
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluating the terminal group switching of responsive monolayer films: examining structural rearrangements via molecular simulations and experimental wetting measurements†
Nicholas C.
Craven
 a,
Allison
Cordova-Huaman
b,
Christopher R.
Iacovella‡
b,
G. Kane
Jennings
b and
Clare
McCabe
*bc
a,
Allison
Cordova-Huaman
b,
Christopher R.
Iacovella‡
b,
G. Kane
Jennings
b and
Clare
McCabe
*bc
aMaterials Science Program, Vanderbilt University, Nashville, TN, USA
bDepartment of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN, USA
cSchool of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, UK. E-mail: c.mccabe@hw.ac.uk
First published on 30th May 2025
Abstract
Monolayers with terminal groups that are driven by thermodynamics to respond to changes in solvent are designed with the Molecular Simulation Design Framework (MoSDeF). Specifically, atomistic simulations of amorphous silica surfaces functionalized with alkylsilane chains terminated with competing hydrophobic and hydrophilic moieties are studied. We show that such moieties can achieve a dynamic switching response through molecular reorientation of the individual hydrophobic/hydrophilic groups when changing from a polar (water) to a non-polar (n-hexane) solvent. This responsiveness occurs more strongly with half-functionalized monolayer films containing a second, shorter alkylsilane chain that provides film stability and free volume for the terminal groups to transition between bistable states via the switching mechanism. Experiments are performed to measure the contact angles of polar and non-polar liquids on a subset of synthetically viable versions of the broader simulated data set. The simulations and experiments show that the interface is responsive to the nature of the solvent, and that the degree of switchability is affected by both the relative length of the terminal groups and the identity of the polar group.
1. Introduction
Interfacially mediated properties are crucial to a wide range of advancing technologies, such as microfluidic devices and anti-wear/anti-stiction coatings for nano/micro electromechanical systems.1–7 Wetting properties are also of fundamental interest in the design of fouling resistant surfaces,8–12 including polymer membranes for dehydration and nanofiltration,13–17 as well as recent work on energy-independent unidirectional liquid transport.18 These applications can be further enhanced by dynamic or responsive control of the interface, enabled by tuning either surface chemistry or structure. Such surfaces are a class of smart materials that are the next generation of interfacial design. Coatings with the ability to respond to environmental factors such as pH, temperature, and ionic or biological solutes, among other stimuli,19–24 have been reported in the literature, with polymeric systems being widely investigated.9,25–27The reported responsive nature of hierarchical block copolymer grafts has been attributed to a rearrangement favouring a single polymer block type exposed to the solid–liquid interface.19,28,29 It is proposed that such rearrangement, termed the molecular “switching” effect, is solely driven by minimizing interfacial free energy with the environment and may be accompanied by a reduction in surface energy. However, while responsive functionality is observed, the timespan for the switchability is on the order of minutes.30,31 An alternative methodology to design more rapidly responsive films would be monolayer films that exhibit similar functional group exchangeable exposure as block-copolymer coatings but significantly faster dynamics. Since the surface properties of these monolayers are mainly governed by the outermost 0.5 nm of the film, their responsive adaptation requires the conformational change of only the end group, surpassing the responsive rate of more commonly studied responsive polymer films.32,33 Additionally, self-assembled monolayers (SAMs) are a well-studied tool for surface modification, providing an expansive set of possible chemical groups that could impart dynamic properties and tune surface energies.
A knowledge gap exists in the current literature relating relevant theories that could predict or inform the design of such monolayer films, making bottom-up design approaches practically difficult. Specifically, while certain films have been reported to exhibit switchable behavior, a direct comparison of different functional group switchabilities has not been performed. Additionally, classical wettability theories, such as the long-standing Fowkes equation, fail for complex, heterogeneous interfaces.34,35 The need to address these gaps was recently brought to attention by Ilgen et al. who discuss the need to incorporate molecular-scale understanding of interfaces into continuum-scale models for predicting interfacial behavior.36
Here, we propose a novel approach to designing films that can simultaneously demonstrate low interfacial free energy in both polar and non-polar solvents and will be referred to as switchable surfaces. This approach relies upon designing the chain terminus in the monolayer with groups that contain both hydrophobic and hydrophilic moieties, such that thermodynamics will drive the rearrangement of the surface moieties to minimize interfacial energy. In essence, these surfaces are designed to self-adapt in both polar and non-polar environments. Because the environmental adaptation is simply a reorientation of a surface group, these systems offer an instantaneous response to minimize interfacial free energy upon exposure to a new environment. The responsiveness of the monolayer films is examined using atomistic molecular dynamics (MD) simulations and related to experimental contact angles. The objective of this manuscript is to observe monolayer films under different environments, namely water as a polar solvent, and n-hexane, and α-bromonaphthalene as non-polar solvents, respectively. These surfaces are synthesized experimentally and switchability is examined through contact angle measurements, which capture changes in surface energy under polar- and non-polar probe liquids. To better ascertain the molecular basis for local switchability and changes in wettability, simulations are performed on these surfaces to understand the atomic level changes in the terminal group structures. Finally, we propose a novel categorization for switchable film states and relate the behaviour of films that fall under these states to observed contact angle measurements.
2. Methods
2.1 Simulation methodology
The structures considered in this work were designed in stages and shown visually in the diagrams of Fig. 1. An amorphous silica crystal substrate was coated with alkylsilane chains, following procedures developed by Summers et al.37 At full coverage, 4.0 chains per nm2 were packed onto the surface to form a dense monolayer designed to mimic experimental coverage, which is estimated to be 3–5 chains per nm2.38 The functionalized alkylsilane chains are ten carbons in length and were chosen to coincide with experimental synthesis candidates. An amide linker chemistry was used to attach different switchable terminal groups to the functionalized alkylsilane chains. 50% of the surface was coated with a CH3-terminated chain of ten carbons in length, termed to as the backfill chains. The backfill chains are used to ensure a tightly packed base film while providing sufficient spacing between the switchable terminal groups. Surfaces with backfill molecules are referred to as “half-functionalized”, while homogenous surfaces with no backfills are termed fully-functionalized. | ||
| Fig. 1 Diagram of switchable films considered in this study. (a) Representation of the film structure with functionalized and backfill chains. T is the terminal group chemistry on the functionalized chains and represents both the polar (P) and non-polar (NP) groups of the terminal group. The pivot carbon is at the intersection of the P and NP functional groups, and an amide group connects the terminal group to the rest of the functionalized chain. The chains are bonded to the silica substrate, which are made up of both functional chains with terminal groups, and backfill chains terminated by CH3. (b) P and NP groups studied, which differ in size and polar moiety (i.e., hydroxyl or carboxyl) and are combined to form the 18 different terminal groups considered herein. The groups are labeled as Pn and NPn, where n is the length of the longest chain of heavy atoms in the group. (c) Illustration of an example of a P4:NP4 molecule. | ||
The switchable terminal groups are defined by a polar group (P), composed of an alkyl chain that terminates in a polar carboxyl or hydroxyl moiety, and a non-polar alkyl group (NP), both of varying lengths. The P and NP groups chosen were combined to form the 18 different terminal groups studied in the simulations and are labeled based on their length as shown in Fig. 1b. An example terminal group would be a carboxyl P4:NP4, which is broken down into P4 and NP4 components as shown in Fig. 1c. The NP4 group is an n-butyl functional group that is 4 carbons in length bonded to the central pivot carbon, which is not considered part of the “4” atoms that identify this group. The P4 group is also bonded to the pivot carbon and is built from: two alkyl carbons, a carbon as part of the carboxyl, and a carboxyl oxygen atom. Since both oxygens in the P group do not contribute to the “length” of the P group, the hydroxyl oxygen is selected to determine the length.
2.2 Simulation workflow
The systems studied were constructed using the mBuild39 Python package with custom generated recipes to create the surfaces and are made publicly available from the MoSDeF tool suite.40 Specifically, a 5 × 5 nm2 substrate was carved from bulk amorphous silica,41 pruned to an experimentally established hydroxyl density of 5 OH per nm2 following previous work,42 and bonded to functionalized alkylsilane chains at the outermost layer of available oxygen reactive sites. These bonding sites were then filled at random by creating a 10 × 10 mesh-grid and identifying the closest points on the carved surface for chain bond formation. A second randomized surface was generated as a top surface, inverted and placed to ensure a 4 nm separation between the two films (see Fig. S1†). The solvent molecules of either n-hexane or water were then packed into the space between the two surfaces at bulk density, resulting in simulation boxes of 20–30k atoms.The OPLS-AA forcefield41,43–45 was used to describe the interactions within the monolayer, sourced from the Foyer46 python package within MoSDeF.44 An OPLS compatible silica forcefield from Summers et al. was used for the surface to monolayer interactions.37 Charge discrepancies of 0.01 electron charges were distributed evenly to ensure the functionalized chains were neutral. The same forcefield was used for the parameters of the hexane molecules and the TIP-3P forcefield47 used to model the water molecules. A table of parameters used for a sample system can be found in Table S1 in the ESI,† and machine-readable files are available in the GitHub repository as detailed in Section S6.†
To enforce a solid silica substrate, the outer 5 Å of both substrates were fixed in place and uncoupled to any given thermostat. This arrangement contains the solvent in the region between the two films and maintains a pseudo-periodic 2D boundary condition. The initial structures were simulated using LAMMPS48 (August 3 2022 version) and run for 50k time steps under the NVE ensemble with a maximum positional step size of 0.001 Å and a 0.1 fs timestep to remove large energetic overlaps. The system was then simulated in GROMACS49 (2022 version) using the steepest descent energy minimizer, followed by 2 ns of NVT relaxation at 298 K using the velocity rescaling thermostat with stochastic coupling50 with τ set to 10 ps and a 0.5 fs timestep. Once relaxed, the remaining steps were run with a 2 fs timestep. The solvent profile was then evaluated between the two monolayers to ensure that the center of mass was halfway between the two layers, the two edges had the same approximate densities, and the middle densities were approximately the bulk average (as determined from bulk density simulations). If any one of these conditions was not met, the two surfaces were compressed together using a Newton–Raphson approximation (eqn (S1)†) at a constant rate of 2 nm fs−1 (each surface is moved at 1 fs) towards a target distance. After this distance was reached, a brief 0.5 ns NVT equilibration was run at 298 K, and the density conditions re-evaluated for the solvent. These two steps were looped until the compression stage was completed. An annealing step was then performed where the temperature was increased from 298 K to 350 K and then decreased back down to 298 K over 5 ns, with the goal of ensuring molecules were not trapped in configurations resulting from the initialization scheme. Finally, simulations were run in NVT at 298 K with a 10 chain Nosé–Hoover thermostat for 10 ns with the monolayer distance fixed. Fig. S2† plots the energy, hydrogen bonds, profiles, and other switching metrics of the simulations over the production simulation, demonstrating that in all cases a steady state was rapidly achieved over the first 1 ns; as such, the last 8 ns were used for data analysis. Triplicate simulations of each monolayer film chemistry were performed, where the placement of the chains was randomized for each replicate, as well as the starting momenta of the trajectory. The trajectory was written out at a sampling rate of 40 ps per frame during the production stage and errors were calculated from the standard deviations across averaged values of six simulated surfaces (three replicates, each with a top and bottom surface).
2.3 Data analysis
Analysis of surface structure was performed using the MDTRAJ51 and MDAnalysis52,53 Python packages. The following statistics were calculated for each frame: film thickness, solvent accessible surface area (SASA), and the intrafilm hydrogen bonds. These metrics were averaged and 95% confidence intervals determined from samples of six different simulated surfaces (i.e., a top and bottom surface from each replicate simulation). Film thickness was determined from the positions of the oxygen atoms bonded between the alkylsilane chain and the outermost surface atoms. The average Z position of these atoms was compared to the maximum Z position of the chains, with the difference tabulated as the thickness. SASA was determined using the Shrake–Rupley method54 provided in MDAnalysis. The central 3 × 3 nm2 area of the surface was considered to only account for the SASA exposed to the test liquid used. Lastly, the surface hydrogen bonds were calculated using the MDAnalysis Hydrogen Bond Analysis tool.55 A 120° angle and 0.3 nm distance cutoff were used to generate hydrogen bonds at each frame. All oxygen and nitrogen atoms were considered as hydrogen bond acceptors, and all hydrogen atoms bonded to these atoms were considered donors. Parameters specific to a given P or NP group, Zgroup, θgroup, and ρgroup were calculated with custom MDAnalysis scripts and are discussed in more detail in the results section. Additional details on these analysis methods, as well as the switching metrics discussed in the Results Section, can be found in Section S3 of the ESI† with the data shown in Tables S2–S6.† Diagrams that define the methods can be found in Section S4.†2.4 Experimental materials
10-Undecenyltrichlorosilane, 97% (10-UTS) and n-octyltrichlorosilane, 97% (OTS) were purchased from Gelest Inc. Anhydrous toluene, 99.8%, anhydrous tetrahydrofuran, 99.8% (THF), and dimethyl sulfoxide, 99.9% (DMSO) were purchased from Sigma-Aldrich. Sodium hydroxide, >95% (NaOH), hydrogen peroxide, 30% (H2O2), sulfuric acid, >95% (H2SO4), hydrochloric acid, 36.5–38.0% w/w (HCl), potassium permanganate, >99% (KMnO4), sodium periodate, >99.8% (NaIO4), thionyl chloride, >99% (SOCl2), and α-bromonaphthalene, 97%, were purchased from Thermo Fisher. Potassium carbonate, >99% (K2CO3) was purchased from Alfa Aesar. Sodium bisulfite, 97% (NaHSO3) was obtained from Acros Organics. L-Alanine, 99%, glycine, 95%, L-leucine, 99%, L-valine, 99%, and (±)-2-amino-1-butanol, 97% were purchased from Thermo Fisher. 3-Amino heptanoic acid, (R)-(–)-2-amino-1-hexanol, 97%, DL-2-amino-3-methyl-1-butanol, 97%, ethanolamine, 98%, and (S)-(+)-2-amino-1-propanol, 98% were purchased from Sigma-Aldrich. DL-2-Amino-1-pentanol, 98% and 3-amino pentanoic acid, 98% were purchased from Santa Cruz Biotechnology. All reagents were used as received. Single-side polished, boron-doped p-type silicon wafers (100) were purchased from University Wafer (1–10 Ω cm) and Pure Wafer (0.01–0.02 Ω cm).2.5 Experimental monolayer preparation
Silicon wafers were used as flat (1–10 Ω cm) and porous (0.01–0.02 Ω cm) silicon substrates in this study. Porous silicon (PSi) substrates were fabricated by anodic etching in a 15% HF solution in ethanol. First, a sacrificial PSi layer was etched with a current density of 100 mA cm−2 for 100 s and then removed with a NaOH solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 1 M NaOH(aq) to ethanol). The sacrificial layer ensures large pores openings at the surface for the etching of the PSi film.56 After the removal of the sacrificial layer, the substrate was etched in a 15% HF solution in ethanol with a current density of 100 mA cm−2 for 100 s to form the PSi thin film. The fabricated PSi films were then thermally oxidized at 800 °C for 1 h using a Thermolyne Type 48000 Furnace.
:9 1 M NaOH(aq) to ethanol). The sacrificial layer ensures large pores openings at the surface for the etching of the PSi film.56 After the removal of the sacrificial layer, the substrate was etched in a 15% HF solution in ethanol with a current density of 100 mA cm−2 for 100 s to form the PSi thin film. The fabricated PSi films were then thermally oxidized at 800 °C for 1 h using a Thermolyne Type 48000 Furnace.
Flat silicon substrates were cleaned by sonication in ethanol for 30 min, rinsed with ethanol, and dried in a stream of N2. PSi wafers were only rinsed with ethanol before drying in a stream of N2. To prepare for silanization, both types of substrates were immersed in piranha solution (H2SO4/H2O2, 7:3, v/v) for 30 min and then immersed in water, rinsed with water and ethanol, and then dried thoroughly with N2.
Single-component vinyl- and methyl-terminated monolayers (10-UTS and OTS, respectively) and half-functionalized monolayers (10-UTS + OTS) were prepared on piranha-treated substrates following the same silanization procedure. Single-component, packed monolayers were prepared by immersing the substrates into freshly prepared 1 mM solutions of 10-UTS or OTS in toluene at room temperature. Half-functionalized monolayers were prepared from toluene solutions containing 10-UTS and OTS at a 1:1 molar concentration. A previous study has shown that mixed monolayers prepared from vinyl- and methyl-terminated trichlorosilanes achieve a composition mirroring the initial solution stoichiometry, so mixed monolayers are expected to have a 1:1 composition of vinyl- and methyl-terminated adsorbates.57 After 3 h of silanization, the samples were removed from the solution, rinsed with toluene and water, and dried using a stream of N2.
Monolayers were generated from fully- and half-functionalized vinyl-terminated silane monolayers in a three-step process, as shown in Fig. S5.† First, the interfacial vinyl groups were oxidized to –CO2H by a single-step oxidation reaction in a permanganate–periodate solution, as described by Wasserman et al.58 Briefly, vinyl-terminated surfaces were placed in an aqueous oxidizing solution composed of 0.5 mM KMnO4, 19.5 mM NaIO4, and 1.8 mM K2CO3 (pH 6.5) for 24 h. After this time, the samples were rinsed consecutively in 0.3 M NaHSO3, water, 0.1 N HCl, water, and ethanol and dried under a stream of N2. Next, the –CO2H groups were activated by exposing the film to a solution of 5 mM SOCl2 in THF for 24 h to create acid chloride-rich surfaces. Finally, the samples were rinsed in THF and immediately placed into a 5 mM DMSO solution of the bifunctional amine for 24 h. Upon removal, the samples were rinsed with DMSO, water, and ethanol and dried with N2.
2.6 Experimental monolayer characterization
The wetting properties of the functionalized monolayers were characterized by contact angle goniometry. Water (surface tension 72.8 mJ m−2) and α-bromonaphthalene (surface tension 43.9 mJ m−2) were utilized as probe liquids dominated by polar and dispersive components, respectively. Sessile contact angles were measured on static drops using a Ramé–Hart manual goniometer. The reported values represent the average and standard deviations from at least two independently prepared samples, with measurements of three drops on each surface. All the contact angle values reported in this work were measured immediately after the drop was placed on the samples. These values remained constant during the time span of the measurements, which was approximately 10 min per each sample, reported in Tables S8 and S9 of the ESI.†The chemical composition of the films was analyzed by attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy, shown in Fig. S6.† Infrared spectra were taken using a Thermo Nicolet 6700 FT-IR spectrometer equipped with a Smart iTR™ ATR attachment with a diamond crystal plate. Spectra were collected in the region of 4000–650 cm−1 at 2 cm−1 resolution over 1024 scans and processed using the OMNIC™ software.
3. Results
3.1 Contact angles
Contact angle measurements allow for probing the wettability of an interface and through comparison across probes can shed light on the switchable nature of the terminal groups. Fig. 2 shows contact angles for water and α-bromonaphthalene (αBN) on monolayers with six hydroxyl Pn:NPn and six carboxyl Pn:NPn terminal groups. Water and αBN were chosen as probe liquids due to their relatively high surface tensions dominated by polar and non-polar dispersive components, respectively. Fully-functionalized monolayers containing OH P2 groups (Fig. 2a) show that without the backfill chains, the water contact angle decreases with increasing non-polar length from NP0 (–H) to NP2 and remains constant from NP2 to NP4. In contrast, the water contact angle on half-functionalized monolayers increases to a maximum for NP2 and isopropyl, and then decreases as the chain length increases to NP3 and NP4. This trend of apparent symmetry shows that functional molecules with long NP groups become more wettable to water, reaching the surface properties of a film with no NP group as the OH groups are better exposed. The αBN contact angle for half-functionalized monolayers shows a similar peak for isopropyl but only a slight decrease for NP3 and NP4. These differing responses of contact angle to the half and fully-functionalized monolayers when changing the size of the NP group indicate that the simple argument of adding a larger non-polar moiety to the film does not yield a consistent change to the surface energy. The presence of the backfill molecules must provide an inherent change in surface structure that modifies the response to increasing the NP group length. A possible explanation is a greater mobility of the NP groups in the half-functionalized films, which would allow the NP groups to shield themselves from the water molecules, and better expose the hydroxyl groups. It is possible then that the larger NP groups better fill the gaps left by the shorter backfill chains and provide better overall film stability, resulting in more homogenous exposure of the hydroxyl layer. This exposure of P and NP groups under water and αBN, respectively, is shown to be instantaneous as the contact angle values do not vary over the timespan of the measurements.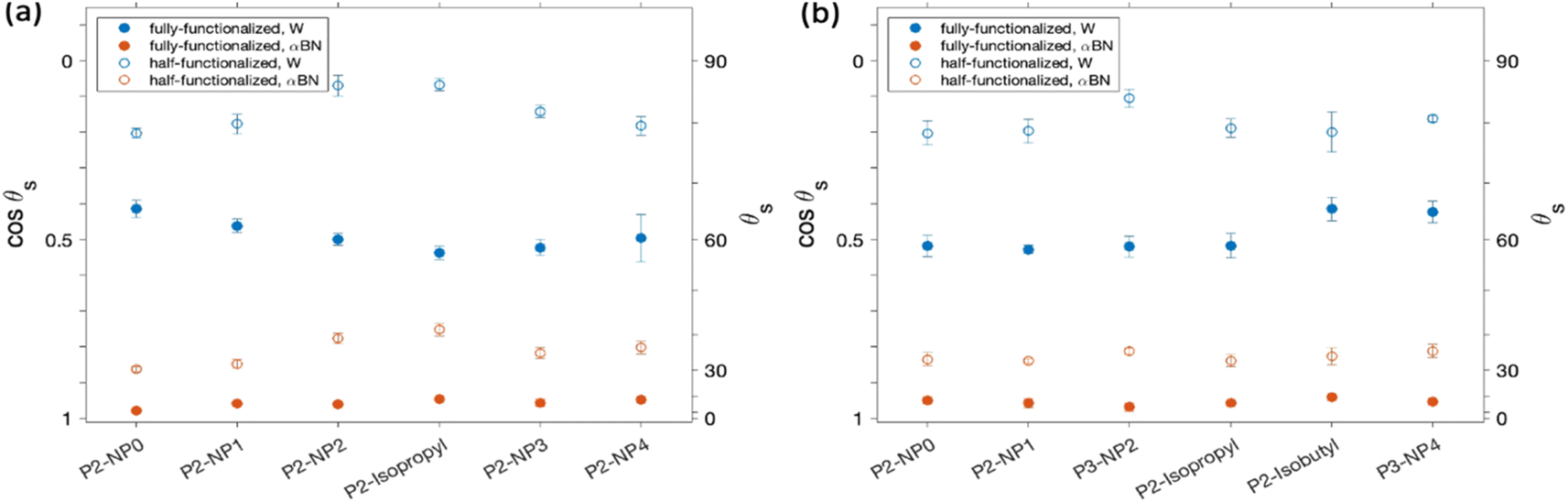 | ||
| Fig. 2 Sessile contact angles on functionalized monolayers with (a) –OH and (b) –COOH as the P group. Fully- and half-functionalized compositions are shown with filled (●) and open symbols, (○) respectively. Contacting liquids are water (blue) and α-bromonaphthalene (red). | ||
Fig. 2b shows the same contact angles with carboxyl-based functional groups. The water contact angle of fully-functionalized monolayers is found to be insensitive to the NP group length until chain lengths of isobutyl and NP4 groups. The half-functionalized carboxyl monolayers follow a muted trend as compared to that of the half-functionalized hydroxyl systems, with a maximum observed for NP2 groups. The half-functionalized films do show the expected overall increase in contact angles compared to the fully functionalized films, but also show greater contact angle dissimilarity across terminal group lengths. Again, this could be evidence of the NP group mobility allowing competitive exposure of the NP group to the nonpolar αBN probe liquid.
3.2 Simulation switching metrics
As it is difficult to separate the contribution of the P and NP group sizes to the contact angle measurements, we turn to molecular simulations of these films to determine the contributions individual P and NP groups make to the interfacial interactions. First, we explore three possible ways to measure the switchability of the groups as illustrated in Fig. 3: Zgroup, which is the group distance from the substrate; θgroup, the angle from the pivot carbon to the group terminal atom relative to the plane of the substrate; and ρgroup, the solvent density in a 5 Å sphere around the terminal atom of the group, where the solvent atoms are either hexane or water as representative non-polar and polar solvents respectively. The central column provides a visual diagram of these metrics, which can be uniquely measured for both the polar (P) and non-polar (NP) groups of half-functionalized monolayers. Fig. 3 demonstrates this measure for a single P4:NP4 carboxyl-based functional group and plots histograms of the surface chains across the simulations. These three switching measures provide an understanding of the surface favorability under both hexane (left column) and water (right column). Under hexane, the NP groups favorably switch upwards since ZNP − ZP, or ΔZ under hexane is significantly greater than 0 (2.0 ± 0.7 Å). A value greater than 0 is reflected in both the θNP − θP = Δθ = 27 ± 5° and ρNP − ρP = Δρ = 15.4 ± 2.6 atoms per nm2 parameters as well. Under water, the switching behavior is weaker across the three measures: ΔZ = −0.7 ± 0.6 Å, Δθ = −16 ± 6°, and Δρ = −4.7 ± 3.2 atoms per nm2. These values are provided in Tables S2–S4† for the other 17 terminal groups simulated. The P4:NP4 surface under water seems to be only slightly favored toward the P group, while under hexane the NP group is better able to expose itself. These parameters certainly indicate an induced change in the interfacial positions, orientations, and interactions of the terminal groups from the liquid exchange. As expected, the ΔZ values for all the switchable groups studied, become more negative in response to water due to the ability of the P group to shift towards the water molecules. This switching phenomenon is most strongly indicated across all films by the metric ρgroup, which perhaps should most closely correlate with the intermolecular interactions that often dominate the overall property of surface tension, since ρgroup measures which ends of the terminal groups are forming greater solvent interactions. If the solvent interactions are greater with the P group, then the surface and film will appear more polar. If the solvent interactions are greater with the NP group, the interface will be more non-polar. Moving forward in this work, we focus on ρgroup as the metric to best understand the interfacial switching behavior and Zgroup as a visual tool to identify where in the film the terminal groups are interacting with the solvent atoms.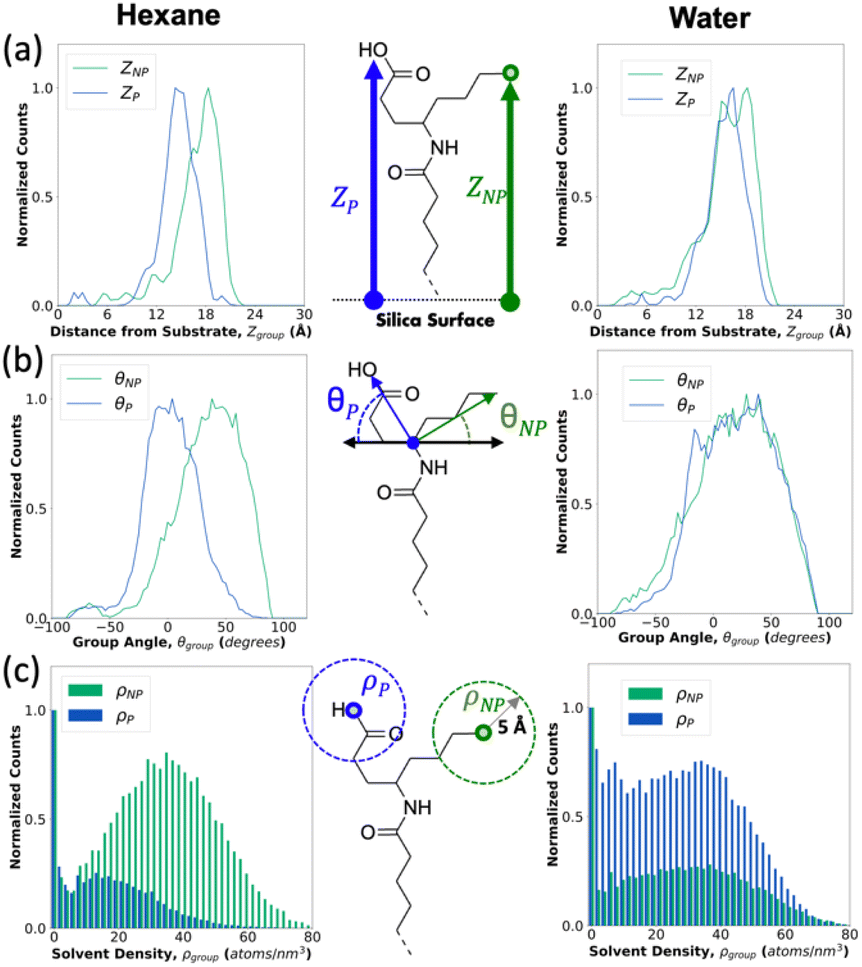 | ||
| Fig. 3 Switching parameter distributions for each P and NP group in a carboxyl-based P4:NP4 terminal group under hexane (leftmost column) and water (rightmost column). The central column shows diagrams for each metric: (a) Zgroup, which is the group distance from the substrate; (b) θgroup, the angle from the pivot carbon to the group terminal atom relative to the plane of the substrate; and (c) ρgroup, which is the count of solvent atoms in a 5 Å spherical radius around the group. | ||
3.3 Carboxyl vs. hydroxyl group switching
To examine the chemical effects of switching, a P4:NP4 film was considered with hydroxyl-based chemistry to compare directly to the carboxyl-based P4:NP4. Fig. 4 compares these two surfaces using heatmaps of Zgroup and ρgroup values. These are plotted under hexane (top row) and water (bottom row) for the P and NP groups for both hydroxyl and carboxyl-type chemistry. Two distinct regions can be seen in the heatmaps: a region of low to mid Zgroup values where ρgroup is 0, and a second region of mid to high Zgroup values and with ρgroup > 0. The first region is indicative of the groups that are caught within the film and therefore cannot extend away from the film enough to form solvent interactions. The second region indicates groups that are found at a large enough Zgroup to start forming interfacial interactions that increase as they reach out further into the solvent. The heat map is an illustrative way to examine the extent to which the P or NP groups are composing the interface in each solvent, as indicated by the intensity and position of the second interfacial region. By comparing the interfacial region of the P and NP groups with a single solvent, we can compare whether the surface would appear more polar or non-polar to that solvent. The carboxyl-based terminal groups show a greater NP group interface in hexane and a greater P group interface in water, indicating a switching response. The hydroxyl-based terminal groups reflect the same behavior caused by the expected group switching. Furthermore, we can compare the same group in both water and hexane to get an indication as to which group is more responsive to the change of solvent. Comparing the carboxyl P group in hexane to water shows an increase in the number of P groups in water in the interfacial region, indicating that some of the groups are responding to the water molecules by switching upwards and outwards. That same behavior is magnified for the hydroxyl P group, which shows even more groups collected in the top right region of the plot in water than for the carboxyl P group, providing evidence that the hydroxyl groups switch up more than the carboxyl groups. In comparison, the switched down state in hexane for the hydroxyl P group and the carboxyl P group look similar. For both carboxyl and hydroxyl terminal groups, the NP group seems to switch to the same degree from water to hexane, which signifies that the NP group can act somewhat independently from the responsiveness of the P group.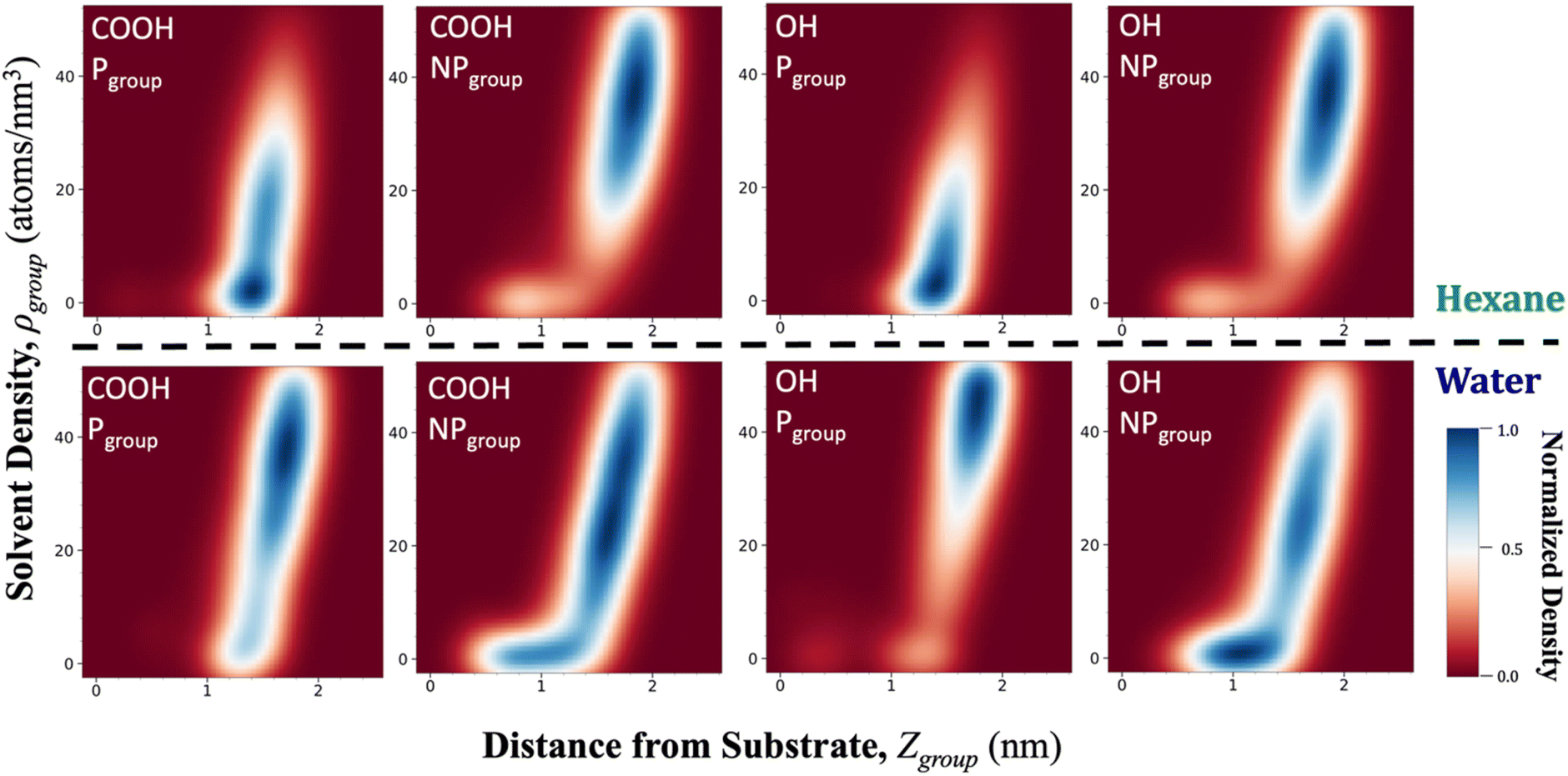 | ||
| Fig. 4 Heatmaps of the distance from the solvation shell density (ρgroup) versus substrate (Zgroup) for P4:NP4 with COOH and OH polar groups in hexane (above dashed line) and water (below dashed line). | ||
3.4 Tether length switching
Fig. 5 demonstrates the effects of adding or subtracting two carbons from the tether lengths of the hydroxyl P or NP groups in both solvents. The six different films shown on the plot consider the average ρgroup value of all chains in the films under both water and hexane. We expect that a switchable surface will exhibit ρNP < ρP in water and ρNP > ρP in hexane due to the dominance of the given group with the corresponding solvent. The left half of the figure shows the results of increasing the NP group size from two to six non-hydrogen atoms with the P group length held constant. This results in ρP decreasing in both solvents and ρNP increasing in hexane only. The right half of the figure shows that increasing the P group size with the NP group held constant results in ρNP decreasing in both solvents and ρP increasing in water only. As can be seen from the figure, all the hydroxyl Pn:NPn groups show a greater P exposed surface in water (i.e., the P group solvent density is higher for water) and NP exposed surface in hexane, indicating the hydroxyl terminal groups show favorable switching. This behavior is not seen for carboxyl terminal groups (Fig. S3†), which show that the curves overlap as the NP group length increases, indicating that these films are more likely to be dominated by the large NP group in both solvents. The two poorest performing hydroxyl surfaces, P4:NP2 and P6:NP4, are both under hexane when the P group is two atoms longer than the NP group and ρP converges towards ρNP. Overall, the difference between ρP and ρNP is larger on average for water than for hexane, indicating a greater preference for the hydroxyl P group to be driven upwards to interact with water than for the NP group to form interactions with hexane, which seems reasonable when considering the energetic gains from polar versus non-polar interactions. Finally, the sensitivity in ρgroup to even two atom changes in chemistry, such as from P4:NP4 to P6:NP4 suggests that small chemical differences could impact switchability.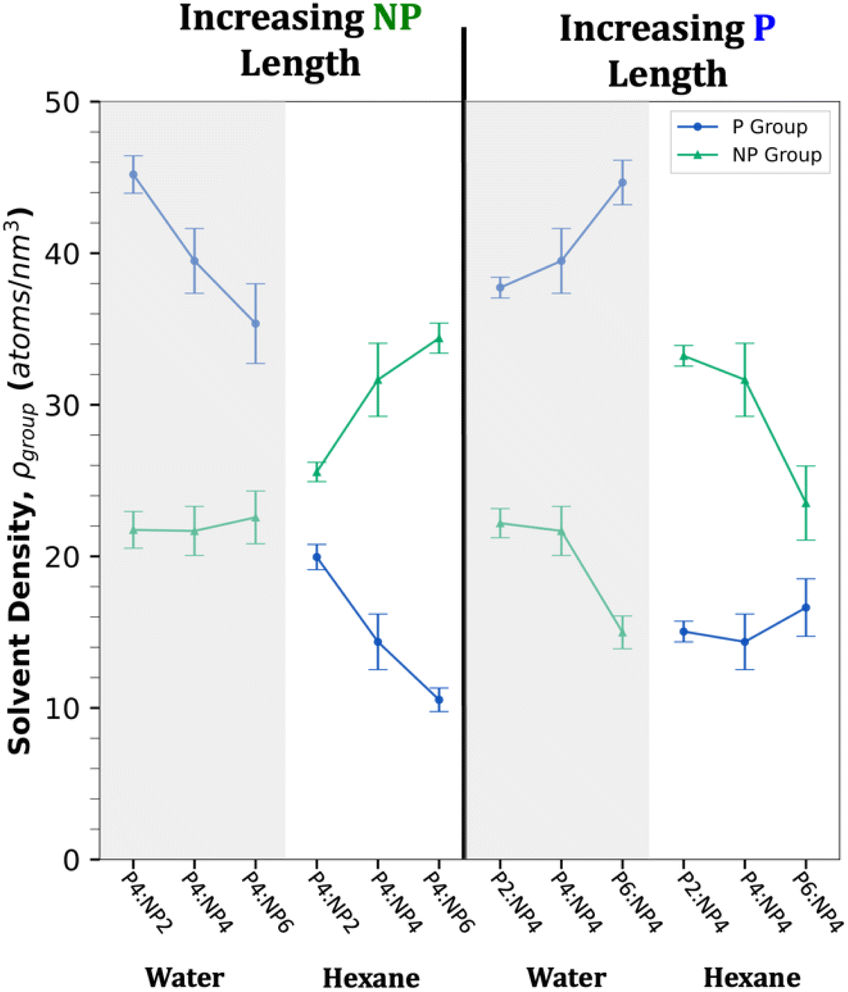 | ||
| Fig. 5 Comparing the effect on ρgroup of changing the relative atomic length of P and NP groups for hydroxyl Pn:NPn systems. The left half is plotted with increasing length of NP group, while the right is plotted with increasing length of P group. The same sets of three systems are compared under water (grey) and hexane (white). Error bars are a 95% confidence interval across six simulated surfaces. | ||
3.5 Chain packing and backfill groups
Turning to the effect of surface packing, we compare in Fig. 6 the carboxyl P4:NP4 terminated film with no backfill chains in the leftmost column to the same film with half the chains as backfill chains (present in all films up to this point) in the center column. These will be respectively referred to as “fully-functionalized” and “half-functionalized” films. The fully-functionalized film shows that the majority of the P groups are not exposed to either water or hexane, indicating that the terminal group is trapped, and minimal switching is observed. This can be compared to the half-functionalized film, which shows a distinct increase in the P group interactions at the water interface. The solvent densities for the fully-functionalized surface are reported in Table S6 in the ESI,† with the difference in surface composition under water (Δρ = ρNP − ρP = 3.9 ± 1.6 atoms per nm3) and under hexane (Δρ = 5.1 ± 0.8 atoms per nm3) appearing statistically the same, as compared to the half-functionalized surface, which measures water Δρ = −4.6 ± 3.3 atoms per nm3 and hexane Δρ = 15.4 ± 2.6 atoms per nm3. The lack of responsiveness in the fully-functionalized films is attributed to the overpacking of the bulky functionalized chains, which have large headgroups dominated by steric repulsions that limit the mobility of the groups, even if an enthalpically more favorable state might exist. The right column of Fig. 6 shows a film that is fully functionalized (no backfills), but with one half the total number of chains (i.e., 2 chains per nm2) packed on the surface, termed “half-packed”, which therefore has the same number of functionalized chains as the half-functionalized film. While the P groups of the half-packed film appear to switch upwards, the solvent clearly penetrates the film, as represented by a broad interfacial regime from 8 to 20 Å. Fig. S4† demonstrates that this effect occurs as well with the NP groups. Because the half-packed film is less densely packed together, the terminal groups can separate and allow other moieties of the chain backbone to dominate the interfacial interactions. The backfill molecules are therefore a necessity to provide a cohesive film that prevents solvent atoms from filling the exposed void between chains.45,59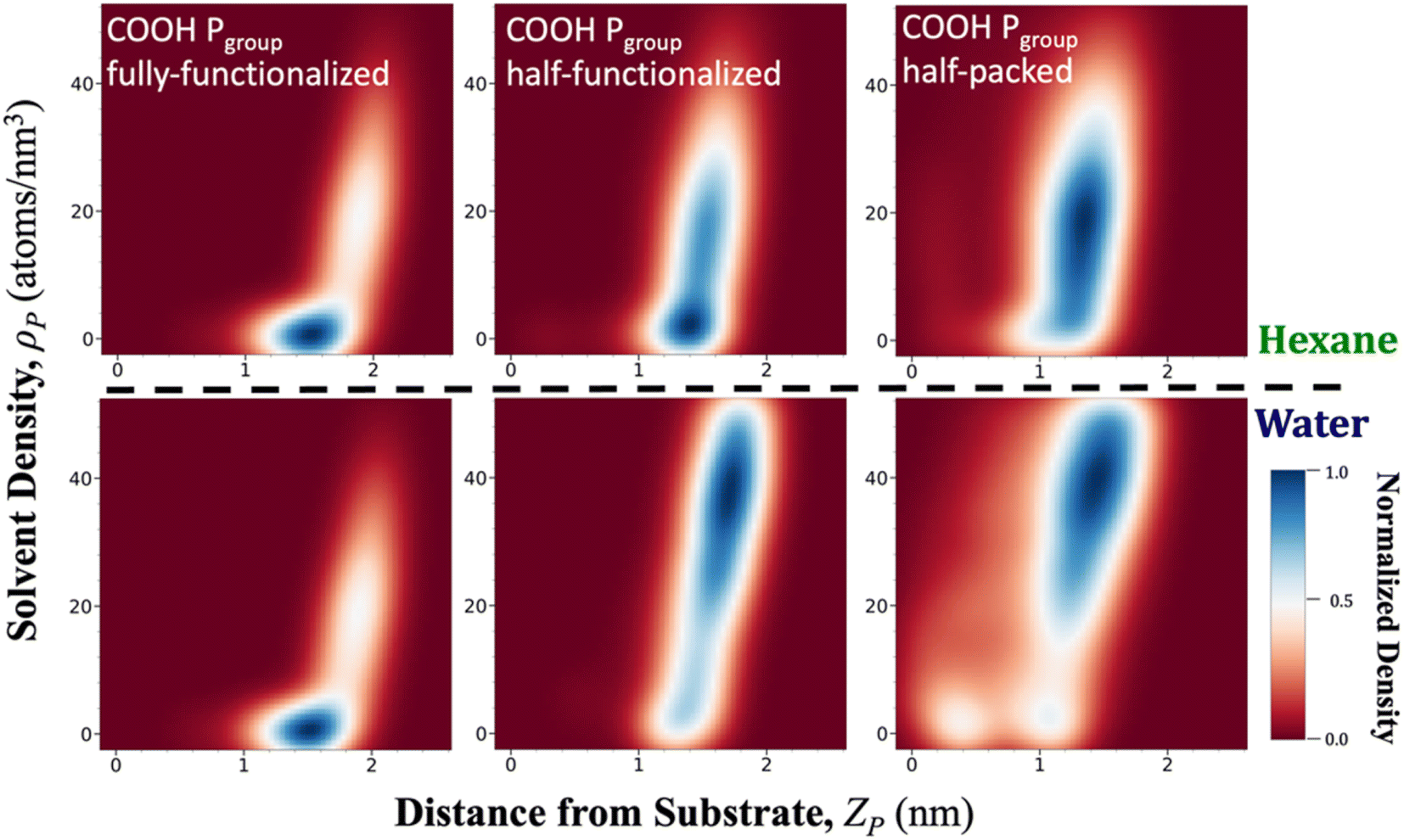 | ||
| Fig. 6 Heatmaps showing the solvent density (ρP) versus distance from the substrate (ZP) for the carboxy P4:NP4 system in hexane (top) and water (bottom). Three variations of the carboxy P4:NP4 surfaces are shown: 4 chains per nm2 density with no backfill chains and only functionalized chains (fully-functionalized, left), 4 chains per nm2 density with half backfill chains and half functionalized chains (half-functionalized, middle), and 2 chains per nm2 density with no backfill chains and only functionalized chains (half-packed, right). The backfill chains are 10-carbon alkylsilane chains in length and randomly placed between the functionalized chains. | ||
4. Discussion
To characterize the responsiveness of the surfaces, the types of surfaces that could result from the switchable terminal groups are first defined in Fig. 7. There are two axes of importance: the magnitude of responsiveness (i.e., the relative change in the interface with the terminal group when the environment is changed) and the individual character of the interface under each environment. The “fixed” class of surfaces are unable to switch, i.e., there is a low responsive nature to the solvent. In this instance, either only one group dominates the interface, or the group contributions are mixed, acting with some intermediate interface energy. For example, the no-backfill surface studied in Fig. 6 and S4† has an NP group dominated interface in both water and hexane, and so would be characterized as a fixed nonpolar-type surface. As previously discussed, these surfaces have low interfacial energy under dispersive environments and high interfacial energy in polar environments.60–62 The “fixed mixed” type surface looks like a heterogeneous film, but with no ability for either group to monopolize the environmental interactions (i.e., the orientation of P and NP groups is unchanged in a polar or dispersive environment). These surfaces have interfacial energies that are generally characterized by a geometric average of the contributing chemistry.63 | ||
| Fig. 7 Schematic defining the reference states (x-axis) of ideal monolayer films in dispersive versus polar type environments (y-axis) as identified by the primary exposed group(s) that dominates the interfacial interactions (indicated by upwards group orientation). The polar (P) and non-polar (NP) chemistries, when exposed, either define the interface, or the surface exists in some heterogeneous state. Fixed surfaces (left) are static for differing environments, but responsive surfaces (right) change their chemical interactions. (☆) indicates a responsive switchable surface, which is primarily polar or non-polar under the corresponding environments. | ||
The responsive type surfaces are shown on the right in Fig. 7. Such surfaces adapt to the contacting solvent by reorienting their switchable groups. This reorientation could favor one of the functional groups and so have a surface that is predominantly non-polar or polar under the aligned environment while under the opposing environment, exhibiting mixed character (i.e., responsive non-polar or responsive polar). This type of switching is demonstrated in the first two columns of Fig. 4 where the carboxyl films do not favor either group under water but are primarily non-polar under hexane, as such they would be categorized as responsive non-polar. Finally, the responsive switchable surface is the ideal case, where the NP chemistry dominates under a dispersive environment, and the P chemistry dominates under a polar environment. The last two columns of Fig. 4 also demonstrate this with the hydroxyl P4:NP4 system favoring the requisite P group under water and NP group under hexane. While the design of surfaces that show symmetrical switching in both environments seems optimal, applications may require the design of a responsive polar or responsive non-polar surface to prioritize the switching in a single environment.
For the classes of switchable coatings examined herein, a metric is developed to classify the surface states under each solvent and allow the type of switching that may occur to be quantified. This metric is calculated from the fraction of the interface considered non-polar or polar in both environments using the weighted ρgroup of each individual chain across the surface. While this metric is not a direct measure of the interfacial energy, it does inform the extent of switching due to the chemistry of the terminal group. Because these groups are designed with P and NP groups, we can reasonably expect that greater interfacial contribution from either group would lead to a likewise polar or non-polar monopolized interface. As such, a %P or %NP value greater than 60% is used to classify a surface as P or NP dominated. Table 1 provides the %P group in water and %NP group in hexane values and classifies the switchability for each terminal group using the six possible states defined in Fig. 7. The ideal films labeled as responsive switchable are mostly composed of the hydroxyl P2 and hydroxyl P4 type chemistry. In fact, the hydroxyl films appear to interact more closely with water than the carboxyl films with a range of values for the interface of 56–75%P in water, as opposed to a range of 46–67%P for the carboxyl systems. The size effect is also clear, just a two-carbon increase in a terminal group length is enough to influence the ability of that group to orient at the surface by up to 20%; i.e., comparing carboxy P6:NP2 to carboxy P6:NP4, the interface goes from 44% to 64%NP in hexane. Finally, all the terminal groups reported here show at least some responsive nature, with the worst performer being the carboxy P6:NP2 surface that is 33%NP under water but 44%NP under hexane, representing just an 11% total change across the solvents. However, whether such responsive structural changes in the surface groups are enough to result in responsive surface properties of these films is still not well understood. To evaluate the validity of these %P and %NP metrics as indicators of a responsive surface, we have compared these values with the contact angle measurements reported in Fig. 2.
| P group | NP group | %P in water | %NP in hexane | Switching type |
|---|---|---|---|---|
| Carboxy-P2 | NP2 | 51 | 62 | Responsive NP |
| Carboxy-P2 | NP4 | 50 | 69 | Responsive NP |
| Carboxy-P2 | NP6 | 46 | 73 | Fixed NP |
| Carboxy-P4 | NP2 | 62 | 56 | Responsive P |
| Carboxy-P4 | NP4 | 54 | 66 | Responsive NP |
| Carboxy-P4 | NP6 | 49 | 67 | Responsive NP |
| Carboxy-P6 | NP2 | 67 | 44 | Fixed P |
| Carboxy-P6 | NP4 | 57 | 64 | Responsive NP |
| Carboxy-P6 | NP6 | 54 | 64 | Responsive NP |
| Hydroxy-P2 | NP2 | 62 | 60 | Responsive switchable |
| Hydroxy-P2 | NP4 | 63 | 69 | Responsive switchable |
| Hydroxy-P2 | NP6 | 60 | 72 | Responsive switchable |
| Hydroxy-P4 | NP2 | 68 | 56 | Responsive P |
| Hydroxy-P4 | NP4 | 65 | 69 | Responsive switchable |
| Hydroxy-P4 | NP6 | 61 | 77 | Responsive switchable |
| Hydroxy-P6 | NP2 | 74 | 50 | Responsive P |
| Hydroxy-P6 | NP4 | 75 | 59 | Responsive P |
| Hydroxy-P6 | NP6 | 56 | 65 | Responsive NP |
 | ||
| Fig. 8 Comparing measures of surface energy derived from experimental contact angles and molecular simulation %P and %NP values. Filled circles (●) indicate surfaces fully functionalized with the indicated terminal groups, open circles (○) indicate surfaces that are half-functionalized with the given multifunctional terminal group, and triangles (▲) are control surfaces functionalized without a switchable group. The experimental values were normalized by γLV for water,64 which is 72.8 mJ m−2. A 95% confidence interval is denoted with error bars. The dashed line denotes a line of best fit with a Spearman's correlation coefficient65 of 0.83. | ||
This comparison can be done using Young's equation,66 which provides the relationship between three phase interfacial tensions for a sessile water drop on a surface, as shown in eqn (1):
| γwLVcosθw = γwSV − γwSL | (1) |
| γαLVcosθα = γαSV − γαSL | (2) |
The two following assumptions allow for a simplified and approximate relationship between the switching metrics and contact angles. The first is that γwSV = γαSV, which reasons from the fact that the state of a given surface outside of the droplet is exposed to air regardless of the droplet itself and would therefore have equivalent energy. Combining eqn (1) and (2) with this assumption gives:
| γwLVcosθw − γαLVcosθα = γαSL − γwSL | (3) |
Second, there is a negative correlation between the switching metrics and γSL for the polar and non-polar liquids. This is assumed due to the predominant intermolecular interactions for water and α-bromonaphthalene, which are polar (and hydrogen bonding) and dispersive, respectively. A surface with higher %P under a polar medium would be generally expected to have a lowered γSL value, and likewise, a surface with higher %NP will have a lowered γSL value when exposed to a non-polar medium, assuming other physical properties of the film are kept consistent.67 Under this stated assumption, an approximate correlation between the related but distinct metrics studied herein (i.e., the simulation metrics that characterize the surface structure changes via the %P or %NP values and the experimental contact angles) is given as:
| γαSL − γwSL ∝ %Pw − %NPhexane | (4) |
This result is plotted for eight surfaces in Fig. 8. These are carboxy P2:NP4 fully-functionalized and half-functionalized, hydroxy P2:NP2 half-functionalized, and hydroxy P2:NP4 half-functionalized. Also included are four controls for simplified comparisons: two pure polar surfaces, carboxy (COOH) and hydroxy (OH), a pure non-polar surface (CH3), and a mixed film with half COOH and half CH3 terminated chains on the surface. The pure CH3 surface can be found in the lower left of the plot. This system has purely non-polar groups, thus %P − %NP is −100%, and has an experimental 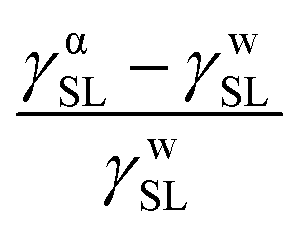 value of −0.65, indicating that the interfacial free energy under water is larger than that under α-bromonaphthalene. On the other hand, a pure polar OH system would again have %P − %NP = 100%, but an experimental difference in interfacial energy of 0.28, indicating that the interfacial tension with α-bromonaphthalene is larger than it is with water. The switchable films and mixed COOH–CH3 surface all range between these two numbers, as they all act partially polar and partially non-polar. The clustering of these values in the middle of the plot results from a canceling out effect: if a surface is more polar in polar conditions and switches to be more non-polar in non-polar conditions, then both %P and %NP will increase, offsetting each other. Likewise, the experimental interfacial tensions will both decrease, resulting in the same cancellation effect. Nominally, these surface energy metrics only capture the trade-off between non-polar and polar behavior, but the correlation (Spearman's correlation coefficient65 is found to be 0.83) across these films between the experimental contact angles and simulation data, as shown in Fig. 8, supports the use of %P and %NP, and therefore ρgroup, as structural parameters that can capture the switching behavior.
value of −0.65, indicating that the interfacial free energy under water is larger than that under α-bromonaphthalene. On the other hand, a pure polar OH system would again have %P − %NP = 100%, but an experimental difference in interfacial energy of 0.28, indicating that the interfacial tension with α-bromonaphthalene is larger than it is with water. The switchable films and mixed COOH–CH3 surface all range between these two numbers, as they all act partially polar and partially non-polar. The clustering of these values in the middle of the plot results from a canceling out effect: if a surface is more polar in polar conditions and switches to be more non-polar in non-polar conditions, then both %P and %NP will increase, offsetting each other. Likewise, the experimental interfacial tensions will both decrease, resulting in the same cancellation effect. Nominally, these surface energy metrics only capture the trade-off between non-polar and polar behavior, but the correlation (Spearman's correlation coefficient65 is found to be 0.83) across these films between the experimental contact angles and simulation data, as shown in Fig. 8, supports the use of %P and %NP, and therefore ρgroup, as structural parameters that can capture the switching behavior.
The extremes in Fig. 8 are set by the pure COOH, OH, and CH3 surfaces, while the three 50% functionalized surfaces with backfills are clustered around (−0.4, 0%). As expected, the three surfaces with the backfill chains are represented similarly in Fig. 8 to a standard heterogeneous two chain film, i.e., the fully-functionalized COOH–CH3 surface. Notably, the carboxyl P4:NP4 fully-functionalized film is offset from these more switchable surfaces for both simulation and experiment, where both demonstrate a more polar behavior. Specifically, from simulations, we report that this surface acts as a fixed polar film with %P = 74% and %NP = 29% and the experiments agree by showing a higher interfacial energy with αBN than with water. Thus while the %P/%NP metric is not able to corroborate/differentiate the switchability determined in simulations, it does indirectly support the observed behavior by providing agreement between the observed polar and nonpolar character from simulation and experiment across the films studied. New experimental techniques are needed to observe the dynamic nanoscale structures of these interfaces in order to achieve direct comparison.
Fig. 8 also indicates some clear sources of discrepancy of the %P and %NP model; the values for %P − %NP are equal for both pure hydroxyl and pure carboxyl films, but there clearly is a difference between these contact angles and more importantly, in the interfacial free energies of these films. Because the model identifies a structural component to the interfacial switching, it considers how much of the P or NP group forms the interface. The model fails to differentiate between P and NP group identities, necessitating consideration of group characteristics on a continuum for accurate weighting and prediction of realistic interfacial energies and switchability. It is important to note that de-protonation could also play a role for the carboxyl systems under water, which was not included in the simulated surfaces. Finally, it is clear that the presence of the backfill allows for more free volume and flexibility of the terminal groups. However, this could also be achieved with small variations of packing density, which is held constant for the simulated surfaces, but could vary during the monolayer formation process, and could also be dependent on the size of the terminal group used.
5. Conclusion
Molecular dynamics simulations were used to identify structural changes in molecular films when exposed to environments of different polarities: water and n-hexane. The 18 terminal groups considered were rationally designed to exhibit a responsive effect due to competing P and NP moieties that are both able to interact with the exposed environment. Following thermodynamic principles, the film interface can undergo structural modifications to form a more stable interface by mimicking the composition of the liquid medium. A metric, ρgroup, is developed that measures the total interactions of the P or NP groups with the different solvent molecules and demonstrates which of the terminal groups may switch more favorably. The groups considered in these films allowed the effects of a tightly packed film structure, competing lengths of the relative ends of the terminal group, and the molecular differences of using hydroxyl or carboxyl type P moieties to be characterized. Excessive packing of terminal groups on the surface was found to hinder physical rearrangements at the interface, which necessitated the use of a secondary backfill chain to provide sufficient free volume for a transition to occur. Another important aspect of the terminal group design was apparent when the P or NP group was much longer (>4 carbons) than its counterpart group and caused the longer group to monopolize the interface in both solvents. This suggests a need to balance the size of the P and NP groups to achieve favorable switching states in both solvents. Finally, the hydroxyl P groups were shown to better switch outwards in response to water, possibly due to reduced interchain hydrogen bonding, as compared to the carboxyl P groups. For the carboxyl P groups, the hydrogen bonding resulted in stable interactions within the film itself, which limited the energetic gains to be realized by substituting these self-interactions with water molecules. These insights are key for the construction of further candidate materials that can exhibit environmentally driven dynamic behavior.The ability to ascertain what plays a role at the molecular scale in the switching process is pivotal for the effective design of responsive surfaces. Molecular methods provide the ability to explore the mechanisms that dictate the interface and understand the precise way a given molecular group contributes to the overall surface energy. Furthermore, by using a computational approach, many functional groups and compositions can be explored to identify key candidates for experimental testing. For environmentally responsive films, the %P and %NP metrics can be obtained from inexpensive simulation methods and used to quantify the relative quantity of P and NP group interactions with a polar or non-polar environment of interest. As a result, the difficulties and the time associated with synthesizing monolayer films with varying compositions for the exploration of possible responsive surfaces can be reduced through computational screening, enabling improved design of future responsive coatings.
Data availability
Data for this article, including Python signac workflows, simulation .gro, .top, and .xtc files, and analysis scripts are available on Zenodo at https://zenodo.org/records/15176320, and the project structure and analysis code can be accessed from GitHub at https://github.com/PTC-CMC/Switchable_Projects.git. The analyzed tables of data used in the generation of the figures for this article have been included as part of the ESI.†Author contributions
Nicholas Craven: data curation, formal analysis, investigation, methodology, software, validation, visualization, writing – original draft, writing – review and editing, Allison Cordova-Huaman: formal analysis, investigation, methodology, visualization, writing – review and editing, Chris Iacovella: conceptualization, methodology, resources, project administration, funding acquisition, supervision, writing – review and editing, G. Kane Jennings: conceptualization, project administration, resources, funding acquisition, supervision, writing – review and editing, Clare McCabe: conceptualization, project administration, resources, funding acquisition, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the contributors to the MoSDeF project, which provided the open-source tools necessary for this work. This work was funded through the National Science Foundation under Grant No. CBET-2052438.Notes and references
- T. Mouterde, A. Keerthi, A. R. Poggioli, S. A. Dar, A. Siria, A. K. Geim, L. Bocquet and B. Radha, Molecular streaming and its voltage control in ångström scale channels, Nature, 2019, 567, 87–90 CrossRef CAS PubMed.
- Z. Yi, X. Wang, W. Li, X. Qin, Y. Li, K. Wang, Y. Guo, X. Li, W. Zhang and Z. Wang, Interfacial friction at action: Interactions, regulation, and applications, Friction, 2023, 11, 2153–2180 CrossRef.
- C. Cottin-Bizonne, J. L. Barrat, L. Bocquet and E. Charlaix, Low-friction flows of liquid at nanopatterned interfaces, Nat. Mater., 2003, 2, 237–240 CrossRef CAS PubMed.
- S. B. Ramisetti and A. Yadav, Insights from molecular simulations on liquid slip over nanostructured surfaces, J. Mol. Model., 2022, 28, 1–13 CrossRef PubMed.
- B. Bhushan, M. Cichomski, E. Hoque, J. A. Derose, P. Hoffmann and H. J. Mathieu, Nanotribological characterization of perfluoroalkylphosphonate self-assembled monolayers deposited on aluminum-coated silicon substrates, Microsyst. Technol., 2006, 12, 588–596 CrossRef CAS.
- M. Cichomski, M. Prowizor, D. A. Kowalczyk, A. Sikora, D. Batory and M. Dudek, Comparison of the Physicochemical Properties of Carboxylic and Phosphonic Acid Self-Assembled Monolayers Created on a Ti-6Al-4V Substrate, Materials, 2020, 13, 5137 CrossRef CAS PubMed.
- N. A. Dudukovic, E. J. Fong, H. B. Gemeda, J. R. DeOtte, M. R. Cerón, B. D. Moran, J. T. Davis, S. E. Baker and E. B. Duoss, Cellular fluidics, Nature, 2021, 595, 58–65 CrossRef CAS PubMed.
- P. Lin, T. L. Chuang, P. Z. Chen, C. W. Lin and F. X. Gu, Low-Fouling Characteristics of Ultrathin Zwitterionic Cysteine SAMs, Langmuir, 2019, 35, 1756–1767 CrossRef CAS PubMed.
- J. Yang, H. Chen, S. Xiao, M. Shen, F. Chen, P. Fan, M. Zhong and J. Zheng, Salt-Responsive Zwitterionic Polymer Brushes with Tunable Friction and Antifouling Properties, Langmuir, 2015, 31, 9125–9133 CrossRef CAS PubMed.
- C. Blaszykowski, S. Sheikh and M. Thompson, Surface chemistry to minimize fouling from blood-based fluids, Chem. Soc. Rev., 2012, 41, 5599–5612 RSC.
- C. Boo, S. Hong and M. Elimelech, Relating Organic Fouling in Membrane Distillation to Intermolecular Adhesion Forces and Interfacial Surface Energies, Environ. Sci. Technol., 2018, 52, 14198–14207 CrossRef CAS PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Antifouling Coatings: Recent Developments in the Design of Surfaces That Prevent Fouling by Proteins, Bacteria, and Marine Organisms, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- M. Thomas, B. Corry and T. A. Hilder, What Have We Learnt About the Mechanisms of Rapid Water Transport, Ion Rejection and Selectivity in Nanopores from Molecular Simulation?, Small, 2014, 10, 1453–1465 CrossRef CAS PubMed.
- C. I. Lynch, S. Rao and M. S. P. Sansom, Water in Nanopores and Biological Channels: A Molecular Simulation Perspective, Chem. Rev., 2020, 120(18), 10298–10335 CrossRef CAS PubMed.
- A. L. Ahmad, A. A. Abdulkarim, B. S. Ooi and S. Ismail, Recent development in additives modifications of polyethersulfone membrane for flux enhancement, Chem. Eng. J., 2013, 223, 246–267 CrossRef CAS.
- W. R. Bowen, A. W. Mohammad and N. Hilai, Characterisation of Nanofiltration Membranes for Predictive Purposes-Use of Salts, Uncharged Solutes and Atomic Force Microscopy, J. Membr. Sci., 1997, 126(1), 91–105 CrossRef CAS.
- A. Deshmukh, C. Boo, V. Karanikola, S. Lin, A. P. Straub, T. Tong, D. M. Warsinger and M. Elimelech, Membrane distillation at the water-energy nexus: limits, opportunities, and challenges, Energy Environ. Sci., 2018, 11, 1177–1196 RSC.
- Y. Wan, Z. Yang, G. Zhang and Y. Wang, Peristome-Mimetic Surfaces Fabricated by Nanosecond Laser for Unidirectional Liquid Spreading, Adv. Eng. Mater., 2024, 26, 2302240 CrossRef.
- D. Bai, Z. Ibrahim and G. K. Jennings, pH-Responsive Random Copolymer Films with Amine Side Chains, J. Phys. Chem. C, 2007, 111, 461–466 CrossRef CAS.
- H. Nandivada, A. M. Ross and J. Lahann, Stimuli-responsive monolayers for biotechnology, Prog. Polym. Sci., 2010, 35, 141–154 CrossRef CAS.
- H. M. Zareie, C. Boyer, V. Bulmus, E. Nateghi and T. P. Davis, Temperature-responsive self-assembled monolayers of oligo(ethylene glycol): control of biomolecular recognition, ACS Nano, 2008, 2, 757–765 CrossRef CAS PubMed.
- F. Guo and Z. Guo, Temperature-responsive self-assembled monolayers of oligo(ethylene glycol): control of biomolecular recognition, RSC Adv., 2016, 6, 36623–36641 RSC.
- B. C. Bunker, Reversible switching of interfacial interactions, Mater. Sci. Eng., R, 2008, 62, 157–173 CrossRef.
- X. Bai, X. Gou, J. Zhang, J. Liang, L. Yang, S. Wang, X. Hou and F. Chen, A Review of Smart Superwetting Surfaces Based on Shape-Memory Micro/Nanostructures, Small, 2023, 19, 2206463 CrossRef CAS PubMed.
- M. Liu, B. Jin and M. Liu, Integrating the Stimuli-Responsiveness of Microparticles via Matrix Embedding for Smart Soft Materials with Customized Switchable Properties, Adv. Funct. Mater., 2024, 34, 2404803 CrossRef CAS.
- Y.-j. Lee, Y.-j. Jung and Y.-b. Lim, Adaptable Self-Assembly of a PEG Dendrimer-Coiled Coil Conjugate, ChemPlusChem, 2024, 89, e202400114 CrossRef CAS PubMed.
- X. Fu, L. Hosta-Rigau, R. Chandrawati and J. Cui, Multi-Stimuli-Responsive Polymer Particles, Films, and Hydrogels for Drug Delivery, Chem, 2018, 4, 2084–2107 CAS.
- Z. Ma, Y. Liu, K. Feng, J. Wei, J. Liu, Y. Wu, X. Pei, B. Yu, M. Cai and F. Zhou, "Brush-like” Amphiphilic Polymer for Environmental Adaptive Coating, ACS Appl. Mater. Interfaces, 2022, 14, 18901–18909 CrossRef CAS PubMed.
- Y. Xu, C. M. Thurber, T. P. Lodge and M. A. Hillmyer, Synthesis and Remarkable Efficacy of Model Polyethylene-graft-poly(methyl methacrylate) Copolymers as Compatibilizers in Polyethylene/Poly(methyl methacrylate) Blends, Macromolecules, 2012, 45, 9604–9610 CrossRef CAS.
- X. Zhou, S. Yu, J. Zang, Z. Lv, E. Liu and Y. Zhao, Colorful nanostructured TiO2 film with superhydrophobic–superhydrophilic switchable wettability and anti-fouling property, J. Alloys Compd., 2019, 798, 257–266 CrossRef CAS.
- S. Minko, M. Müller, M. Motornov, M. Nitschke, K. Grundke and M. Stamm, Two-Level Structured Self-Adaptive Surfaces with Reversibly Tunable Properties, J. Am. Chem. Soc., 2003, 125, 3896–3900 CrossRef CAS PubMed.
- P. E. Laibinis, C. D. Bain, R. G. Nuzzo and G. M. Whitesides, Structure and wetting properties of ω-alkoxy-n-alkanethiolate monolayers on gold and silver, J. Phys. Chem., 1995, 99, 7663–7676 CrossRef CAS.
- C. D. Bain and G. M. Whitesides, Depth sensitivity of wetting: Monolayers of ω-mercapto ethers on gold, J. Am. Chem. Soc., 1988, 110, 5897–5898 CrossRef CAS.
- F. M. Fowkes, Attractive forces at interfaces, Ind. Eng. Chem., 1964, 56, 40–52 CrossRef CAS.
- D. K. Owens and R. C. Wendt, Estimation of the surface free energy of polymers, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- A. G. Ilgen, E. Borguet, F. M. Geiger, J. M. Gibbs, V. H. Grassian, Y. S. Jun, N. Kabengi and J. D. Kubicki, Bridging molecular-scale interfacial science with continuum-scale models, Nat. Commun., 2024, 15, 1–14 Search PubMed.
- A. Z. Summers, C. R. Iacovella, P. T. Cummings and C. Mcabe, Investigating Alkylsilane Monolayer Tribology at a Single-Asperity Contact with Molecular Dynamics Simulation, Langmuir, 2017, 33, 11270–11280 CrossRef CAS PubMed.
- K. Kojio, A. Takahara, K. Omote and T. Kajiyama, Physisorption, Interfacial Films, Membranes-Molecular Aggregation State of n-Octadecyltrichlorosilane Monolayers Prepared by the Langmuir and Chemisorption Methods, Langmuir, 2000, 16, 3932–3936 CrossRef CAS.
- C. Klein, J. Sallai, T. J. Jones, C. R. Iacovella, C. McCabe and P. T. Cummings, A Hierarchical, Component Based Approach to Screening Properties of Soft Matter, Foundations of Molecular Modeling and Simulation: Select Papers from FOMMS 2015, ed. R. Q. Snurr, C. S. Adjiman and D. A. Kofke, Singapore, 2016, pp. 79–92 Search PubMed.
- A. Z. Summers, J. B. Gilmer, C. R. Iacovella, P. T. Cummings and C. McCabe, MoSDeF, a Python Framework Enabling Large-Scale Computational Screening of Soft Matter: Application to Chemistry-Property Relationships in Lubricating Monolayer Films, J. Chem. Theory Comput., 2020, 16, 1779–1793 CrossRef CAS PubMed.
- J. E. Black, C. R. Iacovella, P. T. Cummings and C. McCabe, Molecular Dynamics Study of Alkylsilane Monolayers on Realistic Amorphous Silica Surfaces, Langmuir, 2015, 31, 3086–3093 CrossRef CAS PubMed.
- R. Hartkamp, B. Siboulet, J. F. Dufrêche and B. Coasne, Ion-specific adsorption and electroosmosis in charged amorphous porous silica, Phys. Chem. Chem. Phys., 2015, 17, 24683–24695 RSC.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- C. Quach, J. Gilmer, D. Pert, A. Mason-Hogans, C. Iacovella, P. Cummings and C. McCabe, High-throughput screening of tribological properties of monolayer films using molecular dynamics and machine learning, J. Chem. Phys., 2022, 156, 15 CrossRef PubMed.
- J. E. Black, A. Z. Summers, C. R. Iacovella, P. T. Cummings and C. McCabe, Investigation of the Impact of Cross-Polymerization on the Structural and Frictional Properties of Alkylsilane Monolayers Using Molecular Simulation, Nanomaterials, 2019, 9, 639 CrossRef CAS PubMed.
- C. Klein, A. Z. Summers, M. W. Thompson, J. B. Gilmer, C. McCabe, P. T. Cummings, J. Sallai and C. R. Iacovella, Formalizing atom-typing and the dissemination of force fields with foyer, Comput. Mater. Sci., 2019, 167, 215–227 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 14101 CrossRef PubMed.
- R. T. McGibbon, K. A. Beauchamp, M. P. Harrigan, C. Klein, J. M. Swails, C. X. Hernández, C. R. Schwantes, L. P. Wang, T. J. Lane and V. S. Pande, MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories, Biophys. J., 2015, 109, 1528–1532 CrossRef CAS PubMed.
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, J. Domański, D. L. Dotson, S. Buchoux, I. M. Kenney and O. Beckstein, MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulation, Conference: proceeding of the 15th python in science conference, 2016, pp. 98–105 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, MDAnalysis: A toolkit for the analysis of molecular dynamics simulations, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- A. Shrake and J. A. Rupley, Environment and exposure to solvent of protein atoms. Lysozyme and insulin, J. Mol. Biol., 1973, 79, 351–371 CrossRef CAS PubMed.
- P. Smith, R. M. Ziolek, E. Gazzarrini, D. M. Owen and C. D. Lorenz, On the interaction of hyaluronic acid with synovial fluid lipid membranes, Phys. Chem. Chem. Phys., 2019, 21, 9845–9857 RSC.
- R. Layouni, M. Dubrovsky, M. Bao, H. Chung, K. Du, S. V. Boriskina, S. M. Weiss and D. Vermeulen, High contrast cleavage detection for enhancing porous silicon sensor sensitivity, Opt. Express, 2021, 29, 1 CrossRef CAS PubMed.
- P. Silberzan, L. Léger, D. Ausserré and J. J. Benattar, High contrast cleavage detection for enhancing porous silicon sensor sensitivity, Langmuir, 1991, 7, 1647–1651 CrossRef CAS.
- S. R. Wasserman, Y. T. Tao and G. M. Whitesides, Structure and Reactivity of Alkylsiloxane Monolayers Formed by Reaction of Alkyltrichlorosilanes on Silicon Substrat, Langmuir, 1989, 5, 1074–1087 CrossRef CAS.
- B. D. Booth, N. J. Martin, E. A. Buehler, C. McCabe and G. K. Jennings, Tribological Characterization of Gradient Monolayer Films from Trichlorosilanes on Silicon, Colloids Surf., A, 2012, 412, 57–63 CrossRef CAS.
- Y. Arima and H. Iwata, Effect of wettability and surface functional groups on protein adsorption and cell adhesion using well-defined mixed self-assembled monolayers, Biomaterials, 2007, 28, 3074–3082 CrossRef CAS PubMed.
- L. H. Dubois and R. G. Nuzzo, Synthesis, structure, and properties of model organic surfaces, Annu. Rev. Phys. Chem., 1992, 43, 437–440 CrossRef CAS.
- R. Colorado and T. R. Lee, Wettabilities of self-assembled monolayers on gold generated from progressively fluorinated alkanethiols, Langmuir, 2003, 19, 3288–3296 CrossRef CAS.
- C. D. Bain and G. M. Whitesides, A Study by Contact Angle of the Acid-Base Behavior of Monolayers Containing ω-Mercaptocarboxylic Acids Adsorbed on Gold: An Example of Reactive Spreading, Langmuir, 1989, 5, 1370–1378 CrossRef CAS.
- N. B. Vargaftik, B. N. Volkov and L. D. Voljak, International Tables of the Surface Tension of Water, J. Phys. Chem. Ref. Data, 1983, 12, 817–820 CrossRef CAS.
- C. Spearman, The Proof and Measurement of Association between Two Things, Am. J. Psychol., 1904, 15, 72 CrossRef.
- T. Young, III. An essay on the cohesion of fluids, Philos. Trans. R. Soc., 1805, 95, 65–87 CrossRef.
- X. Zhang, F. Shi, J. Niu, Y. Jiang and Z. Wang, Superhydrophobic surfaces: from structural control to functional application, J. Mater. Chem., 2008, 18, 621–633 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00366k |
| ‡ Present Address: Memorial Sloan Kettering Cancer Center, 1275 York Avenue, New York, NY 10065, USA. |
| This journal is © The Royal Society of Chemistry 2025 |