
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DMC matters: the role of dimethyl carbonate in SEI formation on oxygen functionalized anodes†
Bikram Kumar
Das
 *a,
Henry A.
Cortés
a,
Mauricio
Rincón Bonilla
a,
Menghao
Yang
b,
Javier
Carrasco
cd and
Elena
Akhmatskaya
ad
*a,
Henry A.
Cortés
a,
Mauricio
Rincón Bonilla
a,
Menghao
Yang
b,
Javier
Carrasco
cd and
Elena
Akhmatskaya
ad
aBCAM – Basque Center for Applied Mathematics, Bilbao, E-48009, Spain. E-mail: bdas@bcamath.org
bShanghai Key Laboratory for R&D and Application of Metallic Functional Materials, Institute of New Energy for Vehicles, School of Materials Science and Engineering, Tongji University, Shanghai, P. R. China
cCentre for Cooperative Research on Alternative Energies (CIC energiGUNE), Basque Research and Technology Alliance (BRTA), Alava Technology Park, Albert Einstein 48, Vitoria-Gasteiz, 01510, Spain
dIKERBASQUE, Basque Foundation for Science, Bilbao, 48009, Spain
First published on 16th July 2025
Abstract
Understanding the decomposition mechanisms of electrolyte components on functionalized graphite anodes is critical for optimizing solid electrolyte interphase (SEI) formation and enhancing Li-ion battery performance. This study employs first principles calculations and reactive force field (ReaxFF) simulations to examine the thermodynamic and kinetic feasibility of dimethyl carbonate (DMC) decomposition on four functionalized graphite surfaces (–CO, –COH, –CHO, and –COOH functional groups) during the early stages of battery operation. Our findings reveal that three distinct Hydrogen Atom Transfer (HAT) mechanisms play a key role in DMC decomposition. Among the studied functional groups, –COH exhibits the highest reactivity, followed by –COOH, enabling multiple favorable decomposition pathways. Besides the well-known SEI organic components such as CH3OLi and CH3OCOOLi, we predict the formation of less-reported species, including CH4, CH3OC(OH)OLi, CH3OCHO, CH3OCH3, LiHCO3, and Li2C(OH)O2. Notably, we identify strong competition between DMC and ethylene carbonate/fluoroethylene carbonate decomposition, particularly on –COH and –COOH surfaces, which should profoundly impact SEI formation and evolution. ReaxFF simulations further reveal that inorganic species like LiHCO3 and Li2C(OH)O2 act as precursors for the formation of Li2CO3, a key inorganic SEI component. Moreover, organic decomposition products are found to detach and diffuse away from –COH, –CHO, and –COOH functionalized surfaces, supporting a bottom-up SEI formation mechanism. Conversely, –CO strongly binds organic species via Li+ ions, potentially leading to surface poisoning over extended battery operation. These insights provide a comprehensive understanding of how functional groups influence DMC decomposition and general SEI evolution, offering valuable guidance for designing more stable and efficient anode materials for Li-ion batteries.
1. Introduction
Lithium-ion batteries (LIBs) are one of the core technological components of today's society,1,2 and much effort has been devoted to improving their cost, efficiency, and cycle life.3–5 A critical success factor lies in the solid electrolyte interphase (SEI), which forms in situ on the negative electrode during the initial cycles of battery operation due to the decomposition of electrolyte components.6,7 Once formed, the SEI blocks the electron transfer between the electrode and electrolyte while allowing Li+ ion transfer and preventing continuous electrolyte degradation, thereby ensuring the long-term electrochemical functionality of the battery.7–10Upon initial charging, battery electrolytes undergo electrochemical reduction at potentials below ∼1.0 V vs. Li/Li+ on the negative electrode (anode).9,11 The formation of the solid electrolyte interphase (SEI) has been described through both top-down and bottom-up mechanisms.12–15 In the top-down approach, electrolyte reduction occurs in the bulk solution, generating SEI components that subsequently deposit onto the anode. Conversely, the bottom-up mechanism involves direct reduction of electrolyte molecules at the electrode surface, followed by diffusion of decomposition products away from the surface. Both mechanisms may operate during different stages of SEI growth: the bottom-up process is likely dominant during the initial stages of charging when anode surface reactions primarily contribute to SEI formation, whereas the top-down mechanism may become more relevant as the SEI layer develops.12,16–22 The reduction of the electrolyte leads to the formation of inorganic species such as Li2CO3 and LiF, along with organic components like lithium ethylene dicarbonate (LEDC), lithium methyl carbonate and alkoxides.7,9,10,23 Several mechanistic pathways have been proposed for solvents like ethylene carbonate (EC), dimethyl carbonate (DMC), and additives such as fluoroethylene carbonate (FEC) and vinylene carbonate (VC), including one-electron and two-electron reduction mechanisms, often followed by radical recombination or further reduction steps.10,18,24–28 In the one-electron reduction pathway, a single Li+ ion from the electrolyte salt (e.g., lithium hexafluorophosphate (LiPF6)) participates, primarily contributing to the formation of organic SEI components. When the electron transfer is fast, a two-electron reduction process can occur yielding a CO32− (from EC or DMC) or simultaneously producing two CH3O− (from DMC) which can concurrently react with two Li+ ions to form Li2CO3 or two CH3OLi molecules.18,27,28 Much of the SEI forms during the initial charging, though electrolyte decompositions continue to take place until the SEI is fully developed. These reactions result in a nanometer-thick, passivating SEI layer.9,10
In a typical LIB with a graphite anode, the electrolyte phase contains EC and a linear carbonate (or a mixture of linear carbonates), as well as lithium salts such as LiPF6. DMC has been the most commonly used linear carbonate in this regard.10,13,19,20,29–39 DMC is reported to be the most prominent member of the Li+ solvation shell after EC.20,37–39 Furthermore, the presence of DMC in the electrolyte reportedly leads to higher ionic conductivity compared to other linear carbonates.36 These favorable properties have established DMC as the most widely used linear carbonate in the LIB electrolyte. EC and DMC are generally present in the same volume ratio (vol%: 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50), while LiPF6 is dissolved at a concentration of 1–1.2 M.10,20,29–31,37,40 The graphite surface is naturally enriched with oxygen-containing functional groups (FGs),2,4,6,8,9,41,42 but their specific abundance can be fine-tuned through various modification techniques.9,41,42 Typical SEI characterization involves ex situ conditions due to the challenges associated with probing SEI initiation in operando through standard experimental techniques.8,10,29,43 Yet, such ex situ experiments have established that a fully formed SEI comprises two layers: one adjacent to the anode and composed of inorganic products (e.g., Li2CO3, Li2O, LiF), and a porous upper layer containing organic molecules (such as CH3OLi, CH3OCOOLi).7,9,10,23 However, the mechanisms underlying the initiation and evolution of the SEI continue to be a hotly debated topic and could arguably be considered as “the most important, but least understood”6,8,12 feature of LIBs due to the complexity of the SEI formation reactions.
:50), while LiPF6 is dissolved at a concentration of 1–1.2 M.10,20,29–31,37,40 The graphite surface is naturally enriched with oxygen-containing functional groups (FGs),2,4,6,8,9,41,42 but their specific abundance can be fine-tuned through various modification techniques.9,41,42 Typical SEI characterization involves ex situ conditions due to the challenges associated with probing SEI initiation in operando through standard experimental techniques.8,10,29,43 Yet, such ex situ experiments have established that a fully formed SEI comprises two layers: one adjacent to the anode and composed of inorganic products (e.g., Li2CO3, Li2O, LiF), and a porous upper layer containing organic molecules (such as CH3OLi, CH3OCOOLi).7,9,10,23 However, the mechanisms underlying the initiation and evolution of the SEI continue to be a hotly debated topic and could arguably be considered as “the most important, but least understood”6,8,12 feature of LIBs due to the complexity of the SEI formation reactions.
The formation of SEI components is initiated by the reduction of electrolyte species at the anode surface.9,12,16–22,32,39,42 In addition to surface-driven processes, outer-sphere electron transfer reactions are also important, especially for species weakly interacting with the anode surface. Despite extensive research, the reduction mechanisms of electrolyte species at the graphite anode interface remain a subject of fundamental interest, as these processes both govern the initial formation and contribute to the long-term stability of the SEI. In this quest, considerable effort has been put into studying electrolyte decomposition, SEI growth and composition using atomistic models.8,9,13,14,16–18,20,24,32,44–46 For instance, classical molecular dynamics (CMD) simulations have offered insights into the first stages of SEI formation, suggesting that gases such as ethane (C2H6), carbon dioxide (CO2), carbon monoxide (CO), and methane (CH4) are spontaneously produced during the first few picoseconds.9,13,14 These gas molecules could generate cracks in the SEI, leading to continuous electrolyte decomposition during the battery life. The adsorption and reduction of LiPF6, EC and DMC at the graphite surface have also been explored through ab initio molecular dynamics (AIMD). While EC has been established as the dominant contributor to SEI formation, DMC is postulated to be a significant source of several common SEI products, including Li2CO3, CH3OCOOLi, CH3OLi, and gas molecules like CO and CH4.9,10,13,19,26,32,44
For DMC, two types of reduction mechanisms, namely the one-electron pathway:
 | (1) |
| CH3OCOOCH3 + 2Li+ + 2e− → Li2CO3 + C2H6 or 2CH3OLi + CO, | (2) |
This article aims to fill that gap by combining AIMD, DFT and the Climbing Image Nudged Elastic Band (CI-NEB) method to study the thermodynamics and kinetics of DMC decomposition in the presence of –CO, –COH, –CHO and –COOH functionalized graphite in vacuum. We focus on isolating the early-stage decomposition chemistry of DMC during SEI formation. To further examine the stability of the predicted products in the complexity of the electrolyte medium, implicit solvent DFT calculations and CMD simulations with a reactive force field were performed, offering the first complete picture of how the decomposition of DMC in the presence of common graphite surface functionalizations can fundamentally define the nature and evolution of the SEI.
This paper is organized as follows: first, we provide a detailed discussion of the computational methodology employed in this study. Next, we present the thermodynamic analysis of DMC decomposition reactions on various FGs and examine the thermodynamics of DMC decomposition product formation, followed by a kinetic barrier analysis of the thermodynamically favorable reactions. We then compare the thermodynamic and kinetic spontaneity of the decomposition processes to identify the most reactive FGs and their corresponding DMC decomposition pathways. Finally, we comprehensively analyze the long-term stability of the DMC decomposition products in an explicit solvent environment.
2. Methods
2.1 Model system
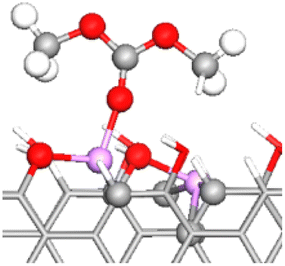
The graphite anode was described by four layers of ABAB-stacked graphite with four facets oriented along the z-axis. The (11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0) plane, representing the graphite armchair edge, was selected for surface functionalization in all calculations (Fig. 1). This specific edge plane was chosen due to its high stability and spontaneous saturation of edge dangling bonds by oxygen-containing FGs.24,47 A significant fraction of the SEI forms before the initiation of Li+ intercalation within graphite during the initial charging cycle.6,7,9,12,48 Consequently, we considered a model graphite anode without intercalated Li+ ions. Four different types of oxygen-containing FGs (–CO, –COH, –CHO and –COOH) were used to functionalize the graphite edge (right panel in Fig. 1). H atoms are frequently found around these FGs, partially passivating graphite edges.41 Thus, the FGs were only added to every alternate edge C atom (48 in total). Throughout this study, we use OC and OE to denote the carbonyl oxygen and the two ether oxygens in a DMC molecule, respectively, and CC to refer to the carbonyl carbon (see the inset in Fig. 1b).
0) plane, representing the graphite armchair edge, was selected for surface functionalization in all calculations (Fig. 1). This specific edge plane was chosen due to its high stability and spontaneous saturation of edge dangling bonds by oxygen-containing FGs.24,47 A significant fraction of the SEI forms before the initiation of Li+ intercalation within graphite during the initial charging cycle.6,7,9,12,48 Consequently, we considered a model graphite anode without intercalated Li+ ions. Four different types of oxygen-containing FGs (–CO, –COH, –CHO and –COOH) were used to functionalize the graphite edge (right panel in Fig. 1). H atoms are frequently found around these FGs, partially passivating graphite edges.41 Thus, the FGs were only added to every alternate edge C atom (48 in total). Throughout this study, we use OC and OE to denote the carbonyl oxygen and the two ether oxygens in a DMC molecule, respectively, and CC to refer to the carbonyl carbon (see the inset in Fig. 1b).
 | ||
| Fig. 1 Structure of the graphite anode models with –COH graphite functionalization from two perspectives: (a) xz plane, and (b) yz plane. The inset in (b) shows a DMC molecule with three types of atoms labeled: the carbonyl oxygen (OC), the ether oxygens (OE), and the carbonyl carbon (CC). Analogous systems to those in (a) and (b) were constructed for each analyzed functionalization (listed in the panel on the right). Red: oxygen, white: hydrogen, gray: carbon. | ||
The lattice parameters along the x and y directions of the simulation cell are 13.34 Å and 12.81 Å, respectively. To prevent spurious interactions between periodic images, a 15 Å vacuum slab was introduced along the z direction. This specific thickness was established through careful convergence testing of the ground-state energy for the model systems (Fig. S1†).
We introduced one and two Li+ ions in each simulation cell to investigate the decomposition of DMC via the one and two electron pathways (eqn (1) and (2)), respectively. In lithium-ion batteries, DMC constitutes a substantial fraction of the electrolyte components that are densely packed in the interfacial layer adjacent to the graphite anode surface.20,39 During the charging process, solvated Li+ ions migrate toward the anode and become partially desolvated near the electrode–electrolyte interface. DMC remains a key constituent in the partially formed solvation shell surrounding the Li+ ions in this region.20,39 Consequently, DMC molecules are expected to interact with the graphite anode via adsorption, often mediated by one or more Li+ ions, and undergo subsequent electrochemical reduction to form SEI components and other decomposition by-products. Importantly, we did not consider the presence of EC and LiPF6 during AIMD and DFT optimization calculations, as the primary objective of this work is isolating and characterizing the role of the graphite surface FGs in DMC decomposition and SEI product formation at the initial stage of battery charging. The inclusion of additional solvent and salt species would substantially increase the computational cost. However, potential additional insights into the decomposition mechanisms of DMC at the anode surface are uncertain, and remain beyond the scope of this work. Instead, after identifying the most favorable DMC decomposition products on each functionalized graphite surface, we investigated their stability in explicit solvent environments using reactive ReaxFF-CMD simulations.
2.2 Computational procedure
To precisely determine the role of each FG on the DMC decomposition reaction routes, the most likely products and the associated kinetic barrier, we followed the steps highlighted in Fig. 2 and described in detail below: | ||
| Fig. 2 Computational procedure followed in this work. In the schematic snapshots, red, white, gray, and purple spheres represent oxygen (O), hydrogen (H), carbon (C), and lithium (Li) atoms, respectively. | ||
Step 1 – AIMD simulations of adsorbed DMC: it is highly unlikely that a graphite – adsorbed DMC molecule spontaneously dissociates within a computationally affordable time span. Therefore, we initiated our AIMD simulations from the optimized adsorbed configurations of DMC and Li+ on graphite in which the C–O bonds were pre-dissociated, following reaction (1) or (2). For each FG and reaction pathway, 10 to 15 different initial configurations were simulated for 5 ps at 450 K. AIMD simulations were conducted at an elevated temperature of 450 K solely to accelerate the bond cleavage and desorption processes, thereby facilitating the observation of relevant DMC decomposition products for subsequent analysis. The DFT calculations at 0 K will then help us discriminate which of the generated pathways are more likely to be thermodynamically and kinetically viable at room temperature (assuming that the entropic contribution to the reaction free energy, −TΔS, is negative). The most favorable reaction mechanisms were identified from the configurations corresponding to the lowest points in the total energy distribution across all the AIMD simulations after energy stabilization (step 1 in Fig. 2).
Step 2 – DFT optimization: the product configurations obtained from the favorable reaction routes were geometrically optimized. From the energy minimization calculations, products formed via an energetically favorable exothermic reaction were identified for subsequent analysis (step 2 in Fig. 2).
Step 3 – calculation of reaction kinetic barriers: the spontaneity of a reaction was further evaluated by calculating the activation energy barrier of the reaction using the CI-NEB49,50 method. This approach identified the kinetically favorable DMC reduction reactions among the thermodynamically favorable ones for each edge-functionalized graphite anode system (step 3 in Fig. 2).
Step 4 – ReaxFF simulations in solvent environments: to assess the long-term stability of the thermodynamically favorable products identified in step 2, CMD simulations were carried out for 50 ps in an explicit solvent environment using the ReaxFF51,52 (step 4 in Fig. 2). Furthermore, the formation of new compounds resulting from bond cleavage and/or formation through interactions between the thermodynamically stable DMC decomposition products and explicit electrolyte molecules was thoroughly analyzed.
2.3 Theoretical methods
All simulations were carried out using the LAMMPS package.74 Initial system configurations were generated with Packmol75 based on DFT-optimized structures replicated into a 2 × 2 × 1 supercell, with the vacuum spaces filled with DMC molecules to achieve an equilibrium density of 1.07 g cm−3. When a mixed explicit solvent is considered, the vacuum spaces were filled with EC and DMC in the same volume ratio along with 1 M LiPF6. To prevent system drift, carbon atoms below the graphite subsurface were fixed. Simulations were run with a 0.20 fs timestep in the NVT ensemble at 298 K, with 2 ps of equilibration followed by 50 ps production runs. The same mixed solvent composition and simulation parameters were employed in additional ReaxFF-CMD simulations to examine the spatial distribution and prevalence of DMC molecules near various functionalized graphite anode surfaces prior to adsorption. In these simulations, the width of the electrolyte region varied between 28 Å and 30 Å, depending on the specific surface functional group. The considered temperature of 298 K for the ReaxFF-CMD simulations is most relevant for practical applications. While varying the ReaxFF-CMD simulation temperature could provide further insights into the temperature-dependent stability of DMC decomposition products, such analysis lies beyond the scope of this work. The graph theory-based reaction network integrator ReactNetGenerator software was used for post-processing trajectory analysis.76
3. Results and discussion
3.1 Binding of Li+ and DMC on functionalized graphite
The adsorption energy, Eads, of an atom or a molecule on the graphite anode surface is computed as:33,63| Eads = EAnode+M − EAnode − EM, | (3) |
First, we considered the adsorption of one and two isolated Li+ ions on functionalized graphite (Fig. 2 (step 1-left)), a necessary step for initiating DMC adsorption and subsequent decomposition on the graphite anode surface, via the one- and two-electron mechanisms in eqn (1) and (2). The Eads values listed in Table 1 indicate that both single and double Li+ adsorptions are exothermic processes (Eads < 0) across all four FGs. Furthermore, Li+ ions exhibited two- or three-way coordination with oxygen atoms in the surface FGs (Fig. S4†). The strongest Li+ adsorption was observed on –CO, while the weakest occurred on –COH. In the latter case, structural relaxation resulted in near Li+ intercalation into the graphite bulk (see Fig. S4b and f†), in agreement with previous simulations predicting a low intercalation barrier through –COH FGs.29
| Functional group | Number of adsorbed Li+ ions | Adsorption energy, Eads (eV) |
|---|---|---|
| –CO | 1 | −4.25 |
| 2 | −7.19 | |
| –COH | 1 | −0.64 |
| 2 | −1.34 | |
| –CHO | 1 | −2.49 |
| 2 | −4.34 | |
| –COOH | 1 | −1.99 |
| 2 | −3.41 |
The primary objective of this study is to investigate the reductive decomposition mechanisms of DMC on the graphite anode surface, with particular focus on the influence of surface functional groups and their role in initiating SEI formation during the early stages of battery operation. For reductive decomposition of weakly surface bound electrolyte molecules, the outer sphere electron transfer processes might be playing a crucial role. However, a detailed investigation of such mechanisms lies outside the scope of the present work and has been investigated elsewhere.23,25
DMC decomposition on the anode surface is expected to compete with other key electrolyte reduction processes, such as EC decomposition. The likelihood of DMC participating in surface reactions depends on its proximity to the anode surface and the availability of reactive sites. In previous studies, for a mixed EC, DMC, and LiPF6 electrolyte, DMC was reported to occupy a major portion of the electrolyte layer closest to the anode surface in both unbiased and biased simulations.20,39 As Li+ ions migrate toward the graphite anode and their solvation shells become progressively disrupted, DMC remains a dominant component in the partially desolvated coordination environment of Li+ near the electrode surface.20,39
In the present work, ReaxFF-CMD simulations were also performed with a mixed electrolyte (EC:DMC = 1:1, 1 M LiPF6) to estimate the spatial distribution and prevalence of DMC molecules near various functionalized graphite anode surfaces prior to adsorption. The time-averaged number of DMC molecules, normalized by the anode surface area, found within 5 Å of the anode surface as determined from ReaxFF-CMD simulations of the electrolyte mixture is shown in Fig. S5.† Consistent with previous studies, DMC molecules were found to persist in significant quantities near the electrode–electrolyte interface. Accordingly, DMC is expected to contribute significantly to SEI formation through adsorption onto the graphite surface followed by reductive decomposition.
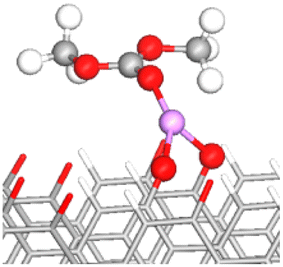
To this end, the adsorption of an isolated DMC molecule was investigated with and without the presence of Li+ on the anode surface. In the absence of Li+, DMC adsorption on the functionalized surfaces is weak and primarily governed by van der Waals interactions (Table S1 and Fig. S6†). In contrast, the presence of one or two Li+ ions on the graphite surface strengthens the binding of DMC, as indicated by the negative Eads values across all four types of functionalized graphite surfaces (see Table 2 for the –CO and –COH FGs, and Table S2† for the –CHO and –COOH FGs).
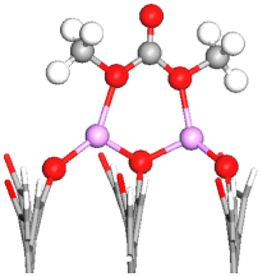
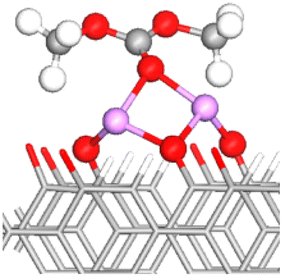
| FG | DMC adsorbed configuration | E ads (eV) | OE–CC bond lengths (Å) (% stretching) | OC–CC bond length (Å) (% stretching) | Snapshot of the most stable configuration |
|---|---|---|---|---|---|
| –CO | OE–1Li–O | −1.63 | 1.382 (3.13%) | 1.206 (−0.74%) |
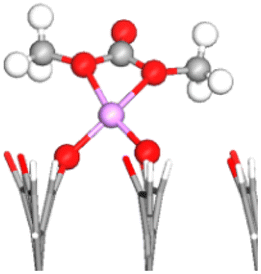
|
| 1.382 (3.13%) | |||||
| OC–1Li–O | −2.38 | 1.319 (−1.57%) | 1.234 (1.57%) |
|
|
| 1.323 (−1.27%) | |||||
| OE–2Li–O | −1.20 | 1.387 (3.51%) | 1.206 (−0.74%) |
|
|
| 1.380 (2.98%) | |||||
| OC–2Li–O | −1.66 | 1.308 (−2.39%) | 1.258 (3.54%) |
|
|
| 1.312 (−2.09%) | |||||
| OC/OE–2Li–O | −2.94 | 1.378 (2.84%) | 1.232 (1.40%) |
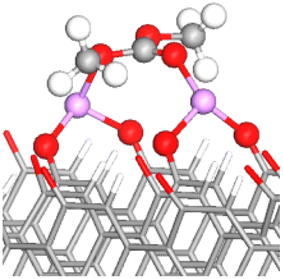
|
|
| 1.311 (−2.16%) | |||||
| –COH | OE–1Li–OH | −1.90 | 1.394 (4.03%) | 1.209 (−0.50%) |
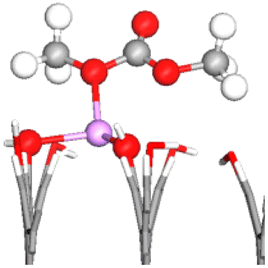
|
| 1.346 (0.45%) | |||||
| OC–1Li–OH | −1.61 | 1.327 (−0.97%) | 1.231 (1.32%) |
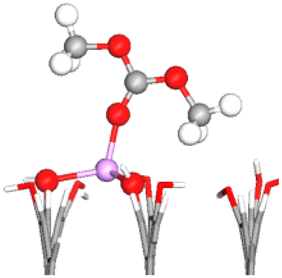
|
|
| 1.320 (−1.49%) | |||||
| OE–2Li–OH | −0.80 | 1.381 (3.05%) | 1.215 (0%) |
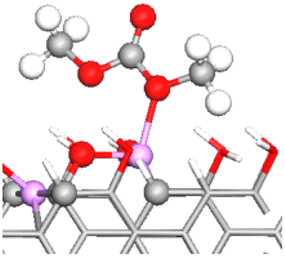
|
|
| 1.353 (0.97%) | |||||
| OC–2Li–OH | −0.89 | 1.332 (−0.59%) | 1.228 (1.07%) |
|
|
| 1.320 (−1.49%) |
To simplify the discussion below, the notation Os–nLi–FG is used to describe Li-mediated binding of DMC. Here, s = C or E indicates the type of interacting oxygen (see Fig. 1b), n is the number of Li+ ions present on the anode surface (1 or 2), and FG is the specific functional group. Moreover, OC/OE is used instead of Os when two types of O atoms in the DMC molecule are simultaneously participating in the binding.
Tables 2 and S2† reveal that exothermicity increases with the fraction of O in the FG. For instance, the average |Eads| is higher for DMC binding on –CO (50% oxygen) and –COOH (50% oxygen) than on –CHO and –COH (both 33% oxygen). DMC adsorption is especially promoted by the presence of –COOH, with −5.69 eV ≤ Eads ≤−4.29 eV (Table S2†). In contrast, the binding strength of DMC on –COH is relatively weak, ranging between −1.9 eV (OE–1Li–OH) and −0.8 eV (OE–2Li–OH). The large differences in binding strength may significantly influence the mechanism behind DMC decomposition: in a –COH-rich surface, top-down SEI formation (i.e., decomposition occurs in the bulk electrolyte and products subsequently deposit onto the anode surface) may play a greater role than in a –COOH-rich surface, in which the bottom-up mechanism (where surface binding precedes electrolyte decomposition) may be dominant.
3.2 Thermodynamics of DMC decomposition on functionalized graphite
We first followed the procedure described in Section 2.2 (Fig. 2, step 1 (mid and right panel)). For each type of functionalized graphite, AIMD simulations began with pre-dissociated DMC molecules, where the ester linkages had already been cleaved according to the reactions in eqn (1) and (2) (examples of these configurations are presented in Fig. S7 and S8†). During the high-temperature (450 K) AIMD simulations, the dissociated DMC fragments interacted with each other and with the graphite surface, yielding several low-energy products that included common organic and inorganic SEI components, as well as several uncommon species as detailed in Fig. S9.† Importantly, the presence of two Li+ ions can facilitate both one- and two-electron mechanisms in eqn (1) and (2), respectively, whereas a single Li+ ion can only lead to the one-electron mechanism.Each resulting decomposition product was geometrically optimized using DFT (Fig. 2, step 2). The thermodynamic favorability of each decomposition process in a vacuum was assessed by comparing the ground-state energy of the DMC-adsorbed configuration to that of its corresponding decomposition products. The energy difference, ΔE, indicates whether a reaction is exothermic (ΔE < 0), and thus expected to occur spontaneously, or endothermic (ΔE > 0), with the likelihood of occurrence approximated ∼e−ΔE/kT, where k is the Boltzmann constant and T is the temperature.77,78
The energy difference between reactants and products was also estimated considering an implicit solvent medium. We considered two different implicit solvent environments, characterized by the dielectric constant, ε, of a solvent medium with an equimolar mixture of EC and DMC (ε-mixed = 20.5)79 and pure DMC – only (ε-DMC = 3.1).31 The predicted values for ΔE, ΔEε-mixed, and ΔEε-DMC are represented in Fig. 3, while Fig. 4 depicts the most likely thermodynamically favorable products (i.e., those for which all ΔE, ΔEε-mixed and ΔEε-DMC are negative). The exo/endothermic nature of the formation of each of the DMC decomposed products, evaluated in a vacuum as well as in the implicit solvent models, across all four FGs is listed in Table S3.†
 | ||
| Fig. 3 Representation of ΔE (vacuum), ΔEε-mixed (effective dielectric medium of 1:1 EC/DMC), and ΔEε-DMC (effective dielectric medium with pure DMC) for DMC decomposition reactions on (a and b) –CO, (c and d) –COH, (e and f) –CHO and (g and h) –COOH FGs with one and two Li+ ions. The wide bars containing the product names represent ΔE in vacuum. The narrow pink and yellow bars represent the ΔEε-DMC and ΔEε-mixed values, respectively. In the product formulae, an adsorbed configuration of molecule X is denoted as X*. | ||
 | ||
| Fig. 4 Structure of the thermodynamically stable DMC decomposition products (ΔE < 0, ΔEε-mixed < 0, and ΔEε-DMC < 0). The type of graphite surface functionalization on which these products were found is mentioned in parentheses. “All” indicates that the product was formed on all four types of functionalized graphite. Descriptions of previously unreported or rarely reported DMC decomposition products are highlighted in green. Adsorbed molecules are indicated with an asterisk (*) (red: O, white: H, gray: C, purple: Li). | ||
In all cases involving one Li+ ion (Os–1Li–FG), exothermic reactions observed in vacuum remained exothermic in implicit solvent environments (Fig. 3a, c and g). The degree of stabilization was greater for the mixed EC + DMC solvent compared to stand-alone DMC. For reactions involving two Li+ ions, the exothermic nature of the reactions was largely maintained, with two exceptions: the formation of 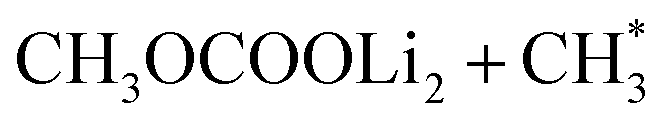 (Fig. 3b and f), where the reaction became slightly endothermic by less than 0.07 eV.
(Fig. 3b and f), where the reaction became slightly endothermic by less than 0.07 eV. 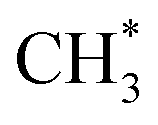 represents an adsorbed
represents an adsorbed 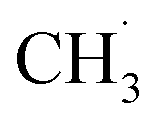 . Following standard notations, an adsorbed configuration of molecule X will be denoted as X*. In addition, a few reactions that were endothermic in vacuum became exothermic in the implicit solvent (in both the mixed and DMC media):
. Following standard notations, an adsorbed configuration of molecule X will be denoted as X*. In addition, a few reactions that were endothermic in vacuum became exothermic in the implicit solvent (in both the mixed and DMC media): 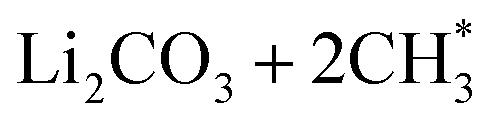 (Fig. 3b) and CH3OCOOLi2 + CH3OH (Fig. 3h). These reactions arise from the –CO and –COOH FGs, respectively, with differences between ΔE and ΔEε-mixed/ΔEε-DMC of approximately 0.6 eV. While this difference is significant, it is worth noting that ΔE in vacuum was less than 0.1 eV, indicating only mild endothermicity. Among all the reactions inspected, the change of the reaction type (from endothermic to exothermic) by a considerable amount of ΔE (>0.15 eV) was consistently observed with both types of implicit solvents in only those two cases. This indicates that vacuum calculations can, in general, predict the correct thermodynamic directionality for a large portion of the reaction landscape. Therefore, vacuum estimates can be considered a reasonable predictor of the thermodynamic driving force for DMC decomposition reactions in a solvent environment. Nonetheless, for slightly endothermic reactions in the vacuum, the implicit solvent can lead to significant differences in ΔE.
(Fig. 3b) and CH3OCOOLi2 + CH3OH (Fig. 3h). These reactions arise from the –CO and –COOH FGs, respectively, with differences between ΔE and ΔEε-mixed/ΔEε-DMC of approximately 0.6 eV. While this difference is significant, it is worth noting that ΔE in vacuum was less than 0.1 eV, indicating only mild endothermicity. Among all the reactions inspected, the change of the reaction type (from endothermic to exothermic) by a considerable amount of ΔE (>0.15 eV) was consistently observed with both types of implicit solvents in only those two cases. This indicates that vacuum calculations can, in general, predict the correct thermodynamic directionality for a large portion of the reaction landscape. Therefore, vacuum estimates can be considered a reasonable predictor of the thermodynamic driving force for DMC decomposition reactions in a solvent environment. Nonetheless, for slightly endothermic reactions in the vacuum, the implicit solvent can lead to significant differences in ΔE.
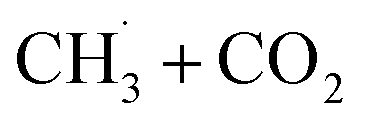 or converted into CH3OCHO.
or converted into CH3OCHO.
CH3OCOOLi and CH3OLi are among the most prominent organic components of the SEI, typically comprising the outer organic layer. CH3OCOOLi can further react with an additional Li+ ion from the electrolyte to form Li2CO3 and more 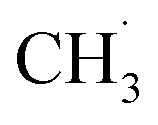 radicals.10,25 In electrolytes containing a mixture of linear carbonates, CH3OLi has been reported to facilitate transesterification reactions, such as the conversion of ethyl methyl carbonate to DMC.80 In the present study, the formation of CH3OCOOLi is accompanied by the simultaneous formation of a
radicals.10,25 In electrolytes containing a mixture of linear carbonates, CH3OLi has been reported to facilitate transesterification reactions, such as the conversion of ethyl methyl carbonate to DMC.80 In the present study, the formation of CH3OCOOLi is accompanied by the simultaneous formation of a 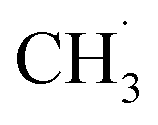 . The radical is either adsorbed on the graphite anode surface or converted into CH4 through hydrogen atom transfer. In our simulations, subsequent
. The radical is either adsorbed on the graphite anode surface or converted into CH4 through hydrogen atom transfer. In our simulations, subsequent 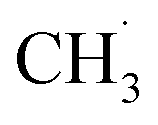 radical interactions are limited to the graphite surface or dissociation fragments of DMC. As reported by Weddle et al., the
radical interactions are limited to the graphite surface or dissociation fragments of DMC. As reported by Weddle et al., the  radicals may also undergo reactions between themselves or other electrolyte elements (such as EC, FEC) to produce gas molecules such as CH4.25
radicals may also undergo reactions between themselves or other electrolyte elements (such as EC, FEC) to produce gas molecules such as CH4.25
In addition to the well-established organic SEI components, several unique DMC decomposition products were predicted to be favorable (Fig. 4, green-labeled molecules). These include: (i) methanol (as a free molecule detached from the surface, i.e., CH3OH), on the –COH FG (Fig. 3d) or attached to the surface via a Li+ ion (i.e., CH3OHLi) on the –COH (Fig. 3c and d) and –COOH (Fig. 3h) FGs; (ii) methyl formate (CH3OCHO) on the –COH and –COOH FGs (Fig. 3c, d and h); and (iii) lithium methoxy acetate (CH3OCOOHLi) on the –COOH FG (Fig. 3g and h). In particular, methanol is one of the major products in the pyrolysis and photolysis of DMC,30,81,82 but formate and acetate products were only observed in trace amounts during the course of photolysis and high temperature thermal decomposition of DMC, respectively.30,81
The formation of C2H6, which is reported to be unique to the two-electron reduction of DMC,10,26–28 was observed only on –COH and –COOH functionalized surfaces (Fig. 3d and h). Interestingly, the evolution of C2H6 on the –COH FG was observed in systems with two Li+ but via an effectively one-electron pathway involving only one Li+ ion in the reaction. Another gaseous product, CO, unique to the two-electron DMC reduction, was never found among the thermodynamically favorable reactions. This supports the observation by Leißing et al.83 that the main source of CO during SEI formation is EC, instead of DMC.
The prediction of formaldehyde (HCHO) and dimethyl ether (CH3OCH3) formation was unexpected, as there are few reports of their detection.27 In particular, CH3OCH3 is one of the main products of the high temperature pyrolysis and photolysis of DMC.30,81 A reason for their absence in SEI characterization experiments may be that, although both compounds are gases at room temperature, they are moderately soluble in carbonate solutions.85,86 Notably, HCHO remained bound to the anode via one Li+ ion on the –CO decorated surface (shown as HCHOLi in Fig. 3a, b and 4. See the schematic in Fig. S10a†). In contrast, CH3OCH3 was bound to one and two Li+ ions when produced in the presence of –CO and –CHO, respectively (shown as CH3O(Li)CH3 and CH3O(Li2)CH3 in Fig. 3a and f, respectively and Fig. S10b and c†), but released from the surface when produced in the presence of –COOH (Fig. S10d†). These results imply that not only do the products promoted by each FG may differ, but the abundance of common products in the electrolyte and the interface may also vary depending on the relative population of these FGs. Therefore, the detection of unusual products like HCHO and CH3OCH3 may be strongly tied to the specific oxygen containing groups on the graphite anode.
 are the final products). In configurations involving OE–Li+ interactions, these CC–OE bonds are likely to relax upon the formation of low-energy products, creating a greater ΔE between reactants and products in the OE–nLi–FG case. In other words, the stretched CC–OE bonds may be considered to be in an “activated” state for OE–nLi–FG adsorbed configurations and are likely to be easily broken during the decomposition process resulting in various different end products.
are the final products). In configurations involving OE–Li+ interactions, these CC–OE bonds are likely to relax upon the formation of low-energy products, creating a greater ΔE between reactants and products in the OE–nLi–FG case. In other words, the stretched CC–OE bonds may be considered to be in an “activated” state for OE–nLi–FG adsorbed configurations and are likely to be easily broken during the decomposition process resulting in various different end products.
Interestingly, all the DMC decomposition processes departing from an Os–1Li–CHO adsorbed configuration (s = C or E) are endothermic (Fig. 3e). Moreover, only two such reactions are exothermic for Os–2Li–CHO (Fig. 3f). Thus, –CHO is the least effective FG at promoting DMC decomposition on the graphite anode surface. Similar endothermic reaction energies were also observed for EC and FEC decomposition on the –CHO FG.24 In contrast, the –COH FG was found to promote the largest number of exothermic decomposition reactions (Fig. 3c and d). In particular, DMC decomposition reactions departing from Os–2Li–COH configurations were found to be highly spontaneous (ΔE < −2.0 eV, Fig. 3d).
3.3 Kinetic barrier analysis of the DMC decomposition reactions
Thermodynamic drive is not the sole indicator of product abundance. Large activation energy barriers (EA) may lead to some exothermic reactions to never occur within experimental timespans. This section aims to elucidate the kinetic barriers for the thermodynamically favorable products (ΔE < 0) using CI-NEB, as described in step 3 of Section 2. Further refinement using additional CI-NEB calculations was performed near the reaction steps involving bond cleavage or atom transfer. In the majority of cases, supplementary CI-NEB calculations did not identify any transition states with higher energy, thereby confirming the accuracy of the transition states and activation barriers obtained from the initial CI-NEB calculations. The resulting predictions of EA are provided in Fig. 5 and S11† for reactions with two and one Li+ ions on the FGs, respectively. The thermodynamic analysis revealed that two slightly endothermic reactions in the vacuum become highly exothermic in the presence of implicit solvents (formation of on –CO (Fig. 3b) and CH3OCOOLi2 + CH3OH on –COOH (Fig. 3h)). The activation barriers for these two DMC decomposition reactions were also computed and are presented in Fig. S12.†
on –CO (Fig. 3b) and CH3OCOOLi2 + CH3OH on –COOH (Fig. 3h)). The activation barriers for these two DMC decomposition reactions were also computed and are presented in Fig. S12.†
 | ||
| Fig. 5 Activation energy barriers (EA) for exothermic reactions following DMC adsorbed configurations (a) OE–2Li–CO, (b) OC–2Li–CO, (c) OE–2Li–COH, (d) OC–2Li–COH, (e) OE–2Li–CHO, (f) OC–2Li–CHO, (g) OE–2Li–COOH, and (h) OC–2Li–COOH. | ||
We observed that reactions with a lower ΔE (more exothermic) tended to have a lower EA. In particular, the lowest EA values (≤1.01 eV) were identified for the highly stable products formed on the –COH FG with two Li+ ions (Fig. 5c and d), except for the formation of LiHCO3 + C2H6, which exhibited EA of 2.01 eV (Fig. 5c).
 free radicals. The presence of CH3OCO˙ free radicals or adsorbed CH3OCO* at the TS led to significantly high EA (e.g., Fig. 6f and S13e†), rendering these reactions kinetically restricted on the graphite anode surface. The formation of the CH3OCO˙ radical via DMC decomposition in the bulk electrolyte is expected to occur more frequently, owing to the reported lower reaction barrier compared to that on the graphite surface.25
free radicals. The presence of CH3OCO˙ free radicals or adsorbed CH3OCO* at the TS led to significantly high EA (e.g., Fig. 6f and S13e†), rendering these reactions kinetically restricted on the graphite anode surface. The formation of the CH3OCO˙ radical via DMC decomposition in the bulk electrolyte is expected to occur more frequently, owing to the reported lower reaction barrier compared to that on the graphite surface.25
 | ||
Fig. 6 TS configurations and Bader charge distribution for reactions (a) OE–2Li–COOH → CH3OCOOHLi + CH4 (EA = 2.00 eV), (b)  , (c) , (c)  , (d) OC–2Li–COH → Li2C(OH)O2 + 2CH4 (EA = 0.80 eV), (e) OE–2Li–CHO → CH3O(Li2)CH3 + CO2 (EA = 3.05 eV), and (f) OE–2Li–COOH → CH3OCHO + CH3OHLi (EA = 2.34 eV). The Bader charge values are in the unit of an electronic charge. (red: O, white: H, gray: C, purple: Li). , (d) OC–2Li–COH → Li2C(OH)O2 + 2CH4 (EA = 0.80 eV), (e) OE–2Li–CHO → CH3O(Li2)CH3 + CO2 (EA = 3.05 eV), and (f) OE–2Li–COOH → CH3OCHO + CH3OHLi (EA = 2.34 eV). The Bader charge values are in the unit of an electronic charge. (red: O, white: H, gray: C, purple: Li). | ||
The Bader charge analysis showed that the OC and the carbonyl oxygen atoms that emerged from the breakage of OE–CH3 bonds (denoted as  ) held more negative charge than OE atoms, irrespective of whether the carbonyl oxygens were free or made part of a chemical or hydrogen bond (e.g., Fig. 6c). For instance, the charge differences within the OC–Li+ bond in Fig. 6a and S13a† are 2.11e and 2.07e, whereas the differences within the OE–Li+ bond in Fig. 6a and S13a† are 1.90e and 1.93e. Thus, the OC–Li+ or
) held more negative charge than OE atoms, irrespective of whether the carbonyl oxygens were free or made part of a chemical or hydrogen bond (e.g., Fig. 6c). For instance, the charge differences within the OC–Li+ bond in Fig. 6a and S13a† are 2.11e and 2.07e, whereas the differences within the OE–Li+ bond in Fig. 6a and S13a† are 1.90e and 1.93e. Thus, the OC–Li+ or  interactions are more polar in nature than the OE–Li+ interactions, stabilizing the TS and reducing the overall EA for the reaction.
interactions are more polar in nature than the OE–Li+ interactions, stabilizing the TS and reducing the overall EA for the reaction.
3.4 DMC decomposition reaction mechanisms
 | ||
Fig. 7 Examples of type A, B and C Hydrogen Atom Transfer (HAT) processes. (a) Type A:  , (b) type B: , (b) type B:  and (c) type C: and (c) type C:  . The green and black encircled areas show the specific hydrogen donor and acceptor fragments involved in the HAT process, while R–/R′– denote the “rest of the molecule” for the participating molecules/free radicals (red: O, white: H, gray: C, purple: Li). . The green and black encircled areas show the specific hydrogen donor and acceptor fragments involved in the HAT process, while R–/R′– denote the “rest of the molecule” for the participating molecules/free radicals (red: O, white: H, gray: C, purple: Li). | ||
(A) HAT from the methyl group of DMC to the surface FGs: this process was only observed on –CO and –CHO FG surfaces (see Fig. 7a for an example). This HAT mechanism is a key pathway leading to the formation of HCHO.
(B) HAT within different parts of the decomposed DMC molecule: this process was only observed on –CO, –COH, and –CHO FGs, resulting in the simultaneous production of CH4 and HCHO (see Fig. 7b for an example). On –CO graphite with two Li+ ions, this process may also lead to the formation of Li2C(OH)O2 and  .
.
(C) HAT to the DMC molecule from the surface FGs: this process was only observed on –COH and –COOH FGs, producing CH3OH, CH3OCHO, CH3OCOOHLi, LiHCO3, Li2C(OH)O2, and CH4. An example of such a HAT process producing CH4 is presented in Fig. 7c.
The formation of CH4 predominantly occurs via the type-C HAT process, with occasional contributions from the type-B HAT pathway. All observed HAT processes proceed without significant energy barriers. In contrast, CH4 formation via HAT between  radicals and EC/FEC molecules within the bulk electrolyte involves comparatively higher reaction barriers, as reported by Weddle et al.25 Therefore, CH4 generation during SEI formation and evolution is expected to occur primarily through surface-mediated HAT mechanisms.
radicals and EC/FEC molecules within the bulk electrolyte involves comparatively higher reaction barriers, as reported by Weddle et al.25 Therefore, CH4 generation during SEI formation and evolution is expected to occur primarily through surface-mediated HAT mechanisms.
Similar H-loss reactions were reported for DMC in high voltage decomposition reactions on a LiCoO2 cathode.33 Only for the formation of CH3OH on the –COOH FG (Fig. 3h) did we observed the transfer of OH− from the surface FG to the free  radical (
radical ( , see Fig. S14† for further details). This reaction is slightly endothermic in a vacuum (ΔE ∼ 0.1 eV), but exothermic in implicit solvent environments.
, see Fig. S14† for further details). This reaction is slightly endothermic in a vacuum (ΔE ∼ 0.1 eV), but exothermic in implicit solvent environments.
 radical, all subsequent processes, including the bending of adsorbed species, additional bond cleavages, and HAT, were found to proceed without a significant energy barrier. The reaction routes involving two Li+ ions in the formation of the final products (i.e., following a two-electron pathway) are highlighted in blue color.
radical, all subsequent processes, including the bending of adsorbed species, additional bond cleavages, and HAT, were found to proceed without a significant energy barrier. The reaction routes involving two Li+ ions in the formation of the final products (i.e., following a two-electron pathway) are highlighted in blue color.
 | ||
| Fig. 8 Overview of the DMC reduction reaction mechanisms on the investigated FGs following OE/OC–nLi–FG DMC adsorbed configurations. Reactions producing products involving two Li+ ions (two-electron pathway) are represented in blue color. Panel (a) represents reactions R1–R3 from OE–1Li–CO, R4–R6 from OE–2Li–CO, R7 from OC–2Li–CO, R8 and R9 from OE–1Li–COH, and R10 from OC–1Li–COH. Panel (b) represents reactions R11–R14 from OE–2Li–COH, R15 and R16 from OC–2Li–COH, R17 from OE–2Li–CHO, R18 from OC–2Li–CHO, and R19–R21 from OE–1Li–COOH. Panel (c) represents reactions R22 from OC–1Li–COOH, R23–R26 from OE–2Li–COOH, and R27 and R28 from OC–2Li–COOH. | ||
Reaction mechanisms involving type (A) and type (B) HAT processes on the –CO FG are outlined in reactions R1 and R4/R5, respectively. DMC decomposition routes on the –COH FG (R8 to R16) and on the –COOH FG (R19 to R28, except for R21) involve the type (C) HAT process. Neither of the thermodynamically favorable reactions on –CHO FG (reactions R17 and R18) involves the HAT process.
 | ||
Fig. 9 Activation energy barrier (EA) vs. reaction energy (ΔE) for the reactions illustrated in Fig. 8. DMC decomposition reactions via the one and two electron pathways are represented with “●” and “★” symbols, respectively. The same applies for the slightly endothermic reactions (Rendo) on –CO and –COOH FG which are represented by the “▲” symbol. The EA and ΔE values from ref. 24 for the most favorable EC oligomerization decomposition pathway via the S1 mechanism (⊕), and the FEC oligomerization decomposition pathway via the S1 (⊞) and S2 ( ) mechanisms on functionalized graphite are also shown. Data points for reactions on –CO, –COH, –CHO and –COOH FGs are displayed with red, blue, orange and green colored symbols respectively. To accommodate the EC decomposition reactions exhibiting very high EA and ΔE values, axis breaks were introduced along the vertical axis in the ranges of 3.20 to 6.70 eV and 6.85 to 7.4 eV, and along the horizontal axis in the ranges of 0.35 to 1.20 eV, 1.55 to 4.30 eV and 4.55 to 7.1 eV. ) mechanisms on functionalized graphite are also shown. Data points for reactions on –CO, –COH, –CHO and –COOH FGs are displayed with red, blue, orange and green colored symbols respectively. To accommodate the EC decomposition reactions exhibiting very high EA and ΔE values, axis breaks were introduced along the vertical axis in the ranges of 3.20 to 6.70 eV and 6.85 to 7.4 eV, and along the horizontal axis in the ranges of 0.35 to 1.20 eV, 1.55 to 4.30 eV and 4.55 to 7.1 eV. | ||
The TS is expected to be more stable in a solvent environment, similar to the reduction in ΔE observed in the presence of implicit solvents (Fig. 3). Moreover, external electric fields and variations in FG coverage may further reduce activation energy barriers.89,90 Even with these attenuating factors at play, reactions with estimated EA values residing in the darker regions of Fig. 9 are likely to require prohibitively high energy to proceed. For all the exothermic reactions with ΔE < 0 eV, spontaneity is expected. However, reactions that are only mildly to moderately exothermic (ΔE > −1.0 eV) may display a greater tendency towards reversibility.78 Considering these factors, a dividing vertical line was drawn at ΔE = −1 eV in the exothermic region in Fig. 9 to distinguish between reactions with higher and lower propensities for reversibility.
Conversely, the vertical color gradient in Fig. 9 represents variations in activation barriers, with reactions in the lighter regions being kinetically more favorable than those in the darker regions. The reactions in the darker region with −1 eV < ΔE < 0 can theoretically occur, but constitute the rarest type of DMC reduction process. Thus, the unique products from these reactions are likely difficult to detect. In contrast, reactions in the lighter region with ΔE < −1 eV are the most spontaneous reactions. These reactions are essentially irreversible.
| Formula of the product | FG | Decomposition pathway |
|---|---|---|
| CH3OCOOLi | –COH | One-electron |
| CH3OCOOHLi (⁑) | –COOH | One-electron |
| CH3OH | –COH | One-electron |
| CH3OHLi | –COH | One-electron |
| CH3OCHO (⁑) | –COH | One-electron |

|
–COOH | One-electron |
| CH4 (⁑) | –COH | One-, two-electron |
| CO2 | –COH | One-electron |
| C2H6 | –COOH | Two-electron |
| CH3OCOOLi2 | –COH | Two-electron |
| Li2C(OH)O2 (⁑) | –COH, –COOH | Two-electron |
Products in the lighter region for −1 eV < ΔE < 0 are also likely, but their abundance is impacted by the reversibility of the decomposition reactions.
Nonetheless, the spontaneity of a reaction is highly sensitive to the surface functionalization of graphite. For identical end products, completely different kinetic barriers have been calculated depending on the surface FG. For instance, we identified the formation of one of the least reported products, CH3OCH3, to be thermodynamically favorable on the graphite anode. However, its formation requires a relatively lower activation barrier to be overcome on the –COOH FG (EA = 1.92 eV, reaction route R21), whereas much higher activation energy barriers (exceeding 3 eV) were observed for the –CO and –CHO FGs (reaction routes R2 and R17). A similarly large EA had been reported previously for the gas-phase decomposition of DMC into CH3OCH3 and CO2.81 Another example of this scenario is CH3OCHO, which is formed exothermically on the –COH and –COOH FGs (reaction routes R11 and R26), but is only kinetically favorable on the –COH FG. These findings underscore that the DMC decomposition on graphite anodes is highly dependent on the relative abundance of the different FGs on the surface. In particular, –COH (promoting both the one- and two-electron pathways) is the most reactive surface FG followed by the –COOH FG.
As anticipated, reactions predominantly lead to the formation of organic SEI components such as CH3OCOOLi, CH3OCOOHLi, CH4, and  in the early stages of battery charging without Li+ intercalation. However, the formation of inorganic products, including Li2C(OH)O2 (on –COH and –COOH) and LiHCO3 (on –COH), is also observed, which may represent the initial steps towards the formation of well-established inorganic SEI products like Li2CO3 from DMC. Interestingly, scarcely reported products such as CH4, CH3OCOOHLi, CH3OCHO and Li2C(OH)O2 are produced in highly spontaneous DMC decomposition reaction routes, which should compete alongside the reactions producing organic SEI components.
in the early stages of battery charging without Li+ intercalation. However, the formation of inorganic products, including Li2C(OH)O2 (on –COH and –COOH) and LiHCO3 (on –COH), is also observed, which may represent the initial steps towards the formation of well-established inorganic SEI products like Li2CO3 from DMC. Interestingly, scarcely reported products such as CH4, CH3OCOOHLi, CH3OCHO and Li2C(OH)O2 are produced in highly spontaneous DMC decomposition reaction routes, which should compete alongside the reactions producing organic SEI components.
Notably, only EC decomposition on the –CO FG falls within the region of the most spontaneous reactions (lighter region of Fig. 9 with ΔE < −1 eV). Although FEC reduction via both the S1 and S2 mechanisms on –CO and –COH FGs exhibits a low EA, these reactions may be limited by their tendency toward reversibility, as indicated by the mild exothermicity (−1.0 eV < ΔE < 0 eV). In contrast to EC and FEC decomposition, the –COOH FG demonstrates high selectivity for promoting DMC decomposition reactions.
3.5 Long-term stability analysis of the DMC decomposition products in explicit solvent
In the previous sections, we identified the thermodynamically and kinetically favorable decomposition pathways of DMC through both one- and two-electron reduction mechanisms.However, the long-term fate of the predicted products remains uncertain, as conducting AIMD simulations over extended timescales with explicit solvent molecules is computationally prohibitive. To address this shortcoming, we assessed the long-term stability of the thermodynamically favorable products using CMD simulations with a reactive force field, ReaxFF, in the presence of explicit solvent molecules (Section 2.2, step 4). A (2 × 2 × 1) supercell of the original model system from Fig. 1 was considered, with the vacuum region filled with solvent DMC molecules until the desired density (1.07 g cm−3) was reached (see simulation details in Section 2.3). Thus, the simulation cells contained four times the number of each product found in the DFT calculations. A snapshot of the ReaxFF-CMD simulation cell for  products on the –CO FG is shown in Fig. 10.
products on the –CO FG is shown in Fig. 10.
 | ||
Fig. 10 An example of the ReaxFF-CMD simulation cell featuring the product  on –CO FG. The supercell dimensions are twice those of Fig. 1 in the lateral directions and identical in the vertical direction. The vacuum gap is filled with explicit solvent molecules (red: O, white: H, gray: C, purple: Li, blue: product molecules). on –CO FG. The supercell dimensions are twice those of Fig. 1 in the lateral directions and identical in the vertical direction. The vacuum gap is filled with explicit solvent molecules (red: O, white: H, gray: C, purple: Li, blue: product molecules). | ||
The fraction of product i that did not undergo secondary reactions till the end of the simulation run is given by N50i/N0i, where N0i is the initial number of product i molecules (free or adsorbed), and N50i the corresponding number of i molecules after 50 ps. The values of N50i/N0i for different products on each FG, involving one or two Li+ ions, are presented in Fig. 11 (top panel). Clearly, as the products continue to react during the simulation, additional by-products are generated, making it valuable to track their formation. To this end, the bottom panel of Fig. 11 depicts the fraction of by-product j produced from product i relative to the total number of by-product molecules generated from this same product at the end of the simulation run,  . Each column of by-products is placed under the product i (top panel) it originates from. From the results in Fig. 11, the following observations can be extracted regarding the long-term stability of different classes of DMC decomposition products:
. Each column of by-products is placed under the product i (top panel) it originates from. From the results in Fig. 11, the following observations can be extracted regarding the long-term stability of different classes of DMC decomposition products:
 | ||
Fig. 11 The top panel shows N50i/N0i values obtained from ReaxFF-CMD simulations for each thermodynamically favorable product identified in the DFT calculations. The initial adsorbed configuration, nLi–FG, is indicated above, with the binding DMC oxygen (Os) omitted for clarity. The bottom panel depicts the fractional amounts of side products  formed during the ReaxFF-CMD simulations (different side products originating from the same initial state are shown in different colors within the same bar). “Pol” indicates polymerization reaction products. formed during the ReaxFF-CMD simulations (different side products originating from the same initial state are shown in different colors within the same bar). “Pol” indicates polymerization reaction products. | ||
(A) Gaseous products
• Gaseous species such as CO2, CH4, and C2H6 remained intact throughout the ReaxFF-CMD simulations. Additionally, new CO2 molecules were formed via the dissociation of Li2C(OH)O2, CH3OCOOLi, and CH3OCOOHLi on –CO, –COH, and –COOH.
• CH3OCH3 remains largely intact when detached from the anode surface in 1Li–COOH and 1Li–CO. However, the CH3OCH3 formed from 1Li–CHO (see Fig. S10† for details) quickly dissociates into CH3OLi and  . As the stability of CH3OCH3 depends on the number of coordinating Li+ ions, its presence in the LIB electrolyte is expected to be limited due to the abundant presence of Li+ ions.
. As the stability of CH3OCH3 depends on the number of coordinating Li+ ions, its presence in the LIB electrolyte is expected to be limited due to the abundant presence of Li+ ions.
• HCHOLi, only found on the –CO FG via reaction routes R1 and R5, remained stable in 50% of the cases, while the remainder underwent polymerization reactions.
(B) Adsorbed radicals
• The adsorbed radicals  and
and  remained stable throughout the ReaxFF-CMD simulations. Moreover, additional
remained stable throughout the ReaxFF-CMD simulations. Moreover, additional 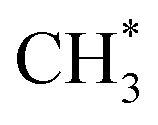 was formed through the dissociation of CH3OCH3 (on –CO and –CHO), CH3OCOOLi (on –COH) and CH3OCOOHLi (on –COOH) in the ReaxFF-CMD runs.
was formed through the dissociation of CH3OCH3 (on –CO and –CHO), CH3OCOOLi (on –COH) and CH3OCOOHLi (on –COOH) in the ReaxFF-CMD runs.
(C) Organic products
• CH3OCHO, produced via reaction routes R9, R11, and R26 on the –COH FG, was stable throughout the ReaxFF-CMD simulations. However, CH3OCHO was quickly transformed into CH3OCHOH through a type (C) HAT process on the –COOH FG or underwent a polymerization process. When stable, the CH3OCHO molecules remained in close proximity to the surface.
• CH3OLi molecules detached from the anode surface and moved into the electrolyte bulk when arising from the –COH and –COOH FGs. In contrast, they remained attached to the anode surface on the –CO and –CHO FGs. Interestingly, CH3OLi formation was abundant through side reactions on each type of functionalized graphite during the ReaxFF-CMD simulations.
• CH3OHLi, formed via thermodynamically favorable reactions on the –COH and –COOH FGs (reaction routes R9, R13, and R26), was found to be unstable and transformed into CH3OLi via a type (B) HAT process very quickly.
• CH3OCOOLi and CH3OCOOLi2 were identified as thermodynamically stable products via reaction routes R6–R8, R10, R12, R16, R18, R22, and R24. However, they were found to be the least stable compounds during the ReaxFF-CMD simulations on the –COH FG, and quickly transformed or dissociated into products such as CH3OCOOH, CH3OCOOHLi, LiOH, 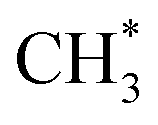 , CO2, and Li2CO3. In contrast, CH3OCOOLi was found to be relatively more stable when originating from –CO, –CHO, and –COOH. At the end of the ReaxFF-CMD simulations, CH3OCOOLi remained attached to the anode surface only in the presence of the –CO FG.
, CO2, and Li2CO3. In contrast, CH3OCOOLi was found to be relatively more stable when originating from –CO, –CHO, and –COOH. At the end of the ReaxFF-CMD simulations, CH3OCOOLi remained attached to the anode surface only in the presence of the –CO FG.
(D) Inorganic products
• Li2C(OH)O2, which emerged from the –CO, –COH, and –COOH FGs via reaction routes R4, R15, and R25, transformed into inorganic SEI components Li2CO3 or LiHCO3 during the ReaxFF-CMD simulations. Transformation to LiHCO3 was abundant on all FGs, whereas transformation to Li2CO3 was only observed on the –COOH FG. These inorganic carbonates remained close to the anode surface.
• LiHCO3, which formed on the –COH FG through reaction route R14, remained stable during the ReaxFF-CMD simulations. Only a small fraction transformed into LiCO3via a (B) HAT process. On the other hand, LiHCO3 was produced abundantly from Li2C(OH)O2 on the –CO, –COH, and –COOH FGs.
As shown in Fig. 9, the DMC decomposition pathways R11, R12, R13, R15, and R16 exhibit the most favorable energetics, characterized by both lower ΔE and EA values compared to other decomposition reaction routes. All these reactions take place on the –COH functionalized graphite surface. The stability of the products formed via these pathways was further assessed using ReaxFF-CMD simulations in an explicit solvent environment containing EC and DMC in the same volume ratio, and 1 M LiPF6 molecules. The fraction of products remaining stable after 50 ps of simulation, along with any by-products formed, is presented in Fig. S16.† The observed trends are generally consistent with those in Fig. 11, with a few notable exceptions. Products that detached from the anode surface, such as gaseous species (CH4 and CO2) and organic molecules (CH3OH and CH3OCHO), remained stable throughout the ReaxFF-CMD simulations. Li2C(OH)O2, formed via reaction route R15, primarily converted to LiHCO3. However, in contrast to the DMC-only solvent environment, a small fraction was further decomposed into CO2 and LiOH. Additionally, all the CH3OHLi (formed via reaction route R13) converted into CH3OLi via a type-A HAT process. The intermediate species HCHOLi, previously observed under DMC-only solvent conditions, was not detected in the mixed-solvent simulations.
The transformation of products observed in the classical simulations was completed within a short timescale of 2 ps. Notably, the longest transformation time, approximately 8 to 15 ps, was associated with the conversion of CH3OCOOLi2/Li2C(OH)O2 into Li2CO3. This likely signals the onset of inner inorganic layer formation during the initial charging process of the LIB. The estimated transformation time is defined as the duration required for the initial DMC decomposition product to undergo bond cleavage and/or formation resulting in the emergence of a chemically distinct molecule. As expected, the thermodynamically favorable products Li2C(OH)O2 and LiHCO3 act as precursors to the formation of known inorganic SEI components, as seen from their transformation to LiHCO3, Li2CO3, and LiCO3 during the course of the ReaxFF-CMD simulations.
Organic products such as CH3OCHO and CH3OCOOH were found to detach from the anode surface. Notably, organic SEI products like CH3OLi and CH3OCOOLi tended to move away from the surface, except on the –CO FG, supporting a bottom-up SEI formation mechanism. The strong attachment of these organic SEI products to the –CO FG populated graphite surface is likely due to stronger O–Li interactions, as indicated in Table 1.
Our results indicate that higher surface coverage of the graphite anode with –CO FGs leads to surface poisoning through the adsorption of free radicals, which reduces the number of active sites. The –COH and –COOH FGs are the ones that favor the formation of inorganic SEI components during the ReaxFF-CMD simulations. In the long-term, these FGs, especially the –COH FG, will be the most active ones to promote DMC decomposition reactions on the graphite anode surface producing both organic and inorganic SEI components following the bottom-up mechanism.
4. Conclusions
In this study, we explored the decomposition mechanisms of dimethyl carbonate (DMC) on functionalized graphite anode surfaces during the early stages of Li-ion battery (LIB) operation. Using a combined first principles and classical reactive force field (ReaxFF) approach, we identified key processes and reaction pathways that contribute to SEI formation and long-term battery performance.Our findings highlight the critical role of Li+ ions in initiating DMC chemisorption and facilitating charge transfer, both of which are essential for subsequent surface mediated decomposition reactions. During the early stages of battery operation, DMC decomposition predominantly leads to the formation of organic products, regardless of the number of Li+ ions present on the functionalized graphite anode surfaces. Notably, Hydrogen Atom Transfer (HAT) mechanisms, classified into three distinct types, emerge as pivotal drivers of these processes.
–COH exhibited the highest reactivity among the investigated FGs, with –COOH following closely. This enables both one- and two-electron DMC decomposition pathways, whereas –CHO demonstrated the lowest activity. Our simulations predicted the formation of well-known SEI components such as CH3OCOOLi and CH3OLi, as well as several less commonly reported products, including CH4, CH3OCOOHLi, CH3OCHO, CH3OCH3, LiHCO3, and Li2C(OH)O2. Furthermore, competitive decomposition of other key electrolyte components, such as ethylene carbonate (EC) and fluoroethylene carbonate (FEC), was particularly pronounced on graphite surfaces enriched with –COH and –COOH groups.
Long-term stability analysis using ReaxFF-CMD revealed that the predicted gas-phase decomposition products remain stable after detaching from the anode surface. However, the facile C–H transfer processes occurring on OH-rich surfaces, which generate CH4, could compromise long-term performance by inducing microcracks within the SEI layer. Additionally, organic DMC decomposition products were observed to detach and diffuse away from –COH and –COOH surfaces, supporting a bottom-up SEI formation mechanism. In contrast, the stronger attachment of these products to –CO functionalized surfaces may lead to surface poisoning, reducing battery efficiency over prolonged cycling. The onset of Li2CO3 formation, a key SEI inorganic component, was detected within around 8 to 15 ps on –COH and –COOH functionalized surfaces, underscoring their sustained reactivity and contribution to SEI growth throughout battery operation.
The insights gained from this study provide a deeper understanding of the intricate interplay between functional groups, Li+ ions, and electrolyte decomposition. This knowledge offers valuable guidance for the rational design of more stable and efficient LIB anodes with tailored surface functionalization to optimize SEI composition, enhance long-term stability, and mitigate side reactions. In particular, controlling the density and distribution of –COH and –COOH groups could help balance the benefits of efficient SEI formation against the risks of gas evolution and surface degradation.
Future research should explore: (i) the decomposition mechanisms of other key electrolyte components, such as ethylene carbonate, ethyl methyl carbonate, and diethyl carbonate and their mixtures on the negative electrode surface, following the methodology established in this study; (ii) the influence of partially and fully Li+-intercalated graphite anodes on the pathways and energetics of electrolyte decomposition; (iii) the synergistic effects of mixed functional groups on electrolyte decomposition and SEI growth; (iv) the impact of electrolyte additives designed to suppress detrimental side reactions while promoting stable SEI formation; (v) the influence of extended simulation timescales and larger system sizes to capture long-term SEI evolution and morphological changes; and (vi) experimental validation of the predicted reaction pathways and by-products through well-defined in situ spectroscopy and advanced microscopy techniques. By addressing these avenues, it will be possible to develop advanced electrode–electrolyte interfaces that support higher energy densities, longer cycle life, and improved safety for next-generation LIBs.
Data availability
The data supporting this article have been included as part of the main text and ESI.†Author contributions
Bikram Kumar Das: conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing – original draft. Henry A. Cortés: formal analysis, methodology, software, validation, writing – review & editing. Mauricio Rincón Bonilla: conceptualization, project administration, resources, supervision, visualization, writing – review & editing. Menghao Yang: writing – review & editing. Javier Carrasco: project administration, supervision, writing – review & editing. Elena Akhmatskaya: funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. K. Das acknowledges financial support from the Juan de la Cierva postdoctoral grant JDC2022-049793-I funded by MICIU/AEI/10.13039/501100011033 and by European Union NextGenerationEU/PRTR. This research was supported by MICIU/AEI/10.13039/501100011033 and by ERDF A way for Europe under Grant PID2022-136585NB-C22; by the Basque Government through the ELKARTEK Program under Grants CICe2024, KK-2024/00062: “Profundizando en el conocimiento para el diseño y desarrollo de nuevos electrolitos y para tecnologias de baterías post-litio-ion.” and through the BERC 2022-2025 program. We acknowledge the financial support from the Ministry of Science and Innovation through BCAM Severo Ochoa accreditation CEX2021-001142-S/MICIU/EI/10.13039/501100011033 and “PLAN COMPLEMENTARIO MATERIALES AVANZADOS 2022–2025”, PROYECTO No. 1101288. The authors acknowledge the financial support received from the grant BCAM-IKUR, funded by the Basque Government by the IKUR Strategy and by the European Union NextGeneration EU/PRTR. M. R. Bonilla acknowledges the support from Diputación Foral de Bizkaia through MODEL2RESIST (6/12/TT/2024/00003) and the Spanish research agency through RYC2022-036500-I. This work has been possible thanks to the support of the computing infrastructure of the i2BASQUE academic network, Barcelona Supercomputing Center (RES, QHS-2025-1-0027), DIPC Computer Center and the technical and human support provided by the IZO-SGI SGIker of UPV/EHU.References
- T. Kim, W. Song, D.-Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li and N. Tavajohi, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS.
- P. Nzereogu, A. Omah, F. Ezema, E. Iwuoha and A. Nwanya, Appl. Surf. Sci. Adv., 2022, 9, 100233 CrossRef.
- S. M. Abu, M. Hannan, M. H. Lipu, M. Mannan, P. J. Ker, M. Hossain and T. I. Mahlia, J. Cleaner Prod., 2023, 394, 136246 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- H. Adenusi, G. A. Chass, S. Passerini, K. V. Tian and G. Chen, Adv. Energy Mater., 2023, 13, 2203307 CrossRef CAS.
- S. Rustam, N. N. Intan and J. Pfaendtner, J. Chem. Phys., 2021, 155, 134702 CrossRef CAS PubMed.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood III, Carbon, 2016, 105, 52–76 CrossRef CAS.
- S.-P. Kim, A. C. Van Duin and V. B. Shenoy, J. Power Sources, 2011, 196, 8590–8597 CrossRef CAS.
- V. A. Agubra and J. W. Fergus, J. Power Sources, 2014, 268, 153–162 CrossRef CAS.
- Z. Liu, P. Lu, Q. Zhang, X. Xiao, Y. Qi and L.-Q. Chen, J. Phys. Chem. Lett., 2018, 9, 5508–5514 CrossRef CAS PubMed.
- X. Tan, M. Chen, J. Zhang, S. Li, H. Zhang, L. Yang, T. Sun, X. Qian and G. Feng, Adv. Energy Mater., 2024, 14, 2400564 CrossRef CAS.
- P.-C. Hsu, Y.-C. Lin, W.-H. Wu, C.-W. Pao and C.-H. Chen, J. Electrochem. Soc., 2022, 169, 120520 CrossRef CAS.
- N. Takenaka, Y. Suzuki, H. Sakai and M. Nagaoka, J. Phys. Chem. C, 2014, 118, 10874–10882 CrossRef CAS.
- P. Ganesh, P. Kent and D.-e. Jiang, J. Phys. Chem. C, 2012, 116, 24476–24481 CrossRef CAS.
- K. Leung and J. L. Budzien, Phys. Chem. Chem. Phys., 2010, 12, 6583–6586 RSC.
- K. Leung, Chem. Phys. Lett., 2013, 568, 1–8 CrossRef.
- G. Gachot, S. Grugeon, M. Armand, S. Pilard, P. Guenot, J.-M. Tarascon and S. Laruelle, J. Power Sources, 2008, 178, 409–421 CrossRef CAS.
- M. J. Boyer, L. Vilčiauskas and G. S. Hwang, Phys. Chem. Chem. Phys., 2016, 18, 27868–27876 RSC.
- Q. Peng, H. Liu and S. Ye, J. Electroanal. Chem., 2017, 800, 134–143 CrossRef CAS.
- M. Nie, D. Chalasani, D. P. Abraham, Y. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 1257–1267 CrossRef CAS.
- E. W. C. Spotte-Smith, R. L. Kam, D. Barter, X. Xie, T. Hou, S. Dwaraknath, S. M. Blau and K. A. Persson, ACS Energy Lett., 2022, 7, 1446–1453 CrossRef CAS.
- N. N. Intan and J. Pfaendtner, ACS Appl. Mater. Interfaces, 2021, 13, 8169–8180 CrossRef CAS.
- P. J. Weddle, E. W. C. Spotte-Smith, A. Verma, H. D. Patel, K. Fink, B. J. T. de Villers, M. C. Schulze, S. M. Blau, K. A. Smith and K. A. Persson, Electrochim. Acta, 2023, 468, 143121 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
- G. V. Zhuang, H. Yang, P. N. Ross, K. Xu and T. R. Jow, Electrochem. Solid-State Lett., 2005, 9, A64 CrossRef.
- S. Leroy, H. Martinez, R. Dedryvère, D. Lemordant and D. Gonbeau, Appl. Surf. Sci., 2007, 253, 4895–4905 CrossRef CAS.
- T. A. Pham, K. E. Kweon, A. Samanta, M. T. Ong, V. Lordi and J. E. Pask, J. Phys. Chem. C, 2020, 124, 21985–21992 CrossRef CAS.
- Y. Fernandes, A. Bry and S. De Persis, J. Power Sources, 2019, 414, 250–261 CrossRef CAS.
- O. Borodin and G. D. Smith, J. Phys. Chem. B, 2009, 113, 1763–1776 CrossRef CAS.
- T. Husch and M. Korth, Phys. Chem. Chem. Phys., 2015, 17, 22799–22808 RSC.
- L. Huai, Z. Chen and J. Li, ACS Appl. Mater. Interfaces, 2017, 9, 36377–36384 CrossRef CAS.
- X. Teng, C. Zhan, Y. Bai, L. Ma, Q. Liu, C. Wu, F. Wu, Y. Yang, J. Lu and K. Amine, ACS Appl. Mater. Interfaces, 2015, 7, 22751–22755 CrossRef CAS.
- S.-H. Pyo, J. H. Park, T.-S. Chang and R. Hatti-Kaul, Curr. Opin. Green Sustainable Chem., 2017, 5, 61–66 CrossRef.
- H. Lee, S. Hwang, M. Kim, K. Kwak, J. Lee, Y.-K. Han and H. Lee, J. Phys. Chem. Lett., 2020, 11, 10382–10387 CrossRef CAS PubMed.
- M. H. C. Peiris, S. Brennan, D. Liepinya, H. Liu and M. Smeu, Colloids Surf., A, 2023, 674, 131831 CrossRef CAS.
- B. Ravikumar, M. Mynam, S. Repaka and B. Rai, J. Mol. Liq., 2021, 338, 116613 CrossRef CAS.
- J. Vatamanu, O. Borodin and G. D. Smith, J. Phys. Chem. C, 2012, 116, 1114–1121 CrossRef CAS.
- I. Azcarate, W. Yin, C. Méthivier, F. Ribot, C. Laberty-Robert and A. Grimaud, J. Electrochem. Soc., 2020, 167, 080530 CrossRef CAS.
- T. Baba, K. Sodeyama, Y. Kawamura and Y. Tateyama, Phys. Chem. Chem. Phys., 2020, 22, 10764–10774 RSC.
- J. Collins, G. Gourdin, M. Foster and D. Qu, Carbon, 2015, 92, 193–244 CrossRef CAS.
- I. Temprano, J. Carrasco, M. Bugnet, I. T. Lucas, J. Zhou, R. S. Weatherup, C. A. O'Keefe, Z. Ruff, J. Xu and N. Folastre, Energy Storage Mater., 2024, 73, 103794 CrossRef.
- K.-S. Yun, S. J. Pai, B. C. Yeo, K.-R. Lee, S.-J. Kim and S. S. Han, J. Phys. Chem. Lett., 2017, 8, 2812–2818 CrossRef CAS PubMed.
- D. Bedrov, G. D. Smith and A. C. van Duin, J. Phys. Chem. A, 2012, 116, 2978–2985 CrossRef CAS PubMed.
- H. G. Lee, S. Y. Kim and J. S. Lee, npj Comput. Mater., 2022, 8, 103 CrossRef CAS.
- Z. Jiang, K. Klyukin and V. Alexandrov, ACS Appl. Mater. Interfaces, 2018, 10, 20621–20626 CrossRef CAS PubMed.
- J. Wu, M. Ihsan-Ul-Haq, Y. Chen and J.-K. Kim, Nano Energy, 2021, 89, 106489 CrossRef CAS.
- D. Sheppard, P. Xiao, W. Chemelewski, D. D. Johnson and G. Henkelman, J. Chem. Phys., 2012, 136, 074103 CrossRef PubMed.
- D. Sheppard and G. Henkelman, J. Comput. Chem., 2011, 32, 1769–1771 CrossRef CAS PubMed.
- M. M. Islam, V. S. Bryantsev and A. C. Van Duin, J. Electrochem. Soc., 2014, 161, E3009 CrossRef CAS.
- T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik and H. M. Aktulga, npj Comput. Mater., 2016, 2, 1–14 CrossRef.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- S. I. Nosé, Mol. Phys., 2002, 100, 191–198 CrossRef.
- W. G. Hoover, Phys. Rev. A:At., Mol., Opt. Phys., 1985, 31, 1695 CrossRef.
- B. Oudot and K. Doblhoff-Dier, J. Chem. Phys., 2024, 161, 054708 CrossRef CAS.
- F. Fasulo, A. B. Muñoz-García, A. Massaro, O. Crescenzi, C. Huang and M. Pavone, J. Mater. Chem. A, 2023, 11, 5660–5669 RSC.
- S. Debnath, V. A. Neufeld, L. D. Jacobson, B. Rudshteyn, J. L. Weber, T. C. Berkelbach and R. A. Friesner, J. Phys. Chem. A, 2023, 127, 9178–9184 CrossRef CAS PubMed.
- S. Svelle, C. Tuma, X. Rozanska, T. Kerber and J. Sauer, J. Am. Chem. Soc., 2009, 131, 816–825 CrossRef CAS.
- E. G. Leggesse, K.-H. Tsau, Y.-T. Liu, S. Nachimuthu and J.-C. Jiang, Electrochim. Acta, 2016, 210, 61–70 CrossRef CAS.
- C. Liu, Z. Lu, J. Duan, H. Dou, Z. Cao, X. Xu, X. Zhang, Z. Chen and W. Xiao, J. CO2 Util., 2024, 84, 102846 CrossRef CAS.
- M. Bin Jassar, C. Michel, S. Abada, T. De Bruin, S. Tant, C. Nieto-Draghi and S. N. Steinmann, ACS Appl. Energy Mater., 2023, 6, 6934–6945 CrossRef CAS.
- X. Yan, C. Zhu, W. Huang and Y.-J. Zhao, J. Mater. Chem. A, 2024, 12, 24401–24408 RSC.
- F. Castro-Marcano, A. M. Kamat, M. F. Russo Jr, A. C. Van Duin and J. P. Mathews, Combust. Flame, 2012, 159, 1272–1285 CrossRef CAS.
- J. Gao, R. He, P. Wu and K. H. Luo, J. Energy Storage, 2025, 106, 114919 CrossRef.
- J. Gao, G. Wang and K. H. Luo, Appl. Energy, 2024, 371, 123708 CrossRef CAS.
- Y. Wang, Y. Liu, Y. Tu and Q. Wang, J. Phys. Chem. C, 2020, 124, 9099–9108 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- J. Zeng, L. Cao, C.-H. Chin, H. Ren, J. Z. Zhang and T. Zhu, Phys. Chem. Chem. Phys., 2020, 22, 683–691 RSC.
- L. D. Landau and E. M. Lifshitz, Course of Theoretical Physics, Elsevier, 2013 Search PubMed.
- P. W. Atkins, J. De Paula and J. Keeler, Atkins' Physical Chemistry, Oxford University Press, 2023 Search PubMed.
- Q. Wu, M. T. McDowell and Y. Qi, J. Am. Chem. Soc., 2023, 145, 2473–2484 CrossRef CAS PubMed.
- H. Yoshida, T. Fukunaga, T. Hazama, M. Terasaki, M. Mizutani and M. Yamachi, J. Power Sources, 1997, 68, 311–315 CrossRef CAS.
- P. J. Bugryniec, S. Vernuccio and S. F. Brown, J. Power Sources, 2023, 580, 233394 CrossRef CAS.
- F. Stehmann, E. Wiegmann and S. Scholl, Adsorption, 2017, 23, 341–348 CrossRef CAS.
- M. Leißing, C. Peschel, F. Horsthemke, S. Wiemers-Meyer, M. Winter and S. Nowak, Batteries Supercaps, 2021, 4, 1731–1738 CrossRef.
- H. Ota, Y. Sakata, A. Inoue and S. Yamaguchi, J. Electrochem. Soc., 2004, 151, A1659 CrossRef CAS.
- M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, 2020 Search PubMed.
- P. Tundo, M. Musolino and F. Aricò, Green Chem., 2018, 20, 28–85 RSC.
- E. W. C. Spotte-Smith, T. B. Petrocelli, H. D. Patel, S. M. Blau and K. A. Persson, ACS Energy Lett., 2022, 8, 347–355 CrossRef.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed.
- D. Sengupta, J. Cryst. Growth, 2006, 286, 91–95 CrossRef CAS.
- O. R. Inderwildi, D. Lebiedz and J. Warnatz, Phys. Chem. Chem. Phys., 2005, 7, 2552–2553 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta02003d |
| This journal is © The Royal Society of Chemistry 2025 |