an organic solvent (DCM) and reacted at room temperature for 24 h. When a 3:2 molar ratio was used, rod-like particles were formed (Fig. S6a†), which aligns with previously reported literature.50 At a 3:4 molar ratio, a significant number of nanowires, along with some rod-like particles, were observed (Fig. S6b†). FT-IR spectra revealed that both the rod-like particles and nanowires share a similar chemical structure to TpPa. However, the nanowires also exhibited characteristic peaks associated with Pa (-NH2) and Tp (-CHO), indicating the presence of unreacted monomers or intermediates (Fig. S6c†). In the PXRD patterns (Fig. S6d†), the reflex at 2θ = 4.8° for the rod-like particles is clear, indicating good crystallinity. In contrast, the corresponding reflex for the nanowires was much weaker, indicating lower crystallinity. Additionally, both structures display reflexes that differ from those of traditional TpPa.51 This difference suggests that by adjusting the molar ratio of Pa to Tp, it is possible to produce crystalline rod-like TpPa particles as well as amorphous polymer nanowires. However, the room temperature conditions may likely lack sufficient energy to promote the complete polymerization of monomers and orderly self-assembly. This suggests that, under room temperature reaction conditions, the polymerization rate of the monomers far exceeds the crystallization rate, and an amorphous polymer is first formed and gradually grows and begins to transform into a crystalline TpPa top film similar to the one obtained via liquid–liquid IP.33 Moreover, once a complete polymer film formed, monomer diffusion was restricted, reducing the polymerization rate and causing the thickness of the film to grow slowly until it ceased. Finally, in the organic phase, only a small amount of Pa reacts with a large amount of Tp, forming crystalline rod-shaped particles and amorphous nanowires. Therefore, high temperature is a necessary condition to obtain a highly crystalline TpPa film.
In the aforementioned discussion, the concentration of Pa in water was 20 mg mL−1. To explore the effect of varying Pa concentrations on the formation and morphology of TpPa, several concentrations were studied, including 2, 5, 10, 20, 30, and 40 mg mL−1 (Fig. S7 and S8†). At lower Pa concentrations, the COF nanocrystals grow only on the surface and around the PAN fibers, without forming a complete TpPa top film. As the monomer concentration increased, symbiosis occurred among the nanocrystals, gradually forming a complete TpPa film. The critical Pa concentration that forms a complete top TpPa film was 10 mg mL−1. Altering the concentration of Pa significantly impacted the thickness of the top TpPa film, which increased from ∼150 nm to ∼1150 nm as the concentration was adjusted from 5 to 40 mg mL−1 (Table S3†). Additionally, when the concentration of Pa in water was 40 mg mL−1, a continuous top TpPa film (thickness ∼210 nm) with good crystallinity could be obtained in just 5 min (Fig. S10a–d†). FT-IR spectra confirmed the structure and PXRD patterns indicated high crystallinity in all cases (Fig. S9 and S10e–f†). At the same time, in order to explore the possibility of preparing COF membranes on a large scale using this method, a larger reactor was selected to obtain a circular TpPa/PAN membrane (yield 74%) with an area of 50.24 cm2 (Fig. S11†). This demonstrates that, with this method, the size of the COF membrane is primarily constrained by the dimensions of the reactor, suggesting that larger COF membranes can be produced as long as a larger reactor is used. Furthermore, in this work, the TpPa membranes were prepared significantly faster in comparison to the reported COF membranes (Fig. S12 and Table S4†).
Next, we demonstrate the potential to fabricate a self-standing, robust, crystalline TpPa-COF membrane by selectively removing the template polymer (PAN) from the TpPa/PAN membranes. As illustrated in Fig. 3a, b and S13,† with the removal of PAN fibers, the three-layer TpPa/PAN membrane is transformed into a double-layer self-standing TpPa-COF membrane with a continuous TpPa film (∼500 nm) supported on hollow TpPa fibers randomly laid onto each other. Transmission electron microscopy (TEM) images (Fig. 3c) clearly display the continuous structure of the hollow TpPa fibers, which have an average diameter of 579 ± 245 nm and a hole diameter of 291 ± 98 nm. The average was calculated based on 80 fibers. The large deviation in total fiber diameter and hole diameter is not due to any procedural error, but rather a consequence of the method of preparation itself. First, an electrospun fiber membrane was used as a template. Due to inherent limitations of the electrospinning process, the PAN fibers in the template membrane did not have a uniform diameter. Second, there may be a gradient in the COF shell thickness on the PAN fibers. During COF formation, Tp must diffuse to the lower PAN layers; however, the rapid formation of COF on the surface limits the diffusion distance. As a result, all PAN fiber layers did not have the same coating thickness. Here, we report the average of all fibers in the self-standing COF membranes, as it is not feasible to measure the average fiber diameters for different layers separately. The variation in hole size may be attributed to compression of some fibers or to holes not having a perfectly spherical shape.
 | ||
| Fig. 3 Cross-sectional SEM pictures of self-standing membranes (a and b). TEM images of hollow TpPa fibers (c). FT-IR spectra (d), 13C and 15N CP MAS solid-state NMR spectra (e and f), and PXRD patterns (g) of TpPa/PAN membrane and self-standing TpPa-COF membrane. N2 adsorption–desorption isotherms (h) and pore size distribution (i) of self-standing TpPa-COF membrane. | ||
Combined analyses from FT-IR spectra, 13C and 15N cross-polarized magic angle spinning (CP MAS) NMR spectra, and PXRD patterns confirm the complete removal of PAN, yielding a highly crystalline, self-standing TpPa-COF membrane (Fig. 3d–g). In addition, the surface area and pore size distribution of the self-standing TpPa-COF membrane were determined by N2 physisorption measurements at 77 K. The self-standing TpPa-COF membrane exhibits a type-I isotherm of microporous materials.52 The BET (Brunauer–Emmett–Teller) surface area is 532 m2 g−1 (Fig. 3h and S13†), which is similar to that of TpPa powder obtained through solvothermal synthesis.53 The pore size distribution of the self-standing TpPa-COF membrane calculated by a quasistationary density functional theory (QSDFT) kernel for carbon-based materials and exhibits a pronounced peak at 1.1 nm (Fig. 3i), which is lower than the reported 1.8 nm. This may be due to the intergrowth of TpPa nanoparticles to form staggered stacking, resulting in reduction in pore size.54,55
Large dimension COF membranes possessing good mechanical properties and flexibility are essential for better handling and therefore, their practical utility in real life. The mechanical properties and flexibility of the self-standing TpPa-COF and TpPa/PAN membranes were analyzed by tensile tests and cyclic bending tests (Table S5†). The self-standing TpPa-COF membranes exhibited excellent mechanical properties, with a tensile strength of approximately 23 MPa and an elongation at a break of around 8% (Fig. 4a). When comparing the TpPa COF/PAN membrane to the TpPa membrane, it is evident that the TpPa COF/PAN membrane exhibits significantly higher elongation at break. This behavior can be attributed to the stretching of the supporting PAN substrate fibers before they fracture. The elongation behavior of the TpPa COF/PAN membrane is primarily dominated by the PAN substrate fibers. In contrast, the TpPa membrane demonstrates nearly double the tensile strength at break compared to the TpPa COF/PAN composite membrane. This higher tensile strength is due to the hierarchical structure of the TpPa membrane, where a dense TpPa COF layer is supported by randomly arranged COF hollow fibers. The proportion of the dense membrane in the TpPa COF is greater than that in the TpPa COF/PAN composite, as its total thickness is reduced following the extraction of PAN, compared to the original composite membrane.
 | ||
| Fig. 4 Stress–strain curves of self-standing TpPa-COF, TpPa-50%-2000-cycle, and TpPa-80%-500-cycle membranes (a). Stress–strain curves of PAN fiber, TpPa/PAN, TpPa/PAN-50%-100,000-cycle, TpPa/PAN-80%-100,000-cycle membranes (b). The test of self-standing TpPa membrane under bending and folding states (c), and the bending and recovery process under a compression rate of 50% and 80% (d). | ||
Furthermore, the self-standing TpPa-COF membranes showed excellent flexibility and durability, recovering to their original shape after bending and folding (Fig. 4c), and were able to withstand 2000 cycles of 50% and 500 cycles of 80% bending compression, respectively, without any significant loss in tensile strength (Fig. 4a, d and S15†). And, SEM images reveal that no cracks appeared on the surface of the self-standing TpPa-COF membrane following multiple flexibility tests (Fig. S16†). In contrast, the TpPa/PAN membranes displayed different characteristics, with their tensile behavior primarily influenced by the PAN electrospun membranes. These membranes were able to recover their original shape not only after curling but also after being kneaded (Fig. 4b and S17a†). In addition, the TpPa/PAN membranes showed no change in tensile properties even after 100000 cycles of bending (compression ratios of 50% and 80%, respectively) (Fig. 4b, S16b and S18†). Similarly, no cracks appeared on the surface of the TpPa/PAN membrane after undergoing multiple flexibility tests (Fig. S19†).
In order to show the universality of the method, we first used this method to successfully grow four other different imine-linked COFs (TpPa-SO3H, TpPa-(CH3)2, TpBD and TpBpy) on PAN fiber membranes, named TpPa-SO3H/PAN, TpPa-(CH3)2/PAN, TpBD/PAN and TpBpy/PAN (Fig. S20–S22 and Table S6†). It was observed that the four imine-linked COF membranes were successfully and continuously grown on PAN fiber membranes, all exhibiting good crystallinity. The thickness of these COF membranes ranged from 300 to 500 nm, completely enveloping the PAN fibers and ensuring strong interactions between the COF membranes and the PAN fiber membranes. Secondly, we chose porous substrates with varying surface energies and pore sizes for growing TpPa membranes. Cellulose paper, with an average pore size of 38.8 μm and a water contact angle of 0°, demonstrating super hydrophilicity, was also tested as a substrate in comparison to the PAN electrospun membrane (average pore size of 1.8 μm, water contact angle of 114°) (Fig. S23†). Using the same method, a highly crystalline TpPa membrane (thickness ∼620 nm) was successfully formed in 3 h at 80 °C on cellulose paper (TpPa/cellulose, Fig. S24 and S25†). The results suggest that the method presented in this work possesses a level of versatility, enabling the construction of various COF membranes on different porous substrates and not restricted to only one type with promising potential for applications across multiple fields.
COF membranes with directional nanochannel are favorable for filtration applications.56–59 To probe the effect of TpPa membrane thickness on filtration performance, we began by using methyl blue (MeBe) as a model dye, as shown in Fig. 5a. As the thickness of the top TpPa layer increased from ∼370 nm to ∼1150 nm (Table S3†), the rejection rate of MeBe improved from 96.7% to 99.3%. However, water permeance decreased from 51.2 to 39.1 L m−2 h−1 bar−1. After thorough evaluation, the TpPa/PAN membrane with a ∼530 nm thick top TpPa layer was chosen for the subsequent filtration performance tests. To assess the filtration performance of the TpPa/PAN membrane, four dyes with different molecular weights were used: MeBe, Congo red (CR), Rhodamine B (RhB), and Methylene blue (MB) (Fig. 5b). The TpPa/PAN membrane demonstrated a high rejection rate (above 96%) and impressive water permeance (over 49 L m−2 h−1 bar−1) for all tested dyes. Compared to state-of-the-art membranes, the TpPa/PAN membrane showed superior filtration performance for dye separation (Fig. 5c and Table S6†). Additionally, during continuous filtration of the MeBe aqueous solution for 12 h, both the rejection rate and permeance remained largely consistent (Fig. 5d). Further, an increase in the filtration time to 24 h led to a slightly decreased rejection rate and permeance, emphasizing excellent operational stability at least up to 12–14 h under laboratory experimental conditions (Fig. S26†).
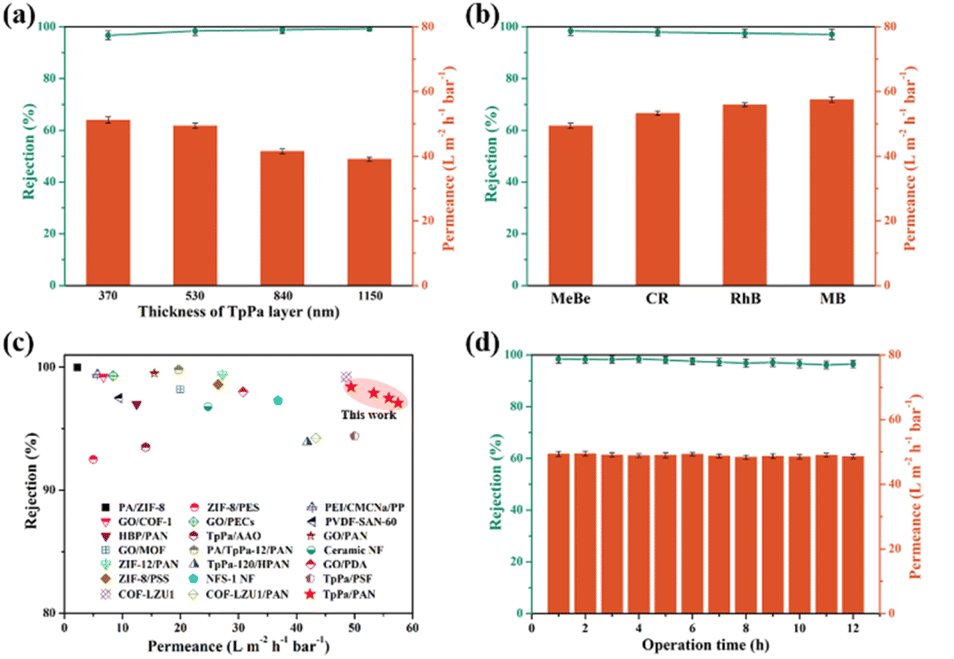 | ||
| Fig. 5 Filtration performance of TpPa/PAN membranes prepared at different Pa concentrations for MeBe aqueous solution (a), and different dyes (b). Comparison of our work with literature-known dye separation membranes, with detailed data provided in Table S6† (c). Long-term filtration performance of TpPa/PAN membrane for MeBe aqueous solutions (d). | ||
Conclusions
In conclusion, we have successfully established liquid–liquid–solid IP technology for the preparation of large-sized, robust, and flexible crystalline COF membranes in high yields (more than 85%) on different porous substrates in a few hours. The membrane thickness showed a clear dependence on the reaction time and initial concentration of the starting materials. Additionally, the porous substrate could be easily extracted to create a self-standing COF membrane. This versatile method can be applied to construct various COF membranes on different porous substrates, addressing issues such as membrane fragility, low crystallinity, and poor substrate selectivity seen in traditional processes. Furthermore, the COF membranes produced using this approach exhibit excellent filtration performance for dye wastewater treatment. This method significantly broadens the scope for COF membrane fabrication, presenting strong potential in membrane technology applications.Data availability
The data that support the findings of this paper are available from the main text, ESI† or from the corresponding author upon reasonable request.Author contributions
Chenhui Ding: conceptualization, data curation, methodology, formal analysis, investigation, visualization, validation, software, writing – original draft. Yingying Du: investigation, data curation, formal analysis, validation, software, writing – review & editing. Tamara Fischer: data curation, writing – review & editing, formal analysis. Jana Timm: data curation, writing – review & editing, formal analysis. Roland Marschall: supervision, resources, writing – review & editing. Jürgen Senker: resources, supervision, writing – review & editing. Seema Agarwal: conceptualization, resources, funding acquisition, supervision, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge Deutsche Forschungsgemeinschaft (DFG) (SFB 1585, 492723217; project A01) for financially supporting the work. Yingying Du thanks the China Scholarship Council (CSC) for financing the research stay in Germany. Prof. Josef Breu is acknowledged for providing the XRD facility. The keylabs ‘Synthesis and Molecular Characterization’ and ‘Electron and Optical Microscopy’ of Bavarian Polymer Institute are thanked for providing the synthesis and SEM, TEM facilities for the present work.Notes and references
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- H. Yang, L. Yang, H. Wang, Z. Xu, Y. Zhao, Y. Luo, N. Nasir, Y. Song, H. Wu and F. Pan, Nat. Commun., 2019, 10, 2101 CrossRef PubMed.
- H. Wang, Y. Zhai, Y. Li, Y. Cao, B. Shi, R. Li, Z. Zhu, H. Jiang, Z. Guo and M. Wang, Nat. Commun., 2022, 13, 7123 CrossRef CAS PubMed.
- L. Liu, L. Yin, D. Cheng, S. Zhao, H. Y. Zang, N. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2021, 133, 15001–15006 CrossRef.
- Y. Cao, M. Wang, H. Wang, C. Han, F. Pan and J. Sun, Adv. Energy Mater., 2022, 12, 2200057 CrossRef CAS.
- A. E. Lakraychi, E. S. Picton, Y. Liang, D. L. Shaffer and Y. Yao, ACS Energy Lett., 2023, 8, 5032–5040 CrossRef CAS.
- Y. Wen, J. Ding, Y. Yang, X. Lan, J. Liu, R. Hu and M. Zhu, Adv. Funct. Mater., 2022, 32, 2109377 CrossRef CAS.
- J. Xu, S. An, X. Song, Y. Cao, N. Wang, X. Qiu, Y. Zhang, J. Chen, X. Duan, J. Huang, W. Li and Y. Wang, Adv. Mater., 2021, 33, 2105178 CrossRef CAS PubMed.
- K. Wang, J. Duan, X. Chen, J. Wang, J. Li, L. Jiang, W. Yan, W. Lyu and Y. Liao, Adv. Energy Mater., 2024, 14, 2401146 CrossRef CAS.
- M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann and D. Trauner, J. Am. Chem. Soc., 2014, 136, 17802–17807 CrossRef CAS PubMed.
- R. Shen, C. Qin, L. Hao, X. Li, P. Zhang and X. Li, Adv. Mater., 2023, 35, 2305397 CrossRef CAS PubMed.
- H. Yang, H. Zhang and D. Zhao, Langmuir, 2022, 39, 20–27 CrossRef PubMed.
- Y. Yang, M. Ratsch, A. M. Evans and K. Börjesson, J. Am. Chem. Soc., 2023, 145, 18668–18675 CrossRef CAS PubMed.
- H. S. Sasmal, A. Halder, S. Kunjattu H, K. Dey, A. Nadol, T. G. Ajithkumar, P. Ravindra Bedadur and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 20371–20379 CrossRef CAS PubMed.
- D. Zhou, X. Tan, H. Wu, L. Tian and M. Li, Angew. Chem., Int. Ed., 2019, 131, 1390–1395 CrossRef.
- J. T. Liu, S. F. Wang, T. F. Huang, P. Manchanda, E. A. Hamad and S. P. Nunes, Sci. Adv., 2020, 6, eabb3188 CrossRef CAS PubMed.
- Z. F. Gao, J. Liu and T. S. Chung, Sep. Purif. Technol., 2022, 294, 121166 CrossRef CAS.
- X. H. Liu, C. Z. Guan, S. Y. Ding, W. Wang, H. J. Yan, D. Wang and L. J. Wan, J. Am. Chem. Soc., 2013, 135, 10470–10474 CrossRef CAS PubMed.
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed.
- H. Fan, A. Mundstock, A. Feldhoff, A. Knebel, J. Gu, H. Meng and J. Caro, J. Am. Chem. Soc., 2018, 140, 10094–10098 CrossRef CAS PubMed.
- H. Fan, M. Peng, I. Strauss, A. Mundstock, H. Meng and J. Caro, Nat. Commun., 2021, 12, 38 CrossRef CAS PubMed.
- Y. Ying, M. Tong, S. Ning, S. K. Ravi, S. B. Peh, S. C. Tan and D. Zhao, J. Am. Chem. Soc., 2020, 142, 4472–4480 CrossRef CAS PubMed.
- M. Wang, P. Zhang, X. Liang, J. Zhao, Y. Liu, Y. Cao, H. Wang, Y. Chen, Z. Zhang, F. Pan, Z. Zhang and Z. Jiang, Nat. Sustainability, 2022, 5, 518 CrossRef.
- J. Yuan, M. Wu, H. Wu, Y. Liu, X. You, R. Zhang, Y. Su, H. Yang, J. Shen and Z. Jiang, J. Mater. Chem. A, 2019, 7, 25641–25649 RSC.
- N. A. Khan, J. Yuan, H. Wu, L. Cao, R. Zhang, Y. Liu, L. Li, A. U. Rahman, R. Kasher and Z. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 28978–28986 CrossRef CAS PubMed.
- J. Du, A. Yao, Q. Sun, L. Liu, Z. Song, W. He, C. Wang, P. Dou, J. Guan and J. Liu, Adv. Mater., 2024, 36, 2405744 CrossRef CAS PubMed.
- S. Wang, L. Yang, K. Xu, H. Chen and N. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 44806–44813 CrossRef CAS PubMed.
- J. Liu, G. Han, D. Zhao, K. Lu, J. Gao and T. S. Chung, Sci. Adv., 2020, 6, eabb1110 CrossRef CAS PubMed.
- L. Cao, H. Wu, Y. Cao, C. Fan, R. Zhao, X. He, P. Yang, B. Shi, X. You and Z. Jiang, Adv. Mater., 2020, 32, 2005565 CrossRef PubMed.
- K. Dey, M. Pal, K. C. Rout, S. Kunjattu H, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- Y. He, X. Lin, Y. Zhou, J. H. Chen, Z. Guo and H. Zhan, Chem. Mater., 2021, 33, 9413–9424 CrossRef CAS.
- A. Giri, G. Shreeraj, T. K. Dutta and A. Patra, Angew. Chem., Int. Ed., 2023, 62, e202219083 CrossRef CAS PubMed.
- N. A. Khan, R. Zhang, H. Wu, J. Shen, J. Yuan, C. Fan, L. Cao, M. A. Olson and Z. Jiang, J. Am. Chem. Soc., 2020, 142, 13450–13458 CrossRef CAS PubMed.
- P. Wang, Y. Peng, C. Zhu, R. Yao, H. Song, L. Kun and W. Yang, Angew. Chem., Int. Ed., 2021, 60, 19047–19052 CrossRef CAS PubMed.
- C. Fan, H. Wu, J. Guan, X. You, C. Yang, X. Wang, L. Cao, B. Shi, Q. Peng, Y. Kong, Y. Wu, N. A. Khan and Z. Jiang, Angew. Chem., Int. Ed., 2021, 60, 18051–18058 CrossRef CAS PubMed.
- H. Fan, M. Peng, I. Strauss, A. Mundstock, H. Meng and J. Caro, J. Am. Chem. Soc., 2020, 142, 6872–6877 CrossRef CAS PubMed.
- E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. Uribe-Romo, G. D. Gutierrez, P. Clancy and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 19416–19421 CrossRef CAS PubMed.
- J. Fu, S. Das, G. Xing, T. Ben, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 7673–7680 CrossRef CAS PubMed.
- D. Hao, J. Zhang, H. Lu, W. Leng, R. Ge, X. Dai and Y. Gao, Chem. Commun., 2014, 50, 1462–1464 RSC.
- C. Zhang, B. H. Wu, M. Q. Ma, Z. Wang and Z. K. Xu, Chem. Soc. Rev., 2019, 48, 3811–3841 RSC.
- H. Wang, Z. Zeng, P. Xu, L. Li, G. Zeng, R. Xiao, Z. Tang, D. Huang, L. Tang, C. Lai, D. Jiang, Y. Liu, H. Yi, L. Qin, S. Ye, X. Ren and W. Tang, Chem. Soc. Rev., 2019, 48, 488–516 RSC.
- H. Fan, J. Gu, H. Meng, A. Knebel and J. Caro, Angew. Chem., Int. Ed., 2018, 57, 4083–4087 CrossRef CAS PubMed.
- C. Ding, M. Breunig, J. Timm, R. Marschall, J. Senker and S. Agarwal, Adv. Funct. Mater., 2021, 31, 2106507 CrossRef CAS.
- H. Hou, J. J. Ge, J. Zeng, Q. Li, D. H. Reneker, A. Greiner and S. Z. Cheng, Chem. Mater., 2005, 17, 967 CrossRef CAS.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853 CrossRef CAS PubMed.
- J. L. Sheng, H. Dong, X. B. Meng, H. L. Tang, Y. H. Yao, D. Q. Liu, L. L. Bai, F. M. Zhang, J. Z. Wei and X. J. Sun, ChemCatChem, 2019, 11, 2313–2319 CrossRef CAS.
- J. Zhang, C. Cheng, L. Guan, H. L. Jiang and S. Jin, J. Am. Chem. Soc., 2023, 145, 21974–21982 CrossRef CAS PubMed.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS.
- W. He, K. Kong, M. Wang, B. Dong, D. Yuan, K. P. Bryliakov and R. Wang, Appl. Catal., B, 2024, 350, 123916 CrossRef CAS.
- Y. Li, B. Pei, J. Chen, S. Bing, L. Hou, Q. Sun, G. Xu, Z. Yao and L. Zhang, J. Colloid Interface Sci., 2021, 591, 273 CrossRef CAS PubMed.
- M. D. Wang, Y. T. Wang, J. Y. Zhao, J. Y. Zou, X. Liang, Z. T. Zhu, J. S. Zhu, H. J. Wang, Y. H. Wang, F. S. Pan and Z. Y. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202219084 CrossRef CAS PubMed.
- Y. Pan, H. Liu, Z. Huang, W. Zhang, H. Gao, L. Liang, L. Dong and H. Meng, Angew. Chem., Int. Ed., 2024, 136, e202316315 CrossRef.
- D. B. Shinde, G. Sheng, X. Li, M. Ostwal, A. H. Emwas, K. W. Huang and Z. Lai, J. Am. Chem. Soc., 2018, 140, 14342 CrossRef CAS PubMed.
- S. Yuan, X. Li, J. Zhu, G. Zhang, P. Van Puyvelde and B. Van der Bruggen, Chem. Soc. Rev., 2019, 48, 2665 RSC.
- J. Wang and S. Zhuang, Coord. Chem. Rev., 2019, 400, 213046 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta02117k |
| This journal is © The Royal Society of Chemistry 2025 |