
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of crystallographic orientation in atom probe analysis of Li-ion battery cathode materials†
Jr-Wen
Lin
af,
Dajie
Xie
af,
Hyewon
Jeong
 af,
Alexander J.
Littlefield
bf,
Timothy
Spila
f,
Beniamin
Zahiri
*af and
Paul V.
Braun
*acdef
af,
Alexander J.
Littlefield
bf,
Timothy
Spila
f,
Beniamin
Zahiri
*af and
Paul V.
Braun
*acdef
aDepartment of Materials Science and Engineering, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA. E-mail: bzahiri@illinois.edu; pbraun@illinois.edu
bDepartment of Electrical and Computer Engineering, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
cDepartment of Chemistry, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
dDepartment of Mechanical Science and Engineering, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
eDepartment of Chemical and Biomolecular Engineering, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
fMaterials Research Laboratory, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
First published on 14th March 2025
Abstract
Determining composition of Li-ion battery (LIB) cathodes at the nanoscale is important to understanding cathode performance. However, in the widely adopted layered transition metal oxide cathode materials, the high crystallographic anisotropy of lithium transport makes characterization particularly challenging due to the potential mobility of Li during characterization. Atom probe tomography (APT) holds promise to provide sub-nanometer, three-dimensional chemical information of such cathode materials, and in particular the lithium distribution within these materials. However, such analysis assumes Li does not migrate under the intense electric field required for APT. Using lithium cobalt oxide (LiCoO2) as a model system, we evaluate the role of crystallographic orientation in APT analysis of anisotropic battery materials. When the crystal orientation favors Li transport, the measured Li/Co ratio is highly dependent upon applied laser pulse energy and ranges from near stoichiometric for a 1 pJ pulse, to as high as 6.4 for a 10 pJ pulse. In contrast, when the orientation impedes ionic transport, Li migration is largely suppressed, and the Li/Co ratio reaches only 1.8 using a 10 pJ laser pulse. Using an extrinsically deposited metallic capping layer, localized Li migration is largely stabilized, the Pearson coefficient is reduced for all evaluated orientations. The results presented here shed light on the impact of and emphasize the necessity to report crystallographic orientation on APT analysis results for materials with fast transport characteristics.
1. Introduction
Rechargeable batteries are growing in importance in society.1,2 Owing to their high energy density and rapidly falling costs, secondary Li-ion batteries (LIBs) are now found in applications spanning from grid-scale energy storage and electric vehicles to consumer electronics.3,4 While already successful, there remains considerable demands for LIBs with even higher energy densities and longer cycle lives. Significant research efforts on next-generation cathode active materials (CAMs) are ongoing with goals including increasing gravimetric energy density, decreasing cost, and increasing cycle life.5–9 Among existing CAMs, the family of lithium layered transition metal oxide (LTMO) materials are the most common due to their desirable rate capability and superior cycle life.10,11 But still, there remains much unknown about these important materials, especially in why and how they lose capacity over cycling.Cathode degradation pathways include local phase transitions,12–15 surface reactions,16–18 and micro-crack formation.19,20 These degradation pathways all have ties to local atomic structures and compositional changes at the nanoscale. Strategies to alleviate capacity loss induced by these effects include surface coating/modification,21–24 elemental doping,25,26 microstructure design,27,28 and electrolyte optimization.16,29 While many successes have been achieved, fundamental insights into the nanoscale origins of cathode degradation and appropriate cathode modification strategies are missing. There is thus a need for characterization techniques that can simultaneously provide (a) nanoscale spatial resolution for structural and compositional analysis, (b) high compositional sensitivity, and (c) high sensitivity to light elements (e.g., Li).
Atom probe tomography (APT) provides sub-nanometer, three-dimensional (3D) resolution and chemical sensitivity at the ppm level for most elements.30 The absence of high-energy electron-beam prevents beam-induced artifacts during analysis31 although one must be careful to not introduce artifacts during sample preparation. These unique characteristics of APT enable it to yield information not provided by other more common techniques. For example, scanning transmission electron microscopy (STEM) and electron energy-loss spectroscopy offers high spatial resolution but can induce beam damage and has limited sensitivity to light elements.32–34 X-ray photoelectron spectroscopy provides chemical compositional sensitivity as low as few tens of ppm but lacks spatial resolution.35 Secondary ion mass spectroscopy (SIMS) has similar compositional sensitivity to APT but not the lateral resolution.36,37 APT presents the unique opportunity to reliably map nanoscale Li distribution within battery materials, which is crucial for understanding cathode performance. APT has been widely applied to provide extensive insights on compositional evolutions at the nanoscale in metals,38–44 however, the use of this technique to understand cathode materials remains limited owing to most cathode materials' brittle and in particular semiconducting nature.45
Compared to metallic materials, the field evaporation behaviors during APT analysis of semiconducting materials such as LTMO materials are much more complicated due to deeper electric field penetration under the required intense applied electric field. Challenges in semiconducting materials include field-induced premature sample fracture,44 molecular ion formation,30,46,47 underestimation of oxygen content due to molecular dissociation,48–50 and inhomogeneous laser heating.51,52 These issues lead to lowering of spatial and compositional resolution. For LTMO materials specifically, Li migration and preferential evaporation under the intense field present as the most significant obstacle.31,53,54 This phenomenon has been termed “in situ delithiation” by Kim et al.55 on account of its similarities to the Li movement during battery operation. In that work, Kim demonstrated the effect of reaction product under atmosphere exposure and a deposited shielding layer to counter the problem. Parikh et al.56 conducted thorough investigations on the effects of laser pulse energy, Ga implantation, field strength on APT analysis result for high-Ni LTMO cathodes. Unexplored are aspects such as crystal anisotropy and transport properties. For LTMOs, the stark difference in Li-ion transport behavior along varying crystallographic orientations has been widely reported,57,58 yet its impact on APT analysis has not been discussed.
By taking advantage of the crystallographically textured nature of our dense, electrodeposited cathodes,59,60 we elucidate the role of laser pulse energy, extrinsically deposited capping layer, and for the first time, crystallographic orientation in APT of LTMO cathode materials. By using the air-stable LiCoO2 (LCO) as a prototypical system representative of the LTMO cathodes, it was found that when the LCO is oriented in such a way that the fast Li-ion conducting channels are aligned parallel to the external electric field, there is a strong, positive correlation between in situ delithiation and applied laser pulse energy. At sufficiently low laser pulse energy (e.g., 1 pJ), Li-ion outward migration can be largely suppressed. In contrast, a less pronounced correlation between pulse energy and in situ delithiation is observed when the Li-ion conduction channels are perpendicular to the electric field. In this orientation, the Li-ion movements are “blocked” and the extent of in situ delithiation is less dependent on laser pulse energy. These findings are summarized in Fig. 1. The results shed light on the largely unexplored critical role that crystallographic orientation plays in APT analysis of battery materials. The contributions of electronic, optical, and thermal anisotropies in LCO to APT analysis are also presented. The effectiveness of extrinsic metallic capping layer on circumventing localized Li migration was evaluated for the case of LCO. This work highlights the importance of understanding and reporting crystallographic orientation in APT of battery materials.
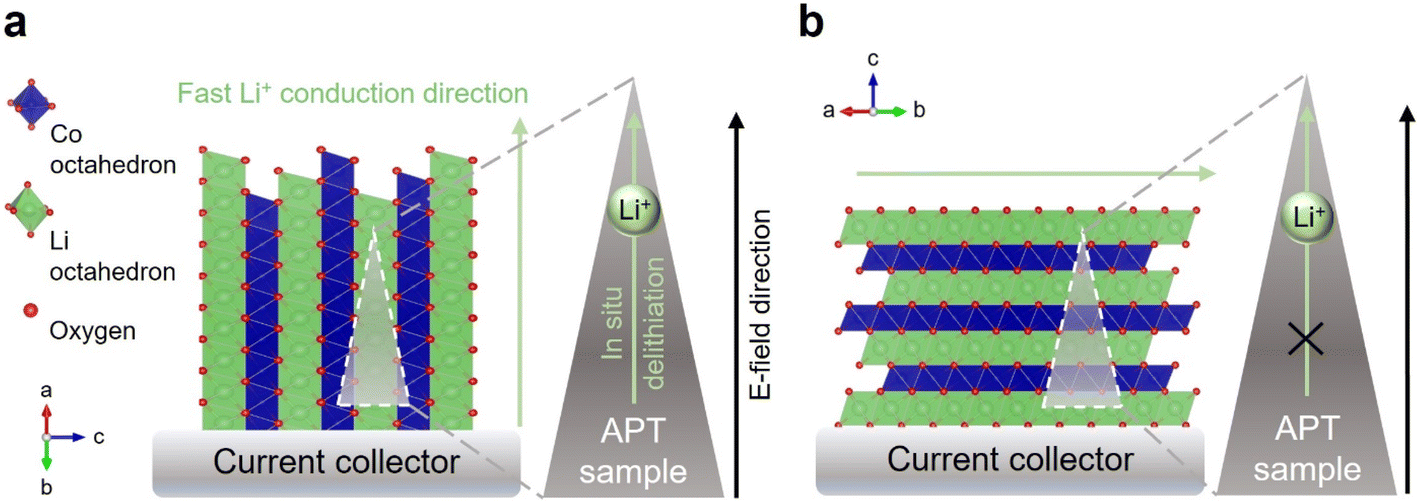 | ||
| Fig. 1 Schematics illustrating the effect of crystallographic orientation on in situ delithiation in APT analysis for LTMOs. (a) When fast Li-ion conducting directions are parallel to the applied field, severe in situ delithiation occurs due to Li+ migration along high mobility directions. (b) When the Li-ion blocking direction is parallel to the applied field, Li+ motion and in situ delithiation is suppressed. | ||
2. Experimental methods
2.1 Materials preparation
The dense, textured LiCoO2 was prepared in thick film form using electrodeposition in a near eutectic molten salt bath primarily composed of KOH and LiOH with Co(OH)2 added as the precursor.59–61 The deposition was conducted in a nitrogen-filled glovebox (oxygen and water level <0.1 ppm) using a three-electrode system with the following setup: Co wire reference electrode (RE), Ni foil (25 micron) counter electrode (CE) and working electrode (WE). To obtain films with different textures, the growth temperature was controlled between 300 and 350 °C. Higher temperature results in the (003) basal planes lying parallel to the substrate, whereas lower temperature leads to the basal planes lying perpendicular to the substrate. At intermediate temperature (325 °C), a mixed orientation sample containing crystals lying in both the aforementioned orientations can be obtained. The deposited sample thickness is controlled with the deposition time and is typically 30–40 microns, corresponding to an areal loading of around 2 mA h cm−2. Subsequently, the as-deposited sample is taken out of the glovebox and washed with deionized water for at least 1 hour to remove residual hydroxides at the sample surface. Finally, the sample is homogenized through annealing at 600 °C for 6 hours in air with a ramp rate of 3 °C min−1.59,612.2 General APT sample preparation
The sharp needle-like APT samples are prepared using a Thermo Scios 2 Dual Beam focused ion beam (FIB). A 300 nm Pt layer is first deposited using ion beam-induced deposition (IBID), followed by a standard lift-out procedure provided by CAMECA Instruments Inc. to obtain wedge-shaped lamella. The lamella is subsequently mounted onto Si posts purchased from CAMECA with Pt welding using IBID on both sides of the wedge. Following that, annular milling steps with 30 kV Ga ion beam (0.3 nA, 30 pA) are performed until the lamella are shaped into pillars with diameters below 300 nm. Then, a 16 kV, 23 pA Ga ion beam is applied to further reduce the diameter of the pillar to below 200 nm in diameter and to reduce Ga ion beam damage. Finally, 5 kV, 7 pA low-energy Ga ion beam milling is applied to remove the surface Ga ion beam damage and to shape the tip apex into sharp needle-shaped geometry with an end radius under 100 nm. Retention of the crystal structure in LCO TEM samples prepared by a similar Ga milling process gives us confidence Ga ion implantation related damage in our samples is minimal.612.3 Orientation-specific APT sample preparation
Surface and cross-sectional morphology of LCOs with different textures are shown in Fig. 2. Fig. 2a and c represent samples with basal planes perpendicular to the substrate. Fig. 2b and d represent samples with basal planes parallel to the substrate. To obtain APT samples with different crystallographic orientation at the tip apex, lift-out sites are carefully chosen to locate the correct facets and position them at the center of the lamellae, and similar milling steps to the previous paragraph are conducted with extra caution to preserve the right portion of the lamellae as the tip apex. The white triangles in Fig. 2c and d visualize the approximate locations of the as-prepared tips with reference to the cross-sectional images. Fig. 2e presents the HAADF-STEM image showing the atomic arrangements at the surface of the facets. The perpendicular alignments of the Co atomic columns to the facets confirm the correspondence between fast Li conducting channels and the facet features at the microscale. Fig. 2f presents the diffraction patterns derived from Fig. 2e. | ||
| Fig. 2 Surface and cross-sectional morphology for electrodeposited LCO and APT tip locations. (a) and (b) Top-view SEM images for (a) F-LCO and (b) B-LCO. The step-like feature in (a) corresponds to the fast Li-ion diffusing facets, hence, they are referred to as faceted-LCO (F-LCO) in this work. The plane-like features in (b) corresponds to the basal planes, which are blocking to Li ion transport. Hence, they are referred to as basal plane-LCO (B-LCO). The orange rectangle marks the top-view location for the cross-sectional images taken. (c) and (d) Cross-sectional focused ion beam SEM images for (c) F-LCO and (d) B-LCO. The white triangles, though not to true scale of the actual APT tips, mark their approximate locations with respect to the locations of the facets and the basal plane. (e) Filtered HAADF-STEM image from selected region from (c), where the blue box signifies the region for the image. (f) Filtered FFT image of (e) with zone axis of [100]. Similar HAADF-STEM images for (d) can be found in Fig. S1.† | ||
2.4 APT analysis
The as-prepared APT samples are transferred into a CAMECA local electrode atom probe (LEAP) 5000XS under air for APT analysis. The laser-assisted mode with a 355 nm UV laser was applied. The analysis base temperature is set at 30 K for all experiments, and detection rates between 0.1 and 0.3% were applied. The laser pulse energy is set from 1 pJ to 10 pJ, and the pulse frequency is set at 200 kHz. Typical dataset size is around 1.5 to 2 million counts. The data reconstruction and analysis are conducted using the CAMECA integrated visualization and analysis (IVAS) software. The frequency distribution analysis is performed by dividing the region of interest into bins, each containing 100 ions. The calculated composition distribution can be plotted alongside the corresponding binomial distribution, which represents a completely randomized distribution for the atoms within the analyzed volume. The deviation of the original distribution from the random, binomial distribution can be used to calculate the Pearson coefficient (μ), which ranges from 0 for a random, uniform distribution to 1 for a non-uniform distribution with possible solute aggregation. As discussed by Singh et al.,62 for Li distribution within battery cathode materials, μ > 0.9 represents non-uniformity possibly induced by in situ delithiation, and μ close to or smaller than 0.5 represents more random, uniform distributions. Note, owing to the normalization process for the calculation of Pearson coefficients, the ability to do additional statistical analysis is limited.3. Results and discussions
3.1 Effect of laser pulse energy on in situ delithiation
The influence of laser pulse energy on APT analysis results for Li-containing battery materials has been extensively discussed by Santhanagopalan et al.63 and Kim et al.55 In this section, we first evaluate the impact of the laser pulse energy, which is directly correlative to peak tip heating condition,52 on the APT analysis results for electrodeposited, dense LCO. Due to their superior electrochemical performance,61,64,65 LCOs with (003) basal planes oriented perpendicular to the substrate are first investigated in this section. They are referred to as faceted LCO (F-LCO) owing to their distinct microstructure.61 Fig. S2† shows the X-ray diffraction (XRD) of the electrodes, confirming the texture of the applied LCOs. The galvanostatic charge–discharge behavior and cycling profile for the F-LCO have been reported by Zhang et al.59 and Zahiri et al.,61 validating the quality and performance of the as-fabricated material. Additionally, inductively coupled plasma mass spectrometry (ICP-MS) confirms the Li/Co ratio to be 1.09, close to the theoretical value of 1.APT analysis results for F-LCO with applied laser pulse energy ranging from 1 to 10 pJ are presented in Fig. 3. In Fig. 3a, for a 10 pJ pulse, the elemental maps corresponding to each LCO component are presented. The tomographic reconstructions visualize the elemental distribution within the material. In this dataset, for all three elements, the results indicate inhomogeneous elemental distributions. The visible “gaps” in the 3D elemental maps can be correlated with the instability observed in the applied voltage history for APT analysis shown in Fig. S3.† Additionally, Fig. 3d displays the one-dimensional composition profile plotted for the dataset in a 15 nm-diameter cylinder; the arrow indicates the direction of the probed portion of the sample moving away from sample surface. As evidenced by the green trace, a severe Li excess accounting for close to 100% of the atomic concentration is observed within a few nanometers of the surface. The bulk composition analysis result, which takes into account the complete dataset and calculates the elemental ratio, indicates a Li/Co ratio of 6.4, far in excess of the theoretical value of 1.0. This lithium excess matches that expected if there is significant in situ delithiation. Occurring is the thermally activated outward electromigration of the mobile Li under laser illumination and heating during APT.55 The semiconducting nature of the LCO results in deeper electric field penetration than if LCO was metallic, which serves as a driving force for Li migration to the surface where it preferentially evaporates. The Li preferential evaporation is owing to the low evaporation field of 14 V m−1 for lithium compared to cobalt (37 V m−1) and oxygen. Though no accurate evaporation field has been reported for oxygen, both atomic and molecular oxygen have higher ionization potential than cobalt, making oxygen even more difficult to evaporate than cobalt.50
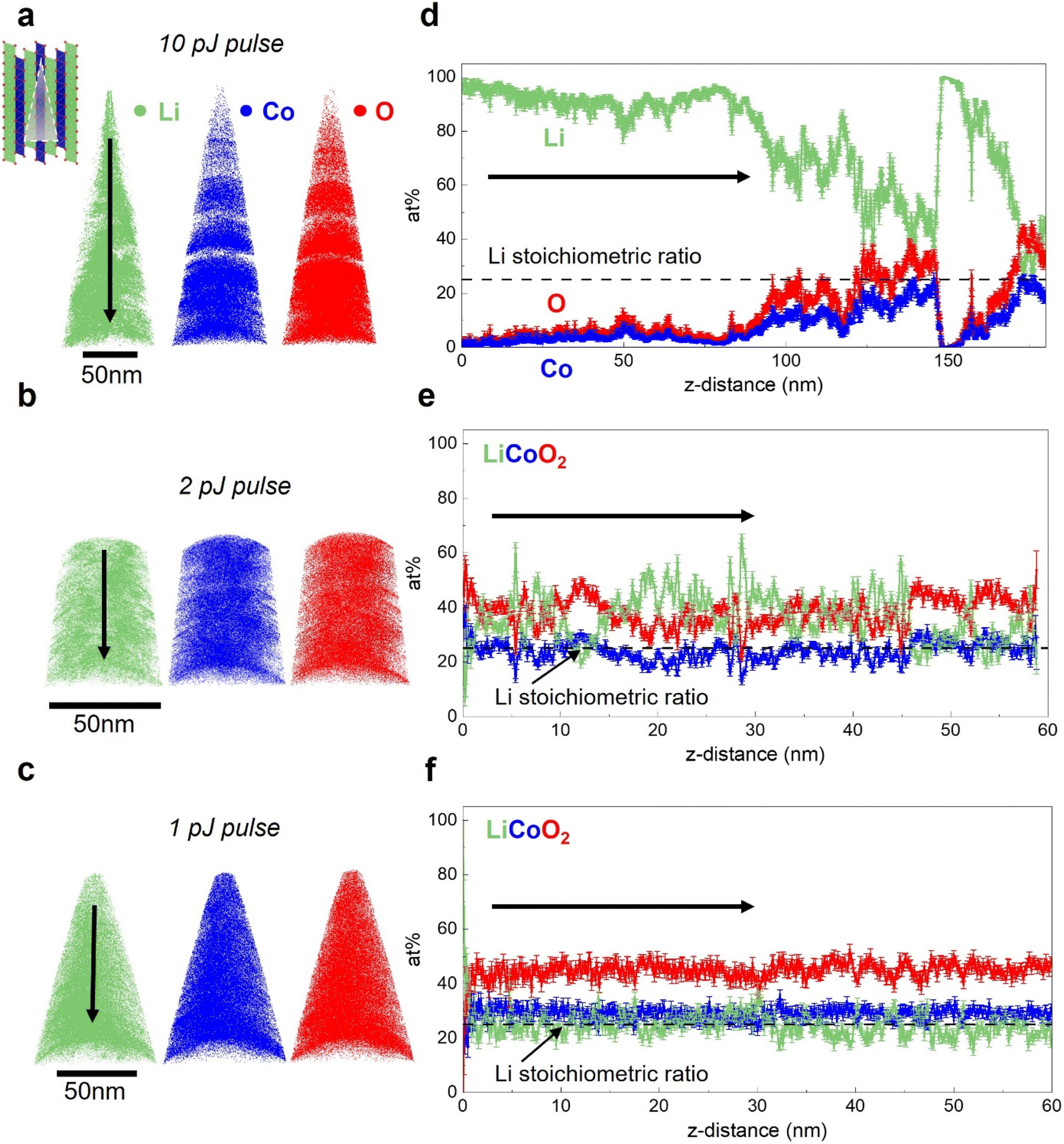 | ||
| Fig. 3 Effect of laser pulse energy on APT results for F-LCO. (a)–(c) Reconstructed 3D ion maps for laser pulse energies of (a) 10 pJ, (b) 2 pJ and (c) 1 pJ. (d)–(f) Analyzed 1-D composition profiles for laser pulse energies of (d) 10 pJ, (e) 2 pJ, and (f) 1 pJ. The illustration included in (a) indicates the crystallographic orientation for all samples. Note, the 1-D composition profiles were calculated from 15 nm diameter cylinder sub-volumes for all cases and the black dashed line in the profiles indicates Li stoichiometric ratio for LCO. The black arrow indicates the depth profiling direction corresponding to the vertical direction in the ion maps. All experiments conducted at a base temperature of 30 K. | ||
APT performed with lower applied laser pulse energies are presented in Fig. 3b, c, e and f. As shown in Fig. 3b, reducing the laser pulse energy to 2 pJ results in improved homogeneity of the elemental spatial distribution and absence of visible gaps in the tomographic reconstruction. In the 1-D composition profile shown in Fig. 3e, significantly less pronounced surface Li excess is observed in comparison to the 10 pJ dataset. However, apparent fluctuations in the Li concentration profile still indicate the influence of in situ delithiation. As can be seen in Fig. 3c and f, when the laser energy is reduced to 1 pJ, which is the lowest commonly used value in literature,66 a comparatively uniform concentration profile agreeing with the expected stoichiometry is obtained. A bulk Li/Co ratio of 1.1 is achieved under this condition, close to the Li/Co ratio of 1.09 obtained via ICP. For LCO of this crystallographic orientation, these results indicate APT analysis is highly sensitive to applied laser pulse energy, and hence, peak tip temperature and that the extent of in situ delithiation can be modulated through tuning the laser heating conditions. Note that although the Li excess artifacts are mostly absent under low pulse energies (low laser heating), small-scale fluctuations in the Li content profile, which will be extensively discussed in Section 3.3, can still be observed which may indicate localized Li migration under the applied field. In the case for the profile shown in Fig. 3f, an underestimated oxygen content can also be observed. This has been commonly attributed to the formation of neutral O2 species that leads to incorrect quantification in APT for oxygen-containing compounds.48–50,67 In summary, it is clear that there is a strong positive correlation between laser pulse energy and Li excess when the LCO is textured with (003) basal planes oriented perpendicular to the substrate.
3.2 Influence of crystallographic orientation
The impact of crystallographic orientation on APT analysis results for LCO cathode materials are further explored in Fig. 4. In the results presented in Section 3.1, the positive correlation between the detected bulk Li excess and laser heating condition is summarized in Fig. 4a. Deviations from the stoichiometric ratio indicate overall Li excess detected for the dataset, which can be caused by the preferential Li evaporation owing to in situ delithiation. Hence, bulk Li/Co ratio calculated from the dataset can be used as a proxy for representing the extent of in situ delithiation observed. A drastic increase from a Li/Co ratio close to unity to 6.4 is observed as the laser power is increased from 1 pJ to 10 pJ. The anisotropic LCO crystal structure plays a critical role in this. As discussed by Zahiri et al.61 and Patra et al.,60 for electrodeposited LTMO cathodes as evidenced by the charge–discharge kinetics, there is a distinct difference in the Li+ and Na+ diffusion kinetics parallel and normal to the (003) planes at room temperature, in agreement with other works on the in-plane and out-of-plane Li+ diffusion kinetics in LTMOs.57,58 In the results reported in the previous section, F-LCO samples were studied. As we note, the F-LCO orientation, which aligns the fast Li-ion conducting channels with the applied electric field direction in APT, is generally favorable for energy storage applications.68 As Fig. 4 summarizes, and as discussed in the following paragraphs, the severe in situ delithiation that occurs at higher laser pulse energy is at least partially induced by the favored Li migration for the F-LCO orientation. | ||
| Fig. 4 Comparison of the Li excess behavior under increasing laser heating for LCOs with orthogonal orientations. (a) and (b) Correlation between measured Li/Co ratio and laser pulse energy for (a) F-LCO and (b) B-LCO. The blue dashed lines represent the theoretical stoichiometric Li/Co ratio of 1. The drastic scaling relationship between the Li excess and the laser pulse energy for F-LCO can be attributed to the favored Li migration in this orientation. Whereas the suppressed correlation for B-LCO is due to the impeded Li transport under the blockage by the basal planes. | ||
To evaluate the influence of crystallographic orientation on in situ delithiation, APT experiments with a different LCO orientation are performed, specifically, with basal planes parallel to the current collector (B-LCO). Use of B-LCO results in an APT sample with orthogonal orientation compared to when F-LCO is used to prepare the APT sample. The B-LCO APT sample has the fast Li-ion conducting direction perpendicular to the applied field. A series of B-LCO APT samples were prepared and analyzed using laser pulse energies from 1 to 10 pJ. The relationship between the Li/Co ratio and laser energy is presented in Fig. 4b. For a 10 pJ pulse, a Li/Co ratio of 1.8 is obtained for B-LCO, which is significantly less than the Li/Co ratio of 6.4 obtained for F-LCO. Though the Li/Co ratio is still positively correlated with pulse energy, the relationship between the Li excess and laser pulse energy is significantly less pronounced for B-LCO. We believe the difference between the B-LCO and F-LCO is due to the strong anisotropy in Li migration in LCO relative to the gradient in the applied electric field (which is either parallel (F-LCO) or perpendicular (B-LCO) to the fast diffusion directions in LCO). Note there is still a Li excess present even for B-LCO under high laser pulse energy, so, even for B-LCO the influence of Li migration cannot be ignored when analyzing APT data.
It is important to consider other potential contributions to the observed Li-excess. There are significant differences between the electronic conductivity perpendicular to and in the (003) plane in LCO.69 The effect of conductivity anisotropy is evident in the applied base voltage history presented in Fig. S4,† where the required voltage to reach the same detection rate for B-LCO is consistently higher than that of F-LCO across various laser heating conditions. The lower electronic conductivity would, in turn, lead to a deeper field penetration depth and result in a more severe case of Li migration. However, this contradicts the observed trend that the in situ delithiation is more severe for the more electronically conductive F-LCO. Since F-LCO shows greater in situ delithiation, we suggest contributions of the ionic transport anisotropy outweigh those imposed by the electronic anisotropy.
The possible effect of anisotropic light absorption and its consequent influence on peak temperature also needs to be considered. As extensively discussed by Vella,52 the laser absorption and heating behavior of a semiconducting, nanometric oxide APT tip can be extremely complicated owing to factors including light diffraction, surface band bending, and the presence of oxygen defect states. Due to the complexity of the situation, we provide only qualitative insights for the case of dense LCO. To estimate laser pulse absorption, micro spectrometer measurements were performed, and calculations based on the data are presented in Fig. S5.† The detailed experimental protocols are elaborated in the ESI.† The results indicate that the extinction coefficient (k) with incident light perpendicular to the (003) planes is near 0.12, which should be the highest among all crystallographic orientations for LCO. Simulation results from prior works52 suggest that the temperature rise difference arising from anisotropic light absorption under this scenario is lower than few tens of degrees and is not significant enough to result in the observed difference in Li migration. These results again suggest ionic transport anisotropy plays a more significant role than anisotropic laser absorption. Finally, it is important to consider the possibility of heat conduction anisotropy. Simulations have shown that the thermal conductivities normal to and in plane with (003) planes in LCO are of the same order of magnitude,70 indicating anisotropy in heat conduction is a relatively insignificant factor.
3.3 The role of extrinsic shielding layer
While reducing the pulse energy even for F-LCO reduces in situ delithiation, even for low pulse energies there remains a non-uniformity in Li distribution for the F-LCO APT datasets. To quantitatively assess the Li distribution uniformity in the APT data, frequency distribution analyses are performed. The details of the analysis method can be referenced from the previous experimental section.39 First, using the 1 pJ pulse F-LCO APT data presented in Fig. 3f, the observed distribution and a theoretically random binomial distribution are plotted together in Fig. 5a. A deviation from random distribution can be reflected by histogram shifting away from the binomial distribution, and the Pearson coefficient (μ) can be extracted from this deviation. A high calculated Pearson coefficient (μ) of 0.98 is obtained for Li, indicating the Li distribution as measured by APT is non-random, which agrees with the small-scale fluctuations observed in Fig. 3f, and indicates the presence of localized Li migration even at low laser pulse energy for F-LCO. Similar behaviors can be observed for both F- and B-LCO across the laser pulse energy range as presented in Fig. 5b–d. Prior work by Kim et al.55 has demonstrated that surface also plays a role in stabilizing Li movement during APT analysis for LTMOs. In the case of LiNi0.8Co0.1Mn0.1O2 (NMC811), during air transfer, there are surface reactions which result in formation of a thin layer of product, later confirmed by APT to be lithium carbonate,66 that shields electric field penetration and improves the quality of the APT data. As LCO is generally inert in air, we apply an alternative strategy involving a thin, extrinsically deposited shielding layer to inhibit even small-scale field-driven Li movement. | ||
| Fig. 5 Effect of in situ deposited metal capping layer on APT analysis result for dense LCO. (a) and (b) Frequency distribution analysis results for F-LCO with and without capping under (a) 1 pJ and (b) 10 pJ laser pulses. (c) and (d) Frequency distribution analysis results for B-LCO with and without capping under (c) 1 pJ and (d) 10 pJ laser pulses. The solid lines represent the observed distributions, and the shaded areas represent the theoretical binomial distributions. The color code red corresponds to the uncapped datasets, and blue corresponds to the capped datasets. The calculated Pearson coefficients represent the degree of deviation of observed distributions from theoretical random distributions. | ||
The application of thin, conformal coatings that provide electronic shielding onto sharp needle-shaped APT samples is challenging. Conventional physical vapor deposition (PVD) is compatible with APT and poses no significant interference to field evaporation.55 But the non-uniform deposition thickness at the nanoscale across the tip apex calls for additional milling steps and is not ideal. Alternatively, Singh et al.62 demonstrated the feasibility of an extrinsically, in situ deposited metallic chromium capping layer through ion beam redeposition in SEM-FIB on stabilizing localized Li migration. Here, following this method, Ni was chosen as the capping layer material owing to its similar evaporation field as Co and strong adhesion to LCO. The corresponding tomographic reconstruction of ion maps and mass spectrum for the capped samples are presented in Fig. S6.† As expected, Ni is initially detected indicating successful capping of the tip. However, the deposited metal layer thickness is significantly thinner than previously reported.62 This could be due to the variation in the sputtering yield for Ni and Cr. Frequency distribution analysis for the capped sample is presented in Fig. 5a. The analysis shows a Li distribution with a μ value 0.36, similar to the value reported for UHV transferred, capped NMC811 tips.62 This indicates capping significantly improves the Li uniformity in the collected dataset, which is evident from the uniform 1-D composition profile presented in Fig. S6.† Exemplary applied base voltage history for both uncapped and capped trials are presented in Fig. S7.† For the uncapped tip, the multiple voltage drops events observable throughout the voltage history signifies unstable field evaporation behavior. In comparison, the monotonic, steadily increasing trend of the applied voltage for the capped case indicates good field evaporation stability, further supporting the value of capping.
To evaluate the effect of the capping layer under extreme laser heating conditions and for samples with different orientations, APT experiments for capped F-LCO and B-LCO under different laser pulse energy are performed. Corresponding frequency distribution analysis results are presented in the remainder of Fig. 5. As plotted in Fig. 5b, for capped F-LCO at 10 pJ, a significant decrease in μ from 0.98 for the uncapped case to 0.23 can be observed, verifying the improved Li uniformity under high laser pulse energy. In Fig. 5c and d, for capped B-LCO at 1 and 10 pJ, similar trends of decreasing μ confirms suppressed localized Li migration by capping for both orientations. Note, capped B-LCO at 1 pJ showed relatively high μ value even after capping. We believe the primary advantage of the applied thin, metallic layer is that it provides consistent electronic shielding that alleviates field propelled Li migration to a certain extent but cannot rule out that capping also improves heat transfer throughout the tip as others have discussed,71,72 with further investigations needed. We suggest the latter to be a more plausible cause for the improved Li uniformity obtained. The impacts of capping on detected Li stoichiometry as a function of laser pulse energy are plotted in Fig. 6. As shown in Fig. 6a, with metal capping, the Li/Co ratio reduces from 6.4 (no cap) to 2 for F-LCO for a 10 pJ pulse. The 1-D concentration profile presented in Fig. S8† further validates the absence of extreme in situ delithiation. The Li/Co ratio remained the same at 1 pJ for capped F-LCO. For B-LCO, similar Li/Co ratio are detected for both the capped and the uncapped case throughout the laser pulse energy range (Fig. 6b). These results showed that (a) capping is also conducive to suppressing large scale Li migration and preferential evaporation with the fast transport direction oriented parallel to the applied electric field, and (b) distinctly different response to metal capping can be observed for F- and B-LCO. The latter further confirms the crucial role of crystallographic orientation in interpreting APT analysis results for anisotropic battery materials. Note though that for both orientations, slight Li excess can still be observed at 10 pJ with capping, indicating capping alone is not sufficient to fully mitigate the effect Li transport in APT studies of air-stable LTMOs.
 | ||
| Fig. 6 Comparison of the measured Li excess for LCOs with and without metal capping. (a) and (b) Measured Li/Co ratio vs. laser pulse energy for (a) F-LCO and (b) B-LCO with and without metal capping. The open red circles represent uncapped data points, and the open blue stars represent capped data points. The blue dashed lines represent the theoretical stoichiometric Li/Co ratio of 1. Values above the stoichiometric ratio indicate an overall Li excess in the dataset caused by the preferential Li evaporation owing to in situ delithiation. | ||
4. Conclusions
Using air-stable, electrodeposited, dense LiCoO2 as a prototypical system, we highlight the critical role of crystallographic orientation in APT for LTMO cathode materials. When fast Li conduction channels (parallel to (003) planes) are aligned with the external applied field, the extent of in situ delithiation is highly dependent on laser heating conditions (laser pulse energy) with the in situ delithation being directly observable in the APT data. When (003) planes are normal to the applied external field, Li ion migration is blocked, and in situ delithiation is significantly suppressed. Contributions to the observed phenomena from electronic, optical, and thermal conduction anisotropies are found to be small relative to the impact of the anisotropic ionic conduction in LCO. It is also found that applying an extrinsically, in situ deposited metallic capping layer significantly improves the uniformity of Li distribution in the acquired dataset even when fast Li conduction directions are parallel to the applied field, which may extend the capability of APT in obtaining nanoscale chemical information for varying sample orientations.The findings here provide insights into how sample orientation impacts APT of battery materials. The significant effects of sample orientation on APT could explain the wide range of optimal APT conditions reported for structurally similar LTMO materials.39,42,44,55,56,73 The inconsistent stoichiometry reported in these works indicates previously unexplored factors at play, such as the effect of crystallographic orientation discussed here. The results presented here apply not only to LTMOs but to any materials with anisotropic, reasonably fast atomic transport characteristics. We hope this work encourages others to consider the importance of crystallographic orientation when interpreting APT data for anisotropic materials. Reporting crystallographic orientations should be standard in APT analysis of energy storage materials and other materials with rapid transport characteristics.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Army Construction Engineering Research Laboratory (W9132T1920008 and W9132T2420002) (APT studies) and the U.S. Department of Energy (SC0020858) (LCO synthesis). The sample preparation and characterizations were carried out in part in the Materials Research Laboratory Central Research Facilities, University of Illinois Urbana-Champaign.References
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 Search PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 Search PubMed.
- T. Kim, W. Song, D.-Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem.A., 2019, 7, 2942–2964 Search PubMed.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 Search PubMed.
- Q. Liu, X. Su, D. Lei, Y. Qin, J. Wen, F. Guo, Y. A. Wu, Y. Rong, R. Kou, X. Xiao, F. Aguesse, J. Bareño, Y. Ren, W. Lu and Y. Li, Nat. Energy, 2018, 3, 936–943 CrossRef CAS.
- L. Wang, B. Chen, J. Ma, G. Cui and L. Chen, Chem. Soc. Rev., 2018, 47, 6505–6602 RSC.
- S. Kalluri, M. Yoon, M. Jo, S. Park, S. Myeong, J. Kim, S. X. Dou, Z. Guo and J. Cho, Adv. Energy Mater., 2017, 7, 1601507 CrossRef.
- D. Andre, S.-J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC.
- Y. Lyu, X. Wu, K. Wang, Z. Feng, T. Cheng, Y. Liu, M. Wang, R. Chen, L. Xu, J. Zhou, Y. Lu and B. Guo, Adv. Energy Mater., 2021, 11, 2000982 CrossRef CAS.
- M. D. Radin, S. Hy, M. Sina, C. Fang, H. Liu, J. Vinckeviciute, M. Zhang, M. S. Whittingham, Y. S. Meng and A. Van der Ven, Adv. Energy Mater., 2017, 7, 1602888 CrossRef.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef PubMed.
- Y. Shao-Horn, S. A. Hackney, A. R. Armstrong, P. G. Bruce, R. Gitzendanner, C. S. Johnson and M. M. Thackeray, J. Electrochem. Soc., 1999, 146, 2404–2412 CrossRef CAS.
- Y. Jiang, C. Qin, P. Yan and M. Sui, J. Mater. Chem. A, 2019, 7, 20824–20831 RSC.
- Y. Zhang, Z. Yang and C. Tian, J. Mater. Chem. A, 2019, 7, 23628–23661 RSC.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- D. Aurbach, B. Markovsky, G. Salitra, E. Markevich, Y. Talyossef, M. Koltypin, L. Nazar, B. Ellis and D. Kovacheva, J. Power Sources, 2007, 165, 491–499 CrossRef CAS.
- S. Liu, D. Liu, S. Wang, X. Cai, K. Qian, F. Kang and B. Li, J. Mater. Chem. A, 2019, 7, 12993–12996 RSC.
- A. Mukhopadhyay and B. W. Sheldon, Prog. Mater. Sci., 2014, 63, 58–116 CrossRef CAS.
- D. J. Miller, C. Proff, J. G. Wen, D. P. Abraham and J. Bareño, Adv. Energy Mater., 2013, 3, 1098–1103 CrossRef CAS.
- L. A. Riley, S. Van Atta, A. S. Cavanagh, Y. Yan, S. M. George, P. Liu, A. C. Dillon and S.-H. Lee, J. Power Sources, 2011, 196, 3317–3324 CrossRef CAS.
- J. W. Kim, J. J. Travis, E. Hu, K.-W. Nam, S. C. Kim, C. S. Kang, J.-H. Woo, X.-Q. Yang, S. M. George, K. H. Oh, S.-J. Cho and S.-H. Lee, J. Power Sources, 2014, 254, 190–197 CrossRef CAS.
- L. Zhu, Y. Liu, W. Wu, X. Wu, W. Tang and Y. Wu, J. Mater. Chem. A, 2015, 3, 15156–15162 RSC.
- M. Bettge, Y. Li, B. Sankaran, N. D. Rago, T. Spila, R. T. Haasch, I. Petrov and D. P. Abraham, J. Power Sources, 2013, 233, 346–357 CrossRef CAS.
- H. Tukamoto and A. R. West, J. Electrochem. Soc., 1997, 144, 3164–3168 Search PubMed.
- L. Sun, Z. Zhang, X. Hu, H. Tian, Y. Zhang and X. Yang, J. Electrochem. Soc., 2019, 166, A1793–A1798 Search PubMed.
- H.-J. Noh, S.-T. Myung, Y. J. Lee and Y.-K. Sun, Chem. Mater., 2014, 26, 5973–5979 CrossRef CAS.
- R. Koerver, W. Zhang, L. de Biasi, S. Schweidler, A. O. Kondrakov, S. Kolling, T. Brezesinski, P. Hartmann, W. G. Zeier and J. Janek, Energy Environ. Sci., 2018, 11, 2142–2158 Search PubMed.
- Y.-M. Song, J.-G. Han, S. Park, K. T. Lee and N.-S. Choi, J. Mater. Chem. A, 2014, 2, 9506–9513 RSC.
- B. Gault, A. Chiaramonti, O. Cojocaru-Mirédin, P. Stender, R. Dubosq, C. Freysoldt, S. K. Makineni, T. Li, M. Moody and J. M. Cairney, Nat. Rev. Methods Primers, 2021, 1, 51 Search PubMed.
- T. Li, A. Devaraj and N. Kruse, Cell Rep. Phys. Sci., 2022, 3, 101188 CrossRef CAS.
- Z. Wang, D. Santhanagopalan, W. Zhang, F. Wang, H. L. Xin, K. He, J. Li, N. Dudney and Y. S. Meng, Nano Lett., 2016, 16, 3760–3767 CrossRef CAS PubMed.
- W. Chen, X. Zhan, R. Yuan, S. Pidaparthy, A. X. Bin Yong, H. An, Z. Tang, K. Yin, A. Patra, H. Jeong, C. Zhang, K. Ta, Z. W. Riedel, R. M. Stephens, D. P. Shoemaker, H. Yang, A. A. Gewirth, P. V. Braun, E. Ertekin, J.-M. Zuo and Q. Chen, Nat. Mater., 2023, 22, 92–99 CrossRef CAS PubMed.
- F. Lin, I. M. Markus, M. M. Doeff and H. L. Xin, Sci. Rep., 2014, 4, 5694 CrossRef CAS PubMed.
- J. Li and A. Manthiram, Adv. Energy Mater., 2019, 9, 1902731 Search PubMed.
- W. Li, A. Dolocan, P. Oh, H. Celio, S. Park, J. Cho and A. Manthiram, Nat. Commun., 2017, 8, 14589 CrossRef PubMed.
- F. Walther, R. Koerver, T. Fuchs, S. Ohno, J. Sann, M. Rohnke, W. G. Zeier and J. Janek, Chem. Mater., 2019, 31, 3745–3755 CrossRef CAS.
- B.-G. Chae, S. Y. Park, J. H. Song, E. Lee and W. S. Jeon, Nat. Commun., 2021, 12, 3814 CrossRef CAS PubMed.
- A. Devaraj, M. Gu, R. Colby, P. Yan, C. M. Wang, J. M. Zheng, J. Xiao, A. Genc, J. G. Zhang, I. Belharouak, D. Wang, K. Amine and S. Thevuthasan, Nat. Commun., 2015, 6, 8014 Search PubMed.
- O. Cojocaru-Mirédin, J. Schmieg, M. Müller, A. Weber, E. Ivers-Tiffée and D. Gerthsen, J. Power Sources, 2022, 539, 231417 CrossRef.
- J. Maier, B. Pfeiffer, C. A. Volkert and C. Nowak, Energy Technol., 2016, 4, 1565–1574 Search PubMed.
- J. Y. Lee, J. Y. Kim, H. I. Cho, C. H. Lee, H. S. Kim, S. U. Lee, T. J. Prosa, D. J. Larson, T. H. Yu and J.-P. Ahn, J. Power Sources, 2018, 379, 160–166 CrossRef CAS.
- D. R. Vissers, D. Isheim, C. Zhan, Z. Chen, J. Lu and K. Amine, Nano Energy, 2016, 19, 297–306 CrossRef CAS.
- S. Choi, B.-N. Yun, W. D. Jung, T. H. Kim, K.-Y. Chung, J.-W. Son, B.-I. Sang, H.-G. Jung and H. Kim, Scr. Mater., 2019, 165, 10–14 CrossRef CAS.
- M. A. Khan, S. P. Ringer and R. Zheng, Adv. Mater. Interfaces, 2016, 3, 1500713 Search PubMed.
- A. Cerezo, C. R. M. Grovenor and G. D. W. Smith, Appl. Phys. Lett., 1985, 46, 567–569 CrossRef CAS.
- T. T. Tsong and Y. Liou, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 4340–4357 CrossRef CAS PubMed.
- M. Bachhav, F. Danoix, B. Hannoyer, J. M. Bassat and R. Danoix, Int. J. Mass Spectrom., 2013, 335, 57–60 CrossRef CAS.
- M. Tsukada, H. Tamura, K. P. McKenna, A. L. Shluger, Y. M. Chen, T. Ohkubo and K. Hono, Ultramicroscopy, 2011, 111, 567–570 Search PubMed.
- A. Devaraj, R. Colby, W. P. Hess, D. E. Perea and S. Thevuthasan, J. Phys. Chem. Lett., 2013, 4, 993–998 CrossRef CAS PubMed.
- R. Kirchhofer, D. R. Diercks, B. P. Gorman, J. F. Ihlefeld, P. G. Kotula, C. T. Shelton and G. L. Brennecka, J. Am. Ceram. Soc., 2014, 97, 2677–2697 Search PubMed.
- A. Vella, Ultramicroscopy, 2013, 132, 5–18 CrossRef CAS PubMed.
- G.-H. Greiwe, Z. Balogh and G. Schmitz, Ultramicroscopy, 2014, 141, 51–55 Search PubMed.
- B. Pfeiffer, J. Maier, J. Arlt and C. Nowak, Microsc. Microanal., 2017, 23, 314–320 CrossRef CAS PubMed.
- S.-H. Kim, S. Antonov, X. Zhou, L. T. Stephenson, C. Jung, A. A. El-Zoka, D. K. Schreiber, M. Conroy and B. Gault, J. Mater. Chem. A, 2022, 10, 4926–4935 Search PubMed.
- P. Parikh, H. Chung, E. Vo, A. Banerjee, Y. S. Meng and A. Devaraj, J. Phys. Chem. C, 2022, 126, 14380–14388 Search PubMed.
- J. Xie, N. Imanishi, T. Matsumura, A. Hirano, Y. Takeda and O. Yamamoto, Solid State Ionics, 2008, 179, 362–370 Search PubMed.
- P. Bouwman, Solid State Ionics, 2002, 152–153, 181–188 Search PubMed.
- H. Zhang, H. Ning, J. Busbee, Z. Shen, C. Kiggins, Y. Hua, J. Eaves, J. Davis, T. Shi, Y.-T. Shao, J.-M. Zuo, X. Hong, Y. Chan, S. Wang, P. Wang, P. Sun, S. Xu, J. Liu and P. V. Braun, Sci. Adv., 2017, 3, e1602427 Search PubMed.
- A. Patra, J. Davis, S. Pidaparthy, M. H. Karigerasi, B. Zahiri, A. A. Kulkarni, M. A. Caple, D. P. Shoemaker, J. M. Zuo and P. V. Braun, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2025044118 Search PubMed.
- B. Zahiri, A. Patra, C. Kiggins, A. X. Bin Yong, E. Ertekin, J. B. Cook and P. V. Braun, Nat. Mater., 2021, 20, 1392–1400 Search PubMed.
- M. P. Singh, E. V. Woods, S. Kim, C. Jung, L. S. Aota and B. Gault, Batteries Supercaps, 2024, 7, e202300403 CrossRef CAS.
- D. Santhanagopalan, D. K. Schreiber, D. E. Perea, R. L. Martens, Y. Janssen, P. Khalifah and Y. S. Meng, Ultramicroscopy, 2015, 148, 57–66 CrossRef CAS PubMed.
- S. Takeuchi, H. Tan, K. K. Bharathi, G. R. Stafford, J. Shin, S. Yasui, I. Takeuchi and L. A. Bendersky, ACS Appl. Mater. Interfaces, 2015, 7, 7901–7911 CrossRef CAS PubMed.
- H. Xia, L. Lu and G. Ceder, J. Power Sources, 2006, 159, 1422–1427 Search PubMed.
- M. P. Singh, S.-H. Kim, X. Zhou, H. Kwak, A. Kwiatkowski da Silva, S. Antonov, L. S. Aota, C. Jung, Y. S. Jung and B. Gault, Adv. Energy Sustainability Res., 2023, 4, 2200121 CrossRef CAS.
- Y. M. Chen, T. Ohkubo and K. Hono, Ultramicroscopy, 2011, 111, 562–566 Search PubMed.
- W. Lu, J. Zhang, J. Xu, X. Wu and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 19313–19318 Search PubMed.
- Y. Takahashi, Y. Gotoh, J. Akimoto, S. Mizuta, K. Tokiwa and T. Watanabe, J. Solid State Chem., 2002, 164, 1–4 Search PubMed.
- J. He, L. Zhang and L. Liu, Phys. Chem. Chem. Phys., 2019, 21, 12192–12200 RSC.
- J. Houard, A. Vella, F. Vurpillot and B. Deconihout, Batter Supercaps, 2011, 84, 033405 Search PubMed.
- F. Vurpillot, J. Houard, A. Vella and B. Deconihout, J. Phys. D Appl. Phys., 2009, 42, 125502 Search PubMed.
- B.-G. Chae, S. Y. Park, J. H. Song, E. Lee and W. S. Jeon, Nat. Commun., 2021, 12, 3814 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00728c |
| This journal is © The Royal Society of Chemistry 2025 |