
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isomer-driven pyrazole frameworks: structural and zwitterionic insights for advanced energetics†
Vikranth
Thaltiri
 a,
Richard J.
Staples
b,
Jessica E.
Burch
c,
Anshuman
Bera
d,
Sivaranjana Reddy
Vennapusa
d and
Jean'ne M.
Shreeve
*a
a,
Richard J.
Staples
b,
Jessica E.
Burch
c,
Anshuman
Bera
d,
Sivaranjana Reddy
Vennapusa
d and
Jean'ne M.
Shreeve
*a
aDepartment of Chemistry, University of Idaho, Moscow, Idaho 83844-2343, USA. E-mail: jshreeve@uidaho.edu
bDepartment of Chemistry, Michigan State University, East Lansing, Michigan 48824, USA
cRigaku Americas Corporation, The Woodlands, TX 77381, USA
dSchool of Chemistry, Indian Institute of Science Education and Research, Thiruvananthapuram, Maruthamala PO, Vithura, Thiruvananthapuram 695551, India
First published on 24th February 2025
Abstract
High-energy density materials (HEDMs) demand innovative molecular strategies for achieving optimized detonation performance, stability, and insensitivity. Now we present an isomer-driven design of two advanced energetic frameworks: a high-energy compound, 5-(3,4-dinitro-1H-pyrazol-5-yl)-3-(trinitromethyl)-1H-1,2,4-triazole (5) and zwitterionic compound 11, N-(5-(5-amino-1,3,4-oxadiazol-2-yl)-4-nitro-1H-pyrazol-3-yl)nitramide (11). The detonation performance was predicted using EXPLO5 (v7.01.01), while thermal stability and sensitivity were evaluated through DSC, and BAM impact and friction apparatus. Additionally, Multiwfn and VMD software were used to visualize ESP maps and LOL-π isosurfaces, providing insights into electronic structure and charge distribution. Compound 5 has an impressive density of 1.926 g cm−3, high detonation velocity of 9206 m s−1, outperforming RDX with an acceptable thermal stability of 163.9 °C. Compound 11 has an excellent density of 1.918 g cm−3, an RDX-like detonation velocity of 8797 m s−1, exceptionally high thermal stability of 242.7 °C, and is insensitive to external stimuli. The outstanding properties of 11 are attributed to its zwitterionic nature, as confirmed by crystal structure analysis, NCI interactions, ESP and aromaticity studies. These findings highlight a paradigm for leveraging positional isomerism and zwitterionic architectures to advance the design of HEDMs with superior performance and stability.
Introduction
The quest for advanced high-energy density materials (HEDMs) has driven innovations in molecular design, offering breakthroughs in density, stability, and detonation properties. Energetic materials (EMs) are indispensable in defence, propulsion, and pyrotechnics, where performance is determined by critical parameters such as density, heat of formation, detonation velocity, detonation pressure, stability, and sensitivity. Traditional EMs like RDX and HMX have served as benchmarks; however, their limitations, including high sensitivity to mechanical stimuli and environmental concerns, necessitate developing safer, more efficient, and environmentally friendly HEDMs.1–8The design and synthesis of high-performance EMs depend critically on the selection of appropriate precursors, as their molecular structure governs reactivity and final properties. Positional isomerism has proven to be a crucial factor in this process, where small structural differences significantly influence the detonation, thermal, and chemical reactivity of the resulting materials.9–15 For example, methyl 4-nitro-1H-pyrazole-3-carboxylate produced a low-performance energetic material (A), while its isomer, methyl 3-nitro-1H-pyrazole-5-carboxylate, led to a high-performing, heat-resistant material (B).16,17 Similarly, 4-azido-3,5-dinitro-1H-pyrazole and its isomeric counterpart, 5-azido-3,4-dinitro-1H-pyrazole, demonstrated distinct chemical reactivities with bases, resulting in structurally and energetically diverse materials (C and D).18,19 Another example includes the 2H,2′H-3,3′-bipyrazole and 1H,1′H-4,4′-bipyrazole isomers, which, despite both yielding high-energy oxidizers with ten nitro substituents, exhibit different thermal stabilities (E and F).20,21 These examples emphasize the profound impact of positional isomerism on achieving tailored properties, making it an anchor for designing advanced HEDMs (Scheme 1(a)).
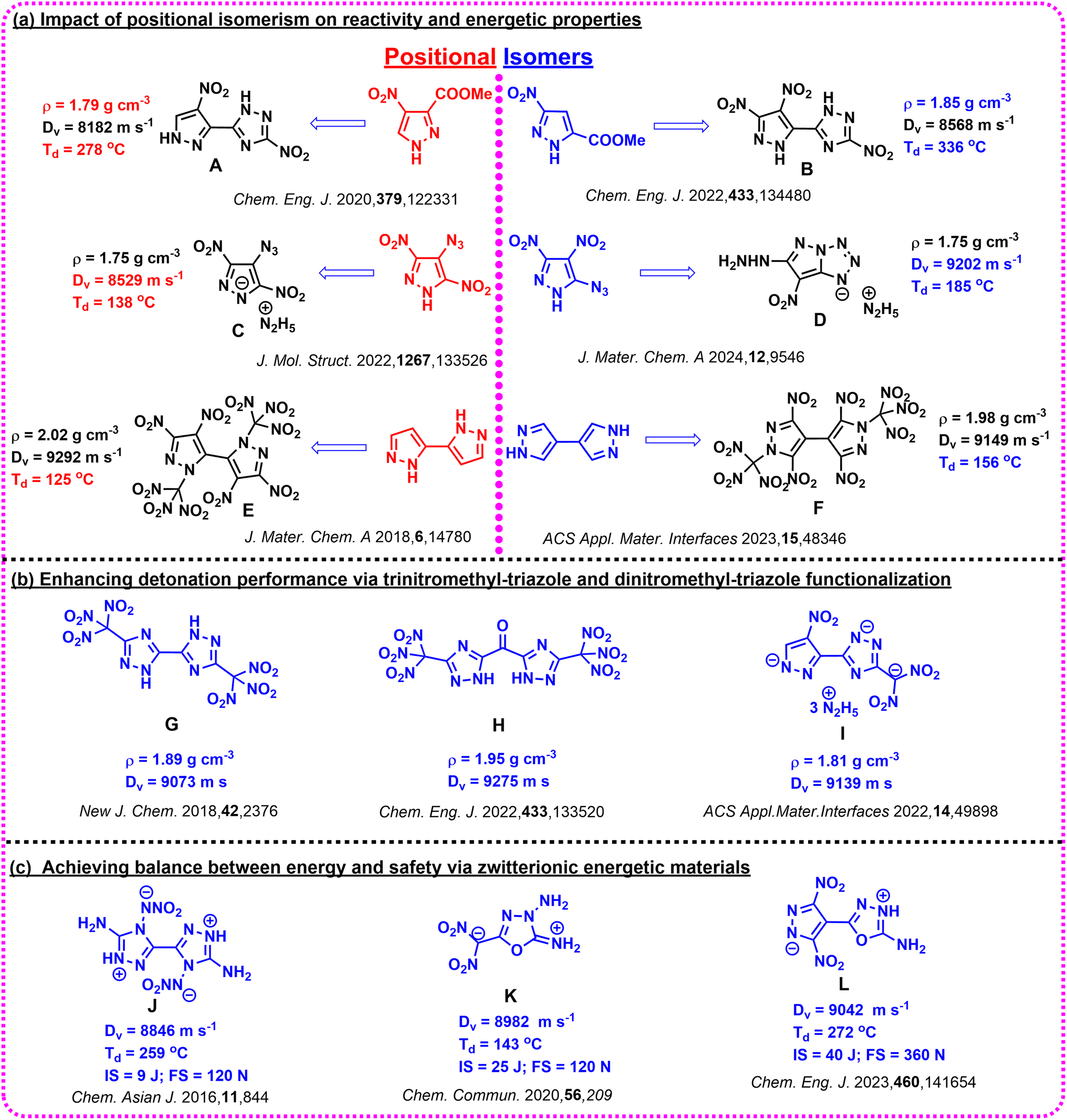 | ||
| Scheme 1 (a) Impact of positional isomerism on chemical reactivity and energetic properties of products obtained which highlights the critical role of precursor selection; (b) selected trinitromethyl-triazole and dinitromethyl-triazole functionalization, which showcase significant improvement in detonation velocity, pressure, and density. (c) Selected zwitterionic compounds for improved density, stability, and energetic properties through charge separation. | ||
Polynitro azoles have recently garnered significant attention for their high performance and reduced sensitivity to impact and friction. Among these, trinitromethyl-triazole and dinitromethyl-triazole frameworks have emerged as prominent candidates for high-performance energetic materials.22–26 These frameworks feature trinitromethyl and dinitromethyl groups, which enhance oxygen balance and density, leading to superior detonation properties, as illustrated in Scheme 1(b). Their exceptional densities, detonation velocities, and pressures make them ideal for next-generation energetic applications. Furthermore, the integration of the –NHNO2 group into azole frameworks has emerged as a key advance. Compounds with –NHNO2 substituents exhibit superior energetic properties due to their high heats of formation and dual functionality as hydrogen donors and acceptors. This enables the formation of strong hydrogen bonds and facilitates proton transfer, which can lead to zwitterionic structures. Such zwitterionic frameworks amplify density, thermal stability, and detonation performance through compact molecular packing and enhanced intra- and intermolecular interactions. Although zwitterionic energetic compounds show immense potential, they are rarely reported in the literature (Scheme 1(c)).27–29 The incorporation of 1,3,4-oxadiazole into zwitterionic frameworks (e.g., compound L)30 leverages the high nitrogen content and electron-deficient nature of the oxadiazole ring, further enhancing oxygen balance, and stability (Scheme 1(c)). These limitations underscore the need for innovative molecular designs to address structural and energetic challenges, and detonation properties. The synergy between pyrazole, –NHNO2 groups, and 1,3,4-oxadiazole represents a significant advance in the design of HEDMs.
Despite significant advances in the design of HEDMs, achieving a balance between detonation performance, thermal stability, and safety remains a persistent challenge. For instance, 5-(4-nitro-1H-pyrazol-5-yl)-3-(trinitromethyl)-1H-1,2,4-triazole (M),31 synthesized from the isomeric derivative of 4-nitro pyrazole precursor, demonstrates excellent detonation performance with moderate density of 1.87 g cm−3. However, its low thermal stability limits its practical applicability, underscoring the need for further functionalization to concurrently enhance both thermal and detonation properties (Scheme 2). Similarly, 5-(3,4-dinitro-1H-pyrazol-5-yl)-1,3,4-oxadiazol-2-amine (N)32 exhibited a low density of 1.72 g cm−3 and suboptimal detonation properties, further emphasizing the necessity of innovative synthetic strategies to optimize both performance and safety.
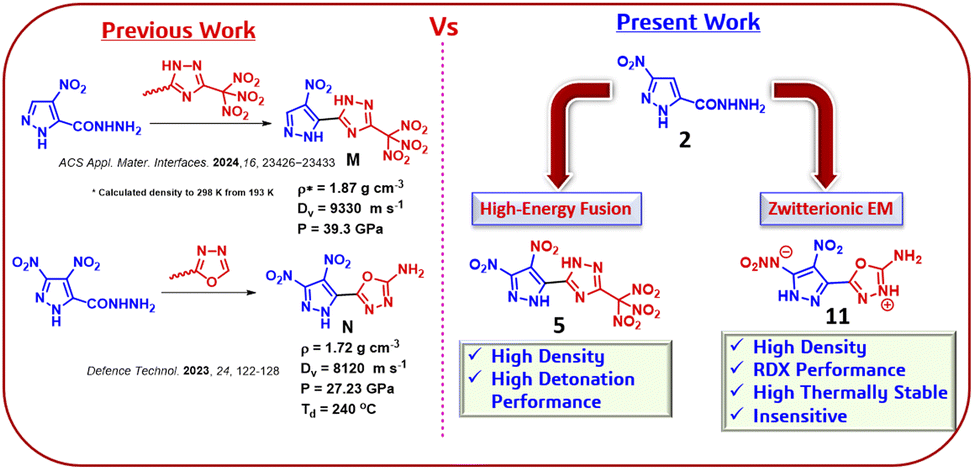 | ||
| Scheme 2 Comparison of previous and present work, highlighting high-energy fusion frameworks and zwitterionic material with superior density and detonation performance. | ||
This work addresses these limitations through an isomer-driven synthetic approach that leverages the structural versatility of isomeric 3-nitropyrazole derivative (2) as a precursor. The introduction of trinitromethyl-triazole framework, combined with the inherent reactivity of compound 2, enabled further nitration at the C4 position, thereby enhancing performance and stability. Additionally, strategic reduction of the C3-nitro group allowed for the incorporation of a –NHNO2 functionality, driving the formation of a zwitterionic structure. These synthetic strategies resulted in two key derivatives: 5-(3,4-dinitro-1H-pyrazol-5-yl)-3-(trinitromethyl)-1H-1,2,4-triazole (5) and N-(5-(5-amino-1,3,4-oxadiazol-2-yl)-4-nitro-1H-pyrazol-3-yl)nitramide (11), as well as energetic salt derivatives of 3-(3,4-dinitro-1H-pyrazol-5-yl)-5-(dinitromethyl)-4H-1,2,4-triazole (7 and 8). Compound 5, featuring high-energy triazole framework, compound 11, exhibiting zwitterionic structure, exemplify the effectiveness of isomer-driven design. These findings highlight the innovative design principles that enable the development of next -generation high-energy materials with enhanced performance and safety.
Results and discussion
Synthesis
The isomer-driven synthetic pathway, illustrated in Scheme 3, methyl 3-nitro-1H-pyrazole-5-carboxylate (1) was reacted with hydrazine hydrate in methanol under reflux to produce hydrazinium 3-nitro-1H-pyrazole-5-carbohydrazide (2), a key versatile precursor for novel energetic materials. For the trinitromethyl-triazole derivative (5), compound 2 was condensed with neutralized 3-ethoxy-3-iminopropanoate in acetonitrile-acetic acid at elevated temperature gave ethyl 2-(5-(3-nitro-1H-pyrazol-5-yl)-1H-1,2,4-triazol-3-yl)acetate (3), which was further hydrolysed and acidified to generate the acetic acid derivative (4). Nitration of compound 4 with mixed acid (HNO3–H2SO4) underwent selective nitration at the C4 position in the pyrazole ring and the carboxyl group, yielding 5-(3,4-dinitro-1H-pyrazol-5-yl)-3-(trinitromethyl)-1H-1,2,4-triazole (5) in 24 hours at ambient temperature. The ability of compound 2 to facilitate C4 nitration highlights the effectiveness and precision of this synthetic approach. | ||
| Scheme 3 Isomer-driven synthesis of compounds 5, 11, and energetic salts 7 and 8 from the key precursor, hydrazinium 3-nitro-1H-pyrazole-5-carbohydrazide (2). | ||
For the 1,3,4-oxadiazole derivative (11), compound 2 was treated with cyanogen bromide in water, yielding 5-(5-amino-1,3,4-oxadiazol-2-yl)-3-nitro-1H-pyrazole (9). Catalytic reduction of 9 using hydrazine hydrate and FeCl3 resulted in the diamine intermediate (10), which was subsequently nitrated with HNO3–CF3COOH to afford N-(5-(5-amino-1,3,4-oxadiazol-2-yl)-4-nitro-1H-pyrazol-3-yl)nitramide (11). This compound crystallized in a zwitterionic configuration, further demonstrating the synthetic versatility of compound 2.
Additionally, nitration of ethyl 2-(5-(3-nitro-1H-pyrazol-5-yl)-1H-1,2,4-triazol-3-yl)acetate (3) produced a dinitroester intermediate (6) at 60 °C, which was treated with aqueous ammonia or hydroxylamine to yield energetic salts (7 and 8). This further underscore the utility of compound 2 as a versatile precursor. All newly synthesized compounds were characterized using multinuclear NMR spectroscopy (1H, 13C, and 14N), IR spectroscopy, and elemental analysis.
Crystal structure analysis
The crystal structures of compounds 5, 8, and 11 were analysed to understand their molecular arrangements and contributions to high density and stability. The crystal structure of 5 was determined by using electron diffraction of a powder sample, demonstrating the utility of this method for characterizing complex energetic materials when single-crystal growth is challenging. Suitable single crystals of 8 and 11 were obtained for SC-XRD analysis through slow evaporation of methanol solutions. The crystallographic information is provided in the ESI.† Compound 5 crystallizes in the monoclinic space group P21/n, with a unit cell volume of 1285.4(12) Å3 and a calculated high density of 1.933 g cm−3 at 298 K. The structure is stabilized by an extensive hydrogen bonding network, with notable interactions such as N3–H3⋯O7 (2.781 Å) and N3–H3⋯N51 (2.872 Å), which significantly enhance molecular packing. Bond angles such as O1–N6–O2 (126.0°) and C5–N5–N4 (104.9°), as well as torsion angles such as O7–N9–C4–C3 (−4°), support minimal strain and effective functional group alignment. The dense packing is primarily stabilized by hydrogen bonding and van der Waals forces, which collectively contribute to the high packing efficiency of the crystal (Fig. 1(b)). | ||
| Fig. 1 (a and c) Thermal ellipsoid plots (50%) of compounds 5 and 8, respectively. (b and d) Molecular packing of compounds 5 and 8, respectively. | ||
Compound 8 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with a unit cell volume of 689.16(8) Å3 and a calculated density of 1.905 g cm−3 at 100 K. Hydrogen bonds, such as N5⋯O4 (3.013 Å) and N5⋯O5 (2.54 Å), play a critical role in enhancing packing efficiency. Bond angles, including C5–N5–N4 (111.2°) and C4–N3–C5 (103.3°), and torsion angles such as O2–N6–C1–N1 (−174.3°), facilitate planar alignment of molecular fragments. The layered arrangement observed in the packing diagram, with interplanar separations of 3.370 Å, supports efficient molecular stacking through a combination of hydrogen bonding and π–π interactions. These structural features contribute to the high packing density of the compound (Fig. 1(d)).
, with a unit cell volume of 689.16(8) Å3 and a calculated density of 1.905 g cm−3 at 100 K. Hydrogen bonds, such as N5⋯O4 (3.013 Å) and N5⋯O5 (2.54 Å), play a critical role in enhancing packing efficiency. Bond angles, including C5–N5–N4 (111.2°) and C4–N3–C5 (103.3°), and torsion angles such as O2–N6–C1–N1 (−174.3°), facilitate planar alignment of molecular fragments. The layered arrangement observed in the packing diagram, with interplanar separations of 3.370 Å, supports efficient molecular stacking through a combination of hydrogen bonding and π–π interactions. These structural features contribute to the high packing density of the compound (Fig. 1(d)).
Compound 11 crystallizes in the monoclinic I2/a space group, revealing a high density of 1.946 g cm−3 at 100 K, the highest among the three compounds. Bond length analysis reveals N3–C4 (1.281 Å), N4–C5 (1.326 Å), and N8–C5 (1.293 Å), indicative of delocalized electron density and partial double-bond character. This bond delocalization is attributed to proton transfer from the nitramine group (N5) to the oxadiazole nitrogen (N4) (Fig. 2), resulting in a zwitterionic configuration. The hydrogen bonding network, involving interactions such as N4⋯O3 (2.816 Å) and N8⋯O3 (2.843 Å), forms a robust zigzag framework that enhances molecular packing efficiency (Fig. 2(b)). Bond angles, including O2–N6–C1 (124.4°) and O3–N6–C1 (115.4°), combined with torsion angles such as C1–C2–N7–O4 (169.7°), ensure minimal steric hindrance and optimal molecular alignment. The layered packing arrangement, with an interlayer distance of 3.347 Å, is stabilized by a combination of π–π stacking interactions and an extensive hydrogen bonding network, contributing to the efficient molecular packing and high density of the crystal (Fig. 2).
 | ||
| Fig. 2 (a) Hydrogen bonding interactions with a maximum D–D distance of 2.9 Å and a minimum angle of 120°. (b) Zigzag hydrogen bonding network. (c) Layered packing with π–π stacking interactions (3.347 Å). (d) Compact lattice showing tight molecular interlocking along the b-axis. | ||
Density and packing efficiency of energetic materials – NCI analysis
Density is a cornerstone property for assessing the performance of energetic materials (EMs) as detonation velocity (DV) and detonation pressure (P) are directly proportional to material density. The densities of the newly synthesized compounds were measured using a gas pycnometer and exhibited exceptional values, with compound 5 at 1.926 g cm−3 and compound 11 at 1.918 g cm−3. These densities surpass those of benchmark materials such as RDX (1.80 g cm−3) and FOX-7 (1.88 g cm−3), demonstrating their high-performance potential.Molecular packing efficiency, a key determinant of density, was evaluated through packing coefficients, which were found to be 73.1% for compound 5 and 77.2% for compound 11 (Fig. 3(c)). The higher packing coefficient of compound 11 correlates with its robust density at 1.918 g cm−3, attributed to extensive hydrogen bonding and strong π–π interactions, as shown by Non-Covalent Interaction (NCI) analysis (Fig. 3(b)).33,34 Interestingly, compound 5, despite weaker π–π interactions (Fig. 3(a)), exhibited a commendable packing coefficient of 73.1%, which contributes significantly to its high density of 1.926 g cm−3—comparable to HMX (1.91 g cm−3) and superior to RDX. Additionally, the hydroxylammonium salt (8) has a density of 1.856 g cm−3, highlighting the structural contributions of its hydrogen bonding network. Meanwhile, the ammonium salt (7) exhibited a density of 1.812 g cm−3, aligning closely with RDX and further emphasizing the impact of molecular design on packing efficiency and density.
 | ||
| Fig. 3 (a) and (b) NCI analysis of 5 and 11; (c) packing coefficient of 5 and 11. | ||
Thermal behavior, energetic performances, and mechanical sensitivities
The thermal stability of an energetic material is pivotal for its practical utility, particularly in applications requiring resilience under high-temperature conditions. Thermal decomposition temperatures (Td) were measured using DSC showing a wide range of stability among the synthesized materials (Table 1). Compound 11, characterized by its zwitterionic nature, exhibited remarkable thermal stability with a decomposition onset of 242.7 °C, surpassing that of RDX (204 °C). This exceptional stability can be attributed to the charge-separated zwitterionic structure, which enhances molecular rigidity through strong hydrogen bonding and π–π stacking interactions. Compound 5, designed with a trinitromethyl group to enhance its energetic properties, has an acceptable decomposition temperature of 163.9 °C. Energetic salts 7 and 8 demonstrate moderate thermal stabilities of 187.6 °C and 158.7 °C, respectively.| Compound | ρ (g cm−3) | D v (m s−1) | P (GPa) | T d (°C) | ΔHfe (kJ mol−1) | ISf (J) | FSg (N) |
|---|---|---|---|---|---|---|---|
| a Density determined by gas pycnometer at 25 °C. b Detonation velocity. c Detonation pressure. d Thermal decomposition temperature (5 °C min−1). e Heat of formation (solid). f Impact sensitivity (BAM drophammer). g Friction sensitivity (BAM friction tester). h Crystal density at 298 K. i Crystal density 100 K. j Ref. 35. k Ref. 36. l Ref. 30. m Data from EXPLO-5 (v 7.01.01). | |||||||
| 5 | 1.926 (1.933)h | 9206 | 38.5 | 163.9 | 367 | 8 | 120 |
| 7 | 1.812 | 8451 | 28.9 | 187.6 | 75.7 | 22 | 240 |
| 8 | 1.856 (1.905)i | 8890 | 34.3 | 158.7 | 190.5 | 16 | 160 |
| 11 | 1.918 (1.946)i | 8797 | 33.2 | 242.7 | 417.7 | 35 | >240 |
| RDXj | 1.80 | 8795 (8836)m | 34.9 (34.2)m | 204 | 92.6 | 7.4 | 120 |
| FOX 7k | 1.88 | 8870 (8737)m | 34.0 (33.0)m | 220 | −130 | 25 | 340 |
| HMXl | 1.91 | 9144 (9380)m | 39.2 (40.3)m | 280 | 74.8 | 7 | 120 |
The heats of formation (HoF) were computed using the isodesmic reaction approach via Gaussian09.37 Compound 5 has an HOF of 367 kJ mol−1, while zwitterionic compound 11 shows the highest HoF of 417.7 kJ mol−1. Compounds 7 and 8 have HoFs of 75.7 kJ mol−1 and 190.5 kJ mol−1, respectively. Detonation parameters, including velocity (Dv) and pressure (P), were calculated by EXPLO5 using experimentally determined densities and theoretical heats of formation (Table 1).38 Compound 5 has a detonation velocity of 9206 m s−1 and a pressure of 38.5 GPa, outperforming RDX (DV: 8795 m s−1; P: 34.9 GPa) and approaching the capabilities of HMX (DV: 9144 m s−1; P: 39.2 GPa). Similarly, compound 11 demonstrated a detonation velocity of 8797 m s−1 and a pressure of 33.2 GPa, rivalling RDX while maintaining superior thermal stability. Among the salts, compound 8 emerged as a strong contender with a detonation velocity of 8890 m s−1 and a pressure of 34.33 GPa, validating the efficacy of its design. The ammonium salt (compound 7) showed slightly lower performance, with a detonation velocity of 8451 m s−1 and a pressure of 28.93 Gpa.
Sensitivity assessments, critical for evaluating safety of EMs, revealed varying responses among the new materials. Compound 5 exhibited impact and friction sensitivities similar to RDX (IS: 7.4 J; FS: 120 N), indicating its suitability for standard applications. Energetic salts 7 and 8 displayed reduced sensitivities due to their hydrogen-bonded networks, with impact sensitivities of 22 J and 16 J and friction sensitivities of 240 N and 160 N, respectively. Compound 11 emerged as the safest material, with negligible sensitivity to impact (>35 J) and friction (>240 N). This remarkable insensitivity is directly linked to its zwitterionic configuration, which enhances intermolecular interactions and molecular stability.
Hirshfeld surface analyses and 2D fingerprint plots, performed using CrystalExplorer 17.5 software,39 provide deeper insights into the molecular interactions underpinning these properties (Fig. 4). Compound 11 displayed a higher percentage of hydrogen bonding interactions (49.1%), underscoring the structural reinforcement provided by these interactions. In contrast, compound 5 exhibited a significant proportion of O⋯O interactions (43.6%), indicative of its sensitivity to external stimuli. These findings emphasize the unique structural attributes of each material and their tailored suitability for diverse energetic applications.
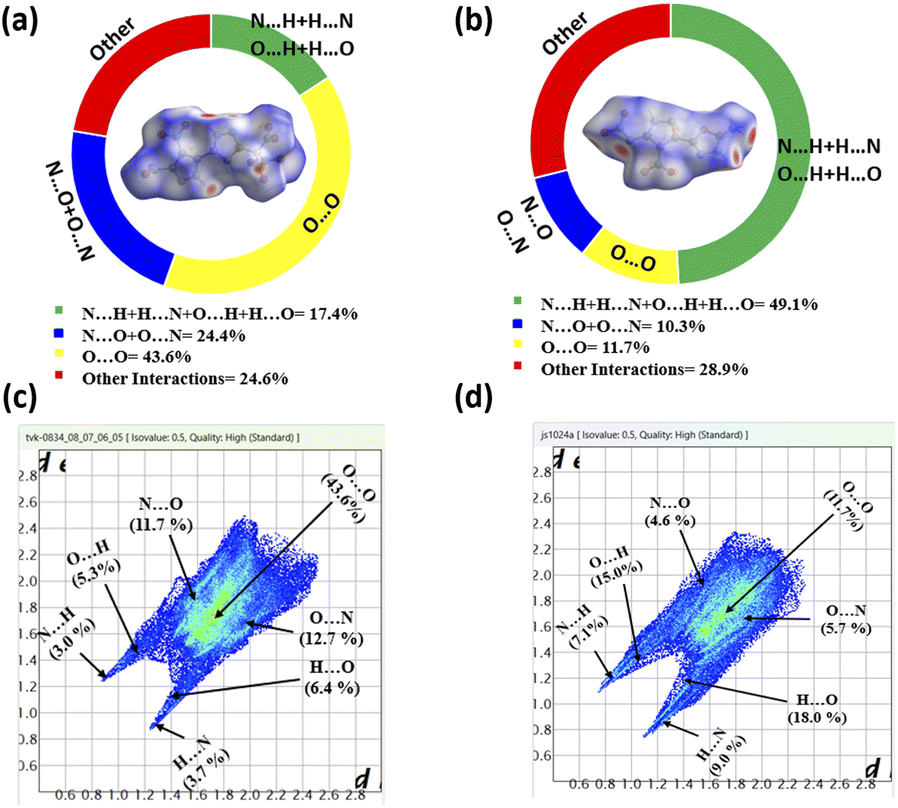 | ||
| Fig. 4 (a and c): Hirshfeld surface graph and 2D fingerprint plots of 5, and (b and d) Hirshfeld surface graphs and 2D fingerprint plots of 11. | ||
Electrostatic potential and aromaticity analysis
The zwitterionic nature and π-electron delocalization of compound 11 were investigated using electrostatic potential (ESP) and aromaticity analyses performed with Multiwfn and Gaussian software.40 The ESP map (Fig. 5(a)) reveals significant charge polarization, with positive regions (red) around hydrogen atoms bonded to nitrogen and negative regions (blue) on oxygen atoms of nitro groups. This distinct charge separation, characteristic of a zwitterionic structure, facilitates strong hydrogen bonding and efficient molecular packing, contributing to the compound's high density and exceptional thermal stability. | ||
| Fig. 5 (a) The electrostatic potential (ESP) maps (isodensity = 0.0004 a.u.) of 11; (b) NICS(0) values at different position of 11; (c) LOL-π isosurface of 11; (d) AICD plot of 11 (vectors moving clock-wise direction is indicated by the appropriate blue arrows). | ||
Aromaticity analysis further confirms the zwitterionic nature and π-delocalization in compound 11. Highly negative NICS values (−9.05 and −11.23, Fig. 5(b)) and a Shannon Aromaticity Index (SAI) of 0.00333 indicate substantial aromatic stabilization. The LOL-π isosurface (Fig. 5(c)) highlights π-electron localization, while the AICD plot (Fig. 5(d)) illustrates diatropic current density vectors, hallmarks of aromatic systems. These analyses demonstrate how zwitterionic charge separation and π-delocalization synergistically enhance density and stability, establishing compound 11 as a promising high-performance energetic material.
Conclusions
This study demonstrates the pivotal role of precursor-driven design and positional isomerism in tailoring molecular architectures for advanced applications. By leveraging the structural versatility of 3-nitro pyrazole, we synthesized two distinct energetic frameworks: compound 5, a pyrazole-triazole-trinitro and compound 11, a multifunctionalized pyrazole-1,3,4-oxadiazole. Compound 5 demonstrates remarkable detonation properties, with a high detonation velocity of 9206 m s−1 and a density of 1.926 g cm−3, outperforming RDX. Compound 11 characterized by its zwitterionic structure, has exceptional thermal stability (242.7 °C) and a density of 1.918 g cm−3, and balanced detonation performance (DV: 8797 m s−1) with enhanced safety (IS: 35 J).The zwitterionic nature of compound 11, supported by charge-separated resonance, hydrogen bonding network and π–π stacking interactions, highlights the significance of molecular architecture in achieving high-performance and stability. This work not only showcases the transformative potential of strategic precursor selection and positional isomerism but also establishes a clear pathway for designing advanced energetic materials that balance detonation performance and stability.
These findings extend beyond EMs, providing insights into the design of advanced materials in catalysis, pharmaceuticals, and materials science. This approach lays a strong foundation for future innovations in the field of HEDMs.
Data availability
All data supporting findings of this study are available within the article and its ESI files.†Author contributions
V. T. investigation, methodology, conceptualization and manuscript writing. R. J. S. and J. E. B. X-ray data collection and structures solving. A. B. and S. R. V. theoretical calculations (ESP, Aromatic analysis). V. T. and J. M. S. conceptualization, manuscript writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Rigaku Synergy S Diffractometer was purchased with support from the National Science Foundation MRI program (1919565). We are grateful for the support of the Fluorine-19 fund.References
- T. M. Klapötke, Chemistry of High-Energy Materials, de Gruyter, Berlin, 6th edn, 2022 Search PubMed.
- J. P. Agrawal, High Energy Materials: Propellants, Explosives and Pyrotechnics, Wiley-VCH, Weinheim, 2010 Search PubMed.
- P. F. Pagoria, G. S. Lee, A. R. Mitchell and R. D. Schmidt, Thermochim. Acta, 2002, 384, 187 CrossRef CAS.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289 CrossRef CAS PubMed.
- M. B. Talawar, R. Sivabalan, T. Mukundan, H. Muthurajan, A. K. Sikder, B. R. Gandhe and A. S. Rao, J. Hazard. Mater., 2009, 161, 589 CrossRef CAS PubMed.
- P. Yin and J. M. Shreeve, Adv. Heterocycl. Chem., 2017, 121, 89 CrossRef CAS.
- V. Thaltiri, K. Chavva, B. S. Kumar and P. K. Panda, New J. Chem., 2019, 43, 12318 RSC.
- P. Y. Robidoux, J. Hawari, G. Bardai, L. Paquet, G. Ampleman, S. Thiboutot and G. I. Sunahara, Arch. Environ. Contam. Toxicol., 2002, 43, 379 CrossRef CAS PubMed.
- Q. Sun, N. Ding, C. Zhao, J. Ji, S. Li and S. Pang, Chem. Eng. J., 2022, 427, 130912 CrossRef CAS.
- Q. Sun, Z. Jiang, N. Ding, C. Zhao, B. Tian, S. Li and S. Pang, J. Mater. Chem. A, 2023, 11, 23228 RSC.
- D. R. Wozniak, B. Salfer, M. Zeller, E. F. C. Byrd and D. G. Piercey, Org. Lett., 2020, 22, 9114 CrossRef CAS PubMed.
- L. M. Barton, J. T. Edwards, E. C. Johnson, E. J. Bukowski, R. C. Sausa, E. F. Byrd, J. A. Orlicki, J. J. Sabatini and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 12531 CrossRef CAS PubMed.
- V. Thaltiri, R. J. Staples and J. M. Shreeve, J. Mater. Chem. A, 2024, 12, 16729 RSC.
- G. Zhang, Z. Yi, G. Cheng, W. Yang and H. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 10594 CrossRef CAS PubMed.
- N. Ding, Q. Sun, X. Xu, Y. Li, C. Zhao, S. Li and S. Pang, Chem. Commun., 2023, 59, 1939 RSC.
- Q. Ma, G. Zhang, J. Li, Z. Zhang, H. Lu, L. Liao, G. Fan and F. Nie, Chem. Eng. J., 2020, 379, 122331 CrossRef CAS.
- J. Cai, C. Xie, J. Xiong, J. Zhang, P. Yin and S. Pang, Chem. Eng. J., 2022, 433, 134480 CrossRef CAS.
- X. Y. Zhang, X. Y. Lin, B. Y. Guo, C. Tan and Y. Han, J. Mol. Struct., 2022, 1267, 133526 CrossRef CAS.
- V. Thaltiri, J. Singh, R. J. Staples and J. M. Shreeve, J. Mater. Chem. A, 2024, 12, 9546 RSC.
- I. L. Dalinger, A. V. Kormanov, I. A. Vatsadze, O. V. Serushkina and T. K. Shkineva, J. Mater. Chem. A, 2018, 6, 14780 RSC.
- J. Meng, T. Fei, J. Cai, Q. Lai, J. Zhang, S. Pang and C. He, ACS Appl. Mater. Interfaces, 2023, 15, 48346 CrossRef CAS PubMed.
- Q. Ma, H. Gu, J. Huang, F. Nie, G. Fan, L. Liao and W. Yang, New J. Chem., 2018, 42, 2376 RSC.
- A. K. Chinnam, J. Singh, R. J. Staples and J. M. Shreeve, Chem. Eng. J., 2022, 433, 133520 CrossRef CAS.
- A. K. Yadav, V. D. Ghule and S. Dharavath, ACS Appl. Mater. Interfaces, 2022, 14, 49898 CrossRef CAS PubMed.
- Y. Tang, C. He, G. H. Imler, D. A. Parrish and J. M. Shreeve, Dalton Trans., 2019, 48, 7677 RSC.
- K. Mohammad, V. Thaltiri, N. Kommu and A. A. Vargeese, Chem. Commun., 2020, 56, 12945 RSC.
- T. M. Klapötke, M. Leroux, P. C. Schmid and J. Stierstorfer, Chem.–Asian J., 2016, 11, 844 CrossRef PubMed.
- T. Liu, S. Liao, S. Song, K. Wang, Y. Jin and Q. Zhang, Chem. Commun., 2020, 56, 209 RSC.
- J. Tang, H. Xiong, G. Zhang, Y. Tang, H. Yang and G. Cheng, Chem. Commun., 2022, 58, 11847 RSC.
- X. Zheng, T. Yan, G. Cheng and H. Yang, Chem. Eng. J., 2022, 460, 141654 CrossRef.
- P. Yi, C. Lin, X. Yi, P. He, T. Wang and J. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 23426 CAS.
- X.-X. Zheng, T.-O. Yan, L. Qian, H.-W. Yang and G.-B. Cheng, Des. Technol., 2023, 24, 122 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- V. Thaltiri, V. Shanmugapriya, T. Yadagiri and P. K. Panda, J. Org. Chem., 2022, 11, e202200487 CAS.
- A. J. Bellamy, Struct. Bonding, 2007, 125, 1 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- M. Suceska, EXPLO5, v. 7.01.01, Brodarski Institute, Zagreb, Croatia, 2013 Search PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00372e |
| This journal is © The Royal Society of Chemistry 2025 |