
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanically and photoelectrochemically stable WO3|BiVO4|NiFeOOH photoanodes synthesised by a scalable chemical vapour deposition method†
George H.
Creasey
 *ab,
Tristan W.
McCallum
b,
Guangrui
Ai
b,
Brian
Tam
b,
John W.
Rodriguez Acosta
a,
Alvia
Mohammad Yousuf
b,
Sarah
Fearn
c,
Flurin
Eisner
d,
Andreas
Kafizas
b and
Anna
Hankin
*a
*ab,
Tristan W.
McCallum
b,
Guangrui
Ai
b,
Brian
Tam
b,
John W.
Rodriguez Acosta
a,
Alvia
Mohammad Yousuf
b,
Sarah
Fearn
c,
Flurin
Eisner
d,
Andreas
Kafizas
b and
Anna
Hankin
*a
aDepartment of Chemical Engineering, Imperial College London, South Kensington, London, SW7 2AZ, UK. E-mail: g.creasey22@imperial.ac.uk; anna.hankin@imperial.ac.uk
bDepartment of Chemistry, The Molecular Science Research Hub, Imperial College London, White City, London, W12 0B, UK
cDepartment of Materials, Imperial College London, South Kensington, London, SW7 2AZ, UK
dSchool of Engineering and Materials Science, Queen Mary University of London, Bethnal Green, London, E1 4NS, UK
First published on 17th March 2025
Abstract
The development of scalable, stable and high performance photoelectrodes remains the major bottleneck in up-scaling photoelectrochemical (PEC) water splitting systems. A photoanode structure of particular promise is WO3|BiVO4, where the formation of staggered heterojunction between nanostructured WO3 and a thin layer of BiVO4 mitigates charge carrier mobility limitations present for BiVO4 alone and suppresses recombination. Although these electrodes remain prone to photo-corrosion, this effect can be mitigated through the application of water oxidation surface co-catalysts. An additional challenge that has rarely been addressed in the literature to date is the need for strong adhesion to the substrate and mechanical stability of these photoelectrodes, so that they can withstand flow-induced shear stress exerted by the electrolyte in continuous flow under operational conditions. Herein, we propose a scalable route to synthesising WO3|BiVO4|NiFeOOH photoanodes entirely by aerosol-assisted chemical vapour deposition (AA-CVD). The mechanical stability of the WO3|BiVO4 heterojunction was optimised by tuning the morphology of the WO3 underlayer and improving its adhesion to the FTO transparent substrate. To address BiVO4 dissolution at the electrode|electrolyte interface, we fabricated a NiFeOOH co-catalyst by a novel AA-CVD method. This suppressed BiVO4 dissolution and enhanced the water oxidation performance of the photoanode, characterised by linear sweep voltammetry (LSV), photoelectrochemical impedance spectroscopy (PEIS) and chopped chronoamperometry. The photoanode materials were physically characterised by X-ray diffraction (XRD), UV-Vis spectroscopy, scanning electron microscopy (SEM), high resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy and time-of-flight secondary ion mass spectrometry (ToF-SIMS). Our optimised photoanodes with 1 cm2 photoactive area delivered a stable photocurrent density of 1.75 mA cm−2 (at 1 simulated sun irradiance and 1.23 VRHE) during 24 hours testing in a continuous PEC flow reactor (operated at 0.5 cm s−1). Our method for growing WO3|BiVO4|NiFeOOH photoanodes is up-scalable, and therefore suitable for producing large-area demonstration devices, providing a pathway to commercial photoelectrochemical hydrogen production.
Introduction
The urgent implementation of green technologies has never been more critical, with 2023 confirmed as the world's hottest year on record and global average temperatures 1.48 °C higher than benchmark 1850–1900 pre-industrial levels.1 As the global energy sector shifts towards electrification, away from traditional solid and liquid fuels, energy storage solutions are required to address mismatches between energy supply and demand. In the UK, fossil fuel-fired electricity generation capacity reached a peak of 71 GW in 2010, which at the time was over 75% of the total. Before this, the share of capacity provided by fossil fuel generators fluctuated between 75 and 80% for decades, but in the years since, it has fallen steadily (to 65% in 2023), due to the increase in renewable energy generation share.2–4 One challenge inherent to an increased share of renewable energy generation is their intermittent availability. Whilst interconnectors between countries have partially addressed the mismatch in renewable energy generation and demand, there is an associated risk with relying on other countries to supply energy, as exposed during the recent crisis in global supply chains of natural gas.5 Therefore, the most viable solution to address the mismatch between peak demand and peak supply of electricity is to consider energy storage during periods when supply exceeds demand. Whilst energy storage solutions including batteries and pumped storage are currently being deployed, options including hydrogen will be needed to achieve the goal of energy security and to deliver energy in peak demand periods.6–8 Unlike batteries which discharge over time, energy can be stored in hydrogen indefinitely, enabling security of energy supply over longer time periods of weeks and months when fossil-fuel driven energy generation is phased out.9Currently, around 96% of hydrogen produced globally is derived from fossil fuels, most commonly by steam methane reforming, which produces around 900 Mt CO2 emissions per year.10 A promising method to produce renewable (green) hydrogen is through water electrolysis, either connected to existing renewable electricity generation infrastructure, or using integrated systems, whereby electrical energy to power the electricity unit is generated in situ via photoactive electrodes.11 Photoelectrochemical cells combine the concepts of photovoltaics (PVs) and electrolysers (EC) into a single device in which the conversion of solar to electrical to chemical energy is co-located. PEC devices are expected to become more competitive with traditional hydrogen production methods over the next decade, for instance to provide decentralised community and domestic energy storage.12
One of the most commonly studied classes of material in photoelectrochemical (PEC) water splitting devices is transition metal oxides (TMOs), which can fulfil many of the key criteria for PEC water splitting, including affordability, durability and ease of synthesis.13 Bismuth vanadate (BiVO4) is one of the most researched TMOs for this purpose. It has a band gap of 2.4–2.5 eV, enabling the absorption of light in the visible range, and giving it a theoretical maximum photocurrent density of ∼7.4 mA cm−2.14 Researchers have employed various strategies to improve the performance of BiVO4-based electrodes, including the use of dopants, nanostructuring, formation of heterojunctions (often with WO3) and addition of surface co-catalysts.15 Some commonly used surface co-catalysts that have been used to enhance the water oxidation performance of BiVO4 based electrodes are nickel and/or iron oxyhydroxide (Ni/FeOOH), cobalt phosphate (CoPi) and cobalt nickel oxide (CoNiO2).15–22 The role of the co-catalyst in water oxidation with BiVO4 is crucial, since unmodified BiVO4 is prone to sluggish water oxidation kinetics and can perform more than one order of magnitude worse than when combined with co-catalysts.17,18,23–25 The role of surface co-catalysts is twofold, to enhance the water oxidation kinetics and to provide a barrier that can mitigate (photo)electrochemical degradation.24,26 Previous studies of NiFeOOH as a co-catalyst for BiVO4 have reported that the addition of NiFeOOH results in an increase in charge carrier lifetimes, making them less prone to recombination.24 Evidence from transient absorption spectroscopy (TAS) suggests that the enhancement in photoelectrochemical performance with the addition of NiFeOOH is through the reduction in the kinetics of surface recombination and reduced recombination losses via fast hole transfer from BiVO4 to the co-catalyst, rather than speeding up the water oxidation kinetics of BiVO4.24,27
To our knowledge, the best recorded performance to date was by Pihosh et al., who reported a photocurrent density of 6.72 mA cm−2 with a WO3|BiVO4 heterojunction photoanode, doped with conductive cobalt and coated with CoPi surface co-catalyst.16 However, superior photoelectrochemical performance does not necessarily correlate with durability. Robustness and stability in long-term continuous operation are principal criteria for candidate PEC materials.13 An obstacle which precludes the use of BiVO4 in photoelectrochemical reactors is its tendency to photo-corrode, which makes the intrinsic material alone unsuitable for long-term operation.
It is seldom acknowledged in the literature that BiVO4 is soluble at typical photoanode characterisation potentials (i.e. 1.23 VRHE), according to thermodynamic predictions.28 According to its Pourbaix diagram, BiVO4 is prone to electrochemical oxidation to bismuth oxides at potentials greater than +1.0 VRHE and chemical leaching of VO4− (in which vanadium ions do not change oxidation state).25,28 Operation in this region therefore requires the implementation of mitigation strategies to prevent chemical and photoelectrochemical degradation. Degradation reportedly occurs not only with increasing anodic bias, but also with decreasing or increasing pH and increasing illumination intensity.28
Two primary mechanisms have been suggested for BiVO4 degradation: chemical leaching of V5+ ions and oxidation of Bi3+ ions.26,28 The formation of a temporary protective BiOx layer can passivate the electrode and prevent total degradation of the material has been reported, but the photoelectrochemical performance can still decrease dramatically (by over 90% within a few hours), with varying reports on BiVO4 degradation rates attributed to different fabrication methods and test conditions.26,28–31
As reported by Lee and Choi, good mechanical stability can significantly decreases degradation rates in BiVO4 electrodes, with highly crystalline and well-ordered structures having improved adhesion to their electrode substrates, leading to improved mechanical stability.32 Forming a heterojunction between WO3 and BiVO4 improves the charge extraction from BiVO4 and reduces hole recombination. These effects improve stability by increasing the tendency of photogenerated charges to be used to oxidise water, rather than bismuth. However, in these cases, it is necessary to also consider the adhesion of the first layer to the electrode substrate and the adhesion of the other layers to each other. In the example of WO3, it has been shown that nanostructured WO3 is much less mechanically stable than planar WO3, which in turn results in inadequate WO3|BiVO4 heterojunction stability.33 The context of this observation is often overlooked. Most up-scaled devices require a flow system, and therefore materials must have sufficient mechanical strength to withstand the shear stress from the flowing electrolyte.34 In these cases, a trade-off must be considered between the improved photoelectrochemical performance of nanostructured materials and their mechanical stability. The electrolyte flow velocity must also be carefully selected to ensure adequate gas bubble removal, while exerting minimal shear stress on delicate system components.34,35
Thermodynamic predictions indicate that BiVO4 is most electrochemically stable at near neutral pH and easily dissolved by strong acids and bases.26,28,31 At near neutral pH, successful stability studies have employed the use of electrolyte tuning and co-catalysts, including cobalt phosphate (CoPi) and nickel iron oxyhydroxides.36–38 In one of the most successful stability studies involving BiVO4-based photoanodes to date, significantly extended stability was demonstrated for 500 hours using a potassium borate (KB) pH 9 buffer electrolyte, enriched with saturated 0.1 M V2O5.32 With an electrolyte saturated with V5+ ions, it was possible to suppress the dissociation of V5+ ions, by virtue of Le Chatelier's principle.32,39 This theory was successfully tested using a low electrode potential of +0.6 VRHE, whereby dual FeOOH/NiOOH oxygen evolution catalysts were employed, enabling a photocurrent of 3 mA cm−2.32
However, it has been argued that the primary mechanism of BiVO4 dissolution is through the photo-oxidation of Bi3+ ions.26 Therefore, the use of a vanadium-enriched electrolyte alone is not enough to suppress BiVO4 dissolution under typical operating conditions. Although V5+ dissolution occurs, ICP-MS electrolyte analysis has shown that under illumination and with electrode potentials between 0.4 and 1.6 VRHE, photoelectrochemically driven dissolution rates of Bi3+ were an order of magnitude higher than V5+ dissolution rates.26 Therefore, although photoelectrochemical oxidation of Bi3+ is accelerated at higher potentials, using an operating potential <1.23 VRHE alone is not an effective method for preventing the degradation of BiVO4. One of the most effective methods to suppress BiVO4 photo-corrosion is through the application of surface water oxidation co-catalysts which can provide very effective protection from degradation, addressing both mechanisms of BiVO4 dissolution (chemical vanadium ion leaching and (photo)electrochemical BiVO4 oxidation). Co-catalysts provide a partial barrier to corrosion at the BiVO4|electrolyte interface and limit the self-oxidation of BiVO4 through providing more active sites for photogenerated holes to oxidise water, which limits BiVO4 self-oxidation.26,40,41 The ability to stabilise BiVO4 by using co-catalysts, and without using a vanadium enriched electrolyte, has been demonstrated, including by Cui et al., who were able to achieve a stable photocurrent density of around 3 mA cm−2 at + 0.8 VRHE for 17 hours.25
Herein, we optimise the photoelectrochemical stability, performance and mechanical robustness of engineered WO3|BiVO4|NiFeOOH photoelectrodes, using a scalable aerosol-assisted chemical vapour deposition (AA-CVD) method. The need to develop PEC materials using low-cost, scalable techniques is as critical as their need to be highly active and stable. AA-CVD is an attractive method for producing photoelectrodes due to its affordability, speed and simplicity. Utilising ambient pressure and moderate temperatures, energy requirements can be minimised by carrying out in situ annealing and sequential coating of individual layers.
To the best of our knowledge, this is the first study to have used scalable AA-CVD to develop both the WO3|BiVO4 heterojunction and NiFeOOH surface co-catalyst. Having demonstrated the fabrication of this material entirely in our purpose-built AA-CVD reactor, one can envisage a processing method in which glass, during its manufacture, is sequentially coated with FTO and then WO3, BiVO4 and NiFeOOH layers using existing commercial infrastructure, decreasing the capital and operating costs of developing PEC materials at a scale commensurate to their application.
Photoelectrode synthesis
Photoanodes were synthesised on TEC-15 FTO glass substrates, cut into 1.5 cm × 2.0 cm pieces. The sequential deposition of WO3, BiVO4 and NiFeOOH co-catalysts was carried out by scalable aerosol assisted chemical vapour deposition (AA-CVD) methods, using a purpose-built reactor (Fig. S1†). Small pieces of cover glass were used to leave an uncoated area of FTO for making electrical connections.Before the deposition of the first layer onto FTO, the glass was pre-treated in a four-step cleaning procedure involving ultrasonication in detergent water, deionised water, acetone and isopropanol, each for 10 minutes, respectively. The WO3 deposition procedures were adapted from Kafizas et al. and Tam et al.33,42 To prepare the WO3 precursor solution, 11.4 mM tungsten hexacarbonyl (Merck, ≥ 97%) was dissolved in 26.7 mL of acetone and 13.3 mL of methanol. The mixture was placed in the ultrasonication bath (VWR ultrasonic cleaner, 30 W, 45 kHz) for at least three minutes until the solids were completely dissolved. The glass substrates were placed on a heated graphitic carbon block in the CVD chamber. For the deposition of planar (flat) WO3, the CVD chamber was heated to 325 °C, while for the deposition of nanostructured WO3, temperatures of 350 or 375 °C were used. Fixed volumes of precursor solution (20 or 40 mL) were aerosolized using an ultrasonic humidifier (2 MHz, Liquifog, Johnson Matthey), carried with nitrogen carrier gas flowrates of between 1.5 and 2.0 L min−1 (MFC, Brooks). Flat and nanostructured WO3 layers were fabricated, along with their combination (i.e. a flat ‘seed’ layer of WO3, followed by a layer of WO3 nanoneedles). To prevent the reaction of the precursor prior to entering the CVD chamber, a water-cooling jacket was placed on the vapour inlet. Following deposition, the samples were annealed for two hours in air at 500 °C.
BiVO4 films were deposited directly onto FTO or onto WO3 layers. To prepare the precursor solution, 5 mM triphenyl bismuth (Alfa Aesar, ≥99%) and 5 mM vanadium(III) acetylacetonate (Merck, ≥97%) were dissolved in 15 mL of acetone and 5 mL of methanol. The mixture was placed in an ultrasonication bath (VWR ultrasonic cleaner, 30 W, 45 kHz) for at least three minutes until the solids were completely dissolved. Similar to the WO3 deposition, the glass substrates were heated in the CVD chamber on the graphitic carbon block, but with a temperature of 400 °C. For the deposition of BiVO4, an air carrier gas flowrate of 1.0 L min−1 was used to carry fixed volumes (20 to 60 mL) of the BiVO4 precursor into the reactor. After deposition, the substrates were annealed in air for two hours at 500 °C.
In the final step, a NiFeOOH co-catalyst layer was deposited on top of the BiVO4 layer. The precursor solution was prepared by dissolving 4 mM nickel(II) acetylacetonate and 1 mM iron(II) acetylacetonate in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of acetone and methanol with various volumes. The mixture was placed in an ultrasonication bath for at least three minutes until the solids were completely dissolved. For the deposition, 20 mL of the precursor was carried into the chamber held at 160 °C using nitrogen, with a flowrate of 1.5 L min−1. After the deposition, the films were annealed for two hours in air at 160 °C to remove impurities.
:1 mixture of acetone and methanol with various volumes. The mixture was placed in an ultrasonication bath for at least three minutes until the solids were completely dissolved. For the deposition, 20 mL of the precursor was carried into the chamber held at 160 °C using nitrogen, with a flowrate of 1.5 L min−1. After the deposition, the films were annealed for two hours in air at 160 °C to remove impurities.
Physical characterisation
X-Ray diffraction measurements were carried out to characterise the crystal structure of the materials using a Bruker D2 Phaser diffractometer with parallel beam optics equipped with a Lynx-Eye detector. X-rays were generated using a Cu source (V = 30 kV, I = 10 mA) with Cu Kα1 (λ = 1.54056 Å) and Cu Kα2 radiation (λ = 1.54439 Å) emitted with an intensity ratio of 2:1. The diffraction patterns were collected at scattering angles between 10° ≤ 2θ ≤ 55°, with a step size of 0.02° every second.
Raman spectra of WO3, BiVO4 and NiFeOOH films were obtained using a Bruker SENTERRA II Raman Microscope. Samples were excited with 532 nm laser excitation of 6.25 mW power over a 50 μm diameter irradiation area. Spectra were recorded with Raman shifts from 50 cm−1 to 3000 cm−1 at 1.5 cm−1 intervals.
The surface morphologies of the materials were characterised by scanning electron microscopy (SEM) using a Zeiss Gemini Sigma 300 FEG SEM. Films were typically characterised at magnifications of 10000 to 100000 with an in-lens detector, using an accelerating voltage of 5 keV and a working distance of between 5 and 6 mm. This analysis was complemented by energy dispersive X-ray analysis (EDX) using the same microscope, with an SE2 detector.
High resolution transmission electron microscopy (HR-TEM) images were captured using a JEOL transmission electron microscope 2100 plus and analysed using Gatan Digital Micrograph software. This analysis was completed by scanning transmission electron microscopy energy dispersive X-ray analysis (STEM-EDX) using a JEOL 2100 plus scanning transmission electron microscope. All HR-TEM analysis is provided in the ESI (Fig. S8 and S13).†
X-Ray photoelectron spectroscopy (XPS) was used to determine the surface composition of the photoelectrode materials and oxidation states of the constituent elements, with a Thermo Fisher K-Alpha Plus XPS spectrometer, equipped with a monochromated Al Kα X-ray source. A flood gun was used for surface charge compensation and binding energies were adjusted to adventitious carbon (C–C) peaks seen in the C 1s binding energy region. The resulting XPS data were processed in CasaXPS.
To characterise optical properties of the photoelectrodes, a Shimadzu UV-2600 UV-Vis spectrometer was used to measure the transmittance and spectral reflectance of the samples over wavelengths between 200 and 1400 nm. These data were subsequently used to calculate the absorbance using the relationship between absorptance, transmittance and reflectance (eqn (1)).
| A (%) = 100 − T (%) − R (%) | (1) |
The optical band gap of the materials was obtained graphically using the method outlined by Chen and Jaramillo.43 The semiconductor materials in this study have absorption coefficient, α, such that the Tauc relation (eqn (2)) applies.44,45
| αhv ∝ (hv − Eg)n | (2) |
To characterise the NiFeOOH co-catalyst layer, a WO3|BiVO4|NiFeOOH sample was analysed by time of flight secondary ion mass spectrometry (ToF-SIMS) using an IONTOF ToF-SIMS V instrument. A 25 keV Bi+ ion beam in high current bunched mode (HCBM) was used to analyse an area of 100 μm2 and negative secondary ions were collected. A 1 keV Cs+ ion beam with a current of 85 nA was used to depth profile the samples over an area of 300 μm2. A flood gun was also used for surface charge compensation during the depth profiling. Subsequent data were analysed using IONTOF Surface lab 7.
To inform the selection of the AA-CVD reaction temperature for the synthesis of NiFeOOH co-catalyst, the thermal decomposition profiles of four precursors, iron(III) chloride hexahydrate, iron(III) acetylacetonate, nickel(II) chloride hexahydrate and nickel(II) acetylacetonate were measured using thermogravimetric analysis (TGA) with a Waters TA Instruments TGA Q500. A fixed 5 mg mass of each precursor was heated from room temperature to 300 °C with a ramp rate of 10 °C min−1 in a nitrogen environment.
Photoelectrochemical
Photoelectrochemical measurements were conducted in back-illumination mode using either a Redox.me Teflon Photo-electrochemical H-Cell for static electrolyte measurements or a Redox.me PTFE Photo-electrochemical Flow H-Cell for electrolyte in-flow measurements. These cells were equipped with Redox.me Pt auxiliary electrodes and Redox.me refillable Ag/AgCl reference electrodes (sat. KCl, 0.197 VSHE at 25 °C). The working electrode potential was controlled using an Autolab PGStat302N potentiostat (Metrohm). For PEC flow measurements, the electrolyte flowrate was regulated using a Redox.me Peristaltic Pump, capable of flowrates between 0.07 and 380 mL per minute. Photoelectrodes were tested using various electrolytes, including 0.1 M potassium phosphate buffer (0.05 M K2HPO4, 0.05 M KH2PO4, pH 7) and 1 M borate buffer (1 M H3BO3, ∼15 g L−1 NaOH titrated to pH 9). Electrolytes were prepared with Milli-Q-water (Millipore Corp., 18.2 MΩ cm at 25 °C).The primary light source used for photoelectrochemical measurements was a Class A Abet Technologies Sun 2000 Solar Simulator with a 550 W Xe lamp and AM 1.5 G filter. A variable working distance was used to provide an equivalent incident light intensity to the reactor of one sun (100 mW cm−2), unless otherwise stated. The light incident on the electrochemical cells was characterised by StellarNet Black Comet UV/Vis (280–900 nm) and Dwarf Star (900–1700 nm) spectrophotometers, interfaced with SpectraWiz software. An example of the measured light source spectrum, compared with the NREL AM 1.5 G reference spectrum, is shown in Fig. S2.†
For IPCE measurements and some PEC stability tests, a PowerMax II 75 W Xe lamp, with KG3 filter and monochromator, was used. The intensity of the light source was also characterised using a ThorLabs PM100D handheld optical power meter equipped with a S120VC sensor. An example of the light source spectrum is shown in Fig. S3.† IPCE was calculated using the following equation:
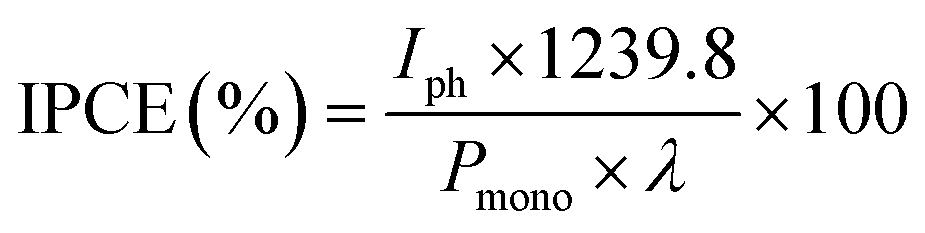 | (3) |
Photoelectrochemical stability measurements were taken by applying a photoanode potential of 1.23 VRHE and recording the photocurrent for up to 24 hours. The PEC stability of the electrodes was quantified by observing photocurrent density as a function of time and comparing the average photocurrent density at 1.23 VRHE, recorded during linear sweep voltammetry before and after the chronoamperometry tests, with typical scan rates of 10 mV s−1. Three linear sweep voltammograms were measured consecutively on each sample under continuous illumination and results from the third scan were used in analysis. This was followed by a scan using chopped light, with a chopping frequency of 1 s−1.
All applied electrode potentials are reported vs. the reversible hydrogen electrode (VRHE in volts), calculated using the Nernst equation.
 | (4) |
 is the standard potential of the reference electrode (all in volts).
is the standard potential of the reference electrode (all in volts).
To complement other photoelectrochemical techniques for the characterisation of electrode kinetics, photoelectrochemical impedance spectroscopy (PEIS) was carried out at various applied potentials. Frequencies of 105 to 10−1 Hz (10 points per decade) were used with electrode potentials of 0.8, 1.0 and 1.23 VRHE. The electrode potential was modulated by 10 mV (±) amplitude. Fitting to the equivalent electrical circuit was carried out using Nova 2.1.7 software (Autolab). The circuit shown in Fig. 1 was selected for this analysis as it gave the lowest pseudo Chi-squared values in the Kramers–Kronig tests of equivalent circuits selected by Hankin et al. in previous work.46
 | ||
| Fig. 1 Equivalent electrical circuit used for fitting PEIS spectra. HF = high frequency, LF = low frequency. | ||
To determine the charge separation and injection efficiencies, linear sweep voltammetry (scan rate 10 mV s−1) was carried out in 1 M borate buffer for water oxidation and 1 M borate buffer with 0.5 M Na2SO3 sacrificial reagent for SO32− oxidation. Surface and bulk charge transfer efficiencies were then determined by the following equations:
 | (5) |
 | (6) |
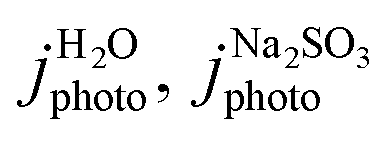 are the measured current densities for H2O and Na2SO3 oxidation, jabs is the theoretical photocurrent density generated when 100% of solar energy (AM 1.5 G at 1 sun power) absorbed by the material is converted into photocurrent, ϕsep is the charge separation efficiency, ϕinj is the charge injection efficiency. To validate the interfacial charge transfer efficiency, transient chronoamperometry measurements were carried out, following a methods adapted from Bedoya-Lora et al.47,48 Photocurrent density was recorded during chronoamperometry at a photoanode potential of 1.23 VRHE. Measurements were carried out for 5 minutes, with alternating 10 seconds periods without illumination (dark current) and with illumination. It was noted that 10 seconds was sufficient to enable stabilisation of the current for the materials tested. The charge injection efficiency was calculated using the following equation.
are the measured current densities for H2O and Na2SO3 oxidation, jabs is the theoretical photocurrent density generated when 100% of solar energy (AM 1.5 G at 1 sun power) absorbed by the material is converted into photocurrent, ϕsep is the charge separation efficiency, ϕinj is the charge injection efficiency. To validate the interfacial charge transfer efficiency, transient chronoamperometry measurements were carried out, following a methods adapted from Bedoya-Lora et al.47,48 Photocurrent density was recorded during chronoamperometry at a photoanode potential of 1.23 VRHE. Measurements were carried out for 5 minutes, with alternating 10 seconds periods without illumination (dark current) and with illumination. It was noted that 10 seconds was sufficient to enable stabilisation of the current for the materials tested. The charge injection efficiency was calculated using the following equation. | (7) |
The faradaic efficiency, ηF, was calculated by interfacing the photoelectrochemical cell with a Pfeifer Vacuum Prisma Pro mass spectrometer and measuring the oxygen production during chronoamperometry measurements. The data was collected using Zilien software and processed using ixdat analysis. The faradaic efficiency was then calculated using the following equation.
 | (8) |
Results and discussion
The stabilities of a range of WO3, BiVO4 and WO3|BiVO4 heterojunction photoanodes were investigated in this work to discover the optimal trade-off between stability and performance to enable future implementation in up-scaled photoelectrochemical reactors. A scheme showing all the photoanodes tested in this study is presented in Table S1.†Preliminary PEC stability testing on planar BiVO4 and nanoneedle WO3|BiVO4 heterojunction photoanodes
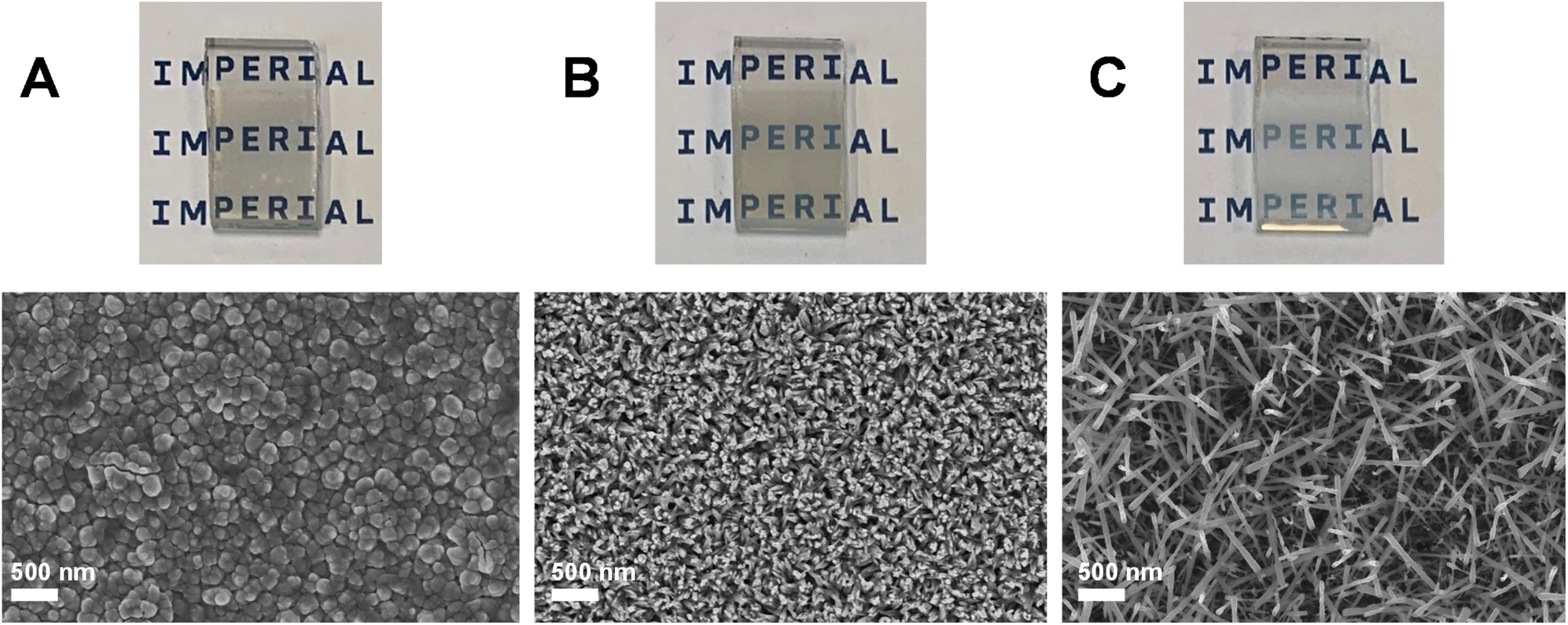
Two photoanodes were investigated during our initial validation and scoping study: (1) FTO|BiVO4 electrodes and (2) FTO|WO3|BiVO4 electrodes prepared by conformally coating BiVO4 on WO3 nanoneedles. Fig. 2 shows SEM images and accompanying photographs of these electrodes, as well as the WO3 nanoneedles prior to coating with BiVO4. | ||
| Fig. 2 SEM images and accompanying photographs of the photoanodes used in the initial scoping and validation study. WO3 nanoneedles on FTO (A), BiVO4 on FTO (B) and BiVO4 conformally coated on the WO3 nanoneedles to form a WO3|BiVO4 heterojunction (C). | ||
All films in this subsection were synthesised by AA-CVD on FTO using methods outlined previously by the authors.42,49 Physical characterisation by XRD and UV-Vis spectroscopy (provided in Fig. S4†) showed that the photoanodes were analogous to those reported in previous studies, with both the WO3 and BiVO4 having monoclinic crystal structures and forming a sharp heterojunction with distinct interfaces between the two layers when combined into one photoelectrode.42
Preliminary photoelectrochemical stability experiments were carried out at pH 7 using 0.1 M KPi buffer as the electrolyte. This decision was taken in consideration with thermodynamic predictions and previous research efforts, which have suggested that the vanadium leaching mechanism that contributes to BiVO4 degradation occurs most readily at high and low pH.28
As Fig. 3 shows, the photoelectrochemical stability during chronoamperometry at 1.23 VRHE in a 0.1 M KPi buffer (pH 7) was very poor for both BiVO4 and WO3|BiVO4 electrodes. The rapid decline in photocurrent for both electrodes was primarily driven by the photo-corrosion of BiVO4, which was expected given that the mechanisms of BiVO4 dissolution were unmitigated. As shown in Fig. 3, the conformal coating of BiVO4 was stripped from the WO3 during the experiment. The irradiated part of the electrode remained slightly opaque after the experiment due to the remaining WO3 on the electrode. The remaining WO3 was likely passivated by the formation of a thin layer of BiOx, which can occur as part of a self-healing mechanism during BiVO4 dissolution, preventing the dissolution of WO3 that would occur in the presence of a pH 7 electrolyte.29 The WO3|BiVO4 electrodes also showed signs of mechanical damage as a result of the mechanically weak WO3 nanoneedles which form the basis of the heterostructure. Previous comparisons between the WO3 nanoneedles and planar WO3 films have shown that the nanoneedles are mechanically unstable and therefore have weak adhesion to the FTO electrode.33 The electrodes can also be damaged easily during handling or cell assembly, which creates operational challenges. Herein, we address this issue, as mechanically stable electrodes are required in up-scaled systems to withstand flow-induced shear stress.
 | ||
| Fig. 3 Top: chronoamperometry stability testing of BiVO4 and WO3|BiVO4 electrodes at 1.23 VRHE in 0.1 M KPi (pH 7) and photograph showing degradation of the irradiated area of the WO3|BiVO4 electrode after the 24 hours test. Below: SEM images of WO3|BiVO4 before and after the stability test. PEC measurements were taken under simulated sunlight (AM 1.5 G, 1 sun). | ||
The influence of a vanadium-enriched electrolyte on the stability of the photoelectrodes was also investigated by preparing 0.1 M KPi buffers with various concentrations of V2O5 and carrying out chronoamperometry stability testing at 1.23 VRHE. However, there were several critical issues which were identified during these experiments. V2O5 has poor solubility at room temperature, at concentrations typically less than 0.005 M in water.50 This poses operational issues since the vanadium component of the electrolyte is prone to precipitation, resulting in fouling of the photoelectrochemical system. We found the solubility of V2O5 in 0.1 M KPi electrolyte to be very limited higher than 0.02 M, despite vigorous agitation, ultrasonication and heating. We also observed precipitation of the vanadium (V5+) salt from the electrolyte following chronoamperometry testing onto the electrode and the electrochemical cell, with this requiring cleaning with concentrated acid before further testing could be carried out. The precipitation occurred with all V2O5 containing electrolytes and became more severe with increasing V2O5 concentration. With increasing V2O5 concentration in the electrolyte, a reduction in the photoelectrochemical performance was also observed, which is likely attributed to the reduced conductivity and transmittance of light through the electrolyte (shown in Fig. 4). Herein, we found that the addition of 0.01 M V2O5 in 0.1 M KPi resulted in the least significant influence on the electrolyte properties, with a conductivity similar to that of 0.1 M KPi, with no precipitation of V2O5 seen and high light transmission at 550 nm and above. However, further increases in the V2O5 concentration resulted in significant decreases in the electrolyte conductivity (up to seven-fold for 0.1 M V2O5) and weaker light transmission at 550 nm and above.
 | ||
| Fig. 4 Conductivity (A) and UV-Vis transmittance (B) of 0.1 M KPi electrolyte (pH 7) with various concentrations of V2O5. Optical pathway of the photoelectrochemical cell used for preliminary PEC testing of WO3|BiVO4 electrodes (C). Photograph of 0.1 M KPi electrolyte containing V2O5 electrolyte with concentrations of 0.01 M, 0.02 M, 0.05 M and 0.1 M from left to right (D). | ||
Given the 2.5 eV (i.e. ∼496 nm) band gap of the WO3|BiVO4 electrodes that we have reported previously,42 it follows that there will be little to no light absorption by the photoanodes when light must pass through a V2O5-containing electrolyte first. This is required with the typical optical pathway of a front-illuminated electrode, and indeed in our own electrochemical cell during back-illumination, whereby light must first pass through a small portion of the electrolyte (as shown in Fig. 4c). The use of V2O5 would also preclude tandem photoanode-photocathode photoelectrochemistry, unless the front-illuminated photoelectrode has a narrow band gap. This is a major issue for future applications and further restricts an already limited pool of suitable candidate photoelectrodes.
The results of photoelectrochemical characterisation of the WO3|BiVO4 electrodes in 0.1 M KPi enriched with V2O5 are summarised in Table 1, with IPCE data presented in Fig. S5.†
| V2O5 concentration in electrolyte/M | Peak IPCE/% | Photocurrent density at 1.23 VRHE/mA cm−2 | Reduction in photocurrent density after 24 hours chronoamperometry testing/% |
|---|---|---|---|
| 0 (baseline) | 23.4 | 1.10 | 77.3 |
| 0.01 | 20.2 | 0.62 | 52.0 |
| 0.02 | 3.1 | 0.10 | n/a |
| 0.05 | 1.1 | 0.03 | n/a |
| 0.1 | 0.0 | 0.00 | n/a |
All electrodes tested in V2O5-enriched electrolytes showed a reduction in peak IPCE, with reduced or zero efficiency in the region beyond 400 nm, as predicted by the UV-Vis transmittance. This resulted in a decrease in photocurrent density with increasing V2O5 concentration in the electrolyte. When light absorbed by V2O5 containing electrolytes was considered (see Fig. S5†), the absorbed photon to current efficiency (APCE) was comparable between the 0.1 KPi baseline and 0.1 M KPi with 0.01 M V2O5. However, with increasing V2O5 concentration, the APCE decreased, as in the IPCE data. Photoanode degradation was somewhat suppressed with the addition of 0.01 M V2O5, with the photocurrent density decreasing by 52% over the course of a 24 hours experiment, compared to 77% without V2O5 (shown in Fig. 5). It should, however, be acknowledged that the final photocurrent after 24 hours was only marginally higher with the addition of V2O5 due to its decrease from a lower initial value.
 | ||
| Fig. 5 Chronoamperometry stability testing of WO3|BiVO4 electrodes at 1.23 VRHE in 0.1 M KPi (pH 7) with and without 0.01 M V2O5. SEM images of the electrodes after the 24 hours test with V2O5 (top) and without V2O5 (bottom) are shown on the right-hand side. PEC measurements were taken under simulated sunlight a 75 W Xe light source equipped with a KG3 filter with equivalent light intensity of 0.42 suns. | ||
The continued degradation of the WO3|BiVO4 electrodes can be attributed to Bi3+ oxidation, which is not inhibited by a V2O5 enriched electrolyte. Further chronoamperometry testing with an electrolyte composed of 0.5 M Na2SO3 hole scavenger and 0.01 M V2O5 (shown in Fig. S6†) sought to remediate both degradation mechanisms in parallel, with the use of the hole scavenger helping to ensure that photogenerated charges were used for sulfite oxidation as opposed to that of Bi3+. Over the 24 hours experiment the photocurrent density decreased by 12.6% but returned to its initial value after surface bubble removal, suggesting that both degradation mechanisms, the vanadium leaching and bismuth oxidation, were successfully suppressed. The SEM images taken after the stability test showed increased agglomeration of the nanoneedles and reduced porosity which is the likely result of the vanadium self-healing mechanism that has previously been reported with vanadium-enriched electrolytes.32 Precipitation of V2O5 was also observed on the electrode and the testing cell, which became more apparent with increasing V2O5 concentration. With a V2O5 concentration of 0.01 M, precipitation of vanadium salt appeared not to significantly impact the photoelectrochemical properties of the material. However, at higher concentrations, the precipitation of V2O5 on the electrode and testing cell caused serious operational issues during PEC experiments. A photograph of precipitation on the electrode after PEC testing is shown in Fig. S7.† The operational issues caused by vanadium salt precipitation, along with the toxicity of V2O5, makes this a relatively undesirable constituent of the electrolyte.
The safety and operability requirements would be exacerbated in an up-scaled system, with stringent safety controls likely needed to address potential loss of containment in a large-volume photoelectrochemical system. Average outdoor temperatures (including night time), where such systems will ultimately be stationed, are likely to be lower than in a laboratory, making the electrolyte more prone to precipitation. For example, in the UK, average daily temperatures have typically only exceeded 10 °C for six months of the year since 2015, meaning that an additional cost for electrolyte temperature control (or another solution) would need to be implemented (especially during overnight downtime) to avoid electrolyte recrystallisation.51
This preliminary study has illustrated the stability issue of BiVO4 and WO3|BiVO4 electrodes in KPi electrolytes. We have also highlighted operational challenges that arise as a result of using vanadium-enriched electrolytes to suppress chemical vanadium leaching. Given the operational challenges and stability issues of the electrolytes tested in the preliminary study, further experiments herein were conducted using sodium borate buffer electrolyte, which has been shown to enhance the stability of BiVO4 electrodes in some previous reports.19,22,25
In the remainder of this study, we address strategies to improve the mechanical stability of WO3 and the (photo)electrochemical stability of BiVO4 and their effects on the WO3|BiVO4 heterojunction, with the objective to develop mechanically and photoelectrochemically stable photoanodes for implementation in up-scaled solar water splitting devices.
Optimisation of the WO3 morphology for improved photoanode mechanical stability
The formation of the WO3|BiVO4 heterojunction has many benefits to the physicochemical properties of the photoanode, including an increased absorption coefficient through nanostructuring and vastly improved charge separation by prolonging charge carrier lifetimes.13,52 However, a challenge to overcome is the poor mechanical stability of nanostructured WO3. Despite their superior photoelectrochemical performance, WO3 nanoneedles are weakly adhered to the FTO substrate and the film can be easily scratched, removed using scotch tape, or wiped off with a wet cloth/towel.33 This in turn results in poor mechanical stability of the WO3|BiVO4 heterojunction, due to the weak adhesion of the underlaying WO3 nanoneedles to the FTO substrates.To decouple the effects of the photo-corrosion of BiVO4 and the mechanical stability of WO3, the optimisation of the stability vs. performance of the WO3 underlayer was studied separately, using a 0.1 M H2SO4 electrolyte (pH 1) in which WO3 is photoelectrochemically stable.53,54 Using different AA-CVD reaction temperatures, three morphologies were synthesised: planar (325 °C), nanoneedles (375 °C) and a hybrid structure (350 °C). The surface topographies, captured by SEM, and photographs of the three types of WO3 photoanodes studied are shown in Fig. 6. The films grown at 325 °C were transparent, with their planar structure enabling a high level of contact between WO3 and FTO that resulted in excellent adhesion. In contrast, the films grown at 375 °C were complex nanostructures with low contact and therefore poor adhesion between WO3 and FTO. The electrodes were hazier in appearance, with SEM images showing complex nanoneedle structures with high porosity. The hybrid structure, grown at 350 °C, had an intermediate transparency in comparison to the planar and nanoneedles structures. The hybrid structure appeared to grow as a more interconnected nanostructured film, as opposed to the nanoneedles, which were more individual in nature, appearing to be stacked on top of each other and were not well-adhered to the FTO electrode. This enabled a higher degree of contact with the FTO compared to the nanoneedles and therefore improved adhesion.
 | ||
| Fig. 6 SEM images of WO3 synthesised at different AA-CVD reaction temperatures. Planar WO3 synthesised at 325 °C (A), hybrid WO3 synthesised at 350 °C (B), nanoneedle WO3 synthesised at 375 °C (C). Accompanying photographs shown in A, B and C insets. | ||
X-ray diffraction (XRD) and Raman analysis (shown in Fig. 7) showed that all the WO3 photoanodes were phase pure and monoclinic (γ-WO3). Peaks associated with cassiterite (i.e. the FTO electrode) are indicated by asterisks. The planar WO3 film yielded additional peaks at around 23° in the XRD pattern compared to the nanoneedle and hybrid WO3 structures, which resemble each other very closely. The additional peaks corresponding to the Miller indices (0 2 0) and (2 0 0) are characteristic of the planar WO3 films, as reported previously.33,55,56 The Raman spectra of all three morphologies include prominent bands at 807 cm−1 and 716 cm−1, which correspond to characteristic O–W – O and W2O6 bond vibrations.57 Weaker bands corresponding to these bond vibrations were also observed at around 328–329 cm−1, 272–275 cm−1, 135 cm−1 and 71–75 cm−1 with some variation between different morphologies.58
 | ||
| Fig. 7 XRD patterns (A) and Raman spectra (B), UV-Vis transmittance (C), UV-Vis absorbance (D) and Tauc plots (E) of WO3 with planar, hybrid and nanoneedle morphologies, deposited on FTO glass. | ||
Fig. 7 also compares UV-Vis data of WO3 electrodes on FTO with different morphologies. Data for the FTO electrode is included in the figure for reference. All WO3 electrodes had their lowest transmission and the highest absorbance in the UV and blue regions. The newly developed WO3 hybrid structure had the highest absorbance of the three morphologies overall and the narrowest band gap, at 2.69 eV (i.e. ∼461 nm). This was attributed to lower transmission because of the formation of a homogenous film of WO3 compared to the nanoneedles that spatially possessed more gaps in their structure, and also the lower specular reflectance of the hybrid film compared to the other morphologies (shown in Fig. S9†). Previous studies have shown that the morphology and thickness of WO3 films has a significant impact on the absorbance and band gap of WO3, with thicker or nanostructured films typically having a narrower band gap than thinner or flatter films.33,49 Our WO3 samples, which were representative of electrodes tested in this study, with band gaps between 2.69 eV and 3.05 eV (i.e. ∼407 nm), were in line with previously published values. The 3.55 eV (i.e. ∼349 nm) band gap of FTO was also consistent with previous reports.59
The photoelectrochemical water oxidation performance of the WO3 photoanodes was examined using linear sweep voltammetry and chronoamperometry. To determine the effect of each morphology on charge transfer efficiencies, sulfite oxidation was evaluated for each type of WO3 electrode. The results of this are summarised in Table 2.
| WO3 morphology | V onset,H2O (VRHE) | j H2O at 1.23 VRHE (mA cm−2) | Ф sep (%) | Ф inj (%) |
|---|---|---|---|---|
| Planar | 0.61 | 0.08 | 10.1 | 71.9 |
| Hybrid | 0.61 | 0.18 | 14.4 | 60.6 |
| Nanoneedles | 0.53 | 0.47 | 17.0 | 94.4 |
Fig. 8 shows the performance and stability of different WO3 electrodes during photoelectrochemical testing. The charge transfer efficiency was highest with the WO3 nanoneedle morphology, with this appreciably higher than the planar and hybrid WO3. Although the charge separation and injection efficiencies were similarly moderate for planar and hybrid WO3, the hybrid samples were able to absorb light much more effectively, enabling higher photocurrents. During chronoamperometry stability testing with flowing electrolyte, the photocurrent produced by the WO3 nanoneedles electrode declined rapidly, falling by ∼80% within two hours. In contrast, the planar WO3 maintained a relatively stable photocurrent over the same period, while the photocurrent generated using the hybrid-structured WO3 fell by ∼30%.
 | ||
| Fig. 8 Performance and stability of WO3 photoanodes in a 0.1 M H2SO4 electrolyte, pH 1 with and without 0.5 M Na2SO3 as a hole scavenger, at 100 mW cm−2 (AM 1.5 G). Linear sweep voltammograms of different WO3 morphologies (A); chronoamperometry testing at 1.23 VRHE with an electrolyte flowrate of 60 mL min−1 (B and C); linear sweep voltammograms of planar (D), hybrid (E) and nanoneedle (F) WO3 morphologies for water and sulfite oxidation. | ||
Overall, the hybrid WO3 samples were considered the most suitable for further investigation with the WO3|BiVO4 heterojunction, taking the benefits and minimising the drawbacks of the planar and nanoneedle structures, with improved photoelectrochemical water oxidation performance compared to the flat WO3 and improved mechanical stability compared to the nanoneedles. Therefore, in the remainder of this study, WO3 films were prepared at 350 °C, to synthesise the hybrid morphology, unless otherwise specified.
Development of a NiFeOOH co-catalyst using a scalable CVD method
Although NiFeOOH has been successfully used to significantly improve the PEC performance of BiVO4 for water oxidation,15,32,60–62 scalable routes to fabricate this co-catalyst require more development. To address this, we have developed an up-scalable AA-CVD fabrication method for NiFeOOH synthesis. To achieve this, it was first necessary to select suitable nickel and iron-based precursors. The leading candidate precursors were nickel(II) chloride, nickel(II) acetylacetonate, iron(III) chloride and iron(III) acetylacetonate. The chloride precursors were identified due to their use in synthesising NiFeOOH by other methods, and the acetylacetonate precursors were identified as organic-based compounds commonly used to synthesise thin films by CVD.15,63–65 To inform the CVD reactor temperature for NiFeOOH formation, thermogravimetric analysis (TGA) was first carried out on the candidate precursors in a nitrogen environment (shown in Fig. 9). | ||
| Fig. 9 TGA profiles of chloride-based (A) and acetylacetonate-based (B) precursors for NiFeOOH synthesis. | ||
The thermal decomposition profile and mass loss of hydrated FeCl3 was consistent with previously reported literature, with the plateau after 150 °C corresponding to the removal of H2O, giving rise to an approximate 50%:50% mixture of FeOCl and β-FeOOH.66 In the hydrated NiCl2, there are two plateau regions between 80 to 160 °C and 210 to 300 °C, which correspond to the removal of H2O and the mass loss is consistent with previous reports.67 The thermal dehydration and decomposition of hydrated NiCl2 yields a mixture of Ni2OCl2, Ni(OH)Cl and NiOOH, which is converted to NiO with increasing temperatures.68 The TGA profile of Ni(acac)2 showed two transitions beginning at around 110 °C and 300 °C, which were consistent with the decomposition of Ni(acac)2 to acetone gas.69 The TGA profile of Fe(acac)3 showed a single transition starting at approximately 180 °C, which corresponded to the decomposition of Fe(acac)3 to acetone and carbon dioxide gases.69
Annealing in air aids the decomposition of unreacted iron and nickel chlorides/acetylacetonates and other precursor byproducts, and the conversion of Ni/Fe oxychlorides to Ni/FeOOH.67,69 Due to the further conversion of Ni/FeOOH at temperatures beyond 150 °C to NiO and Fe2O3, total removal of chloride impurities was not possible by thermal decomposition alone, since this requires temperatures in excess of 300 °C.66–68,70 Consequently, reaction and annealing temperatures of 160 °C were selected to ensure adequate conversion to –OOH, and minimising conversion to NiO and Fe2O3.
To carry out physical characterisation of the co-catalyst, the four candidate precursors and their mixtures were reacted directly over FTO glass, passing an increased volume of 500 mL through the AA-CVD chamber. Photographs of the electrodes after AA-CVD and annealing at 160 °C for two hours are shown in Fig. S10.† Films were successfully deposited using Ni(acac)2, Fe(acac)3 and FeCl3, but no material was deposited using the NiCl2 precursor. This was confirmed by UV-Vis spectra (shown in Fig. S11†), which showed light absorption greater than that of FTO for all combinations except the 1 mM NiCl2. Furthermore, the influence of NiCl2 in the 1:4 mM precursor mixture of NiCl2 and FeCl3 reduced the amount of FeOx deposited, as indicated by lower UV-Vis absorbance. Both iron-based precursors yielded deposits containing FeOOH, suggested by their UV-Vis absorbance edges (both at 490 nm) which is in line with previous reports.71,72 This also confirms that complete conversion of FeOOH to Fe2O3 does not occur at 160 °C, although higher temperature annealing at 500 °C does result in this conversion, evidenced by the shift in the absorbance edge to approximately 575 nm.42,73 Evidence from TGA analysis suggests that the AA-CVD synthesis using Ni(acac)2 precursor results in a mixed deposition containing non-decomposed Ni(acac)2 and some NiOOH. Annealing in air at 160 °C results in the further decomposition of Ni(acac)2 and further hydroxylation of Ni.69 To avoid de-hydroxylation to NiO, the temperature of reaction and heat treatment must be less than 200 °C, although this results in the incomplete removal of any organic impurities from the precursor ligands.69,70 The NiOOH produced from Ni(acac)2 is weakly absorbing and has similar UV-Vis spectral profile to FTO glass, but with a slight reduction in transmittance across all wavelengths. However, in combination with the Fe(acac)3 precursor, the resulting NiFeOOH has an absorption band edge shift to the higher wavelengths compared to that of FeOOH, which provides evidence of the successful incorporation of Ni into the oxyhydroxide co-catalyst.74
To provide evidence of the amorphous structure of the co-catalyst films, X-ray diffraction (XRD) characterisation after annealing was attempted on the films deposited with large (500 mL) volumes of precursor (shown in Fig. S12†). The scattering patterns produced were the same as the bare FTO electrode, showing that the NiFeOOH films produced are highly amorphous.
To carry out further investigation of the co-catalyst material, the NiFeOOH layer was subsequently deposited on the WO3|BiVO4 photoanode with the optimised hybrid WO3 morphology. HR-TEM analysis (shown in Fig. S13†) provided further evidence of a very amorphous NiFeOOH layer on top of the BiVO4, which was easily distinguishable from the as-synthesised BiVO4 films that are highly crystalline. By analysing representative signals generated by time-of-flight secondary ion mass spectrometry (ToF-SIMS), it was possible to distinguish a very thin (to the order of single nanometers) but distinct iron and nickel oxide-based layers on BiVO4. The results from ToF-SIMS (shown in Fig. S14† with representative signals for each layer) provided evidence of intercalation between distinct FTO, WO3, BiVO4 and NiFeOOH layers. The average thicknesses of the NiFeOOH, BiVO4 and WO3 were 5 nm, 60 nm (±5 nm) and 420 nm (±10 nm) respectively. This gave a total thickness of the photoanode of 485 nm (±15 nm).
SEM images of the WO3|BiVO4|NiFeOOH films (shown in Fig. 10) showed that the photoanodes had a nanoporous morphology with the BiVO4 coated uniformly on the hybrid-structured WO3. The resulting morphology of the heterostructure was coral-like, in comparison to the WO3|BiVO4 heterojunction prepared with WO3 nanoneedles, which comprised of more disordered nanorods (shown in Fig. 3, earlier). Due to its low thickness (less than 10 nm according to SIMS analysis) the NiFeOOH was indistinguishable from the BiVO4 film in SEM images.
 | ||
| Fig. 10 SEM images of WO3|BiVO4|NiFeOOH films at 25×k (A) and 50×k (B) magnification. | ||
Physical characterisation of the WO3|BiVO4 films by XRD and Raman spectroscopy (shown in Fig. 11), with and without NiFeOOH, also supported the formation of an amorphous NiFeOOH structure, with no additional peaks when compared to the highly crystalline structure of the WO3|BiVO4 heterojunction. As in the previous sections, the XRD peaks and Raman bands correspond to monoclinic WO3 and monoclinic BiVO4.33,42,55,56,58,75,76
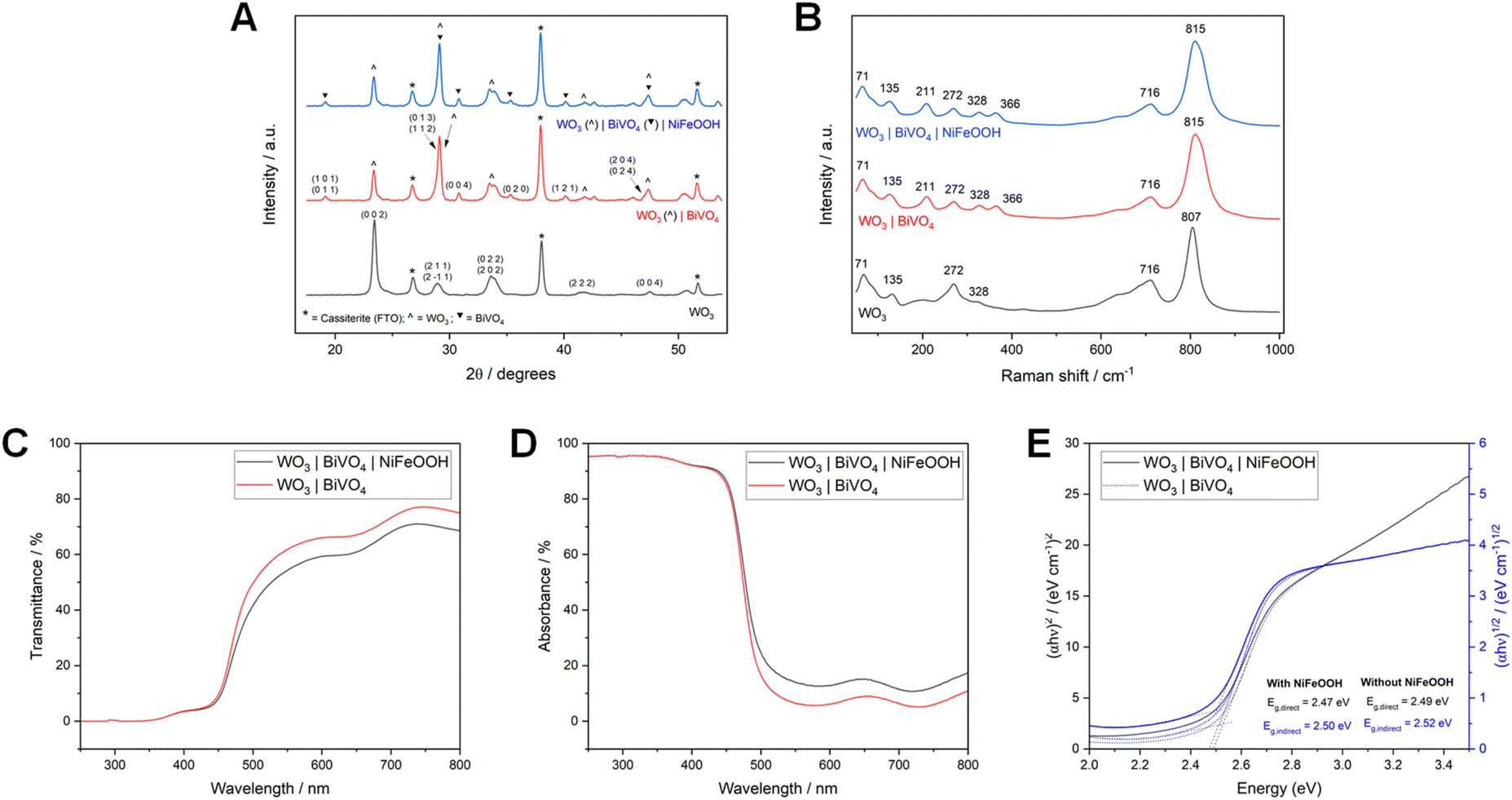 | ||
| Fig. 11 XRD patterns (A) and Raman spectra (B) of WO3, WO3|BiVO4 and WO3|BiVO4|NiFeOOH photoanodes (all of hybrid WO3 nanoneedle morphology), deposited on FTO glass. UV-Vis transmittance (C), absorbance (D) and Tauc plots (E) of WO3|BiVO4 with and without NiFeOOH, deposited on FTO glass. | ||
UV-Vis data of WO3|BiVO4 films, with and without NiFeOOH co-catalyst, are also illustrated in Fig. 11. Up to around 460 nm there is little change in the UV-Vis spectra. Beyond this, the addition of the co-catalyst results in slightly reduced transmission and increased absorption. The direct and indirect band gaps are also shifted slightly, from 2.47 and 2.49 eV (i.e. ∼502 to ∼498 nm) to 2.50 and 2.52 eV (i.e. ∼496 to 492 nm) respectively. The band gaps determined for our materials were most analogous to typically reported band gaps of BiVO4, as has been reported previously for the WO3|BiVO4 heterojunction.42,49,77
XPS analysis of the Bi 4f, V 2p, W 4f and O 1s environments (shown in Fig. S15†) provided evidence that bismuth and vanadium were in the oxidation states +3 and +5, respectively, as expected for pure BiVO4, and tungsten was in the oxidation state +6, as expected for pure WO3.78–81 Analysis of the Ni 2p and Fe 2p environments (shown in Fig. S16†) provided further evidence of the successful loading of NiFeOOH, with the primary oxidation states for nickel and iron +2 and +3 respectively, and peaks characteristic of the –OOH components observed.82–86 XPS analysis is discussed further in the ESI.†
To determine the effect of the co-catalyst on charge transfer, the WO3|BiVO4 electrodes with and without NiFeOOH were characterised during photoelectrochemical water and sulfite oxidation (shown in Fig. 12a–c). The addition of the NiFeOOH co-catalyst resulted in a significant increase in the charge separation efficiency, ηsep, of more than double, from 14.9% to 35.0%, while the charge injection efficiency, ηinj increased slightly from 82.7% to 84.6%. To verify the reproducibility of the synthesis method, the water oxidation performance of three WO3|BiVO4|NiFeOOH electrodes was characterised by linear sweep voltammetry (shown in Fig. S19†).
 | ||
| Fig. 12 Linear sweep voltammograms of WO3|BiVO4 electrodes, with and without NiFeOOH co-catalyst in 1 M borate buffer under AM 1.5 G simulated sunlight (A). Chopped linear sweep voltammetry (chopping frequency = 1 Hz) for water and sulfite oxidation of WO3|BiVO4 electrodes (B) and of WO3|BiVO4|NiFeOOH electrodes (C). Nyquist plots (D) and Bode plots (E and F) from PEIS measurements (0.1 to 1 × 105, under AM 1.5 G 100 mW cm−2 simulated sunlight) for WO3|BiVO4 electrodes with and without NiFeOOH at 0.8 VRHE. | ||
Estimates of the charge transfer efficiency of the WO3|BiVO4 heterojunction with and without the NiFeOOH co-catalyst were also carried out by observing transient photocurrent during chopped chronoamperometry (shown in Fig. S17†), for comparative purposes. The calculated charge transfer efficiencies are shown in Table S2.† Additionally, the onset potential for water oxidation was cathodically shifted by ∼90 mV and the photocurrent density at 1.23 VRHE, increased 2.4 times from 0.7 to 1.7 mA cm−2.
To further evaluate charge transfer effects with the addition of the co-catalyst, photoelectrochemical impedance spectroscopy measurements were carried out over a range of frequencies. A comparison of PEIS data recorded at various potentials is illustrated in Fig. S18.† These are displayed in Nyquist and Bode plots (shown in Fig. 12d–f). The resistance and capacitance values determined from the Nyquist and Bode plots are shown in Table S3.† The addition of NiFeOOH resulted in significant decreases in resistance and capacitance compared to the WO3|BiVO4 electrodes, indicating decreases in surface and bulk charge transfer resistance and capacitance. There was a particularly notable decrease in the bulk resistance (represented by RLF in the equivalent circuit model in Fig. 1), by more than 20 times with the addition of NiFeOOH co-catalyst. Indeed, Zhang et al. showed that charge transfer resistance is notably decreased by the incorporation of the WO3|BiVO4 heterojunction as well as the addition of the oxygen evolution co-catalyst.87 This makes our results particularly impressive due to the additional significant reduction in charge transfer resistance with the addition of NiFeOOH, compared to the bare WO3|BiVO4 heterojunction.
Evaluating the mechanical and photoelectrochemical stability of the fully optimised WO3|BiVO4|NiFeOOH photoanode system
To assess the mechanical and photoelectrochemical stability of the fully optimised WO3|BiVO4|NiFeOOH photoanodes (where WO3 is of the hybrid nanostructure), chronoamperometry tests in flowing borate buffer electrolyte were carried out at a constant potential of 1.23 VRHE. Due to the stability and practical issues with the KPi and vanadium-enriched electrolytes that we have highlighted previously, a 1 M sodium borate buffer (pH 9) electrolyte was selected for further photoelectrochemical testing.To determine the optimal electrolyte flow velocity, flowrates between 40 to 80 mL min−1 (∼0.5 to 1.0 cm s−1) were used in stability tests with three electrodes, shown in Fig. 13. The electrodes tested with flowrates above 40 mL min−1 were found to degrade over the course of the 10 hours experiment, while the electrode tested at 40 mL min−1 maintained a relatively stable photocurrent of around 1.75 mA cm−2. In the range evaluated, the rate of degradation was proportional to the flowrate, with the electrode tested at 60 mL min−1 declining to 75% of its original photocurrent over the course of 10 hours, while the same decrease was seen in less than two hours when the electrolyte flowrate was increased to 80 mL min−1. This effect was also reflected in the visual appearance of the electrodes after stability testing, with damage to the coating of electrodes tested using higher electrolyte flowrates observed (shown in Fig. 13b–d). The decline in photocurrent for the experiments carried out with electrolyte flowrates of 60 and 80 mL min−1 is therefore attributed primarily to mechanical damage, ultimately manifesting in the delamination of the photocatalyst material from the FTO electrode. Following this initial mechanical damage, the degradation of the electrodes was exacerbated through photoelectrochemical bismuth oxidation and chemical vanadium leaching mechanisms, due to the exposure of the BiVO4 layer to the electrolyte.
 | ||
| Fig. 13 Chronoamperometry stability testing at 1.23 VRHE, with various electrolyte flowrates for 10 hours and accompanying photographs of the photoanodes post-stability testing (A, B, C and D); chronoamperometry stability testing at 1.23 VRHE for 24 hours with an electrolyte flowrate of 40 mL min−1 (E) and linear sweep voltammograms (scan rate of 10 mV s−1) in 40 mL min−1 flowing electrolyte before and after the 24 hours stability test (F). All CA and LSV measurements were carried out with 1 cm2 WO3|BiVO4|NiFeOOH electrodes under AM 1.5 G simulated sunlight (100 mW cm−2) in 1 M borate buffer (pH 9). | ||
After the stability test, the irradiated area of the photoanode showed no visible changes (shown in Fig. 13b). Additionally, SEM images captured before and after testing (shown in Fig. S20†) showed that there were no morphological changes to the electrodes during the stability test. Linear sweep voltammetry (shown in Fig. 13f) carried out before and after the 24 hours stability test showed that the photoelectrochemical performance of the material was also unchanged. Mass spectrometry analysis (shown in Fig. 14) carried out with and without the co-catalyst, showed that the addition of NiFeOOH enhanced the faradaic efficiency from 75.6% to 85.5% and resulted in a stable oxygen production rate of 33 pmol s−1, using a ∼0.2 cm2 irradiated by white light (∼100 mW cm−2).
 | ||
| Fig. 14 Normalised oxygen production rate with and without NiFeOOH co-catalyst (A) and cumulative oxygen production over time (B) under white light irradiation (100 mW cm−2). | ||
Comparisons with the literature
It is well known that WO3 is soluble in neutral and alkaline conditions, with its Pourbaix diagram showing that γ-WO3 (s) can only exist in solutions with pH less than 5, with electrode potentials exceeding 0 VRHE.53 However, the formation of the WO3|BiVO4 heterojunction effectively forms a barrier between WO3 and the electrolyte, meaning that WO3 solubility is not an issue unless the BiVO4 layer is damaged. For this reason, it is essential that BiVO4 stability is maintained and also that the coating is conformal, since exposure of the WO3 to non-acidic electrolytes will result in the total and rapid degradation of the photoanode.As shown in numerous studies, BiVO4 degrades easily when the stability issue is unmitigated during photoelectrochemical water oxidation, at virtually any pH and potential.26,28,31,88 However, the rate of degradation can be decreased partially by reducing the photoanode potential, or using electrolytes with near neutral pH, so as to suppress the action of Bi3+ photo-oxidation and chemical leaching of V5+ respectively.26,31,88
The mechanism of BiVO4 dissolution is often misunderstood, with many studies with state-of-the-art stability during water oxidation suggesting that the dissolution is driven primarily by the vanadium leaching, when in fact this process has been proven to be 10 times less significant than photo-corrosion as a result of Bi3+ oxidation.26 The likely reason for this oversight is that most studies use surface co-catalysts to enhance the water oxidation performance of their electrodes, which has the additional advantage of suppressing Bi3+ photo-oxidation by facilitating more efficient use of photogenerated charges for the water oxidation reaction. A common and very effective method to prevent vanadium leaching by virtue of Le Chatelier's principle is through the use of vanadium-enriched electrolytes.32 However, the increased toxicity of these electrolytes should be noted, hence why the use of other methods to suppress this BiVO4 dissolution method should also be considered. Surface co-catalysts can also inhibit V5+ leaching efficaciously, through acting as barrier between BiVO4 and the electrolyte. Given the thickness of such co-catalyst layers, which are often no more than a few nanometres, it is essential for them to be deposited uniformly and conformally on the BiVO4 layer. Combining several methods to mitigate BiVO4 degradation can often prove to be very effective, and several studies, including this work, have shown that stability can be achieved through electrolyte tuning and effective surface co-catalysts.22,25,28,32,88
The importance of the mechanical stability of photoelectrodes in photoelectrochemical systems is rarely considered in the literature. It is also rarer that mechanical degradation from flow-induced shear stress is discussed either, with most articles mentioning mechanical stability in reference to flexible photoelectrodes. In line with our work, the adhesion of WO3 nanoparticles, which are the most analogous to the WO3 nanoneedles prepared in this study, is much weaker than that of films (analogous to our planar and hybrid structures).89–93 In general, nanostructuring reduces the required shear stress to enact mechanical damage, particularly when nanostructures are disordered or poorly adhered to the substrate.94 To improve photoelectrochemical performance of WO3, films are typically annealed in air at moderate temperatures (around 500 °C) to passivate defect states and traps.95 The annealing temperature can also affect WO3 adhesion. Heat treatment at 500 °C, as used to prepare our WO3 electrodes, is optimal in terms of enhancing WO3 mechanical stability.96,97 BiVO4 typically adheres well to FTO and also to WO3, hence why optimising the mechanical stability and adhesion of the WO3 layer was the focus in this work.98,99
There have been relatively few studies of the WO3|BiVO4 heterojunction with NiOOH, FeOOH or NiFeOOH surface co-catalysts.19,20,62,87,100 However, only our study uses the same fabrication method for the WO3, BiVO4 and co-catalyst layers, enabling a future fabrication method of sequential layers in a single process. Moreover, the fabrication of the FTO layer on float glass is carried out using CVD (at ∼600 °C, after the glass ribbon has cooled from its molten state of ∼1600 °C), and therefore, our methodology could theoretically be incorporated with the mass production of FTO electrodes using existing infrastructure. A summary of the key performance indicators of these studies is recorded in Table 3.
| Authors and date | Photoanode material | Electrode photoactive area (cm2) | Fabrication method | Electrolyte | j photo at 1.23 VRHE (mA cm−2) | η sep (%) | η inj (%) | Stability |
|---|---|---|---|---|---|---|---|---|
| a ns = not supplied; for fabrication methods DC = drop casting, ED = electrodeposition, ESC = electro-spray coating, FVD = flame vapour deposition, GLAD = glancing angle deposition, HT = hydrothermal, NL = nanosphere lithography, PD = photo-deposition, SG = sol–gel, PED = photoelectrodeposition, SC = spin-coating. b At 3 suns (data taken from graph). c With an electrolyte flowrate of 40 mL min−1. | ||||||||
| Creasey et al. (2024) (this work) | WO3|BiVO4|NiFeOOH | 1.0 | AA-CVD | 1 M sodium borate buffer (pH 9) | 1.78 | 35.0 | 84.6 | Stable over 24 hours at 1.23 VRHEc |
| Thirumalaisamy et al. (2024)100 | WO3|BiVO4|NiOOH | 1.0 | SC + ED | 0.5 M sodium sulfate (pH 7) | 1.16 | ns | ns | Falls to 25% of original value after 1 hour at 2 VRHE |
| Fang et al. (2022)19 | WO3|BiVO4|NiOOH | ns | SG + PED | 2 M sodium borate buffer (pH 9) | 3.00 | 40.0 | 50.1 | After 10 hours at 1.23 VRHE, 78% of original value |
| Zhang et al. (2022)87 | WO3|BiVO4|NiFeOOH | 1.2 | NL + ED | 0.2 M sodium sulfate (pH 7) | 3.05 | ns | ns | Stable over 10 hours at 0.9 VRHE |
| Ma et al. (2018)20 | WO3|BiVO4|FeOOH | 1.0 | SC + ESC + PD | 0.5 M sodium sulfate (pH 7) | 1.65 | ns | ns | Stable over 10 hours at 1.23 VRHE |
| Cai et al. (2016)62 | WO3|BiVO4|NiFeOOH | 0.26 | FVD + DC + HT | 0.5 M phosphate buffer (pH 7) | 4.50 | ns | 91.0 | Stable over 3 hours at 1.23 VRHE |
| Pihosh et al. (2016)16 | WO3|BiVO4|CoPi | ns | GLAD + ED + PED | 0.1 M phosphate buffer (pH 7) | 6.72 | 67.1b | 98.6b | Stable over 1 hour at 1.0 VRHE at 25 °C and 50 °C |
The highest reported performance of WO3|BiVO4 electrodes in literature remains those produced by Pihosh et. al., with a photocurrent density of 6.72 mA cm−2 at 1.23 VRHE using a CoPi co-catalyst.16 The stability of those electrodes was tested less extensively in their work (∼1 h), with cobalt leaching during longer term measurements reported elsewhere.101,102 Of the studies presented in Table 3, two reported photoelectrochemical stability over at least 10 hours, using pH 7 sodium sulphate electrolytes.20,87 Photoanode stability is commonly characterised at the thermodynamic water oxidation potential of 1.23 VRHE, but some of the studies opted to operate at lower electrode potentials, to suppress the rate of bismuth oxidation and enable water oxidation to be favoured16,87 Grigioni et al.88 also carried out a study of WO3|BiVO4 with a NiFeOOH co-catalyst in various electrolytes. Comparing all these reports, the electrolyte used for photoelectrochemical testing has significant importance, with stability typically poorer in phosphate buffer and better in sodium sulfate or borate buffer.19,20,87,88 Notably, none of the studies in Table 3 employed the use of a vanadium-enriched electrolyte to suppress the vanadium leaching mechanism of BiVO4 degradation, with the surface co-catalyst being able to adequately suppress BiVO4 degradation in their stability studies.20,87
Our photoanodes show exceptional stability in comparison to other WO3|BiVO4 electrodes, delivering a stable photocurrent density of 1.75 mA cm−2 during a 24 hours test at 1.23 VRHE in a 1 M borate buffer electrolyte with a flowrate of 40 mL min−1. Stability is critical to the future scale up of WO3|BiVO4 photoelectrodes, with our electrodes maintaining a stable photocurrent during flow, which was not investigated in the other studies highlighted, and will likely be critical to practicable device operation. Many of the other materials discussed were synthesised using complex or costly fabrication methods, whereas our photoanodes were fabricated by a scalable CVD method, used widely by industry for a number of applications, including the fabrication of FTO electrodes. We can therefore envisage our method being used to produce electrodes for much larger devices than the typical 1 cm2 lab scale investigated herein and will be the subject of future work.
Conclusions
In this study, we sought to address factors that frustrate efforts to translate small-scale photoelectrochemical water splitting cells into up-scaled systems, including: (i) the development of a scalable fabrication method, (ii) maintaining high operational stability and (iii) achieving competitive performance.Using our scalable aerosol-assisted chemical vapour deposition (AA-CVD) method, we successfully fabricated WO3|BiVO4|NiFeOOH, photoanodes, and optimised the fabrication method to achieve both high performance and stability. We believe this to be the first time that WO3, BiVO4 and NiFeOOH have all been synthesised by the same fabrication method. Importantly, the development of a shared AA-CVD fabrication method for the WO3, BiVO4 and NiFeOOH provides the means for the in situ fabrication of multi-layered electrodes and will likely be key to its scalability and future adoption by industry. In a single batch, our AA-CVD fabrication facility is capable of fabricating 16 photoelectrodes, each 2.5 cm × 1.2 cm in size (i.e. total area of ∼48 cm2), all showing consistent and reproducible performance, meaning this method can be translated to the preparation of a larger and singular 5.0 cm × 10.0 cm photoelectrode (i.e. each 50 cm2 in size) for applications in medium-scale prototype demonstrators, and will be the subject of future work.
The mechanical stability of the photoanodes was optimised by varying the deposition temperature of WO3, developing a hybrid structure with both the performance benefits of nanostructuring and the mechanical stability of a flatter film. To address the primary mechanism of BiVO4 dissolution at the electrode|electrolyte interface, namely the photoelectrochemical oxidation of Bi3+ ions, we employed the use of a NiFeOOH co-catalyst to ameliorate water oxidation kinetics, such that photogenerated charges are utilised more exclusively for water oxidation as opposed to photo-corrosion. The addition of NiFeOOH cathodically shifted the onset potential by 90 mV, while increasing the current density at 1.23 VRHE by 2.4 times.
Our optimised WO3|BiVO4|NiFeOOH photoanodes delivered a stable photocurrent density of 1.75 mA cm−2 over 24 hours at 1.23 VRHE, withstanding the mechanical stress of electrolyte flow at velocities that which will be required to efficiently operate up-scaled devices. This work provides a platform from which further research into up-scaled photoelectrochemical water splitting can proceed, with the aim of bringing this technology a step closer to commercial reality.
Data availability
Supporting data for this article is included in the ESI.† Data presented in this article will be uploaded to an online repository prior to the publication on this manuscript.Author contributions
George H. Creasey: conceptualisation, methodology, investigation, formal analysis, writing – original draft, writing – review and editing, visualisation; Tristan W. McCallum: investigation, writing – review and editing; Guangrui Ai: investigation, writing – review and editing; Brian Tam: investigation, writing – review and editing; John W. Rodriguez Acosta: investigation, writing – review and editing; Alvia Mohammad Yousuf: investigation, writing – review and editing; Sarah Fearn: investigation, formal analysis; Flurin Eisner: resources, writing – review and editing, supervision; Andreas Kafizas: resources, writing – review and editing, visualisation, supervision; Anna Hankin: resources, writing – review and editing, visualisation, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. C. thanks the Department of Chemical Engineering at Imperial College London for an EPSRC DTP PhD scholarship (EP/W524323/1). A. K. thanks the Grantham Institute for Climate Change and the Environment for a pump-priming grant and the EPSRC for a Programme Grant (EP/W017075/1). A. H. thanks the Department of Chemical Engineering for the lectureship start-up grant and EPSRC (EP/W033216/1) for funding the PDRA position of J. R. A.References
- Copernicus, Copernicus: 2023 is the hottest year on record, with global temperatures close to the 1.5 °C limit, Reading, 2024 Search PubMed.
- G. Creasey, Capacity of UK Electricity Generation Assets in the 21st Century, 2000 to 2019, London, 2021, DOI:10.25561/101378.
- W. Evans, K. Harris, M. Laycock, N. Cartwright, L. Waters, W. Nye, Z. Clark, V. Martin, G. Creasey, C. Michaels, A. Mettrick, S. Rose, D. Ying, C. Waite, A. Gower, A. Wright and A. Annut, Digest of UK Energy Statistics (DUKES) 2021, London, 2021, DOI:10.25561/101376.
- K. Harris, C. Michaels, E. Chalu, A. Mettrick, A. Heaton, V. Martin, L. Waters, M. Laycock, Z. Clark and W. Evans, Digest of UK Energy Statistics (DUKES) 2023, London, 2023 Search PubMed.
- Department for Energy Security & Net Zero, Powering up Britain, UK Government, London, 2023 Search PubMed.
- Department for Business Energy & Industrial Strategy, UK Hydrogen Strategy, London, 2021 Search PubMed.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CAS.
- M. A. Pellow, C. J. M. Emmott, C. J. Barnhart and S. M. Benson, Energy Environ. Sci., 2015, 8, 1938–1952 CAS.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- International Energy Agency, Hydrogen, Paris, 2022 Search PubMed.
- A. C. Nielander, M. R. Shaner, K. M. Papadantonakis, S. A. Francis and N. S. Lewis, Energy Environ. Sci., 2015, 8, 16–25 RSC.
- I. Holmes-Gentle, F. Alhersh, F. Bedoya-Lora and K. Hellgardt, in Photoelectrochemical Solar Cells, Wiley, 2018, pp. 1–41 Search PubMed.
- B. Moss, O. Babacan, A. Kafizas and A. Hankin, Adv. Energy Mater., 2021, 11, 2003286 CrossRef CAS.
- Y. Park, K. J. McDonald and K.-S. Choi, Chem. Soc. Rev., 2013, 42, 2321–2337 RSC.
- G. Fang, Z. Liu and C. Han, Appl. Surf. Sci., 2020, 515, 146095 CrossRef CAS.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui, D. Fujita, M. Tosa, M. Kondo and T. Kitamori, Sci. Rep., 2015, 5, 11141 CrossRef PubMed.
- G. Fang, Z. Liu, C. Han, P. Wang, X. Ma, H. Lv, C. Huang, Z. Cheng and Z. Tong, ACS Appl. Energy Mater., 2021, 4, 3842–3850 CrossRef CAS.
- G. Fang, Z. Liu, C. Han, X. Ma, H. Lv, C. Huang, Z. Cheng, Z. Tong and P. Wang, Chem. Commun., 2020, 56, 9158–9161 RSC.
- W. Fang, Y. Lin, R. Xv and L. Fu, ACS Appl. Energy Mater., 2022, 5, 11402–11412 CrossRef CAS.
- Z. Ma, H. Hou, K. Song, Z. Fang, L. Wang, F. Gao, Z. Yang, B. Tang and W. Yang, ChemElectroChem, 2018, 5, 3660–3667 CrossRef CAS.
- I. Y. Ahmet, S. Berglund, A. Chemseddine, P. Bogdanoff, R. F. Präg, F. F. Abdi and R. van de Krol, Adv. Energy Sustainability Res., 2020, 1(2), 2000037 CrossRef.
- Y. Kuang, Q. Jia, G. Ma, T. Hisatomi, T. Minegishi, H. Nishiyama, M. Nakabayashi, N. Shibata, T. Yamada, A. Kudo and K. Domen, Nat. Energy, 2016, 2, 16191 CrossRef.
- Y. Zhang, L. Xu, B. Liu, X. Wang, T. Wang, X. Xiao, S. Wang and W. Huang, ACS Catal., 2023, 13, 5938–5948 CAS.
- L. Francàs, S. Selim, S. Corby, D. Lee, C. A. Mesa, E. Pastor, K.-S. Choi and J. R. Durrant, Chem. Sci., 2021, 12, 7442–7452 RSC.
- J. Cui, M. Daboczi, M. Regue, Y. Chin, K. Pagano, J. Zhang, M. A. Isaacs, G. Kerherve, A. Mornto, J. West, S. Gimenez, J. Kim and S. Eslava, Adv. Funct. Mater., 2022, 32, 2207136 CAS.
- S. Zhang, M. Rohloff, O. Kasian, A. M. Mingers, K. J. J. Mayrhofer, A. Fischer, C. Scheu and S. Cherevko, J. Phys. Chem. C, 2019, 123, 23410–23418 CAS.
- T. He, Y. Zhao, D. Benetti, B. Moss, L. Tian, S. Selim, R. Li, F. Fan, Q. Li, X. Wang, C. Li and J. R. Durrant, J. Am. Chem. Soc., 2024, 146, 27080–27089 CAS.
- F. M. Toma, J. K. Cooper, V. Kunzelmann, M. T. McDowell, J. Yu, D. M. Larson, N. J. Borys, C. Abelyan, J. W. Beeman, K. M. Yu, J. Yang, L. Chen, M. R. Shaner, J. Spurgeon, F. A. Houle, K. A. Persson and I. D. Sharp, Nat. Commun., 2016, 7, 12012 CrossRef PubMed.
- X. Yao, X. Zhao, J. Hu, H. Xie, D. Wang, X. Cao, Z. Zhang, Y. Huang, Z. Chen and T. Sritharan, iScience, 2019, 19, 976–985 CrossRef CAS PubMed.
- X. Yao, D. Wang, X. Zhao, S. Ma, P. S. Bassi, G. Yang, W. Chen, Z. Chen and T. Sritharan, Energy Technol., 2018, 6, 100–109 CrossRef CAS.
- S. Zhang, I. Ahmet, S.-H. Kim, O. Kasian, A. M. Mingers, P. Schnell, M. Kölbach, J. Lim, A. Fischer, K. J. J. Mayrhofer, S. Cherevko, B. Gault, R. van de Krol and C. Scheu, ACS Appl. Energy Mater., 2020, 3, 9523–9527 CrossRef CAS PubMed.
- D. K. Lee and K.-S. Choi, Nat. Energy, 2017, 3, 53–60 CrossRef.
- A. Kafizas, L. Francàs, C. Sotelo-Vazquez, M. Ling, Y. Li, E. Glover, L. McCafferty, C. Blackman, J. Darr and I. Parkin, J. Phys. Chem. C, 2017, 121, 5983–5993 CrossRef CAS.
- A. Hankin and F. E. Bedoya-Lora, in Chemical Technologies in the Energy Transition, Royal Society of Chemistry, 2024, pp. 44–90 Search PubMed.
- F. E. Bedoya-Lora, A. Hankin and G. H. Kelsall, Front. Chem. Eng, 2021, 3, 749058 CrossRef.
- J. A. Seabold and K.-S. Choi, J. Am. Chem. Soc., 2012, 134, 2186–2192 CrossRef CAS PubMed.
- L. Chen, F. M. Toma, J. K. Cooper, A. Lyon, Y. Lin, I. D. Sharp and J. W. Ager, ChemSusChem, 2015, 8, 1066–1071 CrossRef CAS PubMed.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 Search PubMed.
- R. G. Mortimer, Physical Chemistry, Academic Press, San Diego, 2nd edn, 2000 Search PubMed.
- M. García-Tecedor, D. Cardenas-Morcoso, R. Fernández-Climent and S. Giménez, Adv. Mater. Interfaces, 2019, 6(15), 1900299 Search PubMed.
- D. Bae, B. Seger, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 CAS.
- B. S. M. Tam, PhD, Imperial College London, 2022 Search PubMed.
- Z. Chen and T. Jaramillo, The Use of UV-visible Spectroscopy to Measure the Band Gap of a Semiconductor, Department of Chemical Engineering, Stanford University, 2017 Search PubMed.
- E. A. Davis and N. F. Mott, Philos. Mag., 1970, 22, 0903–0922 CAS.
- D. L. Wood and J. Tauc, Phys. Rev. B:Condens. Matter Mater. Phys., 1972, 5, 3144–3151 Search PubMed.
- A. Hankin, F. E. Bedoya-Lora, J. C. Alexander, A. Regoutz and G. H. Kelsall, J. Mater. Chem. A, 2019, 7, 26162–26176 CAS.
- F. E. Bedoya-Lora, M. E. Valencia-García, A. Hankin, D. Klotz and J. A. Calderón, Electrochim. Acta, 2022, 402, 139559 CAS.
- F. E. Bedoya-Lora, I. Holmes-Gentle and A. Hankin, Curr. Opin. Green Sustainable Chem., 2021, 29, 100463 CAS.
- C. Reddick, C. Sotelo-Vazquez, B. Tam, A. Kafizas, K. Reynolds, S. Stanley, G. Creasey, A. Hankin, C. Pablos and J. Marugán, Catal. Today, 2024, 437, 114783 CrossRef CAS.
- ILO-WHO, ILO-WHO International Chemical Safety Cards (ICSCs) – VANADIUM PENTOXIDE, https://chemicalsafety.ilo.org/dyn/icsc/showcard.display?p_version=2%26p_card_id=0596, (accessed 20 November 2024).
- Department for Energy Security and Net Zero, Energy Trends: UK Weather - October 2024, London, 2024 Search PubMed.
- S. Selim, L. Francàs, M. García-Tecedor, S. Corby, C. Blackman, S. Gimenez, J. R. Durrant and A. Kafizas, Chem. Sci., 2019, 10, 2643–2652 CAS.
- Y.-J. Lee, T. Lee and A. Soon, Chem. Mater., 2019, 31, 4282–4290 CAS.
- J. M. Spurgeon, J. M. Velazquez and M. T. McDowell, Phys. Chem. Chem. Phys., 2014, 16, 3623 CAS.
- P. Sotelo, M. Moore, M. T. Galante, C. Longo, K. Rajeshwar, L. Suescun and R. T. Macaluso, Polyhedron, 2019, 170, 486–489 CAS.
- P. M. Woodward, A. W. Sleight and T. Vogt, J. Phys. Chem. Solids, 1995, 56, 1305–1315 CrossRef CAS.
- K. Jakubow-Piotrowska, D. Kurzydlowski, P. Wrobel and J. Augustynski, ACS Phys. Chem. Au, 2022, 2, 299–304 CrossRef CAS PubMed.
- W. Song, R. Zhang, X. Bai, Q. Jia and H. Ji, J. Mater. Sci.: Mater. Electron., 2020, 31, 610–620 CrossRef CAS.
- J. A. Spencer, A. L. Mock, A. G. Jacobs, M. Schubert, Y. Zhang and M. J. Tadjer, Appl. Phys. Rev., 2022, 9, 011315 CAS.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- S. Wang, P. Chen, Y. Bai, J. Yun, G. Liu and L. Wang, Adv. Mater., 2018, 30(20), 1800486 Search PubMed.
- L. Cai, J. Zhao, H. Li, J. Park, I. S. Cho, H. S. Han and X. Zheng, ACS Energy Lett., 2016, 1, 624–632 CrossRef CAS.
- H. Yin, Y. Guo, N. Zhang, Y. Wang, S. Zhang and R. Jiang, J. Mater. Chem. A, 2023, 11, 24239–24247 CAS.
- L. Ding, L. Wang, S. Chu, W. Zhai, J. Li, X. Li and Z. Jiao, ACS Appl. Energy Mater., 2024, 7, 2963–2972 CrossRef CAS.
- S. N. Ariffin, H. N. Lim, Z. A. Talib, A. Pandikumar and N. M. Huang, Int. J. Hydrogen Energy, 2015, 40, 2115–2131 CAS.
- S. B. Kanungo and S. K. Mishra, J. Therm. Anal., 1996, 46, 1487–1500 CAS.
- M. K. King and M. K. Mahapatra, Int. J. Thermophys., 2022, 43, 32 CAS.
- S. K. Mishra and S. B. Kanungo, J. Therm. Anal., 1992, 38, 2417–2436 CAS.
- J. Von Hoene, R. G. Charles and W. M. Hickam, J. Phys. Chem., 1958, 62, 1098–1101 CAS.
- P. Pinto, G. Lanza, J. Ardisson and R. Lago, J. Braz. Chem. Soc., 2019, 30(2), 310–317 CAS.
- X. Han, C. Yu, J. Yang, X. Song, C. Zhao, S. Li, Y. Zhang, H. Huang, Z. Liu, H. Huang, X. Tan and J. Qiu, Small, 2019, 5(18), 1901015 Search PubMed.
- J. Hong, N. Suzuki, K. Nakata, C. Terashima, K. Kim, A. Fujishima and K. Katsumata, Renewable Energy, 2021, 164, 1284–1289 CAS.
- M. Mishra and D.-M. Chun, Appl. Catal., A, 2015, 498, 126–141 CAS.
- L. Francàs, S. Corby, S. Selim, D. Lee, C. A. Mesa, R. Godin, E. Pastor, I. E. L. Stephens, K.-S. Choi and J. R. Durrant, Nat. Commun., 2019, 10, 5208 Search PubMed.
- C. Ràfols i Bellés, S. Selim, N. M. Harrison, E. A. Ahmad and A. Kafizas, Sustainable Energy Fuels, 2019, 3, 264–271 RSC.
- S. Wang, P. Chen, J. Yun, Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2017, 56, 8500–8504 CrossRef CAS PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- S. F. Ho, S. Contarini and J. W. Rabalais, J. Phys. Chem., 1987, 91, 4779–4788 CrossRef CAS.
- T. P. Debies and J. W. Rabalais, Chem. Phys., 1977, 20, 277–283 CAS.
- W. E. Morgan, W. J. Stec and J. R. Van Wazer, Inorg. Chem., 1973, 12, 953–955 CAS.
- B. Barbaray, J. P. Contour and G. Mouvier, Environ. Sci. Technol., 1978, 12, 1294–1297 CAS.
- T. Zhou, S. Chen, J. Wang, Y. Zhang, J. Li, J. Bai and B. Zhou, Chem. Eng. J., 2021, 403, 126350 CAS.
- W. Zhang, J. Ma, L. Xiong, H.-Y. Jiang and J. Tang, ACS Appl. Energy Mater., 2020, 3, 5927–5936 CAS.
- J. Gallenberger, H. Moreno Fernández, A. Alkemper, M. Li, C. Tian, B. Kaiser and J. P. Hofmann, Catal.:Sci. Technol., 2023, 13, 4693–4700 CAS.
- A. P. Grosvenor, M. C. Biesinger, R. St. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CAS.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, J. Phys. Chem. C, 2017, 121, 6455–6463 CAS.
- W. Zhang, M. Tian, H. Jiao, H.-Y. Jiang and J. Tang, Chin. J. Catal., 2022, 43, 2321–2331 CAS.
- I. Grigioni, A. Corti, M. V. Dozzi and E. Selli, J. Phys. Chem. C, 2018, 122, 13969–13978 CAS.
- M. Zlamal and J. Krysa, Catal. Today, 2015, 252, 162–167 CAS.
- M. Rodríguez-Pérez, C. Chacón, E. Palacios-González, G. Rodríguez-Gattorno and G. Oskam, Electrochim. Acta, 2014, 140, 320–331 Search PubMed.
- W. J. Lee, P. S. Shinde, G. H. Go and C. H. Doh, Appl. Surf. Sci., 2013, 270, 267–271 CAS.
- P.-K. Chiu, D. Chiang, C.-T. Lee, C.-N. Hsiao, J.-R. Yang, W.-H. Cho, H.-P. Chen and C. L. Huang, IEEE Trans. Magn., 2014, 50, 1–4 Search PubMed.
- W. Wang, X. Wang, X. Xia, Z. Yao, Y. Zhong and J. Tu, Nanoscale, 2018, 10, 8162–8169 RSC.
- Q. Wu, W. Miao, Y. Zhang, H. Gao and D. Hui, Nanotechnol. Rev., 2020, 9, 259–273 CrossRef CAS.
- S. Corby, L. Francàs, A. Kafizas and J. R. Durrant, Chem. Sci., 2020, 11, 2907–2914 RSC.
- M. Rodríguez-Pérez, C. Chacón, E. Palacios-González, G. Rodríguez-Gattorno and G. Oskam, Electrochim. Acta, 2014, 140, 320–331 CrossRef.
- J. Krysa, M. Zlamal, S. Kment and Z. Hubicka, Chem. Eng. Trans., 2014, 41, 379–384 Search PubMed.
- A. Farid Ul Islam, M. Nurul Huda Liton, H. Tariqul Islam, M. Al Helal and M. Kamruzzaman, Chin. Phys. B, 2017, 26, 036301 CrossRef.
- P. S. Archana, Z. Shan, S. Pan and A. Gupta, Int. J. Hydrogen Energy, 2017, 42, 8475–8485 CrossRef CAS.
- L. Thirumalaisamy, Z. Wei, K. R. Davies, M. G. Allan, J. McGettrick, T. Watson, M. F. Kuehnel and S. Pitchaimuthu, ACS Sustain. Chem. Eng., 2024, 12, 3044–3060 CrossRef CAS PubMed.
- R. Zhang, G. van Straaten, V. di Palma, G. Zafeiropoulos, M. C. M. van de Sanden, W. M. M. Kessels, M. N. Tsampas and M. Creatore, ACS Catal., 2021, 11, 2774–2785 CAS.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109–114 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00440c |
| This journal is © The Royal Society of Chemistry 2025 |