
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic oxidation of glycerol with red light employing quinacridone sensitized TiO2 nanoparticles†
Yunshuo
Yang
 ,
Marco
Nalesso
,
Andrea
Basagni
,
Ruggero
Bonetto
,
Raffaella
Signorini
,
Stefano
Agnoli
,
Luka
Đorđević
* and
Andrea
Sartorel
*
,
Marco
Nalesso
,
Andrea
Basagni
,
Ruggero
Bonetto
,
Raffaella
Signorini
,
Stefano
Agnoli
,
Luka
Đorđević
* and
Andrea
Sartorel
*
Department of Chemical Sciences, University of Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: luka.dordevic@unipd.it; andrea.sartorel@unipd.it
First published on 20th May 2025
Abstract
Photocatalytic nanomaterials combining organic dyes and inorganic semiconductor nanoparticles (NPs) are extensively investigated for light-driven production of solar fuels and for conversion of organic feedstocks. However, their applications for the valorization of abundant raw materials by exploiting low-energy visible light remain limited. In this study, we report a facile preparation of TiO2 nanoparticles sensitized with a quinacridone (QA) industrial pigment for the aqueous oxidation of glycerol to glyceraldehyde with red light (λ = 620 nm), reaching 47.5 ± 5.0 μmol gNP−1 h−1 of productivity and 80% selectivity in the presence of TEMPO co-catalyst. The hybrid material outperforms the single components and shows recyclability up to at least 5 additional times under red light while maintaining intact productivity; furthermore, it demonstrates versatility by operating also under green, yellow or white light irradiation. We believe that this work will provide a new avenue for using industrial pigment-sensitized materials in photocatalysis exploiting low energy light, providing novel strategies for the future development of this field.
Introduction
Photocatalysis offers promising solutions for conducting chemical transformations under safe and sustainable conditions, ideally exploiting the inexhaustible power from the sun. Applications of light-catalyzed processes include solar fuel production, removal of pollutants, and development of organic processes.1–9 Photocatalysis can be conducted under homogeneous conditions, at the surface of electrodes or with heterogeneous, photoactive nanomaterials.10 The latter offer significant advantages: (i) the opportunity to integrate in a single component the three main functional modules in photocatalysis, i.e., light absorption, charge separation and redox catalytic routines; (ii) wireless operation, without the application of external bias; (iii) possible photocatalyst recovery and recyclability typical of heterogeneous catalysis.11 Indeed, back in 2014, mixed colloid photocatalysts were considered by H. B. Gray and co-workers as the most viable technology for large scale applications and scalability purposes of photochemical water splitting,12 and since then significant advancement in the field has been reached.13–18In addition to fully inorganic photocatalysts, hybrid nanomaterials combining organic dyes and semiconductor nanoparticles (NPs) can provide further benefits, such as a separate tuning of the two components, with easy modulation of light absorption and of energy levels.19 These hybrid nanocomposites typically exploit visible light and display enhanced charge separation efficiency, due to the location of the charge carriers on two different chemical entities (i.e., the dye and the semiconductor). Moreover, their design takes advantage of the endless arsenal of organic chemistry modulation concerning the dye component. The typical operating principles involve light absorption by the organic dye and electron injection in the conduction band of the semiconductor, inducing the formation of a charge separated state. This may lead to the conduction of the desired redox reactions occurring simultaneously at the surface of the material, possibly taking advantage of co-catalysts.20
To date, organic dye-sensitized NPs have found applications in photocatalytic hydrogen production21–23 and, more recently, in the reduction of carbon dioxide24–26 in the presence of sacrificial electron donors; exploitation of the oxidative route for selective conversion of organic chemicals is less explored.27–29
In 2008, Ma, Zhao and co-workers reported TiO2 NPs sensitized with Alizarin Red S (ARS) dye coupled to a homogeneous 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) free radical co-catalyst for the aerobic oxidation of alcohols to carbonyl compounds in benzotrifluoride solvent and exploiting 400–500 nm light, with concomitant reduction of dioxygen to superoxide anion O2˙−.30 A similar photochemical system for the oxidation of alcohols to carbonyls was reported combining TiO2 with Eosin Y dye.31 A recent breakthrough was reported by Vauthey, Coutsolelos, Odobel and co-workers, on diketopyrrolopyrrole-sensitized TiO2 NPs for the oxidation of benzyl alcohols coupled to hydrogen evolution, exploiting aminoxyl radical and Pt0 co-catalysts, operating in water with green light at 525 nm.28 Other oxidative processes that were targeted with photocatalytic nanomaterials include the oxidation of amines,32 the conversion of sulfides to sulfoxides,33 the activation of sp2 C–H bonds through thiocyanation and cyclization reactions,34 benzylic C–H activation with aerobic oxidation of benzyl ethers,35 and decarboxylation and alkyl addition to tetrahydroisoquinolines.36
From the current state-of-the-art, three main points need to be addressed for further development and application of dye-sensitized semiconductor NPs: (i) the use of cheap and available organic dyes, and their easy integration onto the surface of the semiconductor, possibly avoiding the usage of anchoring functional groups37 that requires additional synthetic steps; (ii) photocatalytic applications in water towards the conversion of abundant raw materials into valuable chemicals, with one relevant example being the oxidation of glycerol as a major by-product of soap manufacturing and of biodiesel processing;38–41 (iii) the use of low-energy visible light, and in particular in the red region of the electromagnetic spectrum,42 a long-sought but still limited goal in photocatalysis.43–46 Besides integrating visible light-absorbing dyes on semiconductors, a strategy to further extend the absorption of low energy radiation is to exploit dyes in aggregated states,47 for which applications in oxidation reactions are even more rare.48–54
With these premises, in this work we selected the quinacridone (QA) industrial pigment as a cheap, non-toxic and versatile tool in photocatalysis applications,55 and sought to incorporate QA into a hybrid TiO2 NP system (QA@TiO2) through an easy protection/deprotection method.56 We applied the photocatalytic QA@TiO2 nanomaterial to the challenging and ambitious selective photocatalytic oxidation of glycerol to high added-value glyceraldehyde in water using low energy photons (up to red light at 620 nm, Scheme 1), and showing recyclability up to five additional cycles.
 | ||
| Scheme 1 Synthetic strategy of QA@TiO2 NPs and their application for photocatalysis in water and glycerol oxidation by exploiting red light, highlighting the figures of merit of this work. | ||
Results and discussion
Preparation of QAL@TiO2 NPs
We sought to first derivatize the parent QA pigment with a transient protecting group, such as tert-butyloxycarbonyl (Boc), to give Boc–QA.56 Then, the increased processability of Boc–QA57 can be used to adsorb the QA dye on NPs, followed by easy deprotection to obtain the dye-doped hybrid. This approach should complement the literature that took advantage of sol–gel deposition or impregnation methods to prepare QA/TiO2 composites onto a silica support.58,59 Advantages of this method are the flexibility and generality of the protocol, being extendable to other dyes and to other materials, while easily controlling the dye loading (vide infra).In the design of the hybrid nanomaterial, we selected commercially available titanium dioxide nanopowder (80%/20% anatase/rutile, 21 nm primary particle size). The sensitization of TiO2 NPs exploits a simple stirring overnight of a suspension of TiO2 NPs in ethyl acetate in the presence of a desired amount of Boc–QA, followed by the elimination of the solvent by evaporation under reduced pressure. This leads to the quantitative physisorption of the Boc–QA dye on the surface of TiO2, to produce bright yellow Boc–QAL@TiO2 NPs, with the subfix L indicating the loading of the dye expressed in μmol per gram of TiO2 (Scheme 2, L between 16 and 624). Heating the Boc–QAL@TiO2 NPs in an oven (150 °C for ca. 30 minutes) allows the removal of the Boc protecting group obtaining the QAL@TiO2 NPs. During this step, the color of the hybrid material clearly changes from yellow to purple (Scheme 2) as a consequence of the Boc–QA → QA transformation upon deprotection. Indeed, diffuse reflectance spectroscopy on the particles shows a shift of the absorption bands from Boc–QA312@TiO2 NPs (broad band between 400 and 550 nm) to QA312@TiO2 NPs (broad band between 450 and 650 nm), see Fig. S3 in the ESI.† We anticipate that the QA312@TiO2 NPs with L = 312 are the most active photocatalyst (vide infra), thus the characterization will refer to this material as a representative case.
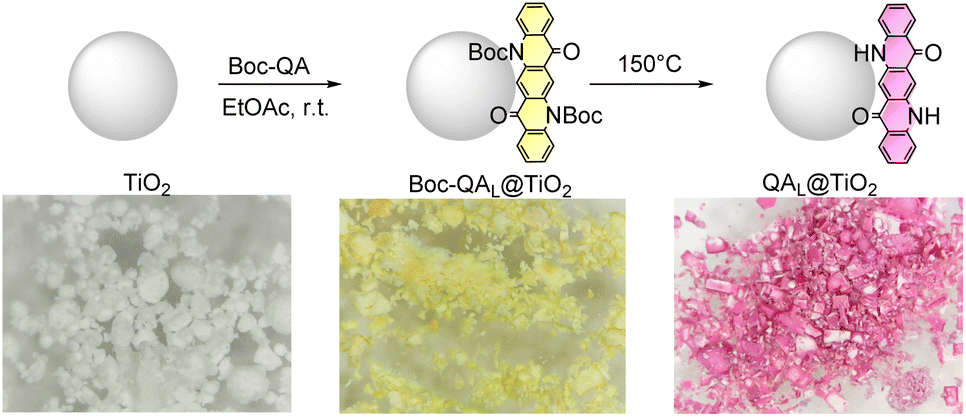 | ||
| Scheme 2 Preparation of quinacridone-sensitized NPs (QAL@TiO2; L = 16–624 μmol gTiO2−1), with digital photographs of the materials at each stage of the protocol. General procedure: 20 mg of TiO2 NPs were stirred overnight in 2 mL of ethyl acetate in the presence of Boc–QA. The solvent was then removed by evaporation under vacuum, and the resulting material heated in an oven at 150 °C for ca. 30 minutes (Boc deprotection). The procedure was also extended to other dyes (indigo, cibalackrot) that were used as photocatalytic materials in control experiments (vide infra). | ||
The loss of Boc protecting group was confirmed by thermogravimetric analysis: this reveals a two-step weight loss for Boc–QA312@TiO2 NPs, the first taking place between 100 and 200 °C (first derivative curve presenting a maximum peak at 153 °C) due to Boc loss,60 and the second mainly occurring between 350 and 500 °C (first derivative curve presenting a maximum peak at 430 °C) due to QA loss (Fig. S4†). In contrast, for QA312@TiO2 NPs only the second weight loss process is observed, being consistent with the nominal loading of QA (Fig. S4†). It is worth mentioning that, different from the protected Boc–QA dye, the QA pigment can form hydrogen bonds23 with the semiconductor surface through the N–H groups,61 thus providing additional supramolecular interactions between the pigment and the semiconductor component. The advantages of this methodology for the preparation of dye-sensitized NPs are (i) the easy control of the relative amount of QA and TiO2, and (ii) the unnecessary introduction on the dye of permanent functional groups for its covalent anchorage to the surface.37,62,63
Characterization of QAL@TiO2 NPs
The hybrid dye-sensitized NPs were characterized by combining several techniques providing complementary information. Transmission electron microscopy (TEM) images of pristine TiO2 NPs and of QAL@TiO2 are reported in Fig. S5,† showing that the functionalization with the QA dye is only slightly impacting the size and shape of the NPs, in line with a spherical model where the QA layer is expected to be lower than 2 nm even at its highest loading (ESI†). We then conducted a scanning transmission electron microscopy (STEM) coupled to energy dispersive X-ray (EDX) tomography to trace the elemental map composition. As can be seen from the secondary electron images (Fig. 1a), the QA312@TiO2 NPs appear smooth and uniform (see Fig. S6† reporting the same analysis on the QA16@TiO2 hybrid and on TiO2 materials; high-angle annular dark-field (HAADF)-STEM images are also reported in Fig. S6†). The homogeneous distribution of the coating of QA is confirmed by the EDX-mapping of C, N, O and Ti (Fig. 1b). | ||
| Fig. 1 Characterization of QA312@TiO2. (a) STEM (scale bar 20 nm). (b) EDX analysis (C, N, O, Ti). (c) XPS (C 1s and N 1s regions) of QA312@TiO2 (top) and of QA (bottom); due to charging effects, the energy scale is reported by setting the chemically shifted C 1s component of C sp2 at 284.6 eV. (d) Diffuse reflectance spectra of QA312@TiO2 reported in Kubelka–Munk units; inset: Tauc plot analysis. | ||
X-ray photoelectron spectroscopy (XPS) spectra of QA312@TiO2 NPs were interpreted in comparison of bare TiO2 and of QA. In the survey scan, only the signals of the expected elements were observed, while the Cu signal from the support was minimal (Fig. S7†). For both QA312@TiO2 and TiO2 NPs, the Ti 2p photoemission line consists of a doublet with a sharper Ti 2p3/2 peak and a broader Ti 2p1/2 peak in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 area ratio (Fig. S8†). No shape changes can be appreciated in the presence of QA, suggesting that there is no surface chemical reaction between the TiO2 and the QA film. In QA312@TiO2, the C 1s spectra was found to be consistent with QA in terms of energy position and full width at half maximum, Fig. 1c. After a multipeak analysis, the C 1s photoemission line revealed the expected three components related to QA. Besides the sp2 C
:1 area ratio (Fig. S8†). No shape changes can be appreciated in the presence of QA, suggesting that there is no surface chemical reaction between the TiO2 and the QA film. In QA312@TiO2, the C 1s spectra was found to be consistent with QA in terms of energy position and full width at half maximum, Fig. 1c. After a multipeak analysis, the C 1s photoemission line revealed the expected three components related to QA. Besides the sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 284.6 eV, another peak due to the carbon in alpha to the nitrogen (C–NH–C) was found at a binding energy of 285.6–285.7 eV, while the chemically shifted component associated with the C of CO groups was found between 286.5 and 286.6 eV (Fig. 1c). The reliability of the deconvolution is also given by the consistency of the N 1s/C 1s ratio across the QA molecule (10.4%) and QA312@TiO2 sample (9.2%), excluding the O–CO contamination.64,65 The N 1s core level is relatively narrow (full width at half maximum = 1.6 eV) and rather symmetrical indicating a single nitrogen type. The peak is centered at 400.0 eV for the QA molecule and at 399.8–399.9 eV for the QA312@TiO2, aligning with the expected binding energies of aromatic amines (Fig. 1c). Similar features are observed in the analysis of QA16@TiO2, except for a lower intensity of the C and N signals due to the lower amount of QA in this material (Fig. S9†).
C at 284.6 eV, another peak due to the carbon in alpha to the nitrogen (C–NH–C) was found at a binding energy of 285.6–285.7 eV, while the chemically shifted component associated with the C of CO groups was found between 286.5 and 286.6 eV (Fig. 1c). The reliability of the deconvolution is also given by the consistency of the N 1s/C 1s ratio across the QA molecule (10.4%) and QA312@TiO2 sample (9.2%), excluding the O–CO contamination.64,65 The N 1s core level is relatively narrow (full width at half maximum = 1.6 eV) and rather symmetrical indicating a single nitrogen type. The peak is centered at 400.0 eV for the QA molecule and at 399.8–399.9 eV for the QA312@TiO2, aligning with the expected binding energies of aromatic amines (Fig. 1c). Similar features are observed in the analysis of QA16@TiO2, except for a lower intensity of the C and N signals due to the lower amount of QA in this material (Fig. S9†).
The diffuse reflectance spectrum of the QA312@TiO2 representative material is reported in Kubelka–Munk units in Fig. 1d (see Fig. S10† for all spectra). The spectra show an absorption feature below 400 nm ascribable to the TiO2 semiconductor and a broad band in the 450–650 nm region, typical of the aggregated states of QA.61 Increasing the amount of QA in the particles leads to an increase in the overall absorption (Fig. S10†),66 together with a redshift of the band. The Kubelka–Munk's analysis reveals an optical band gap comprised between 2.0 and 2.1 eV (inset in Fig. 1d), in agreement with the one observed for QA films on mesoporous surfaces (Fig. S10†).55,61 The combination of the optical band gap with the half-wave potential under anodic scan of 1.23 ± 0.02 V vs. the Reversible Hydrogen Electrode (RHE) for QA films61 indicates a proper energy level alignment to achieve photoinduced electron injection from QA to TiO2 (conduction band at −0.16 V vs. RHE67), Fig. S11.† In particular, while further photochemical characterization on QA@TiO2 NPs is hampered by scattering, aggregated QA films are characterized by lifetimes up to the nanosecond timescale, while photoinduced electron transfer onto TiO2 occurs in a ps timescale in photoelectrode slides.61
Given that we observed aggregated QA in the QAL@TiO2 samples, we performed an XRD analysis and resonant Raman experiments to identify the crystal structure and the possible presence of polymorphs.68,69 The diffractogram of QA312@TiO2 shows the same characteristics of the TiO2 starting material, maintaining the mixture of anatase and rutile phases after sensitization. No shape changes can be appreciated in the presence of the QA film, supporting that it does not affect the bulk crystal structure of the NPs (Fig. S12†). In QA312@TiO2, no signals attributable to QA are detected, probably due to the small amount.
The QA312@TiO2 resonant Raman spectra show the features of both TiO2 (Raman shifts at 138, 395, 514 and 637 cm−1)70 and of the QA pigment (Fig. S13†). Particularly informative is the single peak at 226 cm−1 suggesting the presence of the γ-polymorph of QA, while the β-polymorph shows a pair of bands in this region.68 In QAL@TiO2 hybrids, the appearance of a fluorescence band of the QA pigment is also observed at wavenumbers above 1000 cm−1. Interestingly, this band is much more intense when QA is physisorbed onto SiO2 instead of TiO2 particles, prepared by the same protection–deprotection protocol (Fig. S13†). This indicates a partial quenching of the QA pigment when physisorbed onto TiO2 with respect to SiO2, consistent with the postulated photoinduced QA → TiO2 electron injection. This encouraged us to exploit QAL@TiO2 in photochemical oxidations.
Photocatalysis with QAL@TiO2 nanoparticles
 | ||
| Fig. 2 (a) Light driven oxidation of 0.2 mM TMB with 1 mg per mL QAL@TiO2 NPs in 2 mL of 0.1 M aqueous acetate buffer, pH 5. (b and c) Images of the photoreactor and of the reaction vials over ten minutes containing QA312@TiO2 (right) and TiO2 control (left). (d) Spectral evolution obtained for photochemical oxidation of TMB with QA312@TiO2 NPs. (e) Plot of the absorbance at 652 nm over time for the NPs investigated. The spectroscopic response was recorded with a plate reader, sampling aliquots of 100 μL of the irradiated solution every minute. (f) Control tests in the presence of scavengers (7.5 mM); the absorbance at 652 nm reported in the histograms refers to the QA16@TiO2 particles after 4 minutes irradiation. (g) Apparent Quantum Yield (AQY%) superimposed to the diffuse reflectance spectrum of QA312@TiO2 NPs. | ||
Light-induced oxidation of TMB was then conducted by employing a custom-made photocatalytic reactor with 16 positions (Fig. 2b), emitting visible light (33 mW cm−2) from the bottom of 8 mL glass vials containing the reaction mixtures (2 mL of 0.2 mM TMB in 0.1 M aqueous acetate buffer, pH 5, and 1 mg mL−1 of NPs). Images of reaction vials are reported in Fig. 2c at increasing irradiation time (in a timeframe of 10 minutes), showing the different activity of QA312@TiO2 NPs (right vial) with respect to the TiO2 control (left vial). A typical outcome for QA312@TiO2 NPs is reported in Fig. 2d, which shows the rising of the broad band centered at 650 nm typical of the charge transfer complex as the oxidized product of TMB (formation of the radical cation TMB˙+ was also confirmed by electron paramagnetic resonance spectroscopy,71 see Fig. S14†). The absorbance at 650 nm can be plotted over time to provide the TMB oxidation outcome for the different materials tested, as shown for some traces in Fig. 2e. The results can be discussed as follows: (i) the QAL@TiO2 nanoparticles drive the photochemical oxidation of TMB with a reactivity that depends on the QA loading (compare traces for QA312@TiO2 and QA16@TiO2 in Fig. 2e); (ii) control with bare TiO2 provides a null reactivity; (iii) QA16@SiO2 – employed as a material to elucidate the activity of the sole QA pigment§ – gives a ten times lower reactivity with respect to QA16@TiO2. These experiments support the role of the QA pigment in light absorption and electron injection into the conduction band of TiO2 as the photoinduced events leading to the charge-separated state responsible for TMB oxidation.61 Moreover, the absence of reactivity under a nitrogen atmosphere suggested the involvement of reactive oxygen species (ROS). This prompted us to further investigate the photochemical reactivity in the presence of ROS scavengers: D-mannitol (scavenger for OH˙), L-tryptophan (scavenger for 1O2), sodium 4,5-dihydroxybenzene-1,3-disulfonate hydrate (Tiron monohydrate) and p-benzoquinone (scavengers for O2˙−), Fig. 2f.74 The results show a major abatement of reactivity in the presence of Tiron monohydrate and p-benzoquinone (BQ), suggesting that superoxide O2˙− may be involved in the oxidation process.
Finally, we evaluated the apparent quantum yield (AQY) of the process by running light-driven TMB oxidation with QA312@TiO2 NPs employing light emitting diodes at 525 (green), 590 (yellow) and 620 nm (red), Table S1.† As shown in Fig. 2g, the AQY reaches 0.043% at 525 nm, while decreasing to 0.031% and 0.016% at 590 and 620 nm, respectively, resembling the diffuse reflectance spectrum of QA312@TiO2, and confirming that the particles are photoactive up to 620 nm. A control test employing irradiation at 740 nm, where the particles do not absorb, provides indeed a null reactivity.
 | ||
| Fig. 3 Light driven oxidation of glycerol with QA312@TiO2 NPs co-catalyzed by TEMPO. General conditions: 40 mM glycerol in water, 7.5 mM TEMPO, 3.0 mg photocatalyst in 1.5 mL, irradiation with red light (620 nm, 33 mW cm−2). | ||
An initial screening with white light (200 mW cm−2) showed that the photocatalytic reactivity was dependent on the QA pigment loading and identified QA312@TiO2 as the most active material at an optimal NP loading of 2 mg mL−1 (Table S3 in the ESI†). Under these conditions, glyceraldehyde (GLAD, estimated market value of 177$ per g)79 was formed as a valuable primary oxidation product after 20 h, with a productivity of 58 μmol gNP−1 h−1 and a selectivity of ca. 80% among solution products as revealed by HPLC analysis where also dihydroxyacetone (DHA), glycolic acid (GA) and formic acid (FA) were detected (control tests confirmed that TEMPO was necessary for the reactivity; TiO2 NPs83 showed a GLA productivity of 12 μmol gNP−1 h−1 and a selectivity of 67%). The preferential formation of GLAD results from the oxidation of the secondary position of glycerol typically observed in the case of photocatalysis with aminoxyl radical catalysts.41
The productivity of GLAD with QA312@TiO2 NPs was verified by also exploiting green (525 nm, 120 mW cm−2; GLAD productivity of 38 μmol gNP−1 h−1), yellow (590 nm, 200 mW cm−2; GLAD productivity of 46 μmol gNP−1 h−1) and red light (620 nm, 33 mW cm−2, GLAD productivity of 47.5 ± 5.0 μmol gNP−1 h−1), still maintaining a selectivity among solution products in the range 77–85%, see Fig. 3, S15 and Table S3.† Under these conditions, the reactivity of bare TiO2 NPs is almost entirely suppressed (GLAD productivity of 1.75 ± 0.25 μmol gNP−1 h−1 with red light), Fig. 3. NPs sensitized with other industrial pigments such as indigo312@TiO2 and cibalakrot312@TiO2 provided a lower reactivity than QA312@TiO2 (2.5 and 9.8 μmol gNP−1 h−1, respectively), Fig. 3.
The photocatalytic activity of QA312@TiO2 was maintained up to five additional cycles for a total of 120 h irradiation (Fig. 3), confirming that NPs sensitized with QA pigment combined photocatalytic activity with pigment stability (indeed, XPS spectra of recovered QA312@TiO2 were superimposable with the ones of the pristine material, Fig. S16†). Over these consecutive runs, 20 turnovers were obtained for GLAD production based on the amount of QA in the QA312@TiO2 material. This performance is in line with the value of 50 obtained for diketopyrrolopyrrole-sensitized TiO2 NPs, but was reported for oxidation of benzyl alcohols with green light.28
Conclusions and perspectives
In this work, we introduced an easy and general procedure for enabling the use of the industrial pigment QA to sensitize TiO2 nanoparticles, and showed that the hybrid nanomaterials provide a boosted photochemical reactivity with respect to the separate components, enabling access to red light exploitation (up to 620 nm). We ultimately used these hybrid composites in combination with a TEMPO co-catalyst in the aqueous oxidation of glycerol to glyceraldehyde, as a significant example of conversion of an abundant and cheap raw material into a high added-value chemical. Glyceraldehyde productivity reaches 47.5 ± 5.0 μmol gNP−1 h−1 with 80% selectivity under red light, with recyclability of the hybrid nanomaterial up to further five times without loss of activity. Turnover number for the QA pigment reaches 20, while the materials show versatility, maintaining activity also under green, yellow and white light irradiation.Besides the facility of preparation of the hybrid nanomaterials, their versatile reactivity can be expanded to other target reactions, since TEMPO can catalyze several organic transformations, while the reductive side can be expanded to proton or carbon dioxide reduction by considering the integration of suitable co-catalysts. These strategies are currently under investigation in our groups, together with a nano-engineering of the semiconductive support.
We thus believe that this work can pave the way for new strategies in the development of industrial pigment-based photocatalytic materials taming redox reactions in the absence of sacrificial electron donors or acceptors.
Experimental section
Synthesis of Boc–QA
312 mg (1 mmol) of QA was suspended in 50 mL of CH2Cl2, and 1.06 g (4.8 mmol) of di-tert-butyl carbonate and 254 mg (2.1 mmol) of 4-dimethyl-aminopyridine were added thereto. The reaction mixture was stirred at room temperature for 48 hours under a nitrogen atmosphere. After the specified time, the reaction mixture showed a weak green fluorescence. The mixture was dried by rotary evaporation. Boc–QA was purified by silica gel column chromatography (the eluent gradient was 80:20 petroleum ether/ethyl acetate until pure ethyl acetate) and the product was obtained as a yellow powder (230 mg, yield 45%). 1H-NMR (400 MHz, chloroform-d): δ 8.75 (s, 1H), 8.41 (dd, J = 8.0, 1.6 Hz, 1H), 7.84 (d, J = 8.6 Hz, 1H), 7.72 (ddd, J = 8.7, 7.0, 1.7 Hz, 1H), 7.38 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 1.75 (s, 9H).
Preparation of QAL@TiO2 NPs
The required amount of Boc–QA and 20 mg of titanium dioxide (Merck, product code 718467) were added to an 8 mL vial, and then 2 mL of ethyl acetate was added. After ultrasonication for 1 hour, the suspension was stirred at 400 rpm overnight. The solvent was then evaporated using a rotary evaporator to obtain Boc–QA@TiO2 NPs. These were then placed in an oven at 150 °C for 30 minutes to convert Boc–QA into QA. The same procedure was used to prepare QA16@SiO2 NPs, using SiO2 from Supelco (60737, 40–63 μm particle size, 60 Å pore size).Characterization of NPs
TEM images were recorded with the help of Federico Caicci at the Biology Department of the University of Padova, using an FEI Tecnai Instrument G2 with a side-mounted Olympus Veleta camera and a bottom-mounted TVIPS F114 camera. Particles were suspended in ethanol before being deposited onto the grid.STEM imaging was performed using a Jeol cold-FEG S/TEM F200 facility, at the Department of Chemical Sciences of the University of Padova.
X-ray photoelectron spectroscopy (XPS) spectra of QA, bare TiO2 and QA312@TiO2 NPs were acquired in a custom-designed UHV-system with a 10−10 mbar base pressure, equipped with a dual Al–Mg anode X-ray source, and an Omicron EA125 electron analyzer. Core level photoemission spectra were collected with a non-monochromatic Al Kα X-ray source (1486.6 eV), using an energy step of 0.1 eV, 0.5 s of dwell time, 20 eV pass energy. The data were collected by smearing the powders on the Cu substrate. The multipeak analysis of the C 1s, N 1s, and Ti 2p photoemission lines was performed via subtraction of Shirley backgrounds and linear combination of Voigt functions using the KolXPD software.
Diffuse reflectance spectra were recorded on a Shimadzu UV-2600i equipped with an ISR-2600Plus integrating sphere. Ultrafine BaSO4 powder supplied by Shimadzu Company was used as a reference. Powders of the samples were pressed in holders of ca. 3 cm diameter and 5 mm depth. The powders of the nanoparticles were mixed with the reflectance reference (BaSO4) in a weight ratio of 3:100, before recording the diffuse reflectance spectra. The spectra were recorded between 1000 nm and 200 nm and transformed through the Kubelka–Munk function.
Powder X-ray diffraction was carried out on a Bruker D8 Advance Plus diffractometer, the diffraction data were acquired in Bragg–Brentano geometry by employing the Cu Kα radiation (Cu anode supplied with 40 kV and a current of 40 mA) and a LYNXEYE XE-T detector with 192 measuring channels in 1D mode.
Raman measurements were conducted using a Micro-Raman setup. An argon ion laser emitting a single-line served as the excitation light source, featuring two primary lines at 488 and 514.5 nm (Spectra Physics Stabilite 2017 with an output power of 1 W). The 488 nm radiation was filtered out, and a half-wave plate was employed to control the polarization of the incident light. Optical density filters were strategically placed on a remotely controlled reel to regulate the intensity of light reaching the sample. The laser beam was coupled to a microscope (Olympus BX 40) and directed onto the sample through a 20× or 50× objective (Olympus SLMPL, NA D 0:75), resulting in a typical spot diameter of 3 or ∼1 μm at the focus, respectively. The back-scattered Raman signal, distinct from Rayleigh scattering, was separated using an edge filter and subsequently analyzed through a 320 mm focal length imaging spectrograph (TRIAX-320 ISA) and a liquid nitrogen-cooled CCD camera (Spectrum One, Jobin Yvon). Each Raman spectrum was recorded utilizing the 50× microscope objective, and the spectrograph slit was set at 100 μm. Spectra were generated by averaging ten repeated measurements, each with an acquisition time of 10 seconds (10 seconds × 10 times).
Photocatalysis
Photocatalysis experiments were conducted in 8 mL commercial glass vials under stirring (600 rpm), using: (i) a homemade photoreactor with 16 positions emitting white light (33 mW cm−2 corresponding to 0.33 sun irradiation); or (ii) a homemade photoreactor with LED at different wavelengths: red light (620 nm, 33 mW cm−2); yellow light (590 nm, 200 mW cm−2); green light (525 nm, 120 mW cm−2); or (iii) a Godox-SL600IID lamp as the light source, and the irradiance (radiant power per unit area) was calculated at each wavelength using a calibrated silicon photodiode. The distance to the sample was 3.5 cm (2 sun irradiation, irradiance = 200 mW cm−2). Further details are available in the ESI.†Oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB)
1 mg TMB was dissolved in 1 mL DMSO to prepare a 4 mM stock solution in DMSO, this was then diluted into 19 mL of 0.1 M acetate buffer (pH 5) to get 0.2 mM TMB in acetate buffer. For every reaction, 2 mL of this solution was transferred into 8 mL vials containing 2 mg of nanoparticles, then ultrasonicated for 1 hour. The vial was then subjected to irradiation under white light (0.33 sun intensity), and aliquots of 100 μL were sampled every minute to be analyzed with a UV-vis Infinite 200 Pro plate reader (wavelength range from 300 to 800 nm, step 2 nm, read 10 flashes for every step, settle time 1 ms). The EPR experiment was recorded on a Bruker ESR5000 operating at a microwave frequency of 9.4 GHz. The sample was 1 mM TMB in 0.1 M acetate buffer (pH 5), prepared by adding 100 μL of 4 mM TMB DMSO solution into 300 μL 0.1 M acetate buffer with 1 mg per mL QA16@TiO2 nanoparticles.Oxidation of glycerol
92.1 mg of glycerol (final concentration 40 mM) and 29.3 mg of TEMPO (final concentration 7.5 mM) were added into a 25 mL volumetric flask, filled with water. 1.5 mL of this solution was transferred into an 8 mL vial containing 3 mg of the nanoparticle catalysts (2 mg mL−1), then inserted in an ultrasonic bath for 1 hour. The vial was subjected to irradiation for 20 hours. After the photochemical reaction, the particles were separated by centrifugation, and the solution was analyzed by HPLC.Analysis of the oxidation products of glycerol
For quantification of the products of glycerol oxidation, high performance liquid chromatography (HPLC) was used. The instrument used was an Agilent 1260 Infinity II, equipped with a diode array detector; the instrument was equipped with a Hi-Plex H column 300 × 7.7 mm. Conditions of analysis: [H2SO4] = 5 mM in Milli-Q water as the eluent, isocratic gradient, flow rate 0.7 mL min−1, injected volume 20 μL, temperature 50 °C. Different concentrations of glyceraldehyde, glycolic acid, formic acid, dihydroxyacetone, and glyceric acid aqueous solutions were configured to obtain a standard curve. The solution of the photoreaction product was then centrifuged to separate the nanoparticles obtained. The solution was directly detected using HPLC, and the product concentration was converted between the detection area and the standard curve.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. Y.: investigation, data analysis; M. N.: investigation, data analysis; A. B.: investigation, data analysis; R. B.: investigation; R. S.: investigation, data analysis; S. A.: data analysis, supervision; L. Đ.: conceptualization, supervision, writing, funding acquisition; A. S.: conceptualization, supervision, writing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Italian Ministero dell'Università e della Ricerca (projects “PROMETEO” 2022KPK8WM to A. S.), by the European Union – Next Generation UE (project “PHOTOCORE” P2022ZSPWF to A. S.), and by the European Union (ERC, PhotoDark, 101077698, to L. Đ.). Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. We thank Dr Anna Fortunato, Prof. Francesca Arcudi and Samuel Pressi for support in the diffuse reflectance spectra and in thermogravimetric analysis, and Dr Eric Daniel Glowacki (Brno University of Technology, Czech Republic) for stimulating discussions and for generously providing some samples of dyes.Notes and references
- S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 Search PubMed.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 Search PubMed.
- A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 8–11 CrossRef.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 Search PubMed.
- R. Cao, D. Xiao, M. Wang, Y. Gao and D. Ma, Appl. Catal., B, 2024, 341, 123357 CrossRef CAS.
- D. Spasiano, R. Marotta, S. Malato, P. Fernandez-Ibañez and I. Di Somma, Appl. Catal., B, 2015, 170–171, 90–123 CrossRef CAS.
- M. A. Hassaan, M. A. El-Nemr, M. R. Elkatory, S. Ragab, V. C. Niculescu and A. El Nemr, Principles of Photocatalysts and Their Different Applications: A Review, Springer International Publishing, 2023, vol. 381 Search PubMed.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- D. A. Kader and S. J. Mohammed, RSC Adv., 2023, 13, 26484–26508 RSC.
- C. Bie, L. Wang and J. Yu, Chem, 2022, 8, 1567–1574 CAS.
- N. Manfredi, B. Cecconi, V. Calabrese, A. Minotti, F. Peri, R. Ruffo, M. Monai, I. Romero-Ocaña, T. Montini, P. Fornasiero and A. Abbotto, Chem. Commun., 2016, 52, 6977–6980 RSC.
- L. Zani, M. Melchionna, T. Montini and P. Fornasiero, J. Phys.: Energy, 2021, 3, 031001 CAS.
- X. Li, X. Lv, Q. Zhang, B. Huang, P. Wang, X. Qin, X. Zhang and Y. Dai, J. Colloid Interface Sci., 2018, 525, 136–142 CrossRef CAS PubMed.
- V. Nikolaou, C. Govind, E. Balanikas, J. Bharti, S. Diring, E. Vauthey, M. Robert and F. Odobel, Angew. Chem., Int. Ed., 2024, 63, e202318299 CrossRef CAS.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- V. Nikolaou, P. B. Pati, H. Terrisse, M. Robert and F. Odobel, EES Catal., 2024, 1314–1319 RSC.
- A. E. B. S. Stone, A. Fortunato, X. Wang, E. Saggioro, R. Q. Snurr, J. T. Hupp, F. Arcudi and L. Dordevic, Adv. Mater., 2024, 2408658 Search PubMed.
- D. Romito, C. Govind, V. Nikolau, R. J. Fernandez-Teran, A. Stoumpidi, E. Agapaki, G. Charalambidis, S. Diring, E. Vauthey, A. G. Coutsolelos and F. Odobel, Angew. Chem., Int. Ed., 2024, e202318868 CAS.
- D. Franchi and Z. Amara, ACS Sustain. Chem. Eng., 2020, 8, 15405–15429 CrossRef CAS.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS.
- Y. Zhang, Z. Wang and X. Lang, Catal. Sci. Technol., 2017, 7, 4955–4963 RSC.
- X. Li, H. Hao and X. Lang, J. Colloid Interface Sci., 2021, 581, 826–835 CrossRef CAS PubMed.
- F. Huang, H. Hao, W. Sheng and X. Lang, Chem. Eng. J., 2021, 423, 129419 CrossRef CAS.
- M. Koohgard, Z. Hosseinpour, A. M. Sarvestani and M. Hosseini-Sarvari, Catal. Sci. Technol., 2020, 10, 1401–1407 RSC.
- L. Ren, M. M. Yang, C. H. Tung, L. Z. Wu and H. Cong, ACS Catal., 2017, 7, 8134–8138 CrossRef CAS.
- L. Ren and H. Cong, Org. Lett., 2018, 20, 3225–3228 CrossRef CAS.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- M. R. K. Estahbanati, M. Feilizadeh, F. Attar and M. C. Iliuta, React. Chem. Eng., 2021, 6, 197–219 RSC.
- Y. Xiao, M. Wang, D. Liu, J. Gao, J. Ding, H. Wang, H. B. Yang, F. Li, M. Chen, Y. Xu, D. Xu, Y. X. Zhang, S. Fang, X. Ao, J. Wang, C. Su and B. Liu, Angew. Chem., Int. Ed., 2024, 63, e202319685 CrossRef CAS PubMed.
- D. Bruggeman, A. Laoprte, R. J. Detz, S. Mathew and J. N. H. Reek, Angew. Chem., Int. Ed., 2022, 61, e202200175 CrossRef CAS.
- T. Gobbato, G. A. Volpato, A. Sartorel and M. Bonchio, Chem. Sci., 2023, 14, 12402–12429 RSC.
- H. Zhao, Z. Zhou, X. Feng, C. Liu, H. Wu, W. Zhou and H. Wang, Nano Res., 2023, 16, 8809–8816 CrossRef CAS.
- K. Zhang, X. Dong, B. Zeng, K. Xiong and X. Lang, J. Colloid Interface Sci., 2023, 651, 622–632 CrossRef CAS.
- B. F. Buksh, S. D. Knutson, J. V. Oakley, N. B. Bissonnette, D. G. Oblinsky, M. P. Schwoerer, C. P. Seath, J. B. Geri, F. P. Rodriguez-Rivera, D. L. Parker, G. D. Scholes, A. Ploss and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6154–6162 CrossRef CAS.
- A. H. Schade and L. Mei, Org. Biomol. Chem., 2023, 21, 2472–2485 RSC.
- D. Cappelletti, M. Barbieri, A. Aliprandi, M. Maggini and L. Đorđević, Nanoscale, 2024, 16, 9153–9168 RSC.
- Y. Sheng, W. Li, Y. Zhu and L. Zhang, Appl. Catal., B, 2021, 298, 120585 CrossRef CAS.
- L. Liu, J. Liu, S. Zong and Z. Huang, Int. J. Hydrogen Energy, 2022, 47, 39486–39498 Search PubMed.
- Q. Zhang, L. Jiang, J. Wang, Y. Zhu, Y. Pu and W. Dai, Appl. Catal., B, 2020, 277, 119122 CrossRef CAS.
- Q. Gao, J. Xu, Z. Wang and Y. Zhu, Appl. Catal., B, 2020, 271, 118933 CrossRef CAS.
- J. Wang, W. Shi, D. Liu, Z. Zhang, Y. Zhu and D. Wang, Appl. Catal., B, 2017, 202, 289–297 CrossRef CAS.
- J. Wang, D. Liu, Y. Zhu, S. Zhou and S. Guan, Appl. Catal., B, 2018, 231, 251–261 CrossRef CAS.
- W. Wei, Z. Wei, D. Liu and Y. Zhu, Appl. Catal., B, 2018, 230, 49–57 CrossRef CAS.
- E. Rossin, Y. Yang, M. Chirico, G. Rossi, P. Galloni and A. Sartorel, Energy Adv., 2024, 3, 1894–1904 RSC.
- M. Sytnyk, E. D. Głowacki, S. Yakunin, G. Voss, W. Schöfberger, D. Kriegner, J. Stangl, R. Trotta, C. Gollner, S. Tollabimazraehno, G. Romanazzi, Z. Bozkurt, M. Havlicek, N. S. Sariciftci and W. Heiss, J. Am. Chem. Soc., 2014, 136, 16522–16532 CrossRef CAS.
- M. Forchetta, V. Baia, F. Sabuzi, V. Conte and P. Galloni, Eur. J. Org. Chem., 2023, 26, e202300523 CrossRef CAS.
- H. Ding, H. Sun and Y. Shan, J. Photochem. Photobiol., A, 2005, 169, 101–107 CrossRef CAS.
- C.-J. Lin, Y.-H. Liou, S.-Y. Chen and M.-C. Tsai, Sustain. Environ. Res., 2012, 22, 167–172 CAS.
- T. Marangoni, S. A. Mezzasalma, A. Llanes-Pallas, K. Yoosaf, N. Armaroli and D. Bonifazi, Langmuir, 2011, 27, 1513–1523 CrossRef CAS.
- Y. Yang, G. A. Volpato, E. Rossin, N. Peruffo, F. Tumbarello, C. Nicoletti, R. Bonetto, L. Paoloni, P. Umari, E. Colusso, L. Dell'Amico, S. Berardi, E. Collini, S. Caramori, S. Agnoli and A. Sartorel, ChemSusChem, 2023, e202201980 CrossRef CAS PubMed.
- G. A. Volpato, M. Marasi, T. Gobbato, F. Valentini, F. Sabuzi, V. Gagliardi, A. Bonetto, A. Marcomini, S. Berardi, V. Conte, M. Bonchio and S. Caramori, Chem. Commun., 2020, 56, 2248–2251 RSC.
- G. A. Volpato, E. Colusso, L. Paoloni, M. Forchetta, F. Sgarbossa, V. Cristino, M. Lunardon, S. Berardi, S. Caramori, S. Agnoli, F. Sabuzi, P. Umari, A. Martucci, P. Galloni and A. Sartorel, Photochem. Photobiol. Sci., 2021, 20, 1243–1255 CrossRef CAS PubMed.
- Y. Yu, B. Mao, A. Geller, R. Chang, K. Gaskell, Z. Liu and B. W. Eichorn, Phys. Chem. Chem. Phys., 2014, 16, 11633–11639 RSC.
- S. Chenakin and N. Kruse, J. Catal., 2018, 358, 224–236 CrossRef CAS.
- H. Yoon, D. Kim, M. Park, J. Kim, J. Kim, W. Srituravanich, B. Shin, Y. Jung and S. Jeon, J. Phys. Chem. C, 2018, 122, 12114–12121 CrossRef CAS.
- J. L. Peper, N. E. Gentry, B. Boudy and J. M. Mayer, Inorg. Chem., 2022, 61, 767–777 CrossRef CAS.
- T. Salzillo, A. Rivalta, N. Castagnetti, S. D'Agostino, M. Masino, F. Grepioni, E. Venuti, A. Brillante and A. Girlando, CrystEngComm, 2019, 21, 3702–3708 RSC.
- A. Giunchi, L. Pandolfi, R. G. D. Valle, T. Salzillo, E. Venuti and A. Girlando, Cryst. Growth Des., 2023, 23, 6765–6773 CrossRef CAS.
- H. C. Choi, Y. M. Jung and S. B. Kim, Vib. Spectrosc., 2005, 37, 33–38 CrossRef CAS.
- P. D. Josephy, T. Eling and R. P. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CrossRef CAS.
- (a) F. Arcudi, L. Đorđević, N. Schweitzer, S. I. Stupp and E. A. Weiss, Nat. Chem., 2022, 14, 1007–1013 CrossRef CAS PubMed; (b) L. Đorđević, T. J. Jaynes, H. Sai, M. Barbieri, J. E. Kupferberg, N. A. Sather, S. Weigand and S. I. Stupp, Adv. Mater., 2025, 2418137 CrossRef.
- C. Wang, ACS Sens., 2024, 9, 3808–3809 CrossRef CAS.
- Y. Jiao, L. Đordevic, H. Mao, R. M. Young, T. Jaynes, H. Chen, Y. Qiu, K. Cai, L. Zhang, X. Chen, Y. Feng, M. R. Wasielewski, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 8000–8010 CrossRef CAS PubMed.
- S. Bang, R. Snoeckx and M. S. Cha, ChemSusChem, 2024, 17, e202300925 CrossRef CAS PubMed.
- W. Chen, L. Zhang, L. Xu, Y. He, H. Pang, S. Wang and Y. Zou, Nat. Commun., 2024, 15, 2420 CrossRef CAS PubMed.
- S. Zhong, B. He, S. Wei, R. Wang, R. Zhang and R. Liu, Appl. Catal., B, 2025, 362, 124743 CrossRef CAS.
- Z. Huang, H. Ren, J. Guo, Y. Tang, D. Ye, J. Zhang and H. Zhao, Appl. Catal., B, 2024, 351, 123986 CrossRef CAS.
- H. H. Kuo, T. G. Vo and Y. J. Hsu, J. Photochem. Photobiol., C, 2024, 58, 100649 CrossRef CAS.
- M. R. K. Estahbanati, M. Feilizadeh, F. Attar and M. C. Iliuta, Ind. Eng. Chem. Res., 2020, 59, 22330–22352 CrossRef.
- Y. Liu, B. Zhang, D. Yan and X. Xiang, Green Chem., 2024, 26, 2505–2524 RSC.
- P. Limpachanangkul, P. Nimmmanterdwong, L. Liu, M. Hunsom, K. Pruksathorn, P. Piumsomboon and B. Chalermsinsuwan, Sci. Rep., 2023, 13, 14936 CrossRef CAS.
- M. Liu, H. Liu, N. Li, C. Zhang, J. Zhang and F. Wang, ChemSusChem, 2022, 15, e202201068 CrossRef CAS.
- X. Liu, Y. Zou and J. Jiang, Appl. Catal., B, 2024, 350, 123927 CrossRef CAS.
- E. Tacchi, G. Rossi, M. Natali, L. Đorđević and A. Sartorel, Adv. Sustain. Syst., 2024, 2400538 Search PubMed.
- K. Meng, J. Zhang, B. Cheng, X. Ren, Z. Xia and F. Xu, Adv. Mater., 2024, 36, 2406460 CrossRef CAS PubMed.
- H. Bajpai, T. R. Nivedhitha, E. Dais, S. S. Kanungo and C. S. Gopinath, J. Catal., 2024, 437, 115644 CrossRef CAS.
- H. Bajpai, I. Chauhan, K. N. Salgaonkar, N. B. Mhamane and C. Gopinath, RSC Sustain., 2023, 1, 481–493 RSC.
- T. R. Nivedhitha, H. Bajpai, J. V. Oommen, A. Abraham, I. Chauhan and C. S. Gopinath, ACS Sustain. Chem. Eng., 2024, 12, 14841–14853 CrossRef CAS.
- Y. He, Y. Wang, J. Qian, K. Xu, B. Lu, S. Tang, Y. Liu and J. Shen, Appl. Catal., B, 2025, 361, 124565 CrossRef CAS.
- D. Liu, J. C. Liu, W. Cai, J. Ma, H. B. Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef.
- H. Tateno, S. Y. Chen, Y. Miseki, T. Nakajima, T. Mochizuki and K. Sayama, ACS Sustain. Chem. Eng., 2022, 10, 7586–7594 CrossRef CAS.
- Y. Miao, Z. Li and M. Shao, ChemCatChem, 2024, 16, 1–9 CrossRef.
- Á. Balog, E. Kecsenovity, G. F. Samu, J. He, D. Fekete and C. Janáky, Nat. Catal., 2024, 7, 522–535 CrossRef.
- X. Zhao, J. Yang and J. Cheng, J. Org. Chem., 2023, 88, 540–547 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of calculations and additional tables and figures. See DOI: https://doi.org/10.1039/d5ta01970b |
| ‡ If oxidation of the radical cation of TMB proceeds further towards a second step, formation of the yellow diimine occurs, showing absorption maximum at 450 nm. |
| § We used a QA@SiO2 hybrid as a control test to elucidate the sole role of QA, since this was prepared in the same way as the QA@TiO2 material, while SiO2 is an inert support. Direct use of QA is more challenging given the necessity to properly disperse QA. |
| This journal is © The Royal Society of Chemistry 2025 |