
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of electrode passivation during oxidation of a nitroxide radical relevant for flow battery applications†
Cailin
Buchanan
a,
Nora A.
Shaheen‡
ab,
Caroline K.
Williams
a,
Igor
Messias
a,
Bethany
Dean-Kersten‡
b,
Taewoo
Kim
a,
Justin G.
Connell
 a,
Venkat
Srinivasan
c,
Rohan
Akolkar
b and
Pietro
Papa Lopes
*a
a,
Venkat
Srinivasan
c,
Rohan
Akolkar
b and
Pietro
Papa Lopes
*a
aMaterials Science Division, Argonne National Laboratory, Lemont, IL, USA. E-mail: plopes@anl.gov
bDepartment of Chemical and Biomolecular Engineering, Case Western Reserve University, Cleveland, OH, USA
cArgonne Collaborative Center for Energy Storage Science, Argonne National Laboratory, Lemont, IL, USA
First published on 13th June 2025
Abstract
Nitroxide-radicals such as 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) and their derivatives have gained interest as redox-active organic molecules for applications in grid-scale energy storage. In particular, the higher solubility of 4-hydroxy-TEMPO in aqueous media greatly improves its energy density, but unusual kinetics associated with its surface-mediated electrooxidation have limited further development. Here, the apparent passivation behavior of species formed during 4-hydroxy-TEMPO electrooxidation in concentrated electrolytes is investigated. A combination of surface microscopy, X-ray photoelectron spectroscopy, and quartz-crystal gravimetry confirms the formation of a polymeric-type layer over the electrode surface composed of 4-hydroxy-TEMPO-like subunits, which is otherwise not observed with TEMPO. This study indicates that the design of high energy density and stable TEMPO-based redox molecules must also consider the reactivity that may occur due to the molecular characteristics of solubility-enhancing moieties. It is found that the extent of passivation is dependent on the voltage scan rate and 4-hydroxy-TEMPO concentration, underscoring the importance of studying materials at conditions relevant for their proposed applications. Evidence of incomplete passivation and an electrode self-cleaning process is presented, suggesting a materials design strategy to mitigate surface passivation from side reactions that may occur in redox active materials for energy storage applications.
1. Introduction
The integration of low-cost, high-energy density storage solutions are critical for growing resilient and secure electrical power grids.1 Aqueous-based inorganic redox flow batteries (RFBs) are promising for applications with long discharge duration requirements (>10 hours) given their energy scalability and long lifetimes, as well as the fact that they are less geographically constrained and more modular than pumped hydropower storage.1–3 However, the state-of-the-art all-vanadium RFB (VRFB) has a high cost inherent from the non-abundant nature of the vanadium precursor as well as its relatively low cell voltage and corresponding lower energy density.4 Organic RFBs (ORFBs) represent a promising alternative to the VRFB because they are based on earth-abundant molecules and are tunable for critical factors that affect battery energy and capacity characteristics such as redox potential and solubility.3,5 ORFBs can be aqueous or non-aqueous. Although non-aqueous ORFBs have expanded voltage windows because they are not limited by the electrochemical stability of water,4,6 they can suffer from poor solubility, low power density, and safety concerns.3,5 Aqueous ORFBs represent a superior alternative because they are non-combustible and tend to have higher solubility and ionic conductivity.3,5,6Nitroxide radical-containing molecules like 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) and its derivatives are a promising class of RFB catholyte chemicals on account of their rapid kinetics and relatively high redox potential.7 These TEMPO-based molecules have been explored extensively for use in RFBs in both non-aqueous8–15 and aqueous16–41 electrolytes. Many different functionalization reactions of TEMPO have been reported in an effort to improve its stability, solubility, redox potential, and reduce crossover, including the addition of an oxygen,12 methoxy,10,41 hydroxy (4-OH-TEMPO or HT),21–23 carboxyl,20 amine-based groups such as TMAP-TEMPO,19 TEMPTMA or NMe-TEMPO,24,25,30,31 (TPABPy)Cl3,37 MIAcNH-TEMPO,36 and N2-TEMPO,26 sulfonates,33 Riboflavin,38 and various polymerized TEMPO molecules.11,13–15,18,28,29,39,42 HT has been identified as an especially promising redox-active species for use in aqueous catholytes or posolytes due to its commercial availability, higher solubility in water compared to TEMPO (2.11 M vs. 0.07 M),22,40 and comparable reaction durability.
Previous studies of HT have established it as a plausible aqueous RFB catholyte at modest concentrations (e.g., 0.05–0.5 M), however detailed kinetic studies by Shaheen et al.43,44 observed an unusual electrochemical behavior that must be investigated further if HT is to be used in RFBs. The seminal investigation of HT for use as an RFB catholyte tested its performance at concentrations up to 0.5 M HT in 1.5 M NaCl (its solubility limit at this concentration of NaCl), coupled with methyl viologen as the anolyte, and found that a voltage of 1.25 V and capacity retention of 89% could be achieved after 100 cycles with a current density of 60 mA cm−2.22 Linear sweep voltammetry (LSV) studies revealed that the oxidation of HT was mass transport controlled at a scan rate of 5 mV s−1 at potentials greater than 0.9 V vs. NHE.22 An additional study of HT in a full cell configuration coupled with a Zn anode reported a voltage of 1.42 V and coulombic efficiency of 86.1% for a system of 0.1 M HT in 1 M NaCl at 10 mA cm−2 current density.21 Cyclic voltammetry (CV) behavior of 0.1 M HT in 1 M NaClO4 appeared reversible, and the reported kinetic rate constant suggested a quasi–reversible reaction at the conditions tested.21 Shaheen et al. have previously conducted electrochemical studies of HT in both aqueous and non-aqueous electrolytes, however, that resulted in unusually large anodic charge transfer coefficients in both aqueous and non-aqueous electrolytes. Additionally, in situ electrochemical surface enhanced Raman spectroscopy showed the appearance of new C- and N-related bands during oxidation, and the open circuit voltage relaxation time following an oxidation event of HT was slower than expected for a purely diffusion-limited process.44 Based on these observations, a two-step charge transfer mechanism was proposed wherein HT first undergoes a fast adsorption and electron transfer, followed by a rate-limiting desorption step.43 In this study, however, we explore a new interpretation that would be equivalent with the previously proposed mechanism: passivation of the electrode surface. Irreversible passivation would be detrimental to the performance of an RFB, which requires repeated deep discharge and charge cycling. Importantly, limited experimental information based on concentrations of HT less than 0.5 M and scan rates greater than 5 mV s−1, as used throughout the literature, prevent a deeper understanding of the electrochemical behavior of HT in conditions that are more realistic for RFB applications. Therefore, it is important to work at concentrations close to the solubility limit to fully utilize the potential of the redox-active organic material (ROM), and to understand how the ROM behaves at scan rates that are close to long duration energy storage battery operation.
Herein, we expand the concentration range of HT studied to be between 0.002 and 2 M, and we explore voltametric behavior at scan rates between 0.5 and 1000 mV s−1 across several dissimilar electrodes. We identify that HT passivation indeed occurs at high concentrations and low scan rates and is independent of electrode surface, and that the rate of passivation increases as HT concentration increases or scan rate decreases. Additionally, we further characterize the species present on the electrode surface through electrochemical techniques coupled with X-ray photoelectron spectroscopy (XPS), quartz crystal gravimetry, and electron microscopy. These characterization efforts confirm the formation of a surface passivating film. Furthermore, the presence of the film uniquely forming with HT, but not TEMPO, suggests the significance of the hydroxyl moiety in mediating the passivation reaction. Finally, via rotating disk electrode (RDE), rotating ring disk electrode (RRDE), and in situ inductively coupled plasma mass spectrometry (ICP-MS), we reveal evidence of a possible self-cleaning mechanism of the electrode surface that can be achieved at intermediate HT concentrations which presents a possible path forward for using HT as an RFB catholyte. These findings related to the behavior and composition of the passivation layer have important implications for the use of HT as an active material in an RFB and highlight the importance of studying a promising active material at conditions consistent with the end-use application.
2. Experimental
2.1 Electrolyte preparation
Sodium chloride (NaCl, 99%, Sigma-Aldrich), TEMPO (C9H18NO, 99%, Sigma-Aldrich), 4-hydroxy-TEMPO (HT, C9H18NO2, >98% purity, Alfa Aesar) were used as received. Aqueous electrolytes of TEMPO (0.05 M) and HT (0.002 M–2 M) in 0.5 M NaCl were prepared using Millipore ultrapure (18.2 MΩ cm) deionized water and de-aerated by purging with Ar gas (Airgas, Research Plus grade).2.2 Electrochemical measurements
All electrochemical measurements were performed in a standard three-electrode configuration. For rotating disk electrode (RDE) experiments, the working electrodes were glassy carbon (GC), Pt, or Au. For rotating ring disk electrode (RRDE) experiments, a platinum ring (7.5 mm inner diameter, 8.5 mm outer diameter) that is integrated into the collet that secures the disk electrode along with its PTFE sleeve (u-cup) was used. Prior to each experiment, the disk electrodes were polished sequentially using 1 μm alumina slurry, followed by 0.3 μm, and finally 0.05 μm slurry. After the electrodes were polished, they were ultrasonicated first in ethanol followed by deionized water. For RRDE experiments, the ring was polished with a 0.05 μm Al2O2 slurry on a microfiber polishing pad, then sonicated in isopropanol (once for 5 minutes), followed by two sonication steps in DI water. The counter electrode was a graphite rod (Alfa Aesar, 99.9995% metals basis), and the reference electrode was Ag/AgCl (3 M KCl, Sigma-Aldrich). An AutoLAB PGSTAT302N was used for all electrochemical measurements. Voltammetry using the RDE configuration was performed at rotation rates varying between 0 and 1600 rpm. Appropriate IRΩ-correction was applied to steady state data using RΩ = 40 Ω for a fixed cell geometry and electrode placement. Due to the observed higher resistivity at concentrations at, or greater, than 0.2 M HT, CV data was collected using live IRΩ-compensation, which accounts for 90% of total resistance.For quantification of oxidation side-products (O2, Cl2, and H2O2) using RRDE measurements, the Pt ring electrode was scanned over a potential range of −0.2 to 1.7 V vs. RHE under three distinct experimental conditions, each designed to isolate the current contribution from a specific species. In the first, the electrolyte (0.5 M NaCl) was saturated with O2 (Airgas, 99.9999% Research Plus grade). In the second, 0.001 M of H2O2 was added to the Ar-saturated (Airgas, 99.9999% Research Plus grade) electrolyte. In the third (for Cl2 detection), a mixed electrolyte of 0.1 M H2SO4 + 0.05 M NaCl was employed. In this case, Cl− was electrochemically oxidized to Cl2 using a 6 mm IrO2 disk electrode, which is known for its activity toward chlorine evolution.45 The IrO2 electrode was held at anodic potentials sufficient to generate Cl2, while the Pt ring was continuously scanned to detect the oxidation products.
The selectivity toward O2, Cl2, and H2O2 produced at the glassy carbon electrode in 0.5 M NaCl was evaluated using the RRDE configuration. The disk electrode was swept from 0.2 to 1.55 V vs. Ag/AgCl at a scan rate of 0.5 mV s−1 under hydrodynamic conditions (900 rpm), while the Pt ring was held at three potentials chosen to enable the selective detection of each species. Partial currents corresponding to O2, Cl2, and H2O2 were deconvoluted using eqn (1)–(3), which describe the individual electrochemical responses at the ring. Ring currents were corrected using the theoretical collection efficiency based on the RRDE geometry (N = 0.24), yielding the corresponding product selectivity.
 | (1) |
 | (2) |
 | (3) |
2.3 Quartz crystal microbalance (QCM)
A QCM200 (Stanford Research Systems) was used to collect all in situ gravimetric data. A 5 MHz FilTech Pt quartz crystal was used as the working electrode. The counter and reference electrodes were graphite and saturated Ag/AgCl, respectively. The crystal was cleaned by first rinsing with ethanol followed by de-ionized water, and dried with Ar. The electrolyte was de-aerated with an Ar purge.2.4 Scanning electron microscopy (SEM)
All SEM images were taken on a Zeiss Sigma 300 microscope and collected at 5 kV and 10 kV. Electrode samples were polished using the procedure outlined above. Following electrochemical treatment, the electrodes were rinsed with excess deionized water and dried with an Ar stream to ensure residual electrolyte was removed. Samples were immediately mounted for imaging.2.5 X-ray photoelectron spectroscopy (XPS) measurements
XPS measurements were performed using a Specs PHOIBOS 150 hemispherical energy analyzer using a monochromated Al Kα X-ray source. Survey spectra were measured using a pass energy of 40 eV at a resolution of 0.2 eV per step and a total integration time of 0.1 s per point. Core level spectra were measured using a pass energy of 20 eV at a resolution of 0.05 eV per step and a total integration time of 0.5 s per point. Deconvolution was performed using CasaXPS software with a Shirley-type background and 70–30 Gaussian–Lorentzian peak shapes. Spectra were charge referenced using the deconvoluted position of sp3 carbon at 284.8 eV. Samples were prepared using polycrystalline Pt electrodes and the same rinsing/drying procedure employed for SEM analysis.2.6 Inductively coupled plasma mass spectrometry (ICP-MS)
In situ ICP-MS was coupled with a stationary probe rotating disc electrode (SPRDE) as described in a previous report46 to measure Au (197 amu) dissolution. The system operated with a flow rate of 14 μL s−1, a 6 s delay time, and a working electrode rotation rate of 100 rpm which resulted in a probe collection efficiency of 0.25.3. Results and discussion
3.1 Electrochemical evidence of passivation
We begin by exploring the effect of scan rate on the behavior of the glassy carbon (GC) RDE in the presence of HT in an aqueous solution. CVs were collected in 0.2 M HT/0.5 M NaCl as a function of scan rate at 900 rpm, see Fig. 1. As seen in Fig. 1a and b, at scan rates of 1000 mV s−1 and 100 mV s−1, the CVs are consistent cycle after cycle. There are clear oxidation and reduction peaks, however the reduction peak is not equal in magnitude to the oxidation peak. This difference could be because the use of rotation results in a thinner diffusion layer than there would be without rotation, which means there is less time required for the oxidized product to diffuse away from the surface before rotation convection transports the ions away, which could result in smaller reduction activity. This rotation effect in which the local ion concentration affects the CV shape is a known phenomenon that has been observed for other electrochemical systems.47,48 For the first CV cycle at 10 mV s−1 (Fig. 1c), the current plateaus at potentials greater than 1.2 V vs. Ag/AgCl, indicating mass transport control, and the reduction peak disappears almost entirely. The loss of the reduction peak as a function of scan rate could be due to the effect of rotation limiting the amount of oxidized HT available for reduction or it could suggest the existence of a two-step mechanism in which the oxidation (E) is coupled with a slow chemical step (C).49 Importantly, the overpotential required to reach the current plateau in successive CV cycles increases in Fig. 1c, suggesting that the GC electrode surface is altered with each CV cycle at 10 mV s−1. To further probe this effect, a significantly slower scan rate of 0.5 mV s−1 was explored (Fig. 1d). | ||
| Fig. 1 Series of CVs of GC RDE in 0.2 M HT/0.5 M NaCl solution collected at 900 rpm for the following scan rates: (a) 1000 mV s−1, (b) 100 mV s−1, (c) 10 mV s−1, and (d) 0.5 mV s−1. | ||
In the first CV cycle of Fig. 1d, an oxidation peak is observed on the forward sweep, and there is no obvious reduction peak observed on the reverse sweep, similar to Fig. 1c. A peak current of 39 mA cm−2 is observed at 0.82 V vs. Ag/AgCl. Based on the Levich equation,50 which indicates that the diffusion-limited current, iL, will depend on the rotation rate (but not the scan rate), iL is expected to be 76 mA cm−2 in 0.5 M NaCl, meaning that the observed current is only 51% of the expected iL (see Table S1 in ESI for more details†). In successive CV cycles (see inset of Fig. 1d), the oxidation peaks continuously decrease in magnitude and are significantly diminished relative to the first cycle. A similar observation was reported in a 2018 study on phenol oxidation,51 and was attributed to the formation of a blocking film. As a consequence of such a film passivating the electrode, we also observe an increase in the overpotential required to drive 1 mA cm−2. Between cycles 1 and 2, the overpotential increased by over 400 mV. Passivation clearly becomes more prevalent at lower scan rates. Given that passivation is already a significant problem at 0.5 mV s−1, we did not study even lower scan rates despite their relevance for long discharge duration applications in order to strike a balance between experimentally practical and realistic conditions.
To determine the type of mechanism that is occurring with HT present in solution, we collected electrochemical behavior of 0.2 M HT on two other, dissimilar electrodes: Pt and Au. As shown in Fig. S1,† the electrochemical behavior across CV scan rates is consistent. At both 0.5 mV s−1 and 10 mV s−1, the potential at which activity begins is the same for all three surfaces, after accounting for differences in IRΩ correction. This suggests that the electron transfer that occurs when HT is electrochemically oxidized in 0.5 M NaCl is the same for all surfaces, indicative of an outer-sphere electron transfer (ET). At 0.5 mV s−1 for each of the electrodes (Fig. S1b†), some amount of passivation occurs for each of the electrodes based on the peak in current that is less than the expected iL.
To explain our observations, we suggest the occurrence of a surface passivation mechanism. At conditions where passivation will be sluggish, the electron transfer occurs such that HT is oxidized to the product HT+via an outer-sphere mechanism (Scheme 1a). We propose that a competing surface reaction of passivation becomes more prevalent at slower scan rates. Under such conditions, the oxidized HT+ either self-reacts or reacts with HT (Scheme 1b) to form a blocking film. The rate of this passivation reaction is also expected to be dependent on the amount of HT present in solution. At high applied overpotentials, i.e., high current, the amount of HT+ produced is large and passivation is facile. At low applied overpotentials, and low current, the amount of HT+ produced is small and passivation is sluggish. At low overpotentials (0.65 V vs. Ag/AgCl in Fig. S2†), HT+ self-reaction and surface passivation may still occur. However, because the amount of HT+ produced is still very small, the electrode behaves as a clean electrode. The competitive nature of HT oxidation, proposed to be dependent on both scan rate and HT concentration, highlights the importance of studying electrochemical processes at conditions relevant for their end application, e.g., slow charging rates and high ROM concentrations for RFBs.
 | ||
| Scheme 1 Proposed oxidation mechanisms for HT in 0.5 M NaCl for the following limiting cases: (a) low HT concentration ([HT]), fast scan rates (ν), or (b) high [HT], slow ν. | ||
3.2 Characterizing the passivated layer
Given the influence of HT concentration on the rate of passivation proposed in Scheme 1, ex situ scanning electron micrographs (SEM) were collected to physically characterize the electrode surface following the oxidation of HT at two different concentrations, 0.05 M (Fig. 2a–c) and 0.2 M (Fig. 2d–f). The current transient responses to an applied potential of 1.5 V vs. Ag/AgCl (within the diffusion limited region) are shown in Fig. 2a and d for 0.05 M and 0.2 M HT, respectively. In both cases, the current transient rapidly decays and a heterogeneous organic layer is clearly prevalent on the electrode surface, albeit to different extents. Fig. 2b and c show that at 0.05 M HT, isolated islands of an oxidized product appear on the electrode surface. The SEM of a bare GC electrode is included in the ESI† for comparison, and only the polishing grooves of the pristine electrode are visible (Fig. S3†). At 0.2 M HT, the oxidized product has significantly more coverage over the GC electrode (Fig. 2e and f), highlighting that HT concentration plays a role in extent of passivation. A cross-sectional view of the GC oxidized at 0.2 M HT (Fig. S4†) reveals that the passivated product formed on the GC surface has radial dimensions, potentially indicating that the unpassivated portions of the covered electrode act as a series of microelectrodes experiencing radial diffusion during passivation at these concentration and potential conditions.52 We find that a disperse passivation film also forms for the 0.2 M HT solution at lower potentials (Fig. S5†), highlighting the interplay between HT concentration and potential on the passivation mechanism. Additionally, the effect of the hydroxyl moiety was investigated by comparing HT to TEMPO (Fig. S6†), and the electrochemical and microscopic results suggest that the hydroxyl moiety participates in mediating the surface passivation. | ||
| Fig. 2 For a 0.05 M HT/0.5 M NaCl solution and a GC RDE oxidized for 1500 s at 1.5 V vs. Ag/AgCl: (a) current transient response to the applied potential step, and corresponding SEM images with a (b) top-down view and (c) cross sectional view at an 86.5° angle. For a 0.2 M HT/0.5 M NaCl solution and a GC RDE oxidized for 1500 s at 1.5 V vs. Ag/AgCl: (d) current transient response to the applied potential step, and corresponding SEM images with a (e) top-down view and (f) cross sectional view at an 86.5° angle. | ||
In addition to SEM imaging, in situ gravimetry and ex situ X-ray photoelectron spectroscopy (XPS) were used to confirm the presence of a passivated film in solutions with HT. Fig. 3a shows the gravimetric response to the electrooxidation of HT in an aqueous electrolyte collected on an electrochemical quartz crystal microbalance (eQCM) using a Pt electrode, which is more commonly used for eQCM measurements than GC. As discussed previously, we have shown that the electrochemical behavior of Pt, GC, and Au RDEs are all similar in 0.2 M HT/0.5 M NaCl (Fig. S1†), suggesting the passivation mechanism is still relevant and that findings for the Pt electrode are transferable to the GC electrode.53 In order to increase the amount of mass changes due to the passivation film formation we employed a slower sweep rate of 0.1 mV s−1. As the voltammogram is scanned anodically in Fig. 3a (upper panel), the onset potential is 0.55 V vs. Ag/AgCl. At this potential in Fig. 3a (lower panel), the relative mass of the Pt electrode also increases. As the current density decreases on the reverse scan in Fig. 3a, the relative mass does not return to zero, however. While Pt oxide could be forming at these potentials, we show that at these pH and electrolyte conditions, this does not result in significant changes in mass (see Fig. S7†). On the reverse sweep, there is in fact a mass loss, which has also been reported in previous reports of Pt oxidation and reduction,54,55 and is not what we observe in Fig. 3a. Based on the fact that we observe a significant mass gain in the presence of HT oxidation that is not replicated when HT is not present, we conclude that Pt oxidation is not significantly contributing to our eQCM results and instead, it further confirms the formation of a species blocking the electrode surface. To be consistent with the eQCM study, and because any C 1s core level XPS features would be masked on GC, XPS of an oxidized Pt electrode was explored next.
 | ||
| Fig. 3 For a 0.2 M HT/0.5 M NaCl solution: (a) slow-scan (0.1 mV s−1) voltammetry collected on a Pt eQCM, and the corresponding gravimetric response. Peak-to-peak normalized, high-resolution XPS spectra for (b) C 1s (upper panel) and N 1s (lower panel) collected on a Pt electrode for the following conditions: drop cast HT (dark gray), oxidized for 30 s at 0.05 M HT and 1.5 V vs. Ag/AgCl (light pink line), oxidized for 30 s at 0.2 M HT and 1.25 V vs. Ag/AgCl (pink line), and oxidized for 30 s at 0.2 M HT and 1.5 V vs. Ag/AgCl (dark pink line). | ||
XPS analysis of Pt electrodes exposed to HT under a variety of conditions, including drop casting and oxidation for 30 seconds in the presence of HT at varying concentrations (0.05 and 0.2 M) and oxidation potentials (1.25 and 1.5 V vs. Ag/AgCl) reveal systematic changes in the chemistry of the adsorbed film (Fig. 3b). We note that HT is relatively volatile, and pump-down to ultrahigh vacuum conditions for XPS measurements resulted in low signals related to the HT layer, particularly for drop-cast and 0.05 M HT samples; nonetheless, clear trends in surface chemistry and layer thickness were still distinguishable. In particular, the surface chemistry of electrodes oxidized in the presence of 0.05 M HT is essentially indistinguishable from that of an electrode with pristine HT drop cast onto it, consistent with the relatively thin and discontinuous passivation layers observed in Fig. 2b and c. In contrast, electrodes oxidized in the presence of 0.2 M HT at 1.25 V vs. Ag/AgCl reveal clear shifts to higher binding energy (BE) in the peak envelopes for both the C 1s and N 1s (Fig. 3b) core levels, consistent with the oxidation of HT via the mechanism discussed above. Increasing the oxidation potential to 1.5 V vs. Ag/AgCl yielded increased shifts in these peak envelopes to higher BE, as well as a thicker film as determined by the greater extent of signal attenuation from the underlying Pt electrode (Fig. S8, S9 and Table S2†). The observed changes in the C 1s core level spectrum indicate the formation of a greater fraction of oxidized C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO species (Fig. S10, Tables S3 and S4†), consistent with increasing oxidation of the HT molecule at higher potentials and/or concentration. Although the N 1s signal is relatively weak, there is also a clear shift to higher BE that is attributed to electron density being drawn away from the nitrogen atom in the HT molecule as it is oxidized. Scheme S1 in the ESI† demonstrates a possible mechanism that aligns with these XPS findings as well as SEM evidence of a polymer and the passivation mechanism laid out in Scheme 1. Overall, these results are consistent with increased oxidation of HT at higher potentials, as well as with the thicker passivating films observed in Fig. 2e and f for 0.2 M solutions relative to 0.05 M solutions. The difference in passivated films observed for a 0.05 M and 0.2 M HT solutions observed through SEM and XPS further aligns with our proposed passivation mechanism in Scheme 1 that HT concentration affects the extent of passivation.
O and O–CO species (Fig. S10, Tables S3 and S4†), consistent with increasing oxidation of the HT molecule at higher potentials and/or concentration. Although the N 1s signal is relatively weak, there is also a clear shift to higher BE that is attributed to electron density being drawn away from the nitrogen atom in the HT molecule as it is oxidized. Scheme S1 in the ESI† demonstrates a possible mechanism that aligns with these XPS findings as well as SEM evidence of a polymer and the passivation mechanism laid out in Scheme 1. Overall, these results are consistent with increased oxidation of HT at higher potentials, as well as with the thicker passivating films observed in Fig. 2e and f for 0.2 M solutions relative to 0.05 M solutions. The difference in passivated films observed for a 0.05 M and 0.2 M HT solutions observed through SEM and XPS further aligns with our proposed passivation mechanism in Scheme 1 that HT concentration affects the extent of passivation.
3.3 Effect of HT concentration of passivated layer
Given the evidence from SEM and XPS that in addition to scan rate, HT concentration affects passivation, we studied the effect of HT concentration on the electrochemical behavior. We find that passivation of the GC does not begin until concentrations are greater than 0.01 M HT and that passivation happens faster as concentration increases. Fig. 4a shows CVs of a GC collected at various concentrations of HT in 0.5 M NaCl, ranging from 0.002 M to 2 M HT. These CVs were collected using a rotation rate of 900 rpm and a scan rate of 0.5 mV s−1, which were conditions previously established to result in passivation at 0.2 M HT. At concentrations greater than 0.2 M HT, there is a clear peak in the anodic sweep, consistent with the passivation behavior observed at 0.2 M HT. Successive cycles of each of these samples demonstrated significantly lower current density magnitudes (Fig. S11†), corroborating our conclusion that passivation is occurring at these HT concentrations. The noise incurred in the CV at 1 M and 2 M HT samples is due to current range limitations of the potentiostat. The effect of HT concentration of Au's and Pt's behavior was also explored (Fig. S12†), and it was found that the electrochemical behavior remains the same for all electrodes up to 0.2 M HT and a wide range of scan rates, further supporting the proposed outer-sphere electron transfer behavior. From Fig. 4a and b, it is evident that the oxidation current is mass transport-controlled at HT concentrations equal to or less than 0.02 M HT. While there is no evidence of passivation for the 0.002 M HT solution, the 0.02 M HT solution reveals irreversible behavior given the decrease in activity on the negative scan that could still suggest passivation is occurring, albeit at slower rates.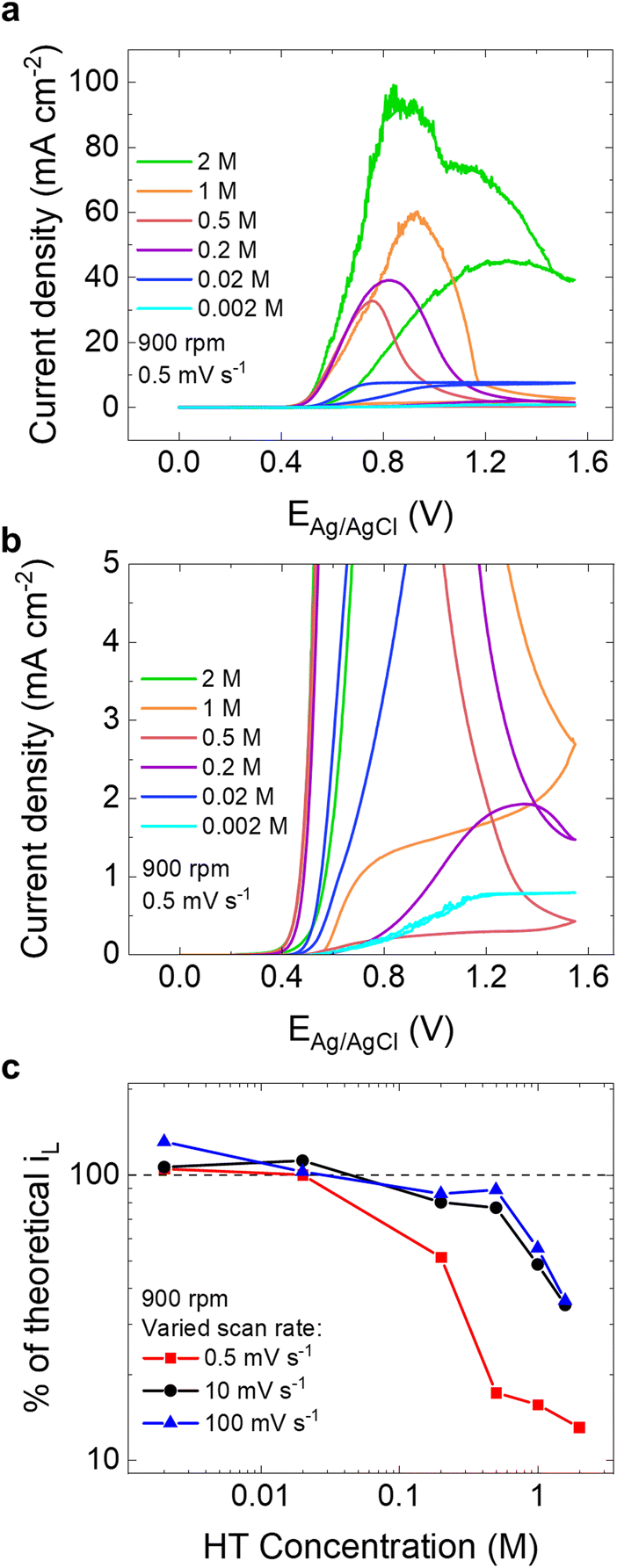 | ||
| Fig. 4 (a) CVs of GC RDE in 0.5 M NaCl collected at a rotation rate of 900 rpm and a scan rate of 0.5 mV s−1 at a variety of HT concentrations ranging from 0.002 M to 2 M. (b) Zoomed in inset of CVs collected at 900 rpm, 0.5 mV s−1, and 0.002–2 M HT. (c) Percentage of theoretical limiting current density, iL, as a function of HT concentration at 900 rpm and varied scan rates (0.5 mV s−1, red squares; 10 mV s−1, black circles; 100 mV s−1, blue triangles). | ||
To better understand the extent of passivation as a function of concentration and scan rate, the peak current achieved for each HT concentration is compared to the expected theoretical diffusion-limited current, iL, in Fig. 4c at several scan rates. Details regarding the theoretical iL are included in Table S1.† The percentage of observed current relative to iL are most likely greater than 100% at 0.002 M and 0.02 M HT because the diffusion coefficient (D) and viscosity (μ) values used in the Levich equation were based on a more concentrated HT solution, where D is expected to be larger and μ is expected to be smaller (see ESI for more details†). At 0.5 mV s−1, there is a significant drop in observed current relative to iL from 100% at 0.02 M HT to 51% at 0.2 M HT. The observed current drops even more dramatically at 0.5 M HT, meeting only 17% of the expected limiting current. As the HT concentration is further increased, the percentage of theoretical iL met continues to decrease, although at a slower rate. It is evident that at a scan rate of 0.5 mV s−1, passivation becomes more prevalent as HT concentration increases. Compared to the electrochemical behavior at 0.5 mV s−1, passivation proceeds more slowly at 10 mV s−1 and 100 mV s−1, with more than 75% of iL still being achieved at 0.5 M HT for both scan rates. At concentrations greater than 0.5 M HT, however, the drop off in observed current relative to iL becomes sharper, suggesting that the rate of passivation is controlled by an interplay of scan rate and concentration. Clearly, when assessing active materials for RFB applications, it is critical to study the electrochemical behavior of the material at realistic operating conditions, e.g., slow discharge or charge rates and high concentrations. If HT is to be considered as a ROM for a RFB, the reality of its passivation behavior must be contended with.
Curiously, studying the CV cycles at the intermediate concentration of 0.02 M HT revealed an interesting electrochemical behavior that could be exploited to address the issue of HT passivation through a self-cleaning strategy. In Fig. 5a, a series of sequential CV cycles completed on one GC RDE in 0.02 M HT/0.5 M NaCl and a fresh GC RDE in 0.5 M NaCl without HT are included. The oxidation on the first cycle with HT is mass-transport controlled and achieves the expected iL, however, there is a reduced level of electrochemical activity on the return sweep, unlike the steady state behavior observed at 0.002 M HT. It is notable that on the second CV cycle in Fig. 5a, the oxidation current density for the 0.02 M HT solution is reduced and the overpotential required to oxidize the material is increased relative to the first cycle (see Fig. S13† for overlapping CV cycles), both of which suggest that the GC is already partially passivated. Surprisingly, there is an uptick in activity at approximately 1.20 V vs. Ag/AgCl, which aligns with the potential at which the GC surface appears to undergo an oxidation reaction on the second cycle even without HT present (lighter colored traces in Fig. 5a). Note that the dashed vertical lines in Fig. 5a were identified simply based on when the uptick in activity occurs for the system with HT present and then compared to the bare GC potential profile. While we refer to it as the onset potential for carbon oxidation, it is important to emphasize that our choice reflects the correlation between the carbon oxidation voltammetry traces observed for bare GC surface and the recovery of the HT oxidation activity approaching the expected diffusion limit. It is understood that the choice of onset potential does not have a standard accepted definition,56 and the correlation observed here serves to give context to the processes underlying the sudden changes in activity observed during oxidation of lower concentrations of HT. On the reverse sweep, the activity for the solution with HT present is higher than in the forward, anodic scan direction. In the third CV cycle, the oxidation current activity with 0.02 M HT is still lower, but the uptick that aligns with oxidation of the GC surface without HT present occurs at an earlier potential, 1.12 V vs. Ag/AgCl. The current density on the return sweep is once again elevated and is comparable to the current observed on the return sweep in cycle 2. Although this shift of onset potential to lower voltages might seem counterintuitive given that more GC is being blocked by passivated product, there are other factors at play such as roughening of the GC surface or the ease of carbon oxidation increasing once it is activated, which we in fact observed (see lighter colored traces in Fig. 5a). This unexpected electrochemical behavior suggests that the passivation mechanism can be mitigated via removal of a surface layer that effectively cleans the passivating film, as we discuss next.
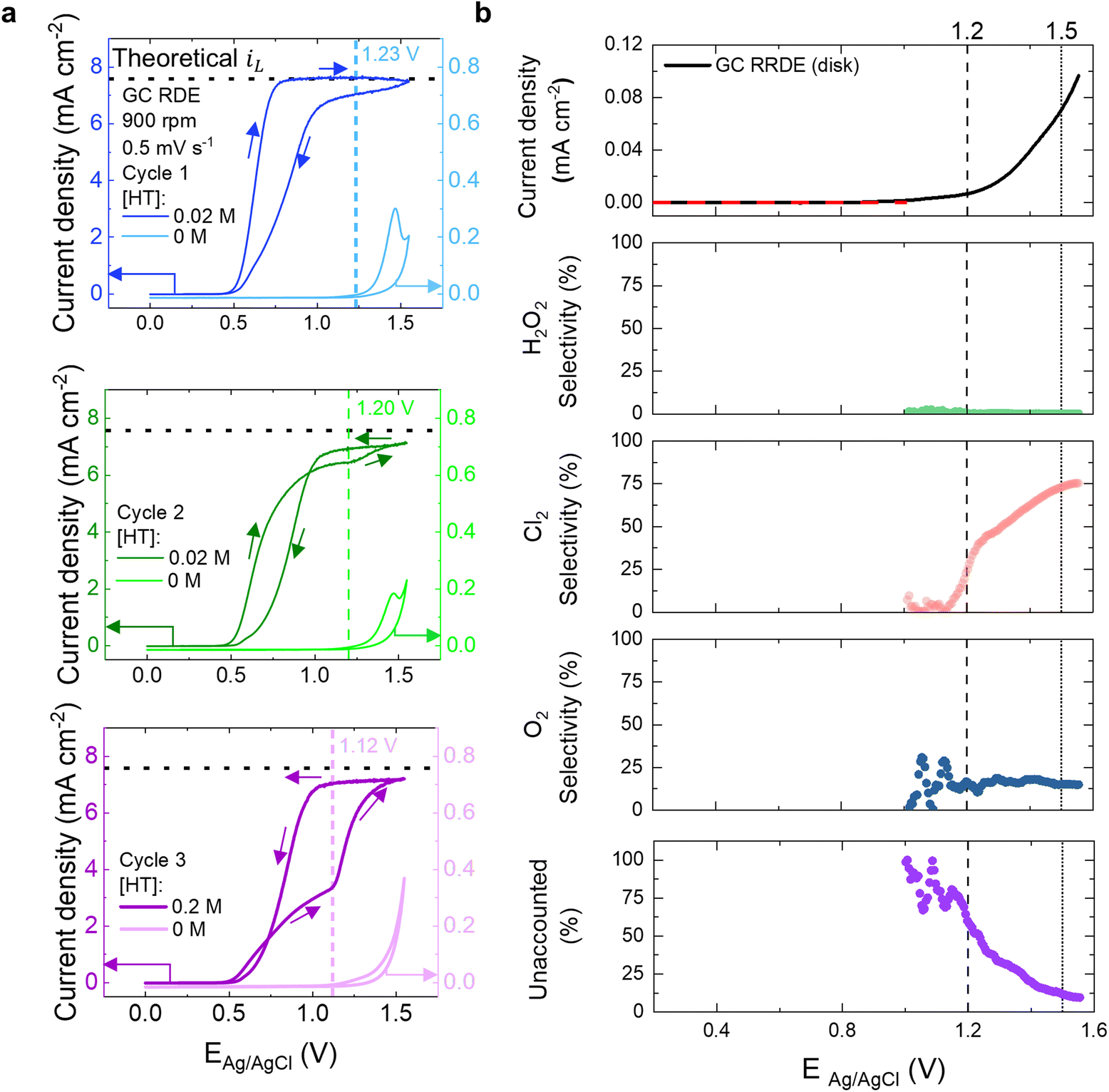 | ||
| Fig. 5 (a) Comparison of a series of CVs of GC RDE in 0.5 M NaCl with and without 0.02 M HT collected at a rotation rate of 900 rpm and a scan rate of 0.5 mV s−1. (b) Simultaneous monitoring of GC electrode current (top, black) (the red dotted lines indicate the potential range excluded from the calculations), H2O2 selectivity (second plot from the top), Cl2 selectivity (third plot from the top), O2 selectivity (second plot from the bottom), and amount of activity unaccounted for from ring (bottom plot) during a CV experiment on GC in 0.5 M NaCl electrolyte. The scan rate for RRDE was 0.5 mV s−1 and the rotation rate was 900 rpm. | ||
To align with the observed electrochemical behavior at 0.02 M HT, we propose an addition to our mechanism outlined in Scheme 1 that is relevant at intermediate HT concentrations on GC, referred to as “self-cleaning”. We propose that at intermediate HT concentrations like 0.02 M, incomplete passivation of the GC surface occurs, which is corroborated by the fact that oxidation activity is still observed on the reverse sweep in the first cycle of Fig. 5a. The incomplete passivation results in exposed areas of GC that are capable of being oxidized, which has been observed previously for GC in neutral media.57 An alternative mechanism could be that oxygen evolution is occurring, however, it would not be thermodynamically favorable in neutral media.57 Additionally, as described in more detail below, our rotating ring disk electrode (RRDE) measurements indicate that a process like carbon corrosion could be occurring at these early electrode potential oxidation conditions.
To further probe the possible products formed during oxidation and identify possible mechanisms that happen during the observed self-cleaning, we used RRDE measurements since it can be used to quantify products formed at the disk electrode surface by reacting them at the ring.58 As demonstrated by Fig. 5b, a Pt ring was used to quantify the production of O2, H2O2, and Cl2 at the GC disk surface (see methods section and Fig. S14† for protocol used to establish the relevant potentials that the ring should be held at). At negative ring potentials (−0.1 V vs. RHE, see ESI†), where all three species are electrochemically reducible, the total faradaic efficiency (FE) reaches a maximum of ∼90%. This suggests that a portion of the anodic current measured at the GC disk does not correspond to detectable species at the Pt ring, likely side reactions such as carbon corrosion (see “Unaccounted” panel in Fig. 5b). To isolate the contribution from Cl2, the ring potential was held at 0.9 V vs. RHE, where only Cl2 reduction occurs. Under these conditions, a FE of about 25% towards Cl2 was observed at 1.2 V vs. Ag/AgCl, which is where the onset of the self-cleaning begins in the first cycle, and the FE for H2O2 was negligible at below 1%. Additionally, 16% of the activity was attributed to O2. Thus, at 1.2 V vs. Ag/AgCl, 58% of the activity goes towards an unknown product, which could be assigned to carbon corrosion. At 1.5 V vs. Ag/AgCl, a FE towards Cl2 of approximately 73% was observed, indicating that Cl2 is the primary product of electrolyte oxidation at high voltages. Again, the FE for H2O2 was negligible, and O2 has an FE of 15%. At 1.5 V vs. Ag/AgCl, 12% remains for an unknown process such as carbon corrosion. Based on the data presented in Fig. 5b, it is reasonable to assume that carbon corrosion occurs to varying degrees at the potentials relevant to self-cleaning between 1.0 and 1.5 V vs. Ag/AgCl and could be driving the self-cleaning mechanism. It is also plausible that the self-cleaning mechanism involves oxidation of the passive layer by the formation of reactive oxidation species such as H2O2 or Cl2, which ultimately lead to removal of the passivation layer formed during HT oxidation.
From Fig. 5a, the potential required to oxidize the GC without HT present decreases with each successive cycle. While the product being formed through GC corrosion could not be directly measured through RRDE, we expect it would be a standard carbon oxidation product such as CO and CO2.59 We propose that the oxidized carbon interferes with the passivated layer, for instance by preventing adhesion of the passivated layer to the carbon surface. As a result, the amount of passivated area is lessened, and there is carbon surface area available for additional HT oxidation and re-passivation on the reverse sweep, as shown in the second and third CV cycles of Fig. 5a. The high passivation activity on the second scan could be due to the fact that only small amounts of GC are exposed and available for oxidation after the first cycle, but the amount gradually increases. The reduction in oxidation capacity on the forward scan observed from cycle 1 to 3 could indicate that the passivated HT layer that is removed from the electrode surface following the self-cleaning mechanism is destroyed or otherwise altered so that it no longer contributes to the oxidation reaction. Despite this loss in capacity, however, cycles beyond CV number 3 show that the oxidation activity on the anodic sweep eventually stabilize in the presence of 0.02 M HT (Fig. S15†), suggesting that oxidation of the underlying, exposed carbon surface is an effective method of removing the passivated material. By cycle 3, a steady state has been reached in which the GC is partially passivated, cleaned, and then partially re-passivated on the reverse sweep.
In addition to studying the self-cleaning mechanism on GC, Au was used to further probe the possibility of using electrode corrosion to our advantage. In Fig. 6a, the electrochemical behavior of GC and Au (along with Pt) in 1 M HT and a scan rate of 0.5 mV s−1 are shown. While onset potential is similar between all three electrodes, and differences in IRΩ correction are assumed to account for the difference in the slope of the current density increase (see Fig. S12† discussion), the maximum current density achieved by Au is significantly higher than what is achieved by either GC or Pt. Fig. S6b† shows that the trend in percentage of theoretical iL met for Pt is very similar to GC at 0.5 mV s−1, corroborating our conclusion that the passivation is independent of electrode material and is a product of HT itself. For Au, however, the passivation does not become significant at 0.5 mV s−1 until 1 M. We attribute this to a difference in electrode dissolution rates between the electrode surfaces, with Au dissolving more readily. Although the differences in extent of passivation are less pronounced at faster scan rates (10 mV s−1, Fig. 6c and d), Au still has a higher current density, again suggesting dissolution is leading to differences in passivation. In situ Inductively Coupled Plasma Mass Spectrometry (ICP-MS) of the Au electrode (Fig. 6e and f) demonstrates that Au dissolution occurs significantly at potentials greater than 0.72 V vs. Ag/AgCl in 0.5 M NaCl. Over 500 monolayers (!) of the working electrode were dissolved into the electrolyte with each CV scan. The amount of dissolution in the first scan of Fig. 6f corresponded to 0.31C cm−2, which accounts for 22% of the observed coulombs passed in the first CV of Fig. 6e. We attribute the remainder of the current observed in Fig. 6e to other oxidation processes, such as OER or Cl2 production. Based on this evidence, we can conclude that Au dissolution will occur when HT is oxidized on Au. Given the high rate of Au monolayer dissolution in NaCl observed we would expect the self-cleaning mechanism to be even more prevalent for an Au electrode compared to the other electrodes considered (GC, Pt).
 | ||
| Fig. 6 (a) First CV cycle of GC RDE (solid, red), Pt RDE (dotted, green), and Au RDE (dashed, blue) at 900 rpm, 1 M HT/0.5 M NaCl, and 0.5 mV s−1. (b) Percentage of theoretical limiting current density, iL, as a function of HT concentration at 900 rpm and 0.5 mV s−1 for GC (red squares, solid line), Pt (green triangles, dotted line), and Au (blue circles, dashed line) electrodes. (c) First CV cycle of GC RDE (solid, red), Pt RDE (dotted, green), and Au RDE (dashed, blue) at 900 rpm, 1 M HT/0.5 M NaCl, and 10 mV s−1. (d) Percentage of theoretical iL as a function of HT concentration at 900 rpm and 10 mV s−1 for GC (red squares, solid line), Pt (green triangles, dotted line), and Au (blue circles, dashed line) electrodes. For in situ ICP-MS, (e) current density and (f) Au dissolution rate as a function of CV cycle and potential. Measurements collected in 0.5 M NaCl at 10 mV s−1 scan rate. (g) Schematic diagram of the proposed self-cleaning mechanism that at occurs at intermediate HT concentrations ([HT]) on GC and higher concentrations on Au and slow scan rate (ν) conditions. | ||
From our investigation of electrochemical behavior of GC at intermediate HT concentrations and Au at higher HT concentrations (afforded by the faster rate of dissolution), we conclude that a self-cleaning mechanism is occurring in which electrode surface dissolution allows for regained activity by detaching the passivation layer, as we show in Fig. 6b. It is important to note that the electrode oxidation is irreversible and eventually will lead to degradation of the current collector. Additionally, given the cost and low abundance of Au, it should not be considered a practical alternative to carbon, despite its higher resistance to passivation driven by the higher rate of solution. However, this finding is still crucial for the advancement of HT as an active material in RFB applications. It suggests that if we can design an oxidizing layer to promote self-cleaning and it can be remade during the battery open circuit or discharge cycles, we can control and perhaps eliminate HT passivation issues. Achieving this control would thereby allow for reversible, repeated charging and discharging of a battery that contains HT as the cathode material.
4. Conclusions
HT is a promising redox-active organic material for RFBs given its high solubility compared to other TEMPO derivatives, but its electrochemical behavior at realistic RFB operating conditions, e.g., slow discharge and charge rates and high concentrations, had not been investigated previously. Herein, we used cyclic voltammetry to show evidence of the formation of a surface passivating film at slow scan rates (0.5 mV s−1). Gravimetric eQCM data showed a mass increase at potentials corresponding to a current increase, suggesting the presence of an irreversible blocking film. Additionally, micrographs collected immediately following electrochemical measurements show the presence of a polymer-like film on the electrode surface that was present in reactions with HT but not TEMPO. This suggests the hydroxyl moiety plays an important role in passivating the electrode. XPS analysis is consistent with the deposition of an oxidized HT film whose degree of oxidation and thickness increases with oxidation potential and HT concentration. From CVs, the percentage of theoretical diffusion-limited current observed was dependent on scan rate and concentration, with passivation occurring to a larger extent at slower rates and higher HT concentrations. Similar electrochemical behavior is found for GC, Pt, and Au, suggesting a charge transfer that is independent of electrode material. Our findings are consistent with a passivation mechanism. Investigation of the electrochemical behavior at intermediate HT concentrations on GC and higher concentrations of HT on Au revealed the possibility of a self-cleaning mechanism driven by the relative rate of dissolution of the electrode material.The discovery that HT passivates electrode surfaces like GC at high concentrations and slow scan rates during oxidation underscores the importance of conducting experimental investigations at conditions that are consistent with the intended end-use applications. The use of HT at the necessary high concentrations in an RFB will be limited if the passivation mechanism pathway is not addressed, as it will prevent the ability to charge and discharge the battery multiple times. Herein, we identified a possible solution to the passivation of HT through the proposed self-cleaning mechanism observed. We found that incomplete passivation allows for oxidation of the carbon electrode itself, which could lead to the removal of the passivated layer. We further showed that the extent of passivation can be limited by moving to a surface like gold that dissolves more readily at high voltages in chloride containing electrolytes. Future work should investigate this effect further to confirm that electrode dissolution is indeed the source of the changing oxidation activity rather than OER and focus on methods of engineering an oxidizing layer of the electrode material itself to allow for self-cleaning of the passivated layer, particularly at higher HT concentrations. Carbon oxidation may lead to a degraded electrode material with repeated cycling, and so efforts to create a surface layer that can be made in situ and removed during charging will be needed to successfully implement this self-cleaning mechanism. Other avenues of modifying the electrode surface to prevent passivation should also be considered in the future, such as changing the geometry by moving to high surface area systems. Additionally, although this work presents evidence for electrode passivation during electrooxidation of HT, more efforts are needed to fully understand the passivation product and the role of the side functional groups that are often engineered with a focus on solubility and not on side reactions. Developing a holistic understanding of the passivation pathway and product will inform routes towards synthesizing highly soluble and stable redox-active organic molecules for redox flow batteries.
Data availability
All data to support this study is present in this manuscript and related ESI documents.† Any additional request for data or materials should be addressed to the corresponding author.Author contributions
Cailin Buchanan: writing – review & editing, writing – original draft, validation, methodology, formal analysis, data curation. Nora A. Shaheen: writing – review & editing, writing – original draft, methodology, formal analysis, data curation. Caroline Williams: data curation. Igor Messias: data curation. Bethany Kersten: data curation. Taewoo Kim: data curation. Justin Connell: data curation. Venkat Srinivasan: supervision. Rohan Akolkar: supervision, funding acquisition, conceptualization. Pietro Papa Lopes: writing – review & editing, writing – original draft, visualization, supervision, project administration, methodology, funding acquisition, formal analysis, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work completed at Argonne was supported by the Materials Science Division (MSD) under the Joint Center for Energy Storage Research (JCESR), a U.S. Department of Energy, Energy Innovation Hub, and by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists, Office of Science Graduate Student Research (SCGSR) program. The SCGSR program is administered by the Oak Ridge Institute for Science and Education (ORISE) for the DOE. ORISE is managed by ORAU under contract #DE-SC0014664. Investigation of HT was supported by Breakthrough Electrolytes for Energy Storage (BEES)—an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award #DE-SC0019409. Additionally, C. B. acknowledges the Argonne Laboratory Directed Research and Development (LDRD) fund via the Walter Massey Fellowship. P. P. L. further acknowledges the support from the Department of Energy Office of Science, Materials Science and Engineering Division, Materials Chemistry Program via the Early Career Research Project Award. All opinions expressed in this paper are the author's and do not necessarily reflect the policies and views of DOE, ORAU, or ORISE. We acknowledge the help from Dr Kaline Nascimento da Silva and Dr Ronnie Emmons during the review process.Notes and references
- A. Hollas, A. Tuan, V. Viswanathan and I. Ragazzi, Adoption Readiness Level Assessment of Redox Flow Batteries, Pacific Northwest National Laboratory Technical Report, Richland, Washington, 2024 Search PubMed.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. Liu, J. Appl. Electrochem., 2011, 41, 1137–1164 CrossRef CAS.
- P. Leung, A. A. Shah, L. Sanz, C. Flox, J. R. Morante, Q. Xu, M. R. Mohamed, C. Ponce de León and F. C. Walsh, J. Power Sources, 2017, 360, 243–283 CrossRef CAS.
- M. L. Perry, K. E. Rodby and F. R. Brushett, ACS Energy Lett., 2022, 7, 659–667 CrossRef CAS.
- Z. Li, T. Jiang, M. Ali, C. Wu and W. Chen, Energy Storage Mater., 2022, 50, 105–138 CrossRef.
- G. Yang, Y. Zhu, Z. Hao, Y. Lu, Q. Zhao, K. Zhang and J. Chen, Adv. Mater., 2023, 35, 2301898 CrossRef CAS PubMed.
- T. Suga, Y. J. Pu, K. Oyaizu and H. Nishide, Bull. Chem. Soc. Jpn., 2004, 77, 2203–2204 CrossRef CAS.
- X. Wei, W. Xu, M. Vijayakumar, L. Cosimbescu, T. Liu, V. Sprenkle and W. Wang, Adv. Mater., 2014, 26, 7649–7653 CrossRef CAS PubMed.
- Z. Li, S. Li, S. Liu, K. Huang, D. Fang, F. Wang and S. Peng, Electrochem. Solid-State Lett., 2011, 14, A171 CrossRef CAS.
- K. Takechi, Y. Kato and Y. Hase, Adv. Mater., 2015, 27, 2501–2506 CrossRef CAS PubMed.
- J. Winsberg, S. Muench, T. Hagemann, S. Morgenstern, T. Janoschka, M. Billing, F. H. Schacher, G. Hauffman, J. F. Gohy, S. Hoeppener, M. D. Hager and U. S. Schubert, Polym. Chem., 2016, 7, 1711–1718 RSC.
- S. K. Park, J. Shim, J. H. Yang, K. H. Shin, C. S. Jin, B. S. Lee, Y. S. Lee and J. D. Jeon, Electrochem. Commun., 2015, 59, 68–71 CrossRef CAS.
- T. Sukegawa, I. Masuko, K. Oyaizu and H. Nishide, Macromolecules, 2014, 47, 8611–8617 CrossRef CAS.
- G. Hauffman, Q. Maguin, J. P. Bourgeois, A. Vlad and J. F. Gohy, Macromol. Rapid Commun., 2014, 35, 228–233 CrossRef CAS PubMed.
- G. Hauffman, J. Rolland, J. P. Bourgeois, A. Vlad and J. F. Gohy, J. Polym. Sci., Part A:Polym. Chem., 2013, 51, 101–108 CrossRef CAS.
- J. Winsberg, C. Stolze, S. Muench, F. Liedl, M. D. Hager and U. S. Schubert, ACS Energy Lett., 2016, 1, 976–980 CrossRef CAS.
- T. Janoschka, C. Friebe, M. D. Hager, N. Martin and U. S. Schubert, ChemistryOpen, 2017, 6, 216–220 CrossRef CAS PubMed.
- J. Winsberg, T. Janoschka, S. Morgenstern, T. Hagemann, S. Muench, G. Hauffman, J. F. Gohy, M. D. Hager and U. S. Schubert, Adv. Mater., 2016, 28, 2238–2243 CrossRef CAS PubMed.
- Y. Liu, M. A. Goulet, L. Tong, Y. Liu, Y. Ji, L. Wu, R. G. Gordon, M. J. Aziz, Z. Yang and T. Xu, Chem, 2019, 5, 1861–1870 CAS.
- B. Liu, C. W. Tang, H. Jiang, G. Jia and T. Zhao, ACS Sustain. Chem. Eng., 2021, 9, 6258–6265 CrossRef CAS.
- A. Orita, M. G. Verde, M. Sakai and Y. S. Meng, J. Power Sources, 2016, 321, 126–134 CrossRef CAS.
- T. Liu, X. Wei, Z. Nie, V. Sprenkle and W. Wang, Adv. Energy Mater., 2016, 6, 1501449 CrossRef.
- P. Navalpotro, N. Sierra, C. Trujillo, I. Montes, J. Palma and R. Marcilla, ACS Appl. Mater. Interfaces, 2018, 10, 41246–41256 CrossRef CAS PubMed.
- T. Janoschka, N. Martin, M. D. Hager and U. S. Schubert, Angew. Chem., 2016, 55, 14427–14430 CrossRef CAS.
- B. Hu, Y. Tang, J. Luo, G. Grove, Y. Guo and T. L. Liu, Chem. Commun., 2018, 54, 6871–6874 RSC.
- B. Hu, M. Hu, J. Luo and T. L. Liu, Adv. Energy Mater., 2022, 12, 2102577 CrossRef CAS.
- M. Pan, L. Gao, J. Liang, P. Zhang, S. Lu, Y. Lu, J. Ma and Z. Jin, Adv. Energy Mater., 2022, 12, 2103478 CrossRef CAS.
- T. Hagemann, J. Winsberg, M. Grube, I. Nischang, T. Janoschka, N. Martin, M. D. Hager and U. S. Schubert, J. Power Sources, 2018, 378, 546–554 CrossRef CAS.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- J. Luo, B. Hu, C. Debruler and T. L. Liu, Angew. Chem., Int. Ed., 2018, 57, 231–235 CrossRef CAS PubMed.
- M. Pan, Y. Lu, S. Lu, B. Yu, J. Wei, Y. Liu and Z. Jin, ACS Appl. Mater. Interfaces, 2021, 13, 44174–44183 CrossRef CAS PubMed.
- J. Winsberg, C. Stolze, A. Schwenke, S. Muench, M. D. Hager and U. S. Schubert, ACS Energy Lett., 2017, 2, 411–416 CrossRef CAS.
- M. Gao, M. Salla, Y. Song and Q. Wang, Angew. Chem., Int. Ed., 2022, 134, e2022208223 Search PubMed.
- Z. Chang, D. Henkensmeier and R. Chen, ChemSusChem, 2017, 10, 3193–3197 CrossRef CAS.
- Z. Chang, D. Henkensmeier and R. Chen, J. Power Sources, 2019, 418, 11–16 CrossRef CAS.
- H. Fan, B. Hu, H. Li, M. Ravivarma, Y. Feng and J. Song, Angew. Chem., Int. Ed., 2022, 61, e2022115908 Search PubMed.
- S. Hu, L. Wang, X. Yuan, Z. Xiang, M. Huang, P. Luo, Y. Liu, Z. Fu and Z. Liang, Energy Mater. Adv., 2021, 2021, 9795237 Search PubMed.
- G. S. Nambafu, K. Siddharth, C. Zhang, T. Zhao, Q. Chen, K. Amine and M. Shao, Nano Energy, 2021, 89, 106422 CrossRef CAS.
- T. Hagemann, M. Strumpf, E. Schröter, C. Stolze, M. Grube, I. Nischang, M. D. Hager and U. S. Schubert, Chem. Mater., 2019, 31, 7987–7999 CrossRef CAS.
- E. Pedraza, C. de la Cruz, A. Mavrandonakis, E. Ventosa, R. Rubio-Presa, R. Sanz, S. T. Senthilkumar, P. Navalpotro and R. Marcilla, Adv. Energy Mater., 2023, 13, 2301929 CrossRef CAS.
- R. Yang, Y. Zhang, K. Takechi and E. J. Maginn, J. Phys. Chem. C, 2018, 122, 13815–13826 CrossRef CAS.
- K. Ehtiati, I. Anufriev, C. Friebe, I. A. Volodin, C. Stolze, S. Muench, G. Festag, I. Nischang, M. D. Hager and U. S. Schubert, RSC Adv., 2024, 14, 32893–32910 RSC.
- N. A. Shaheen, M. Ijjada, M. B. Vukmirovic and R. Akolkar, J. Electrochem. Soc., 2020, 167, 143505 CrossRef CAS.
- N. A. Shaheen, W. Dean, D. Penley, B. Kersten, J. Rintamaki, M. B. Vukmirovic, B. E. Gurkan and R. Akolkar, J. Electrochem. Soc., 2022, 169, 053511 CrossRef CAS.
- J. G. Vos and M. T. M. Koper, J. Electroanal. Chem., 2018, 819, 260–268 CrossRef CAS.
- P. P. Lopes, D. Strmcnik, D. Tripkovic, J. G. Connell, V. Stamenkovic and N. M. Markovic, ACS Catal., 2016, 6, 2536–2544 CrossRef CAS.
- J. M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- Y. Liu, P. P. Lopes, W. Cha, R. Harder, J. Maser, E. Maxey, M. J. Highland, N. M. Markovic, S. O. Hruszkewycz, G. B. Stephenson, H. You and A. Ulvestad, Nano Lett., 2017, 17, 1595–1601 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- V. G. Levich, Physicochemical Hydrodynamics, New Jersey, Prentice-Hall, 1962 Search PubMed.
- L. Kiss, D. Bősz, F. Kovács, H. Li, G. Nagy and S. Kunsági-Máté, Polym. Bull., 2019, 76, 5849–5864 CrossRef CAS.
- R. G. Compton and C. E. Banks, in Understanding Voltammetry, World Scientific Publishing Europe Ltd, 3rd edn, 2018, pp. 203–218 Search PubMed.
- N. A. Shaheen, Investigation of Mechanisms Governing Charge Transfer in Redox-Active Organic Molecules, PhD thesis, Case Western Reserve University, 2022.
- P. P. Lopes, D. Tripkovic, P. F. B. D. Martins, D. Strmcnik, E. A. Ticianelli, V. R. Stamenkovic and N. M. Markovic, J. Electroanal. Chem., 2018, 819, 123–129 CrossRef CAS.
- L. Xing, M. A. Hossain, M. Tian, D. Beauchemin, K. T. Adjemian and G. Jerkiewicz, Electrocatalysis, 2014, 5, 96–112 CrossRef CAS.
- C. Batchelor-McAuley, Curr. Opin. Electrochem., 2023, 37, 101176 CrossRef.
- Y. Yi, G. Weinberg, M. Prenzel, M. Greiner, S. Heumann, S. Becker and R. Schlögl, Catal. Today, 2017, 295, 32–40 CrossRef CAS.
- I. Messias, J. Kupferberg, A. R. Bielinski, R. Nagao, A. B. F. Martinson and P. Papa Lopes, ACS Catal., 2025, 15, 2750–2759 CrossRef CAS.
- S. G. Ji, H. Kim, C. Park, W. Kim and C. H. Choi, ACS Catal., 2020, 10, 10773–10783 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00481k |
| ‡ Current address: Chemical and Fuel Cycle Technologies Division, Argonne National Laboratory, Lemont, IL, USA. |
| This journal is © The Royal Society of Chemistry 2025 |