
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning hydrocarbon selectivity in electrochemical CO2 reduction via copper-porphyrin immobilization on carbon nanotubes†
Alaleh
Esfandiari
 a,
Maryam
Abdinejad
*bc and
Ali
Seifitokaldani
*a
a,
Maryam
Abdinejad
*bc and
Ali
Seifitokaldani
*a
aDepartment of Chemical Engineering, McGill University, Montreal, Quebec, Canada. E-mail: ali.seifitokaldani@mcgill.ca
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 02139 Cambridge, MA, USA
cDepartment of Energy Conversion and Storage, Technical University of Denmark, Fysikvej, 2800 Kgs Lyngby, Denmark. E-mail: marab@dtu.dk
First published on 19th May 2025
Abstract
Electrochemical CO2 conversion using renewable energy offers a promising pathway for producing value-added chemicals with zero emissions. In this study, copper porphyrin (Cu-TMCPP) molecules are immobilized on multi-wall carbon nanotubes (MWCNTs) to form Cu-TMCPP/CNTs, which serve as a tunable, heterogeneous electrocatalyst for electrocatalytic CO2 reduction (ECR). A systematic comparison of the synthesized catalyst with stacked Cu-TMCPP and a physical mixture of Cu-TMCPP with MWCNTs (Cu-TMCPP + CNT) showed that the Cu-TMCPP/CNT catalyst suppressed the hydrogen evolution reaction (HER) to a large extent and improved selectivity towards hydrocarbon (total FE: 75.68%), mainly CH4 (FE: 37.3%), at a potential of −1.08 V versus the reversible hydrogen electrode (RHE) with a partial current density of 91.8 mA cm−2. The in-depth mechanistic analysis of in situ and ex situ X-ray adsorption spectroscopy (XAS) and electrochemical characterization illustrated the presence of ultrathin copper porphyrin blocks immobilized on CNTs, presumably located at the Helmholtz layer with a high oxidation state and the co-existence of Cu2+/Cu0 under reaction conditions, which are responsible for improved selectivity towards hydrocarbons. This research provides insights into the immobilization impact of molecular catalysts and the advantage of the consequent high capacitance double layer in suppressing the HER, which enhances electron transfer, thereby improving heterogeneous electrocatalytic CO2 performance.
Introduction
The rising risks associated with global warming necessitate the urgent development of effective strategies to reduce atmospheric CO2 concentrations. Electrocatalytic CO2 reduction (ECR) to value-added products is considered a promising solution, enabling carbon-neutral goals and sustainable production of chemicals and fuels using renewable energy sources.1–5 Electrocatalysts are essential to overcome the inherent low reactivity of CO2 and facilitate its bond cleavage to produce valuable reduced products.6,7 However, the industrial scale implementation of ECR systems is hindered by the low selectivity and activity of electrocatalysts.8–10 A major challenge in achieving high selectivity and efficiency in ECR, especially at high current density, is the competing hydrogen evolution reaction (HER).11 Single-atom catalysts (SACs) present a promising approach for ECR because they suppress H–H coupling, thereby mitigating the HER12–14 and improving selectivity.15The synthesis of SACs benefits from the use of molecular structures, which enable tuning of electron distribution and enhanced electron transfer through the modification of functional groups.16 For example, metalated porphyrins have shown considerable promise as efficient electrocatalysts for ECR.17 To date, various transition metal complexes, including Mn,18 Ni,19,20 Co,21,22 Cu,23 and Fe,22,24–26 have been investigated as metal centres of metalated porphyrins. Among them, copper-porphyrins stand out as promising candidates for ECR owing to their vast product distribution;27 however, they have been insignificantly investigated compared with other metal-based systems.
Photo- and electrochemical CO2 conversion studies have demonstrated that the higher oxidation states of metal catalysts, such as copper (Cu+/Cu2+), play a crucial role in enhancing product selectivity toward hydrocarbons during ECR.28–30 The metal centre, as the main active site of porphyrin catalysts, must be active, stable, and earth-abundant for effective CO2 reduction. The higher oxidation states of copper fulfil this requirement, enabling enhanced catalytic performance.26,31–33 In copper-porphyrin systems, the active Cu centre maintains a high oxidation state,23,34 which improves the selectivity of ECR toward hydrocarbon production. Leveraging the high tunability of porphyrins by incorporating electron-withdrawing groups has been shown to stabilize the high oxidation state of copper. This stabilization has been reported to result in higher catalytic activity for ECR and a positive shift in the onset potential.34–37
Traditional metalated porphyrins typically facilitate two-electron transfer processes during ECR, predominantly producing CO and formate as the primary products.19,38,39 In this study, to extend beyond the two-electron transfer limit, we synthesized Cu-SACs with copper as the active site for ECR, integrated into a 5,10,15,20-tetrakis(4-methoxycarbonylphenyl)porphyrin (TMCPP) framework, modified with carbonyl methoxy electron-withdrawing groups. To facilitate multi-electron transfer, the heterogenization of molecular catalysts offers enhanced stability and higher activity than their homogeneous analogs. Thus, to promote efficient electron transfer and improve product selectivity, TMCPP was immobilized onto multi-walled carbon nanotubes (MWCNTs)26,40,41via non-covalent π–π interactions, forming Cu-TMCPP/CNT. This immobilization strategy has been shown to enhance the conductivity, stability, and selectivity, ensuring continuous electron transfer from the current collector to the active site, thereby improving the overall catalytic performance and durability.19,42 To systematically evaluate the impact of this heterogenization, we compared three configurations: Cu-TMCPP alone (without immobilization), Cu-TMCPP physically mixed with MWCNTs (Cu-TMCPP + CNT), and Cu-TMCPP immobilized onto MWCNTs (Cu-TMCPP/CNT). Among the tested configurations, Cu-TMCPP/CNT exhibited better performance by facilitating an eight-electron transfer pathway that enhanced methane (CH4) selectivity while effectively suppressing the HER during ECR. This study highlights the effective role of immobilization in optimizing the metal-electrode interface and provides insights for the development of Cu-porphyrin-based electrocatalysts with improved selectivity, stability, and scalability in the future.
Results and discussion
Preparation and characterization of the catalyst
Fig. 1a illustrates the schematic of the synthesis method for the designed Cu-TMCPP and Cu-TMCPP/CNT electrocatalysts. The synthesis of Cu-TMCPP involved reacting CuSO4·5H2O with TMCPP in a chloroform–ethanol solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) using N,N-diisopropylethylamine (DIPEA) as a base (see ESI† for more information). The mixture was refluxed for 2 hours under vigorous stirring at 60 °C, followed by extractions with chloroform and distilled water three times to remove impurities. The purified product was then dried under vacuum and heated at 80 °C for 2 hours in the oven to remove any residue. The successful metalation of TMCPP with copper was confirmed via mass spectrometry and UV-Vis spectroscopy (Fig. S1a–e†), which demonstrated a characteristic shift in the Soret band (from 420 nm to 414 nm) and the appearance of two Q-bands instead of four, indicating the proper insertion of the copper center into the porphyrin macrocycle. The results agree with those of previous studies.19,22 To immobilize the Cu-TMCPP molecule onto MWCNTs, a mixture of Cu-TMCPP and MWCNTs in a 2:1 ratio was dispersed in dimethylformamide (DMF) and sonicated for 1 hour. The resulting suspension was stirred overnight and then drop-casted onto a glassy carbon electrode (for H-cell studies) or carbon paper gas diffusion layer (for flow-cell experiments) to serve as the working electrode. In this study, an H-cell was used for fundamental electrocatalytic analysis, while a flow cell was employed to assess electrocatalytic performance, enhancing mass transport capabilities and CO2 diffusion.26,43,44
:1) using N,N-diisopropylethylamine (DIPEA) as a base (see ESI† for more information). The mixture was refluxed for 2 hours under vigorous stirring at 60 °C, followed by extractions with chloroform and distilled water three times to remove impurities. The purified product was then dried under vacuum and heated at 80 °C for 2 hours in the oven to remove any residue. The successful metalation of TMCPP with copper was confirmed via mass spectrometry and UV-Vis spectroscopy (Fig. S1a–e†), which demonstrated a characteristic shift in the Soret band (from 420 nm to 414 nm) and the appearance of two Q-bands instead of four, indicating the proper insertion of the copper center into the porphyrin macrocycle. The results agree with those of previous studies.19,22 To immobilize the Cu-TMCPP molecule onto MWCNTs, a mixture of Cu-TMCPP and MWCNTs in a 2:1 ratio was dispersed in dimethylformamide (DMF) and sonicated for 1 hour. The resulting suspension was stirred overnight and then drop-casted onto a glassy carbon electrode (for H-cell studies) or carbon paper gas diffusion layer (for flow-cell experiments) to serve as the working electrode. In this study, an H-cell was used for fundamental electrocatalytic analysis, while a flow cell was employed to assess electrocatalytic performance, enhancing mass transport capabilities and CO2 diffusion.26,43,44
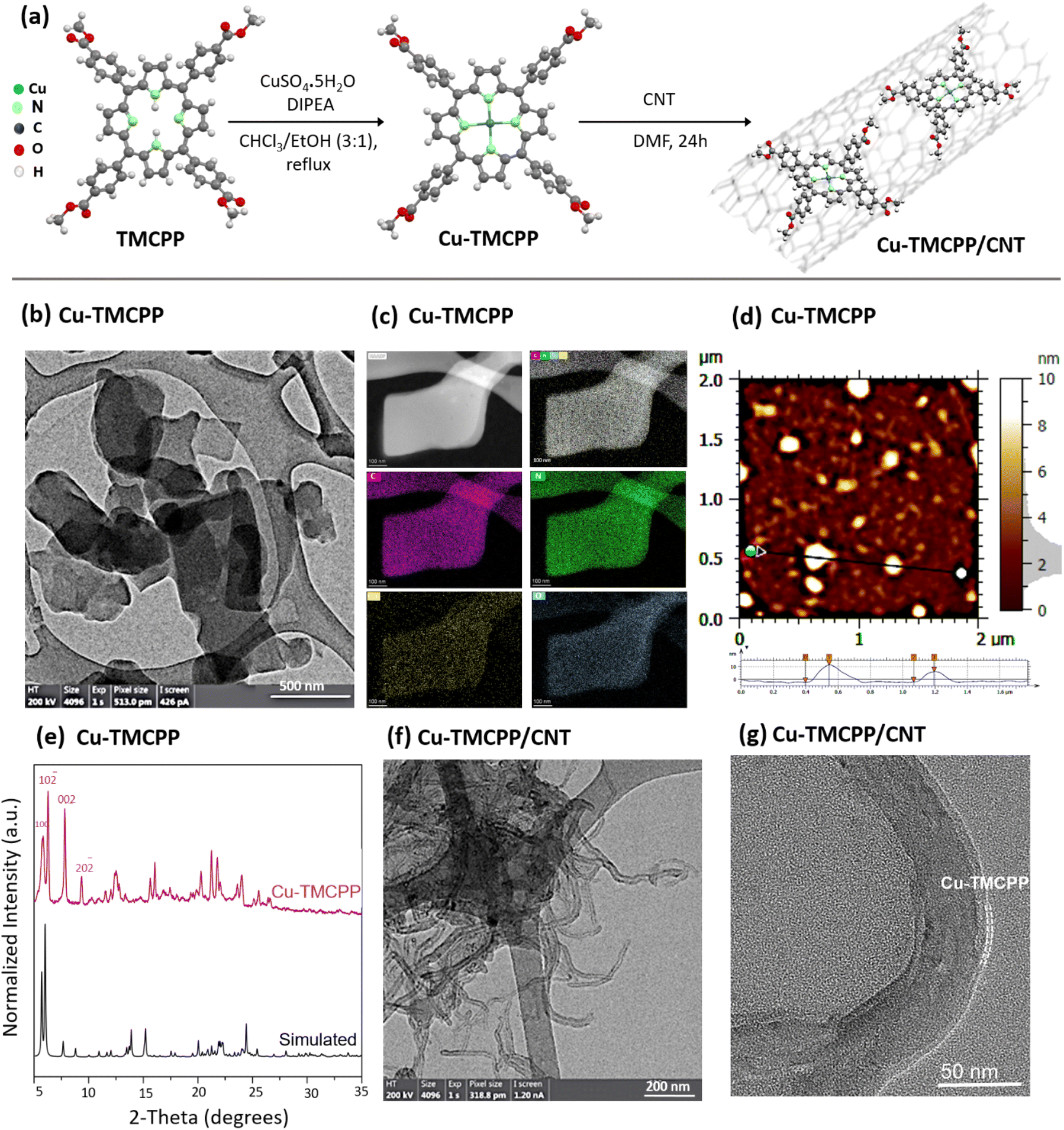 | ||
| Fig. 1 (a) Schematic representation of the synthesis method for copper(II)-5,10,15,20-tetrakis(4-methoxycarbonylphenyl)porphyrin (Cu-TMCPP) and its immobilized counterpart on multi-walled carbon nanotubes (MWCNTs) to form Cu-TMCPP/CNTs. (b) High-resolution transmission electron microscopy (HR-TEM) image of Cu-TMCPP showing its sheet-like morphology. (c) Energy-dispersive X-ray (EDX) elemental mapping of Cu-TMCPP, illustrating the homogeneous distribution of copper atoms. (d) Atomic force microscopy (AFM) of Cu-TMCPP. (e) X-ray diffraction (XRD) patterns of Cu-TMCPP compared with simulated XRD data, confirming its crystalline structure. (f and g) HR-TEM images of Cu-TMCPP/CNTs, highlighting the immobilization of Cu-TMCPP onto MWCNTs via non-covalent interactions. | ||
To gain insight into the structural and morphological properties of the synthesized catalysts, high resolution transmission electron microscopy (HR-TEM), energy-dispersive X-ray spectroscopy (EDX), atomic force microscopy (AFM), and X-ray diffraction (XRD) analyses were conducted. Fig. 1b shows HR-TEM images of Cu-TMCPP, exhibiting a sheet-like morphology probably attributed to the stacking of monomers into a layered structure driven by π–π stacking interactions, which is consistent with previous studies on porphyrin-based systems.45,46 As shown in Fig. 1c, EDX elemental mapping demonstrates the homogeneous distribution of single Cu atoms within the Cu-TMCPP sheets, further supporting the successful synthesis of the material. AFM analysis (Fig. 1d and S2a†) confirms that Cu-TMCPP sheets have a mean diameter of approximately 84.6 nm and an average thickness of 4.8 nm, corresponding to about five stacked Cu-TMCPP layers, as predicted from the proposed molecular structure.46–48 The XRD study (Fig. 1e) further shows the ordered nanosheet structure of Cu-TMCPP crystal, with distinct low-angle peaks.48 Simulated XRD patterns based on the structural model (Fig. S2b†) align well with the experimental XRD results, suggesting that the nanosheets are composed of interconnected single-crystal Cu-TMCPP units.
In the case of the Cu-TMCPP/CNT, TEM images (Fig. 1f and g) show ultra-thin porphyrin layers immobilized onto CNTs, forming wrinkle-like structures that confirm the successful anchoring of Cu-TMCPP onto CNTs via non-covalent π–π interactions.19,49 In contrast, TEM analysis of the physical mixture of Cu-TMCPP and CNTs (Cu-TMCPP + CNT; Fig. S2c–i†) shows aggregated structures lacking the uniformity observed in Cu-TMCPP/CNT. This comparison highlights the structural advantages of immobilization, where Cu-TMCPP layers maintain close contact with CNTs, enhancing their conductive and catalytic properties.
To investigate the chemical and electronic states of the catalysts, X-ray photoelectron spectroscopy (XPS) was performed. Full XPS survey spectrum of the Cu-TMCPP and Cu-TMCPP/CNT shows the presence of Cu, C, N and O elements, as depicted in Fig. 2a. The N 1s XPS spectrum for Cu-TMCPP and porphyrin (TMCPP) presented in Fig. 2b deconvoluted into three peaks related to pyridinic N, pyrrolic N and graphitic N. Pyridinic N attributed to ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– has a positive shift of 0.61 eV after the insertion of Cu stemming from homogenous electron distribution and delocalization in N. It has also been revealed from the N 1s XPS spectrum that the distinct peak related to pyrrolic N attributed to –NH– weakens for Cu-TMCPP, confirming the incorporation of Cu2+ with porphyrin.50
N– has a positive shift of 0.61 eV after the insertion of Cu stemming from homogenous electron distribution and delocalization in N. It has also been revealed from the N 1s XPS spectrum that the distinct peak related to pyrrolic N attributed to –NH– weakens for Cu-TMCPP, confirming the incorporation of Cu2+ with porphyrin.50
 | ||
| Fig. 2 (a) X-ray photoelectron spectroscopy (XPS) survey comparison of Cu-TMCPP, Cu-TMCPP + CNTs, Cu-TMCPP/CNTs and CNTs. (b) XPS spectra of N 1s fitting comparison of TMCPP, Cu-TMCPP, Cu-TMCPP + CNTs and Cu-TMCPP/CNTs. (c) XPS spectra of Cu 2p fitting of Cu-TMCPP, Cu-TMCPP + CNTs and Cu-TMCPP/CNTs. (d) Fourier transform of extended X-ray absorption fine structure (FT−EXAFS) spectra comparison of Cu-TMCPP, Cu-TMCPP + CNTs, Cu-TMCPP/CNTs, CuO, Cu2O and Cu. (e) X-ray absorption near edge structure (XANES) spectra characterization at the Cu-K edge of comparison of Cu-TMCPP, Cu-TMCPP + CNTs, Cu-TMCPP/CNTs, CuO, Cu2O and Cu. (f) First derivative of XANES spectra comparison of Cu-TMCPP, Cu-TMCPP + CNTs, Cu-TMCPP/CNT, CuO, Cu2O and Cu. | ||
The Cu 2p XPS spectrum was deconvoluted for CNT, Cu-TMCPP, Cu-TMCPP + CNT and Cu-TMCPP/CNT, as illustrated in Fig. 2c and S3a.† Cu-TMCPP exhibits 2 main peaks at 935.20 eV and 955.17 eV belonging to 2p3/2 and 2p1/2 peaks, respectively. The two satellite peaks at 944.45 eV and 963.77 eV were attributed to the Cu2+ oxidation state stemming from coordination with nitrogen in the TMCPP structure. The comparison of the Cu 2p XPS spectrum of Cu-TMCPP, Cu-TMCPP + CNT and Cu-TMCPP/CNT revealed a shift to a higher binding energy for Cu-TMCPP/CNT, which can be attributed to π–π interactions between CNT and Cu-TMCPP, causing charge delocalization.51 The deconvolution of C 1s and O 1s are reported in Fig. S3c–j† for CNT, Cu-TMCPP, Cu-TMCPP + CNT and Cu-TMCPP/CNT. It can be observed in the C 1s spectra (Fig. S3e†) that the shake-up (π–π*) transition peak intensity in Cu-TMCPP/CNT decreased compared to its counterpart in CNT, suggesting disturbance of the aromatic C structure owing to immobilization.52,53
Fig. 2d, e and S4a† illustrate both extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge spectroscopy (XANES) of the Cu K-edge for Cu-TMCPP, Cu-TMCPP/CNT and Cu-TMCPP + CNT. Cu, CuO, and Cu2O were used as reference samples to investigate the coordination chemistry. The coordination of copper in Cu-TMCPP with N was confirmed with a signal at ∼1.46 Å, as depicted in Fig. 2d. In these normalized Cu K-edge XANES spectra, there is no peak related to Cu–Cu coordination in all samples, showing single atom copper with a high oxidation state inside the porphyrin structure and confirming the nonexistence of aggregated copper.45 Additionally, there is a shift toward higher energy in the XANES spectrum as depicted in Fig. 2e, which shows a higher oxidation state for the Cu-TMCPP, Cu-TMCPP/CNT and Cu-TMCPP + CNT samples compared with the Cu2O and Cu references. The oxidation states of copper in Cu-TMCPP, Cu-TMCPP/CNT and Cu-TMCPP + CNT samples calculated by linear combination fitting using the derivative of normalized adsorption are Cu1.85+, Cu2.02+ and Cu1.99+, respectively, which shows a higher oxidation state for Cu-TMCPP/CNT compared to the other catalysts. A thorough analysis of the first derivative of XANES, shown in Fig. 2f, for all samples validates the aforementioned oxidation state.16
Additionally, intense absorption in the near-edge shifted to a lower value, showing an improvement of the electron density of the copper in Cu-TMCPP/CNT (Fig. S4a†).34 The higher accumulated electrons are considered to enhance charge transfer for ECR.54 XANES fitting was performed to investigate the coordination environment in each sample, and the corresponding data are presented in ESI Table 1.† The coordination numbers of copper with N are 4.38, 4.35 and 4.54 for Cu-TMCPP, Cu- TMCPP/CNT and Cu-TMCPP + CNT, respectively, which shows the breakage of porphyrin stacks during immobilization, reducing the coordination number of copper in each porphyrin unit. It is expected that the under-coordination condition improves electrocatalytic performance by promoting the hydrogenation of *CO and the production of CH4.55 Fig. S4b–f† shows the wavelet-transform EXAFS (WT-EXAFS) spectrum, which depicts similarity between the local atomic environment of Cu-TMCPP, Cu-TMCPP/CNT and Cu-TMCPP + CNT, indicating Cu2+ in CuN4 in all samples and no trace of metallic Cu, which is evidence for the successful synthesis of the catalysts.
Electrocatalytic CO2 reduction performance
The electrocatalytic performance of the designed catalysts was evaluated in an electrochemical flow cell (Fig. S5a†), as examined by chronoamperometry (Fig. S6†) using an anion exchange membrane, Ni foam as the counter electrode, and drop-cast catalysts on carbon paper with a gas diffusion layer as the working electrode. Solutions of 0.5 M KHCO3 and 0.5 M KOH were used as the catholyte and anolyte, respectively. To quantify the gaseous and liquid products, gas chromatography (GC) and 1H nuclear magnetic resonance (1H NMR) spectroscopy techniques were applied, respectively (Fig. S7†). It is worth mentioning that catalysts with different Cu-TMCPP and CNT loading ratios were examined, and the optimal ratio (2:1) was chosen for the chronoamperometry test (Fig. S6a†). Comparing the faradaic efficiency (FE) for ECR among Cu-TMCPP, Cu-TMCPP + CNT, and Cu-TMCPP/CNT in Fig. 3a–c shows that the major product is CO at lower potentials of ∼−0.6–0.8 V vs. reversible hydrogen electrode (RHE). The electroreduction of CO2 predominantly produced CO, while at a higher potential, CH4 became the major product in Cu-TMCPP/CNT. In contrast, in the case of Cu-TMCPP + CNT, CO remained the dominant product even at a higher potential of −1.11 V vs. RHE. However, for Cu-TMCPP, the selectivity shifted from CO to CH4 at elevated potentials. CH4 production was initiated in the Cu-TMCPP/CNT at around −0.8 V vs. RHE, while in the Cu-TMCPP + CNT system, it was initiated at a more negative potential of −1.0 V vs. RHE. These findings underscore the enhanced CO2 electroreduction performance achieved through immobilization. The Cu-TMCPP/CNT composite exhibited an FE of 37.3% for CH4 at a relatively low potential of −1.08 V vs. RHE, indicating a shift in selectivity toward CH4. This performance showed selectivity alteration compared to previously reported copper porphyrin-based catalysts, where CO and formate are typically the dominant products.31,56,57 Furthermore, immobilization enhances the FE for multi-carbon products, such as ethanol, ethylene, and minor amounts of propanol, while suppressing the HER to less than 21% at −1.03 V vs. RHE. This behaviour was observed neither in non-immobilized Cu-TMCPP nor in the Cu-TMCPP + CNT catalysts, confirming the critical role of the immobilization strategy in leveraging the synergistic properties of molecular catalysts and CNTs to enhance catalytic performance.
 | ||
| Fig. 3 Faradaic efficiency (FE) of hydrocarbons for (a) Cu-TMCPP, (b) Cu-TMCPP + CNTs, and (c) Cu-TMCPP/CNTs examined via chronoamperometry in a flow-cell. (d) In situ X-ray absorption near edge structure (in situ XANES) spectra of Cu-TMCPP/CNTs at potential of OCP, −0.63, −0.83, −0.90, −1.01, −1.03, −1.08 V vs. RHE, and Cu-TMCPP/CNTs before and after experiment (ex situ) at −1.03. (e) First derivative of in situ XANES of Cu-TMCPP/CNTs at potential of OCP, −0.63, −0.83, −0.90, −1.01, −1.03, −1.08 V vs. RHE, and Cu-TMCPP/CNT before and after experiment at −1.03. (f) In situ Fourier transform EXAFS spectra of Cu-TMCPP/CNTs at −1.01 and −1.08 V vs. RHE. Wavelet-transform EXAFS (WT-EXAFS) of (g) Cu-TMCPP/CNTs before experiment, (h) in situ Cu-TMCPP/CNTs under electrocatalytic reaction conditions of −1.03 V vs. RHE. Working electrode: carbon paper gas diffusion layer (GDL); reference electrode: Ag/AgCl; counter electrode: Ni foam in 0.5 M KHCO3 catholyte and 0.5 M KOH anolyte. (i) Ex situ Cu-TMCPP/CNTs after applying −1.03 V vs. RHE in a flow cell. | ||
To investigate the oxidation state and coordination environment of Cu atoms under ECR conditions, in situ X-ray absorption spectroscopy (XAS) was carried out on the most promising catalyst, Cu-TMCPP/CNT, as shown in Fig. 3d–f, S8 and S9a–c.† The XANES spectra at the Cu K-edge for Cu-TMCPP/CNT, shown in Fig. 3d, clearly demonstrate changes in the local oxidation state of copper before, during, and after operating under ECR conditions. Applying the potential results in a negative shift toward a lower energy due to the reduction of Cu2+ (Fig. S9b†). As observed in the first derivative of the in situ XANES spectra (Fig. 3e), the Cu0 peak gradually becomes apparent at higher potentials, indicating the progressive reduction of Cu species. As depicted in Fig. 3d, e and S9a–c,† it can be concluded that some Cu2+ species with a high oxidation state in the Cu–N4 structure are reduced to Cu0 as soon as a negative potential is applied, directly affecting the ECR. Interestingly, the ex situ measurement of the catalyst exhibits nearly identical XANES spectra, suggesting the reversible nature of this phenomenon, which is consistent with previous research findings.58–60 Linear combination fitting using the Cu foil and CuO powder references the estimated oxidation state of Cu under experimental conditions of approximately +1.46, corresponding to 73% Cu2+ and 27% Cu0, respectively, which confirms limited formation of copper cluster under applied potential of 1.03 V vs. RHE. (Fig. S10a†)
The wavelet-transform EXAFS (WT-EXAFS) analyses of Cu-TMCPP/CNT before, during and after reduction under applied potential are depicted in Fig. 3g–i. The WT maximum was observed at approximately 1.45 Å in the ex situ experiment, corresponding to the Cu2+ oxidation state, while the in situ experiment showed the WT maximum at 2.2 Å assigned to Cu0. Similar WT-EXAFS counters of Cu-TMCPP/CNT before and after the ECR experiment (Fig. 3g and i) illustrate the reversibility and stability of the catalyst structure even after the applied potential. The coordination numbers (CNs) of Cu relative to N for different catalysts, illustrated in Table S1 and Fig. S10b,† indicate an under-coordination value of 2.56 under electrocatalytic experimental conditions for Cu2+ within the porphyrin structure. This under-coordination site increases the likelihood of intermediate bonding with copper, thereby enhancing the activity toward the ECR. As shown in Table S1,† the Debye–Waller factor (σ2), which estimates the structural disorder,61 indicates a lower disorder during and after applying the potential for ECR experiments.
As discussed, under the applied reduction potential, metallic copper tends to cluster, as evidenced by the emergence of a peak at 2.1 Å, while the Cu2+ oxidation state peak at 1.6 Å weakens (Fig. S9a†). One of the important factors affecting catalytic selectivity and activity is the coexistence of copper cluster active sites under reduced applied potential. Based on previous reports,62 since the CH4 is the main product in Cu-TMCPP/CNT, clusters are presumably smaller than 10 nm to produce CH4. It has been reported that the cluster size can be estimated based on the cuboctahedral model and its size-dependent Cu–Cu CNs.60 In Cu-TMCPP/CNT, Cu–Cu CNs under an applied potential of −1.03 V are calculated to be 4.18. Based on the aforementioned model,60 it has been anticipated that the copper cluster should be less than 1 nm under reaction conditions, which can justify the dynamic and reversibility of the copper–copper aggregated active sites. However, the precise in situ measurement of the size and morphology of metallic Cu clusters is demanding.58
In Cu-TMCPP/CNT, it can be concluded that C2 products and CH4 are produced as the potential increases (Fig. 3c). According to the in situ XAS and the first derivative of its spectrum demonstrated in Fig. 3d and e, this potential escalation leads to metallic Cu–Cu atom coordination and a higher Cu oxidation state. Therefore, there are two kinds of copper active sites under reaction conditions: one is copper with a high oxidation state, and the other is metallic Cu–Cu atom coordination; each tends to have different product distributions. To investigate the impact of in situ copper oxidation state change on product distribution, the Cu2+/Cu0 ratio is calculated. For example, a closer comparison of FE at −1.01 V and −1.08 V shows that the selectivity toward CH4 is doubled, while ethanol and ethylene FE decrease slightly by increasing the potential to −1.08 V. Based on linear combination fitting, Cu2+/Cu0 ratio at −1.08 V is 8.61, while at −1.03 V, it decreases to 6.81. Additionally, EXAFS and XANES spectra in Fig. 3f and S9c† depict a higher amplitude of Cu2+ in the R = 1–2 Å range and a shift toward higher energy at −1.08 V compared to −1.01 V. We posit that the higher production of CH4 stems from the Cu2+ higher fraction at −1.08 V, while less Cu0 leads to lower C2 products at this potential compared to −1.01 V. This conclusion is in good agreement with the literature reporting that neighbouring Cu–Cu atom coordination favoured ethanol production,58 and the Cu0 cluster may be responsible for increasing the coverage of *CO intermediate; thus, dimerization and C2H4 production occur.62–64 The stable Cu2+ species enhance the adsorption and hydrogenation of the *CO intermediate, facilitating CH4 production while inhibiting the formation of multi-carbon products.65 Additionally, copper clusters may serve as active sites for the HER, offering disadvantages over single atoms located at the centre of porphyrins. Based on the HER FE data presented in Fig. 3a–c, both Cu-TMCPP and Cu-TMCPP + CNT are expected to form Cu0 clusters under negative applied potentials as well. However, in Cu-TMCPP + CNT, the high FE for HER can be attributed to the bare CNT exposed to the electrolyte. Apart from the synergic effect of Cu0 and Cu2+ discussed in this section by analysing in situ XAS, double layer capacitance, considered another contributing factor directly affecting selectivity and activity, is discussed in the subsequent section.
Comprehensive fundamental electrochemical characterization is of great importance for investigating the electrocatalytic characteristics and possible mechanisms of ECR. The intrinsic ECR activity of Cu-TMCPP, Cu-TMCPP/CNT, and Cu-TMCPP + CNT was performed using linear sweep voltammetry (LSV) in an H-cell with 0.5 M KHCO3 under both Ar and CO2 (Fig. S5b†). As shown in Fig. 4a, the onset potentials under CO2 were observed to be −0.5 V, −0.7 V, and −1.2 V vs. RHE for Cu-TMCPP/CNT, Cu-TMCPP + CNT, and Cu-TMCPP, respectively. These results indicate a reduction in the energy barrier upon immobilization of TMCPP onto CNT, highlighting the importance of the immobilization strategy in improving the ECR performance. Turn over frequency (TOF) for hydrocarbon products of Cu-TMCPP/CNT at −1.08 V vs. RHE is 2.47 s−1, which is improved compared to its counterpart of Cu-TMCPP (1.15 s−1), Cu-TMCPP + CNT (0.34 s−1) catalysts and reported TOF in copper-porphyrin-based catalysts so far.16,34,56 Tafel slopes calculated in Fig. 4b suggest boosted kinetics of ECR for Cu-TMCPP/CNT (211 mV dec−1) compared with Cu-TMCPP (274 mV dec−1) and Cu-TMCPP + CNT (218 mV dec−1) catalysts. Consequently, the copper porphyrin inclined toward the production of CH4 can be triggered by immobilization. An ultra-thin layer of Cu-TMCPP on carbon nanotubes showed better ECR activity and kinetics compared to stacked nanosheets with a higher thickness of Cu-TMCPP molecules, which can be attributed to the fact that only the first layer of the molecule stays active for the electrocatalytic CO2 reduction reaction.
 | ||
| Fig. 4 (a) Linear sweep voltammetry (LSV) comparison of Cu-TMCPP, Cu-TMCPP + CNTs and Cu-TMCPP/CNTs under Ar and CO2 in an H-cell. (b) Tafel slope comparison of Cu-TMCPP, Cu-TMCPP + CNTs and Cu-TMCPP/CNTs. (c) Nyquist plots of Cu-TMCPP, Cu-TMCPP + CNTs and Cu-TMCPP/CNTs in H-cell with glassy carbon as the working electrode. (d) Electrochemical double-layer capacitance of Cu-TMCPP, Cu-TMCPP + CNTs and Cu-TMCPP/CNTs. (e) Partial current density of hydrocarbon (CH4 + C2H4 + CO) production for Cu-TMCPP, Cu-TMCPP + CNTs and Cu-TMCPP/CNTs in the flow cell. (f) Stability of Cu-TMCPP/CNTs at 1.08 V vs. RHE examined by chronoamperometry under the following electrocatalytic reaction conditions: working electrode: carbon paper gas diffusion layer (GDL); reference electrode: Ag/AgCl; counter electrode: Ni foam in catholyte of 0.5 M KHCO3 and 0.5 KOH anolyte. | ||
Electrochemical impedance spectroscopy (EIS) was conducted in an H cell. Cu-TMCPP Nyquist plots fitted with Randles' equivalent circuit (Fig. S11a†) show a solution resistance (Rs) of 19.8 Ω and a high charge-transfer resistance (Rct) of 3.74 kΩ, with an insignificant double-layer capacitance (Cdl) of 165 nF. The solution and charge-transfer resistances are also estimated from the plots in Fig. S11b–d† for Cu-TMCPP/CNT and Cu-TMCPP + CNT. A low charge-transfer resistance of about 0.6 Ω and 3.1 Ω attributed to Cu-TMCPP/CNT and Cu-TMCPP + CNT, respectively, demonstrates the impact of CNT introduction to Cu-TMCPP for improving charge transfer. Charge transfer is enhanced even further with immobilization, which reduces activation energy for the ECR, thereby facilitating multi-electron ECR product formation.66 Nyquist plots of Cu-TMCPP/CNT in Fig. 4c show almost vertical slopes at low frequency attributed to capacitive behaviour,23,67 implying a larger active site.68,69 To further evaluate the electrode/electrolyte interface and validate the capacitive behaviour of the Cu-TMCPP/CNT, the Cdl has been calculated (Fig. 4d) by the cyclic voltammetric (CV) plots in the non-faradaic potential region at various scan rates (ranging from 20 to 120 mV s−1) (Fig. S12†). Cu-TMCPP/CNT's Cdl of 660 μF cm−2 is about 300 times higher than Cdl of Cu-TMCPP (2 μF cm−2) and 30 times higher than Cdl of Cu-TMCPP + CNT (29 μF cm−2). Additionally, the electrochemical active surface area (ECSA) has a positive correlation with Cdl. Therefore, Cu-TMCPP/CNT has the highest value of ECSA, which can be attributed to the Cu-TMCPP nano-sheet stacked breakage during immobilization, resulting in the exposure of more Cu-TMCPP blocks to the electrolyte and providing more active sites.
The capacitance double layer directly influences selectivity and catalytic performance.70,71 The high value of Cdl may imply that the major contributing factor for the boosted ECR and higher selectivity of CH4 could be the impact of the electrode/electrolyte interface on the Cu-TMCPP/CNT catalyst. To further investigate the high Cdl of the Cu-TMCPP/CNT catalyst illustrated in Fig. 4d, the electrode/electrolyte interface and Helmholtz layer were analysed for both Cu-TMCPP/CNT and Cu-TMCPP catalysts. At the catalyst surface, the Helmholtz layer consists of two sub-layers: the inner Helmholtz plane (IHP), where the electrochemical reaction occurs, and the outer Helmholtz plane (OHP), where the electrolyte cations accumulate.72,73 The electric field heavily depends on the distance from the current collector and affects ECR performance, particularly in molecular electrocatalysts. It has been reported that the electric field weakens in the diffusion layer compared to the Helmholtz layer.74,75 The Cu-TMCPP active sites located in the diffusion layer do not experience a strong electric field, preventing the formation of an ordered cation accumulation, i.e., outside the OHP. The decay in the electric field and potential disrupts the structured arrangement of cations, making them completely disordered near the active surface of Cu-TMCPP. In contrast, for Cu-TMCPP/CNT, the ultra-thin layer of immobilized Cu-TMCPP facilitates the formation of a well-structured, compact alkali cation layer, which leads to the exceptionally high capacitance observed in this sample, with a large Cdl of 660 μF cm−2. This electric field also increases polarizability, causing strong intermediate adsorption in ECR.76 As previously mentioned, the active surface of Cu-TMCPP does not experience the same potential as the ultra-thin Cu-TMCPP layer immobilized on CNT, which can change both selectivity and activity. This is consistent with the LSV results, which show that a higher potential is required for Cu-TMCPP to be activated for ECR. The same reasoning applies to the Cu-TMCPP + CNT catalyst.
Additionally, the local concentration of H+ and intermediate formation can be affected by OHP. The OHP is partially dehydrated, influencing the selectivity distribution.77 At high potential, the compact OHP can limit the access of active sites to H+, resulting in HER suppression,78 which may explain the low HER activity in Cu-TMCPP/CNT compared to that on Cu-TMCPP. Based on reported computation and experimental results,16,78–80 K+ cations in catholyte assist in the adsorption and stabilization of intermediates, such as *CO2 and *COOH, which serve as the main precursors to CH4 and more complicated products. It has also been suggested that in OHP, H+ couples with *CO to form a different intermediate, thereby inhibiting the release of CO. The cations in the OHP coordinate with dissolved CO2, leading to a high local concentration of CO2 and stabilizing the reaction intermediates.78,81 Therefore, the higher CO2 coverage and shielding of intermediates from desorption in Cu-TMCPP/CNT facilitate dimerization and C–C coupling, leading to the formation of complex products, such as ethylene, ethanol, and propanol. In contrast, for Cu-TMCPP and Cu-TMCPP + CNT, the reduced presence of OHP due to the lower electric field may hinder the formation of multi-carbon intermediates. Consequently, CO and H2 are the primary products because they are released before undergoing further hydrogenation to form CH4 and other complex hydrocarbons at potentials lower than −0.85 V vs. RHE.
Our in situ XAS analysis on Cu-TMCPP/CNT demonstrates the coexistence of Cu0 and Cu2+ under ECR conditions, showing that electron transfer to the catalyst exceeds the amount required for CO2 reduction. This leads to the partial reduction of Cu2+ to Cu0, particularly at more negative potentials. This suggests a strong adsorption of the catalyst's species within OHP, which can improve electron transfer, allowing the catalyst to behave like a metal,82 inhibiting further local oxidation and facilitating the formation of high-electron products that would not otherwise be possible outside of the OHP layer.38 In contrast, for Cu-TMCPP, which remains in direct contact with the electrolyte located outside the OHP, it has been proposed that electron transfer occurs via the metal-porphyrin molecule framework, resulting in a localized positive charge on the molecule due to the electron transfer during CO2 reduction. This behaviour aligns with that of a homogeneous catalyst.82,83
Fig. 4e compares the partial current density of hydrocarbon products over a wide potential range, revealing a value of −90 mA cm−2 for Cu-TMCPP/CNT. This partial current density for hydrocarbon products is higher than the reported values for most copper porphyrin catalysts.23,48,56,80 The enhanced partial current density for hydrocarbon products in Cu-TMCPP/CNT highlights the critical role of CNT immobilization in improving mass transfer efficiency and boosting the partial current density for ECR toward hydrocarbons. Previous studies84 have also recognized that cations in the OHP generate strong electric fields, which influence the dipole moment of intermediates, thereby improving ECR partial current densities and faradaic efficiencies. This effect may contribute to the enhanced partial current density observed for hydrocarbon production in the Cu-TMCPP/CNT catalyst. A detailed reaction mechanism for Cu-porphyrin-based catalysts is beyond the scope of this study; it can be concluded that the strong interaction between the CO2 molecule and the metal centre, along with effective C–O bond cleavage to form intermediates leading to CH4 production,7 is facilitated by catalyst immobilization. This is likely due to the influence of double-layer capacitance and an enhanced electric field. Furthermore, the stability of Cu-TMCPP/CNT was assessed at a constant potential of −1.08 V vs. RHE in a flow cell, as shown in Fig. 4f. The catalyst demonstrated stability for over 7.5 hours, reinforcing the critical role of immobilization in maintaining stable performance. To evaluate the stability of Cu-TMCPP/CNT, the chronoamperometry at −1.08 V vs. RHE was conducted for 9 h. A comparison of the Cu 2p XPS spectra before and after the reaction was performed, as shown in Fig. S13.† Initially, the Cu 2p XPS spectra of Cu-TMCPP/CNT displayed two prominent peaks at 935.20 eV and 955.17 eV, corresponding to the Cu 2p3/2 and 2p1/2 peaks, respectively. After the prolonged reaction, these two peaks shifted to lower binding energies, and additional peaks appeared at 932.60 eV and 951.52 eV owing to the reduction of Cu2+, resulting in electron delocalization, which is commonly observed in electrochemical CO2 reduction reactions.85,86
Conclusions
In this study, we developed and evaluated Cu-TMCPP/CNT as a tunable, heterogeneous electrocatalyst for ECR. A systematic comparison of Cu-TMCPP, Cu-TMCPP + CNT, and Cu-TMCPP/CNT revealed that immobilizing Cu-TMCPP onto MWCNTs significantly enhances ECR performance. The Cu-TMCPP/CNT catalyst exhibited promising selectivity toward multi-electron products, particularly CH4, with an FE of 37.3% at −1.08 V vs. RHE and a partial current density of 91.8 mA cm−2, surpassing many Cu-porphyrin-based catalysts. This enhancement in catalytic performance can be attributed to the strong interaction between the Cu center and CNT, promoting efficient electron transfer while minimizing side reactions, such as HER. Electrochemical analysis, supported by in situ XAS, revealed a dynamic oxidation state of Cu, with the coexistence of Cu2+ and Cu0 species under ECR conditions. The immobilization of Cu-TMCPP on CNTs facilitates the formation of a well-structured alkali cation layer within the OHP, improving electron transfer and shifting product selectivity toward multi-electron products. This highlights the importance of double-layer capacitance and an enhanced local electric field in modulating the reaction pathway. Moreover, the high stability of Cu-TMCPP/CNT, demonstrated by its performance over 7.5 hours at a constant potential, emphasizes the significance of immobilization in maintaining long-term catalytic activity. The results suggest that the immobilization strategy optimizes the catalyst–electrode interface and provides a robust framework for sustainable CO2 electroreduction. This work provides valuable insights into the design of Cu-porphyrin-based electrocatalysts with enhanced selectivity, stability, and scalability, paving the way for future advancements in CO2 conversion technologies.Data availability
All the experimental raw data produced in this research project are available upon request submitted to the first author or the corresponding author.To analyze the data and prepare the figures presented in the manuscript and the ESI,† the following software was used:
• Mercury software for porphyrin visualization.
• VESTA software for molecular structure visualization and bond/plane distance calculations.
• MestReNova software for NMR spectroscopy result processing and analysis.
• ATHENA software package for XAS result analysis.
• Advantage software for XPS result analysis.
• Crystallographic information file (.cif) from Cambridge Structural Database (CSD).
• Microsoft Office package (Word, PowerPoint, and Excel) for document and figure preparation.
All details for using these packages are provided in the ESI.†
Author contributions
Conceptualization: M. A., A. S. and A. E.; methodology: M. A. and A. E.; software: A. E.; validation: M. A., A. S. and A. E.; formal analysis: A. E.; investigation: A. E.; resources: A. S.; data curation: A. E.; writing – original draft: A. E.; writing – review & editing: M. A., A. S. and A. E.; visualization: A. E.; supervision: A. S. and M. A.; project administration: M. A., A. S. funding acquisition: A. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. E. acknowledge the McGill Engineering Doctoral Award (MEDA) and Fonds de recherché du Quebec (FRQNT) Doctoral Scholarship. A. S. would like to acknowledge the NSERC Alliance Mission Grant (ALLRP 577240), Canada Research Chair (950-23288), and Canada Foundation for Innovation (CFI-JELF 39715). The authors would also like to acknowledge the researchers, technicians, and staffs of the Canadian Light Source (CLS) for their support for X-ray absorption spectroscopy (XAS). A. E. would like to thank Dr. Hatem Titi and Andrew Golsztajn for their assistance in analytical analysis.References
- B. Kumar, J. P. Brian, V. Atla, S. Kumari, K. A. Bertram, R. T. White and J. M. Spurgeon, Catal. Today, 2016, 270, 19–30 CrossRef CAS.
- S. M. Jarvis and S. Samsatli, Renewable Sustainable Energy Rev., 2018, 85, 46–68 CrossRef CAS.
- A. Mukherjee, M. Abdinejad, S. S. Mahapatra and B. C. Ruidas, J. Mater. Chem. A, 2023, 11, 9300–9332 RSC.
- M. Abdinejad, C. Ferrag, M. N. Hossain, M. Noroozifar, K. Kerman and H. B. Kraatz, J. Mater. Chem. A, 2021, 9, 12870–12877 RSC.
- M. Abdinejad, M. K. Motlagh, M. Noroozifar and H. B. Kraatz, Mater. Adv., 2022, 3, 1224–1230 RSC.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- S. Gong, X. Han, W. Li, G. Zhao, Y. Zhai, W. Wang, Q. Xia, X. Wang, J. Wu, C. Wu, X. Lv and X. Zhang, Chem. Eng. J., 2025, 507, 160286 CrossRef CAS.
- Y. Yang and F. Li, Curr. Opin. Green Sustainable Chem., 2021, 27, 1–7 Search PubMed.
- S. Gong, W. Wang, X. Han, H. Wang, G. Wang, X. Wang, J. Xie, D. Rao, C. Wu, J. Liu, S. Shao, M. Zhu and X. Lv, Chem Catal., 2024, 4(1) DOI:10.1016/j.checat.2023.100848.
- M. Khalil, G. T. M. Kadja, F. A. A. Nugroho, L. G. Sutanto, P. K. Jiwanti, F. F. Abdi, F. Hussin and M. K. Aroua, Renewable Sustainable Energy Rev., 2024, 206, 114869 CrossRef CAS.
- Y. Zhu, X. Yang, C. Peng, C. Priest, Y. Mei and G. Wu, Small, 2021, 17, 1–24 Search PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Catal. Today, 2017, 288, 74–78 CrossRef CAS.
- T. N. Nguyen, M. Salehi, Q. Van Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 1–24 Search PubMed.
- P. Yu, X. Lv, Q. Wang, H. Huang, W. Weng, C. Peng, L. Zhang and G. Zheng, Small, 2023, 19, 1–7 Search PubMed.
- M. Abdinejad, K. Tang, C. Dao, S. Saedy and T. Burdyny, J. Mater. Chem. A, 2022, 10, 7626–7636 RSC.
- M. Abdinejad, T. Yuan, K. Tang, S. Duangdangchote, A. Farzi, H. P. Iglesias van Montfort, M. Li, J. Middelkoop, M. Wolff, A. Seifitokaldani, O. Voznyy and T. Burdyny, Chem. - Eur. J., 2023, 29, 1–7 CrossRef PubMed.
- M. Abdinejad, L. F. Wilm, F. Dielmann and H. B. Kraatz, ACS Sustainable Chem. Eng., 2020, 9, 521–530 CrossRef.
- H. Veldhuizen, M. Abdinejad, P. J. Gilissen, J. Albertsma, T. Burdyny, F. D. Tichelaar, S. van der Zwaag and M. A. van der Veen, ACS Appl. Mater. Interfaces, 2024, 16, 34010–34019 CrossRef CAS PubMed.
- A. N. Marianov and Y. Jiang, Appl. Catal., B, 2019, 244, 881–888 CrossRef CAS.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X. A. Zhang and H. B. Kraatz, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- K. Kosugi, M. Kondo and S. Masaoka, Angew. Chem., Int. Ed., 2021, 60, 22070–22074 CrossRef CAS PubMed.
- M. Abdinejad, C. Dao, X. an Zhang and H. B. Kraatz, J. Energy Chem., 2021, 58, 162–169 CrossRef CAS.
- S. Gu, A. N. Marianov, T. Lu and J. Zhong, Chem. Eng. J., 2023, 470, 144249 CrossRef CAS.
- Q. Zhao, C. Zhang, R. Hu, Z. Du, J. Gu, Y. Cui, X. Chen, W. Xu, Z. Cheng, S. Li, B. Li, Y. Liu, W. Chen, C. Liu, J. Shang, L. Song and S. Yang, ACS Nano, 2021, 15, 4927–4936 CrossRef CAS PubMed.
- J. Huang, T. Yang, K. Zhao, S. Chen, Q. Huang and Y. Han, J. Energy Chem., 2021, 62, 71–102 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- N. M. Latiff, X. Fu, D. K. Mohamed, A. Veksha, M. Handayani and G. Lisak, Carbon, 2020, 168, 245–253 CrossRef CAS.
- Z. Y. Zhang, H. Tian, H. Jiao, X. Wang, L. Bian, Y. Liu, N. Khaorapapong, Y. Yamauchi and Z. L. Wang, J. Mater. Chem. A, 2023, 12, 1218–1232 RSC.
- P. P. Yang, X. L. Zhang, F. Y. Gao, Y. R. Zheng, Z. Z. Niu, X. Yu, R. Liu, Z. Z. Wu, S. Qin, L. P. Chi, Y. Duan, T. Ma, X. S. Zheng, J. F. Zhu, H. J. Wang, M. R. Gao and S. H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS PubMed.
- H. Jiang, P. Zhao, H. Shen, S. Yang, R. Gao, Y. Guo, Y. Cao, Q. Zhang and H. Zhang, Small, 2024, 20, 1–9 Search PubMed.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y. G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- H. Zou, G. Zhao, H. Dai, H. Dong, W. Luo, L. Wang, Z. Lu, Y. Luo, G. Zhang and L. Duan, Electronic Perturbation of Copper Single-Atom CO2 Reduction Catalysts in a Molecular Way, Angew. Chem., Int. Ed., 2023, 135(6), e202217220 CrossRef.
- K. Kosugi, H. Kashima, M. Kondo and S. Masaoka, Chem. Commun., 2022, 58, 2975–2978 RSC.
- M. Abdinejad, A. Farzi, R. Möller-Gulland, F. Mulder, C. Liu, J. Shao, J. Biemolt, M. Robert, A. Seifitokaldani and T. Burdyny, Nat. Catal., 2024, 2024, 1–11 Search PubMed.
- M. Abdinejad, C. Dao, B. Deng, F. Dinic, O. Voznyy, X. A. Zhang and H. B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 9549–9557 CrossRef CAS.
- J. Liu, K. Yu, Q. Zhu, Z. Qiao, H. Zhang and J. Jiang, ACS Appl. Mater. Interfaces, 2023, 15, 36135–36142 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- M. Abdinejad, C. Dao, X. an Zhang and H. B. Kraatz, J. Energy Chem., 2021, 58, 162–169 CrossRef CAS.
- D. Ma, T. Jin, K. Xie and H. Huang, J. Mater. Chem. A, 2021, 9, 20897–20918 RSC.
- W. Wang, S. Gong, H. Wang, Y. Tan, X. Zhu, X. Wang, J. Liu, W. Yu, G. Zhu and X. Lv, Chem. Eng. J., 2024, 490, 151849 CrossRef CAS.
- Y. R. Wang, M. Liu, G. K. Gao, Y. L. Yang, R. X. Yang, H. M. Ding, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 21952–21958 CrossRef CAS PubMed.
- W. Chen and S. Fukuzumi, Eur. J. Inorg. Chem., 2009, 4, 5494–5505 CrossRef.
- G. Xu, T. Yamada, K. Otsubo, S. Sakaida and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 16524–16527 CrossRef CAS PubMed.
- Y. Zhou, S. Chen, S. Xi, Z. Wang, P. Deng, F. Yang, Y. Han, Y. Pang and B. Y. Xia, Cell Rep. Phys. Sci., 2020, 1, 100182 CrossRef.
- M. Abdinejad, C. Dao, B. Deng, M. E. Sweeney, F. Dielmann, X. an Zhang and H. B. Kraatz, ChemistrySelect, 2020, 5, 979–984 CrossRef CAS.
- Y. Liu, Y. Yang, Q. Sun, Z. Wang, B. Huang, Y. Dai, X. Qin and X. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 7654–7658 CrossRef CAS PubMed.
- Y. Huang, H. Dai, D. Moonshiram, Z. Li, Z. M. Luo, J. H. Zhang, W. Yang, Y. Shen, J. W. Wang and G. Ouyang, J. Mater. Chem. A, 2023, 11, 2969–2978 RSC.
- J. R. Araujo, A. M. Silva, C. P. Gouveâ, E. S. Lopes, R. A. A. Santos, L. A. Terrazos, R. B. Capaz, C. A. Achete and I. O. MacIel, Carbon, 2016, 99, 1–7 CrossRef CAS.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- Z. Geng, X. Kong, Q. Li, J. Ke and J. Zeng, Curr. Opin. Electrochem., 2019, 17, 7–15 CrossRef CAS.
- Z. Xu, C. Peng, G. Luo, S. Yang, P. Yu, S. Yan, M. Shakouri, Z. Wang, T. K. Sham and G. Zheng, Adv. Energy Mater., 2023, 13, 1–8 CrossRef.
- J. X. Wu, S. Z. Hou, X. Da Zhang, M. Xu, H. F. Yang, P. S. Cao and Z. Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- K. Kosugi, H. Kashima, M. Kondo and S. Masaoka, Chem. Commun., 2022, 58, 2975–2978 RSC.
- R. Purbia, S. Y. Choi, C. H. Woo, J. Jeon, C. Lim, D. K. Lee, J. Y. Choi, H. S. Oh and J. M. Baik, Appl. Catal., B, 2024, 345, 123694 CrossRef CAS.
- X. Bai, X. Zhao, Y. Zhang, C. Ling, Y. Zhou, J. Wang and Y. Liu, J. Am. Chem. Soc., 2022, 144, 17140–17148 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- A. Iglesias-Juez, G. L. Chiarello, G. S. Patience and M. O. Guerrero-Pérez, Experimental Methods in Chemical Engineering X-ray Absorption Spectroscopy XAS, Can. J. Chem. Eng., 2021, 100, 3–22 Search PubMed.
- M. Salehi, H. Al-Mahayni, A. Farzi, M. McKee, S. Kaviani, E. Pajootan, R. Lin, N. Kornienko and A. Seifitokaldani, Appl. Catal., B, 2024, 353, 124061 CrossRef CAS.
- Z. Zhang, L. Bian, H. Tian, Y. Liu, Y. Bando, Y. Yamauchi and Z. L. Wang, Small, 2022, 18, 1–24 Search PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 Search PubMed.
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 Search PubMed.
- H. Han, S. Park, D. Jang, S. Lee and W. B. Kim, ChemSusChem, 2020, 13, 539–547 CrossRef CAS PubMed.
- Z. Weng, Y. Su, D. Wang, F. Li, J. Du and H. Cheng, Adv. Energy Mater., 2011, 917–922 Search PubMed.
- A. Sacco, J. CO2 Util., 2018, 27, 22–31 CrossRef CAS.
- J. Wang, Y. Chen, S. Zhang, C. Yang, J. Y. Zhang, Y. Su, G. Zheng and X. Fang, Small, 2022, 2202238 CrossRef CAS PubMed.
- S. Banerjee, C. S. Gerke and V. S. Thoi, Acc. Chem. Res., 2022, 55, 504–515 CrossRef CAS PubMed.
- C. M. Gunathunge, V. J. Ovalle and M. M. Waegele, Phys. Chem. Chem. Phys., 2017, 19, 30166–30172 RSC.
- T. Luo, K. Liu, J. Fu, S. Chen, H. Li, H. Pan and M. Liu, Adv. Energy Sustainability Res., 2023, 4(3) DOI:10.1002/aesr.202200148.
- M. Dunwell, Y. Yan and B. Xu, Curr. Opin. Chem. Eng., 2018, 20, 151–158 CrossRef.
- D. Bhattacharyya, P. E. Videla, J. M. Palasz, I. Tangen, J. Meng, C. P. Kubiak, V. S. Batista and T. Lian, J. Am. Chem. Soc., 2022, 144, 14330–14338 CrossRef CAS PubMed.
- Q. Zhu, C. L. Rooney, H. Shema, C. Zeng, J. A. Panetier, E. Gross, H. Wang and L. R. Baker, Nat. Catal., 2024, 7(9), 987–999 CrossRef CAS.
- J. Le, M. Papasizza, G. Hussain, P. Laura, G. Cabello, J. Cheng and A. Cuesta, Electrochim. Acta, 2019, 327, 135055 CrossRef.
- V. J. Ovalle, Y. S. Hsu, N. Agrawal, M. J. Janik and M. M. Waegele, Nat. Catal., 2022, 5, 624–632 CrossRef CAS.
- B. Pan, Y. Wang and Y. Li, Chem Catal., 2022, 2, 1267–1276 CrossRef CAS.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- Y. Zhang, Q. Zhou, P. Wang, Y. Zhao, F. Gong and W. Y. Sun, ChemSusChem, 2022, 15, 1–7 Search PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. De Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- S. D. Tilley, Nat. Catal., 2022, 5, 359–360 CrossRef.
- C. J. Kaminsky, S. Weng, J. Wright and Y. Surendranath, Nat. Catal., 2022, 5, 430–442 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- X. Su, Z. Jiang, J. Zhou, H. Liu, D. Zhou, H. Shang, X. Ni, Z. Peng, F. Yang, W. Chen, Z. Qi, D. Wang and Y. Wang, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Phys. Chem. Chem. Phys., 2015, 17, 20861–20867 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01319d |
| This journal is © The Royal Society of Chemistry 2025 |