
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient ethane production via SnCl4 Lewis acid-enhanced CO2 electroreduction in a flow cell electrolyser†
Sankeerthana
Bellamkonda
 *ab,
Ian
Brewis
a,
Venkateswara Rao
Gedela
a,
Rana Faisal
Shahzad
a,
Mohamed
Mamlouk
b and
Shahid
Rasul
*a
*ab,
Ian
Brewis
a,
Venkateswara Rao
Gedela
a,
Rana Faisal
Shahzad
a,
Mohamed
Mamlouk
b and
Shahid
Rasul
*a
aSchool of Engineering and Environment, Northumbria University, Newcastle upon Tyne, UK. E-mail: sankeerthana999@gmail.com; shahid.rasul@northumbria.ac.uk; Tel: +44 7471042866 Tel: +91 9445435743
bSchool of Engineering, Newcastle University, Newcastle upon Tyne, UK
First published on 12th March 2025
Abstract
The development of efficient and selective catalysts for electrochemical CO2 reduction (ECR) is critical for advancing sustainable energy solutions. Here, we report a unique catalyst system based on SnCl4 Lewis acid-modified Cu2O, demonstrating enhanced performance in CO2 electroreduction to ethane. The SnCl4 modification introduces chloride ions directly onto the Cu2O surface, creating a synergistic interaction between Sn, Cl, and Cu active sites that optimizes the electronic environment for ECR. The SnCl4 catalyst was deposited on Cu2O coated gas diffusion electrode (GDE) and tested in a flow cell electrolyser, integrating a Fumasep bipolar membrane and platinum (Pt) foil anode. This system achieved a peak faradaic efficiency of 34.8% for ethane production at −1.0 V vs. RHE, along with 11.3% efficiency for ethylene. Electrochemical studies revealed that the SnCl4-modified Cu2O exhibits low charge transfer resistance and high stability during prolonged electrolysis, achieving a total current density of 74.8 mA cm−2 with a Tafel slope of 92.3 mV dec−1 at 0.4 V overpotential. Mechanistic investigations, supported by density functional theory, Raman, XRD, and electrochemical impedance spectroscopy analyses, highlight the critical role of chloride ions in stabilizing CO intermediates and facilitating C–C bond formation, essential for C2 product generation. Operating in a flow cell configuration, the system demonstrated high energy efficiency and selectivity, establishing the SnCl4-modified Cu2O (CTC) as a promising catalyst for ECR. These findings offer a scalable and economically viable pathway for renewable hydrocarbon production, paving the way for practical applications in carbon-neutral energy cycles.
1. Introduction
Concerns about global warming and a potential energy crisis have driven the search for innovative methods to convert CO2, a prominent greenhouse gas emitted by fossil fuel combustion, back into fuels or other valuable molecules.1 Reducing CO2 emissions is critical for addressing the challenges of climate change. Carbon capture, utilization, and storage (CCUS) technologies are gaining traction to achieve net-zero emissions by converting CO2 into useful products.2–4 CO2 electrolysis is particularly promising, using renewable energy to turn CO2 into chemicals and fuels, offering a sustainable route to carbon neutrality. This approach has the potential to significantly reduce emissions and facilitate the transition to a greener future.5 A key strategy for achieving a carbon-neutral energy cycle involves powering electrochemical CO2 reduction (ECR) using renewable energy sources.6 However, ECR faces significant energy barriers due to the strong chemical inertness of CO2 molecules and the complex multi-electron transfer processes involved.7–9 ECR can yield various C1 products, including CO and CH4, as well as C2 products such as ethylene (C2H4), ethanol (C2H5OH), and ethane (C2H6).10,11 Ethane, in particular, stands out due to its high energy density of approximately 1.43 MJ mol−1, making it a highly desirable product for energy storage and other applications.12,13However, achieving high selectivity for ethane in electrochemical CO2 reduction is challenging, primarily due to the intricate control required for C–C bond formation and subsequent hydrogenation processes. The selective production of ethane is particularly difficult because it requires not only the formation of C–C bonds but also precise control over the hydrogenation steps to avoid over-reduction to methane or under-reduction, leading to less valuable products.14,15 Therefore, the development of high-performance electrocatalysts that can steer the reaction pathways towards ethane while minimising the production of undesired byproducts is crucial. Despite significant advancements,16–19 directing reaction pathways toward desirable products like ethane and reducing reaction potentials remain substantial challenges, necessitating the development of advanced electrocatalysts with improved product selectivity and catalytic activity.
One established strategy for enhancing ECR performance is modifying catalyst surfaces with functional additives.19 This can be achieved either by directly altering the electronic properties of the catalyst surface or indirectly by affecting binding strengths through adsorbate–adsorbate interactions. For example, Kim et al.20 demonstrated that the CO selectivity and reduced overpotential of Ag nanoparticles on carbon substrates could be enhanced using different anchoring agents. They suggested that the improved CO selectivity was due to the localized unpaired electrons of the anchoring agent at the surface state of Ag. Additionally, Au surfaces functionalized with imidazolium ions and thiol-tethered ligands have been investigated for their ability to control the selectivity of ECR products.21
Anion adsorption on metal catalysts has also been shown to improve ECR performance. Numerous studies have demonstrated that the presence of chloride anions enhances CO selectivity while suppressing the hydrogen evolution reaction (HER).21–23 Jihun Oh revealed that chloride-adsorbed Au electrodes exhibited four times the CO selectivity of bare Au at −0.39 V.24 Their studies, both theoretical and experimental, elucidated the role of adsorbed chloride on the Au surface in influencing ECR activity and selectivity. Similarly, adsorbed chloride on Ag surfaces has been shown to boost ECR intrinsic activity and selectivity by minimising side reactions associated with the HER.25 Zn-based catalysts26 have also been extensively investigated in chloride anion-containing electrolytes. It has been hypothesized that the increased CO faradaic efficiency results from chloride anion adsorption on the electrode surface, which preferentially facilitates electron transfer from CO2 to intermediates,26 thereby enhancing CO generation selectivity. For instance, a chloride anion-linked Ag nanocoral structure exhibited 32 times greater ECR activity compared to pure Ag.27 While halide adsorption on Cu-based catalysts has been explored, the role of chloride ions in tuning ECR pathways for selective ethane formation remains underexplored, particularly when introduced via direct catalyst modification rather than electrolyte-based approaches.
To address this gap, this study employs a novel surface modification strategy using SnCl4 as a Lewis acid to enhance the ECR performance of Cu2O. Unlike previous studies that introduce halide-based salts into the electrolyte to investigate halide adsorption effects,28–31 this work directly anchors SnCl4 onto the electrode surface rather than relying on dissolved Cl− ions. This approach ensures controlled and localized chloride adsorption, avoiding fluctuations in Cl− concentration associated with electrolyte-based methods, which can lead to uncontrolled adsorption–desorption behavior, unpredictable competitive interactions with reaction intermediates, and potential electrode degradation. By modifying Cu2O with SnCl4, chloride ions are directly bound to the catalyst surface, providing a stable and reproducible environment for systematically investigating their role in CO2 reduction. This method allows for a more precise understanding of the impact of chloride species on catalytic performance, particularly in steering the reaction pathway toward ethane formation.
In this study, we systematically investigate the role of chloride ions on Cu–Sn electrodes (copper tin chloride, CTC) for ECR. First, we experimentally evaluate the influence of adsorbed chloride by functionalizing the Cu2O surface via SnCl4 deposition and analyse its effect on ECR activity and product selectivity. Special attention is given to ethane formation, examining how chloride anions influence C–C bond formation and hydrogenation pathways. We then propose an ECR mechanism highlighting the significance of chloride ions in selectively driving CO2 reduction toward hydrocarbons, particularly enhancing ethane production. The findings from this work provide key insights into catalyst design strategies for optimizing electrochemical CO2 reduction to C2 hydrocarbons, offering a scalable pathway for sustainable fuel synthesis.
2. Experimental section
2.1 Materials
Cuprous oxide (Cu2O, Merck), a carbon gas diffusion layer (Fuel Cell Systems), and tin(IV) chloride pentahydrate (Merck) were used as precursors for the Sn and Cl deposition on the Cu2O-coated gas diffusion electrode. Potassium hydroxide (KOH, Merck) was used to prepare the electrolytes. Deionized water was used to prepare all solutions and to rinse the samples and glassware.2.2 Electrode fabrication
2.3 Electrode characterization
Scanning electron microscopy (SEM, Hitachi S-4800) was used to examine the morphologies of the synthesized catalysts. The elemental composition and mapping of samples were analysed with energy-dispersive X-ray spectroscopy (EDS, Oxford Instruments). X-ray diffraction (XRD) characterization was performed using a Rigaku Miniflex 600 benchtop sixth-generation X-ray diffractometer. The X-ray photoelectron spectroscopy (XPS) spectra were recorded using an ESCA+ (Omicron Nanotechnology, Oxford Instrument, Germany) equipped with a monochromatic aluminum source (Al Kα radiation ℏν = 1486.6 eV), operated at 15 kV and 20 mA. The C 1s binding energy of 284.6 eV is taken as a reference. The Raman measurements were obtained at room temperature using a BRUKER RFS 27 with a 627 nm laser source.2.4 Electrochemical measurements
The electrochemical measurements were performed using an Autolab potentiostat/galvanostat (Metrohm Autolab PGSTAT302N). Ag/AgCl and Pt foil were used as the reference and counter electrodes, respectively. The reference electrode was converted to RHE using the following equation:| ERHE = EAg/AgCl + 0.1976 + 0.0591 × pH |
We investigated the ECR catalysts using a lab-customised 3D-printed electrolyser flow cell, as shown in Fig. S2 in the ESI.† The catalysts were deposited on a carbon paper GDL substrate, with Ag/AgCl and Pt foil serving as the reference and counter electrodes, respectively. CO2 gas was introduced into the gas chamber via a gas inlet and diffused across the GDL to reach the catalyst layer. The CO2 gas flow rate was maintained at a constant 50 mL min−1 using a flow meter (Cole-Parmer TMR1-010462). A Fumasep FBM bipolar membrane was used between the cathodic and anodic compartments to allow the transfer of cations. Platinum foil was used as the anode. After 30 minutes of CO2 electrolysis, the gas products were collected from the gas outlet for GC measurements, and the catholyte was collected for liquid product analysis using ion chromatography. To investigate the ECR behaviour of CTC-65 electrodes, we conducted chronoamperometry (CA) tests for 1 hour at potentials of −0.5 V, −0.65 V, −0.75 V, −0.85 V, and −1.00 V vs. RHE. The faradaic efficiency (FE) of the electrochemical reaction was estimated from the CA measurements by considering the input charge, the duration of the electrochemical process, and the quantification of the gaseous/liquid products and their molar masses.
2.5 CO2 reduction measurements and product analyses
In the CO2 recycling tests, a lab-customized electrochemical cell with three compartments (for the working, reference, and counter electrodes) was used. Each cathodic and anodic compartment contained 6 mL of 1 M KOH electrolyte. The CO2 reduction reaction was performed at various potentials in CO2-saturated 1 M KOH (pH = 13.8, measured with an Accumet AB15 pH meter) for 60 minutes, with the electrolyte remaining static (no peristaltic pump was used). After electrolysis, a 1 mL gas sample was taken through a septum port using a gas-tight syringe and injected into a gas chromatograph (GC) (Shimadzu 2014) to quantify the amounts of CO, H2, and hydrocarbons. Ion chromatography (Thermo Fisher Dionex ICS-1600) was used to analyse the liquid products.The FE was calculated after quantifying the formate as follows:
| FE = (2 × F × n)/Q |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), n is the concentration of formate (mol), and Q is the total charge during the reaction time. The gaseous products were quantified using a gas chromatograph (Shimadzu 2014) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID) with a methanizer. The TCD was used to detect hydrogen (H2), while the FID was used to detect carbon monoxide (CO) and other carbon-based gaseous products. Argon gas was used as the carrier gas. Prior to the CO2 reduction studies, the GC was calibrated with known concentrations of both CO, CH4, C2H4, C2H6 and H2. After quantification of the gaseous products, the faradaic efficiency was calculated using the following equation:32
485 C mol−1), n is the concentration of formate (mol), and Q is the total charge during the reaction time. The gaseous products were quantified using a gas chromatograph (Shimadzu 2014) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID) with a methanizer. The TCD was used to detect hydrogen (H2), while the FID was used to detect carbon monoxide (CO) and other carbon-based gaseous products. Argon gas was used as the carrier gas. Prior to the CO2 reduction studies, the GC was calibrated with known concentrations of both CO, CH4, C2H4, C2H6 and H2. After quantification of the gaseous products, the faradaic efficiency was calculated using the following equation:32| FE (g) = (n × x × F × v × P)/(R × T × I) |
485 C mol−1), θflowrate = volumetric flow rate, P = 101325 Pa, R = gas constant (8.314 J mol−1 K−1), T = 298 K, and I = average current density after reaching a steady state (A).
2.6 Computational methods
First principles, plane wave self-consistent field calculations were executed using the generalized gradient approximation (GGA) method using QUANTUM ESPRESSO.33a The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional with projector-augmented wave type pseudopotentials was used to treat electron core interactions in all the atoms. A kinetic energy and charge density cut-off of 30 Ry and 300 Ry, respectively, were applied for better convergence. The slab models for Cu contained four atomic layers with the bottom two layers fixed, while the slab models for CTC contained 1 Sn atom and 2 Cl atoms on the surface of 5 nuclear layers of Cu with the bottom two layers fixed. In the aperiodic directions, at least 15 Å vacuum space was added to each model to avoid interactions between neighbouring images. The structural coordinates of the relaxed Cu, CTC, CTC–CO and CTC–CO–K slabs are given in the ESI. Complete structural relaxation with a 6 × 6 × 1 k-mesh was performed until the total energy was converged to 10−5 eV and the residual forces on all unconstrained atoms were less than 0.02 eV Å−1. The conducting nature of the system was incorporated through the tetrahedron method in calculating the electronic properties.33b The binding energy of CO and K was computed as the difference between the energy of the composite system and the sum of the energies of the clean surface and uncoordinated adsorbate.3. Results and discussion
3.1 Electrochemical CO2 reduction in an electrolyser flow cell
We analysed loading-dependent ECR performance by drop-casting various amounts (20–80 μL) of SnCl4·5H2O Lewis acid onto Cu2O GD electrodes, and the electrodes are denoted as CTC-20, CTC-42, CTC-65, and CTC-80. The electrodes are named based on the volume of SnCl4 deposited onto Cu2O GDEs. Briefly, the electrochemical performance of the CTC electrodes is determined by bubbling CO2 into the cathode compartment electrolyte (catholyte) at a flow rate of 50 mL min−1, and a constant potential was applied to the SnCl4/Cu2O cathodes with a potentiostat. The effluent gas stream was collected, and the reaction products were analysed every 30 min with gas and ion chromatography.The distribution of the gaseous products obtained during the CO2 electrochemical reduction at cathodic potentials ranging from −0.5 to −1.0 V vs. RHE is shown in Fig. 1. It is evident that only minor differences exist in the faradaic efficiency (FE) of the gaseous products between the electrodes deposited with different loadings of SnCl4. Carbon monoxide (CO), methane (CH4), ethylene (C2H4) and some ethane (C2H6) were gaseous products observed from CO2 electrochemical reduction. CO desorbs from the surface and is a well-known intermediate for the formation of C2H6, C2H4 and CH4. Ethane is the major product among the hydrocarbons for CTC-65 and CTC-80 electrodes with a small variation in the ethane/methane ratio observed. Ethane formation was detected with low FE for CTC-65 investigated at −0.75 V vs. RHE. Fig. 1A demonstrates that as the CTC-65 electrode potential becomes more negative, the FE for C2H4 and C2H6 increases, while the efficiencies for CH4, formic acid (HCOOH), and CO decline or remain relatively constant. Fig. 1B and C further illustrate the decreasing selectivity for CO and HCOOH, respectively, at more negative potentials, emphasizing that lower SnCl4 loading on the Cu2O GDE led to a more pronounced decrease in selectivity. Fig. 1D shows the declining trend of CH4 FE selectivity as the potential becomes more negative, specifically at higher loadings of SnCl4 Lewis acid. In contrast, Fig. 1E demonstrates that C2H4 selectivity increases with more negative potentials, particularly at a SnCl4 loading volume of 65 μL. Finally, Fig. 1F illustrates a decline in ethane selectivity with more negative potentials for both reagent volumes, although selectivity is higher at a lower volume (65 μL). Overall, the optimal loading of SnCl4 for the electrochemical reduction of CO2 is 65 μL, which serves as the basis for the exploration of the CTC-65 GDE. In contrast, the bare Cu2O GDE primarily produces C1 products such as CO and CH4, and the Cu3Sn (Sn deposited Cu2O) GDE shows improved selectivity for HCOOH and CO due to the presence of Sn, which favours formate formation. The FEs of bare Cu2O and Cu3Sn GDEs are given in Fig. S3.†
 | ||
| Fig. 1 Faradaic efficiency of different products (C2H4, C2H6, HCOOH, CH4, and CO) as a function of potential for the electrode CTC-65 during electrochemical reduction using a flow cell electrolyser (A); and faradaic selectivity of CO (B); HCOOH (C); CH4 (D); C2H4 (E); and C2H6 (F) as a function of potential for the electrodes loaded with different volumes of SnCl4 Lewis acid on the Cu2O GDE. | ||
The Cu2O/SnCl4 GDEs are fabricated by drop casting 65 μL of SnCl4·5H2O solution on a Cu2O GDE and named CTC-65. The photographs of the as-fabricated Cu2O, pre-electrolysed CTC-65 and post-CO2 electrolysed CTC-65 electrodes are shown in Fig. 2. The CTC-65 electrode becomes whitish after coating the brick-reddish Cu2O, which reveals the deposition of chloride on the electrode.
 | ||
| Fig. 2 Photographs of the appearance of Cu2O and CTC-65 and post-CO2 electrolysed CTC-65 coated on gas diffusion layer (GDL) electrodes. | ||
3.2 Morphological, structural and electrochemical analysis
As shown in Fig. 3, the SEM images and EDS analysis provide insight into the structural and compositional characteristics of the Cu2O and pre-electrolysed CTC catalysts. The SEM images and EDS analysis of the post-electrolysed CTC catalysts are given in Fig. S4 and S5.† The SEM image of Cu2O reveals a relatively smooth surface morphology, which serves as the initial substrate for further modifications. In contrast, the pre-electrolysed CTC-65 exhibits a more complex, roughened surface with well-defined nanoscale structures, suggesting an increased surface area and enhanced active sites, which are beneficial for catalytic performance. The post-electrolysed CTC morphology in Fig. S4† shows the retention of these structural features, although some aggregation and possible morphology changes are visible, indicating that the catalyst has undergone structural adjustments due to electrochemical CO2 cycling. The elemental mapping from EDS confirms the presence of Cu, Sn, and Cl as the primary components in the fresh CTC-65 catalyst, with high concentrations of Sn and Cl distributed evenly, suggesting successful incorporation of these elements into the catalyst structure. The EDS spectrum further corroborates the composition, showing peaks for Sn, Cl, Cu, and O, consistent with the intended CTC structure. Additionally, the EDS analysis of post-CO2 electrolysed CTC-65 (Fig. S5†) confirms the retention of Sn and Cl species, suggesting the structural stability of the catalyst under electrolysis conditions, reinforcing its suitability for sustained CO2 reduction. | ||
| Fig. 3 Scanning electron microscopy images of Cu2O and pre-CO2 electrolysed CTC-65 and its elemental mapping. | ||
In order to find the crystallization transformation of CTC-65 electrodes, we performed XRD characterization to study the electrode before electrochemical measurements. Fig. 4A shows XRD patterns of CTC-65 before the CO2 electrolysis treatment. The XRD profiles of the bare Cu2O GDE, GDL, and post-CO2 electrolysed CTC-65 are given in Fig. S6.† For Cu2O, the peaks at 29.49°, 36.48°, 42.55°, 61.42°, and 73.69° correspond to (110), (111), (200), (220), and (311) planes, respectively.34–36 After adding SnCl4 onto Cu2O, the peaks corresponding to the (111) and (200) planes of Cu2O diminish and shift to 35.86° and 40.16° with the disappearance of the remaining corresponding Cu2O peaks. This demonstrates that the exposure of the Cu2O layer is decreased due to the addition of SnCl4·5H2O over its surface. Furthermore, the shift in the peaks indicates that there is an interaction between Cu2O and Cl or Sn, which is in accordance with the appearance of additional sharp peaks related to the mixed phases of SnCl2·2H2O, Sn(OH)Cl, Sn2(OH)2O, CuCl and Cu2(OH)3Cl.37–40
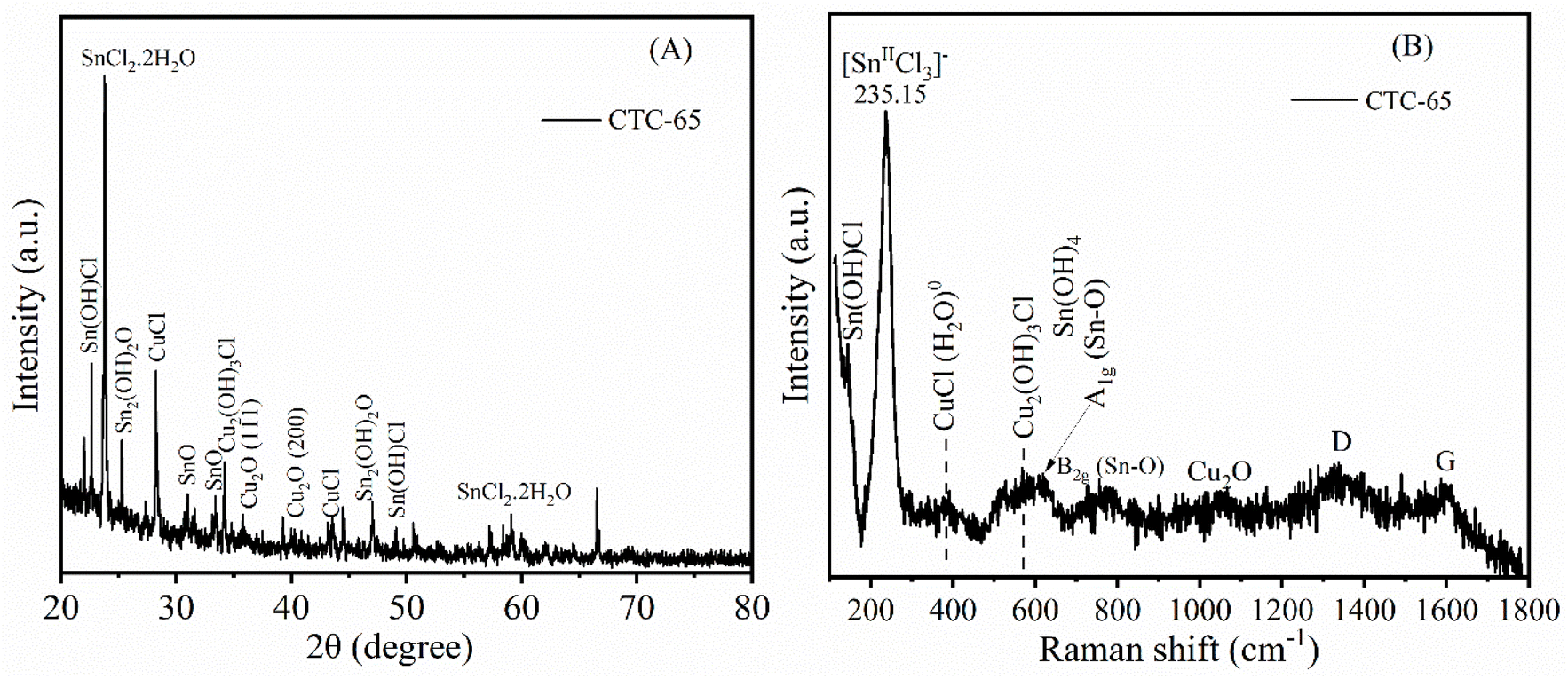 | ||
| Fig. 4 (A) Powder XRD patterns and (B) Raman signals of CTC-65 electrodes recorded at room temperature. | ||
As SnCl4·5H2O is highly hygroscopic, it readily absorbs atmospheric moisture, leading to spontaneous hydrolysis and the formation of hydrolysed tin and chloride species. Upon exposure to air, SnCl4 undergoes hydrolysis, forming compounds such as Sn(OH)Cl3 and Sn(OH)2Cl2. When deposited onto the Cu2O surface, these hydrolysed species interact with Cu, resulting in the formation of Sn–O–Cu and Cu–Cl bonds, which modify the electronic environment of the electrode. This is evident in the XRD patterns, where multiple phases coexist within the same compound, indicating the structural and compositional changes induced by SnCl4 modification. The possible mechanisms leading to the formation of these phases are as follows:
| SnCl4 + H2O → Sn(OH)Cl3 + HCl |
| Sn(OH)Cl3 + H2O → Sn(OH)2Cl2 + HCl |
| Cu2O + 3H2O + Cl → Cu2(OH)3Cl + H+ |
These structural transformations are further confirmed by Raman analysis, as shown in Fig. 4B, indicating the formation of Cu–Cl, Sn–OH–Cl, and Cu–OH–Cl bonds on the surface of the CTC-65 electrode. The prominent peak at 235.15 cm−1 in the Raman spectrum corresponds to the Sn(II)Cl3− species, suggesting the presence of tin chloride complexes that are essential for catalytic performance.38,41 Additional peaks around 400–600 cm−1 are attributed to CuCl and Cu2(OH)3Cl phases,42 as well as Cu2O, indicating that copper species are present in different oxidation states and environments within the CTC-65 GDE. The observed Cu2O signals suggest the incorporation of copper oxides, which may contribute to the stability and activity of the catalyst. The peaks around 800–1000 cm−1 correspond to Sn(OH)4 and Sn–O vibrations,43 demonstrating that tin is integrated in both hydroxylated and oxide forms, which may play a role in facilitating CO2 adsorption and activation. The weak D and G bands observed at higher Raman shifts (∼1300–1600 cm−1) are associated with carbonaceous materials, possibly originating from the carbon GDL. This indicates the presence of multiple active species, including CuCl, Cu2O, and Sn(OH)4, which together create a mixed catalytic surface that supports the electrochemical reduction of CO2 by providing various active sites for intermediate stabilization and electron transfer. The results from powder XRD and Raman confirm that chemical transformations occurred on the surface of the Cu2O GDE immediately after drop casting 65 μL of SnCl4·5H2O Lewis acid. We speculate that due to its hygroscopic nature, SnCl4·5H2O rapidly reacted with nearby species upon contact, resulting in the formation of hydroxychlorides, metal chlorides, and metal hydroxides. The Raman signal at 235.15 cm−1 in the Raman spectrum of the post-CO2-electrolyzed CTC-65 electrode (Fig. S7†) confirms the stability and retention of Cl and Sn on the CTC-65 surface after electrolysis.
The XPS spectra presented in Fig. 5 reveal detailed insights into the chemical state changes of Cu, Sn, Cl, and other elements on the CTC-65 catalysts before and after CO2 electrolysis. Fig. 5A and B show the Cu 2p spectra, where the shift in lower binding energies and the reduced intensity of the satellite peaks after electrolysis indicate the reduction of Cu2+ to Cu0, confirming the transformation of Cu2O to a more metallic Cu state.44–46 This change in the oxidation state suggests enhanced electronic conductivity and improved CO adsorption on metallic Cu sites, promoting C–C coupling. In Fig. 5C, the Sn 3d spectra show minimal shift in binding energy, indicating the stability of Sn species during CO2 reduction.47,48 The presence of Sn in the +4-oxidation state before and after electrolysis suggests that Sn acts as an electronic modulator, maintaining a consistent electronic environment that stabilizes CO intermediates, thereby enhancing C2 product selectivity.49Fig. 5D displays the Cl 2p spectra, where the consistent binding energies before and after electrolysis confirm that chloride ions remain anchored to the catalyst surface, preventing leaching during prolonged CO2 reduction.34,50 This stable Cl environment is crucial for suppressing the hydrogen evolution reaction (HER) and increasing the local CO concentration, favoring C–C coupling. The C 1s spectra in Fig. 5E reveal the presence of C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–F species of the PTFE coated GDL,51,52 and C–F species are from the PTFE coated GDL.53,54
C and C–F species of the PTFE coated GDL,51,52 and C–F species are from the PTFE coated GDL.53,54
 | ||
| Fig. 5 XPS analysis of the CTC-65 electrode before and after CO2 electrolysis, showing the spectra of Cu 2p (A and B), Sn 3d (C), Cl 2p (D), C 1s (E), and K 2s (F). | ||
Fig. 5F shows the K 2s spectra of post-CO2 electrolysed CTC-65, confirming the adsorption of K+ ions from the KOH electrolyte, which likely contribute to electrostatic stabilisation of CO2 reduction intermediates.55 The presence of K+ ions emphasizes the significance of the KOH electrolyte in facilitating CO2 electrolysis using CTC-type halide-based electrocatalysts. These observations collectively demonstrate the synergistic roles of Sn and Cl in stabilising key intermediates and optimising the electronic structure of the CTC catalysts, leading to enhanced selectivity and efficiency for C2 hydrocarbon production. XPS analysis further confirms the successful incorporation of Cu, Sn, and Cl species into the Cu2O/SnCl4 GDE, which is in correspondence with the SEM elemental mapping.
3.3 Electrochemical CO2 reduction activity
To explore the electrochemical properties of the Cu2O and CTC-65 electrodes, cyclic voltammetry was performed in a three-electrode system (Fig. 6A and B). The cyclic voltammograms of Cu2O and CTC-65 were recorded in 1 M KOH electrolyte in the potential range from −2.5 V to +1.0 V (vs. Ag/AgCl) with a scan rate of 50 mV s−1. In Fig. 6B, Cu2O shows two reduction peaks with cathodic peak potential (Epc) values of −0.45 and −0.98 V vs. Ag/AgCl, which can be assigned to the reduction of Cu2+/Cu+ and Cu+/Cu, respectively.56 During the reverse scan, two oxidation peaks at −0.36 and −0.05 V vs. Ag/AgCl were observed. The first anodic peak (Epa − 0.37 V) is associated with the oxidation peak of Cu/Cu+, while the second peak (−0.05 V) is associated with Cu+/Cu2+. For the CTC-65 electrode, more pronounced cathodic peaks related to Cu2+/Cu+ and Cu+/Cu and anodic peaks related to Cu/Cu+ and Cu+/Cu2+ are positively shifted with an additional peak at −0.74 V vs. Ag/AgCl corresponding to reduction of Sn (Sn2+/Sn).56 The positive shift in the redox peaks of Cu in CTC-65 can be ascribed to the interaction of Cl− ions with the Cu electrode.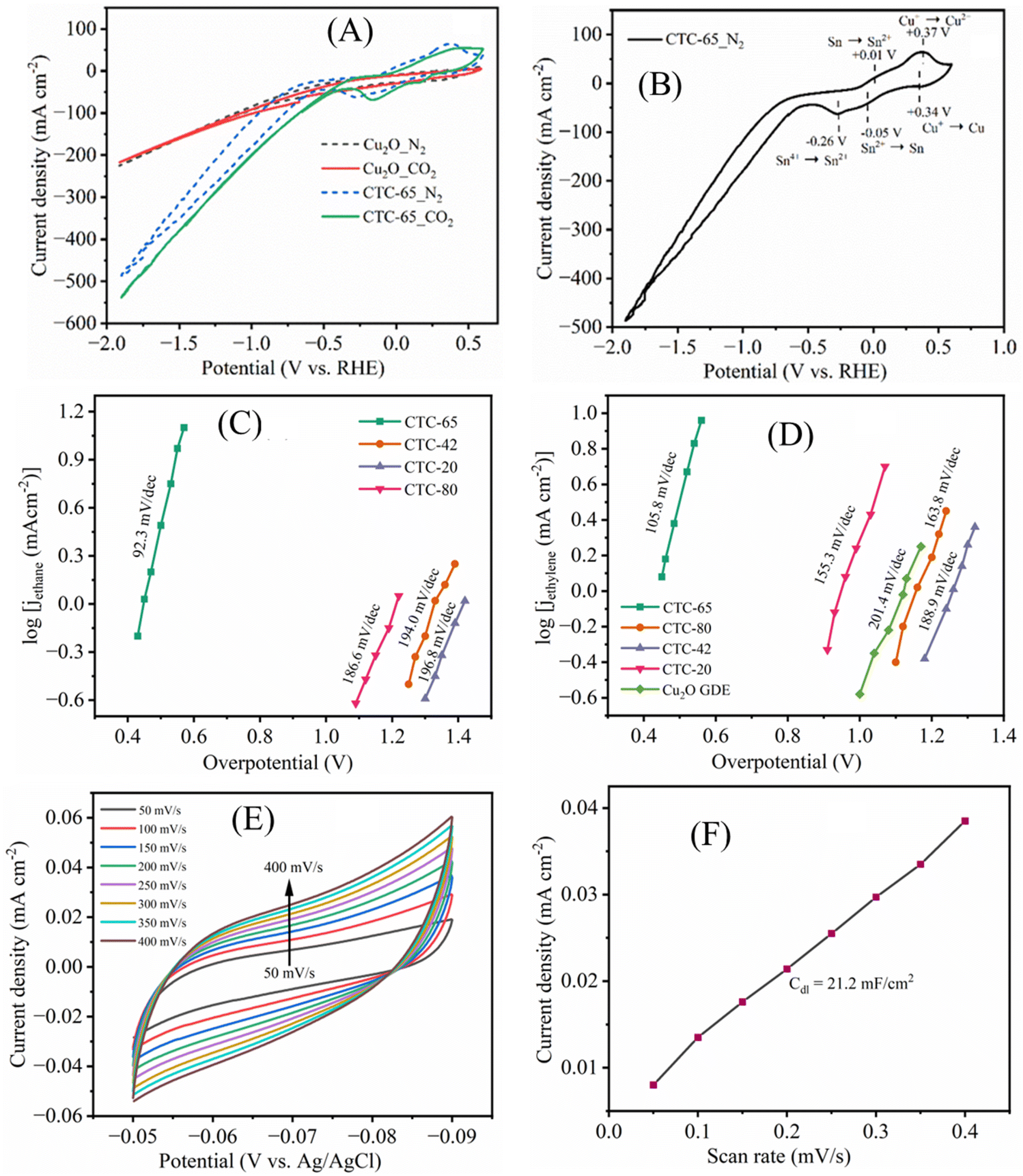 | ||
| Fig. 6 (A) CV polarization curves of Cu2O and CTC-65 electrodes scanned in the potential range of −2.0 to +0.5 VRHE with a 50 mV s−1 scan rate under N2 and CO2 flows in 1 M KOH electrolyte; and (B) cyclic voltammogram of CTC-65 under a N2 flow; (C) and (D) dependence of partial current density of ethane and ethylene on overpotential; (E) electrochemical active surface area of CTC-65; and (F) electrochemical double layer capacity of the CTC-65 GDE. | ||
The electrochemical CO2 reduction activity of CTC-65 was evaluated using CV in 1 M KOH aqueous electrolyte, providing insights into its superior catalytic performance. As shown in the CV curves collected in CO2- and N2-saturated electrolytes (Fig. 6A), CTC-65 exhibits significantly higher current density (232 mA cm−2 at −1.0 VRHE) compared to the Cu2O GDE, demonstrating its enhanced activity for ECR. In the CO2-saturated electrolyte, a pronounced increase in current density is observed for CTC-65, highlighting its efficient catalytic activity toward ECR. The reduction activity was further evaluated as the potential was scanned toward negative values. A reduced current density was initially observed, attributed to the competition between the HER and ECR. However, a sharp increase in current density was recorded at potentials more negative than −0.45 V (vs. RHE), confirming the activation of ECR on the CTC-65 electrode. Notably, the current density significantly increased at potentials beyond −0.75 V (vs. RHE), where electrochemical CO2 reduction becomes dominant, as Cl− ions play a dual role. At lower overpotentials, Cl− suppresses competing reactions such as the HER, delaying the onset of reduction activity, but at higher overpotentials, it enhances the reduction of CO2, enabling the formation of hydrocarbons.
This is corroborated by the low Tafel slopes for ethane and ethylene production (Fig. 6C and D), which indicate efficient charge transfer kinetics and high catalytic activity, aligning well with the faradaic efficiency (FE) results from Fig. 1. The addition of SnCl4 to Cu2O significantly boosts CO2 reduction activity by improving the electronic environment of the catalyst and creating active sites conducive to C–C bond formation. Furthermore, the higher electrochemical double-layer capacitance (Cdl) of 21.2 mF cm−2 (Fig. 6E and F) reflects a larger electrochemical surface area, which facilitates enhanced adsorption and activation of CO2 intermediates. These properties drive the selective production of valuable C2 hydrocarbons, such as ethane and ethylene, confirming the superior performance of CTC-65 as a catalyst for ECR. This systematic progression of activity, selectivity, and surface area contributions provides a cohesive understanding of the role of SnCl4 in transforming Cu2O into an efficient and selective catalyst for CO2 reduction.
Constant potential electrolysis was performed on Cu2O and all CTC electrodes at potentials of −0.5 V, −0.65 V, −0.75 V, −0.85 V and −1.0 V (vs. RHE) in a gastight three-electrode CO2 electrolyser flow cell, with their total current densities (Jtot) presented in Fig. 7A. The electrocatalytic CO2 reduction results for the CTC-65 electrodes at various potentials are shown in Fig. 7B. Initially, the CTC-65 electrodes exhibited a high geometric current density, with a recorded total current density (Jtot) of 74.8 mA cm−2 for 3600 seconds at −1 V vs. RHE, corresponding to the reduction of Cu2O/SnCl4 to CuSn structures. Following this transformation, the electrode achieved a stable current density during electrolysis. At a potential of −0.5 V vs. RHE, the CTC-65 electrode demonstrated a stable Jtot with a FE of 41.8% for CO and 1.6% for HCOOH production. At a more negative potential of −0.65 V vs. RHE, Jtot increased, with the corresponding FEs of 27.6% for CO, 2.2% for HCOOH, and 13.7% for CH4 production. At −0.75 V vs. RHE, ethane production became significant, with an FE of 16.4%, along with FEs of 24%, 22.9%, and 3.7% for CO, CH4, and HCOOH, respectively, at an increased current density. Notably, a peak FE of 34.8% for C2H6 production was observed at −1 V vs. RHE, accompanied by an FE of 11.3% for C2H4 production (Fig. 1A). This corresponds to an overpotential of only 40 mV relative to the equilibrium potential for the CO2/C2H6 reaction (0.554 V vs. RHE). Additionally, the FE for C2H6 production on the CTC-65 electrode remained relatively stable over time, indicating consistent catalytic activity for CO2 reduction. These results highlight the efficiency and stability of the CTC-65 electrode in selectively converting CO2 into C2 hydrocarbons during electrocatalysis.
 | ||
| Fig. 7 (A) Total current density profiles for Cu2O, CTC-20, CTC-42, CTC-65, and CTC-80 electrodes at various potentials; (B) constant potential electrolysis of CTC-65 at various potentials over 3600 seconds; (C) faradaic efficiencies of C2 products at various potentials with their corresponding partial current densities; and (D) electrochemical impedance spectra of the CTC-65 GDE at various potentials after CO2 electrolysis. | ||
Fig. 7C illustrates the FEs of C2 products, including both C2H6 and C2H4, generated during CO2 electrolysis at negative potentials of 0.75 V, 0.85 V, and 1.00 V vs. RHE, along with their corresponding partial current densities. At −1.00 V vs. RHE, the partial current density for C2 products reaches 34.48 mA cm−2, indicating that C2H6 and C2H4 production accounts for nearly half of the total current density (74.8 mA cm−2). This demonstrates that higher negative potentials significantly enhance the formation of C2 products during CO2 reduction on CTC electrodes. These results emphasize the favorable role of more negative potentials in facilitating C–C bond formation, which is crucial for achieving selective and efficient conversion of CO2 into valuable C2 hydrocarbons.
The EIS in Fig. 7D provides insights into the charge transfer resistance (Rct) and interfacial properties of CTC-65 during CO2 electroreduction at various cathodic potentials. The Nyquist plots exhibit a clear trend of decreasing Rct as the applied potential becomes more negative, indicating enhanced charge transfer kinetics facilitated by the presence of surface-bound chloride species. At lower potentials (−0.55 V to −0.65 V), higher Rct values suggest initial limitations in electron transfer, likely due to the adsorbed chloride layer restructuring the electrode interface. As the potential shifts more negative (−0.85 V to −1.00 V), Rct significantly decreases, corresponding to an improved catalytic process where chloride ions stabilize CO2 reduction intermediates, particularly CO, and promote C–C coupling reactions essential for C2 hydrocarbon formation. The equivalent circuit model (inset) includes a constant phase element (CCPE), which accounts for surface heterogeneity introduced by Sn and Cl modifications. The excellent fit between experimental data and the modeled impedance response further confirms that the preloaded chloride layer modulates the electrode–electrolyte interface, reduces hydrogen evolution side reactions, and enhances CO2 electroreduction selectivity toward ethane and ethylene. These findings corroborate faradaic efficiency trends and structural characterization (XRD, XPS, and Raman), reinforcing the role of SnCl4-derived chloride in stabilizing active sites and improving long-term catalytic performance.
3.4 Mechanism involved in CO2 reducing to hydrocarbons
From XRD, Raman, SEM, XPS and EIS analyses, it is confirmed that the CTC electrodes contain Sn and Cl species in the form of metal oxychlorides and metal hydroxychlorides. These chloride ions are physically adsorbed on the surface of CTC-65 electrodes, significantly influencing CO2 electroreduction in an alkaline environment, as shown in Fig. 8. These adsorbed Cl− ions create a more negative electrode environment, facilitating electron transfer to the CO2 molecule and stabilizing the CO2 radical. Additionally, the large ionic radius of Cl− (0.181 nm) effectively inhibits proton adsorption, minimizing hydrogen evolution side reactions.22,57 This aligns with the established role of M–X bonds (X = halogen and M = metal) in enhancing electron transport to CO2 while limiting proton adsorption. This mechanism is further supported by EIS measurements, showing increased charge transfer at varying potentials. In CO2-saturated 1 M KOH electrolyte, Cl− ions promote a nucleophilic reaction with CO2, enhancing local electron density and enabling CO2 activation. According to the double-layer model, Cl− resides on the inner Helmholtz plane, while solvated K+ ions from the KOH electrolyte (Fig. 5F) occupy the outer Helmholtz plane, facilitating unidirectional electron flow from Cl− to CO2 and then to K+. This unique double-layer configuration enhances CO2 activation and reduction, facilitating efficient C–C coupling for hydrocarbon formation. The nucleophilic attack by Cl− weakens the C–O bond, chemically activating CO2 and influencing subsequent electrochemical reduction steps. This strategic modulation of the electronic environment explains the enhanced selectivity towards C2 hydrocarbons such as ethane and ethylene, demonstrating the pivotal role of adsorbed Cl− ions in driving CO2 electroreduction on CTC-based electrodes. | ||
| Fig. 8 Schematic representation of CO activation on CTC catalysts in KOH electrolyte. | ||
The C2 hydrocarbon formation pathway on the CTC catalysts is primarily governed by the CO intermediate, as supported by the faradaic efficiency trends and Tafel slope analysis. The FE results in Fig. 1 reveal that at lower potentials, CO is the dominant product, but its concentration decreases at more negative potentials, coinciding with a rise in C2 hydrocarbon formation, including ethane and ethylene. This trend suggests that CO is consumed as an intermediate in C2 product formation. The simultaneous decrease in CO and increase in C2 hydrocarbons indicates a CO dimerization mechanism, where CO molecules couple to form C2 intermediates that subsequently undergo hydrogenation to produce ethane and ethylene. The constant concentration of HCOOH across all potentials confirms that the HCOOH pathway is independent and does not contribute to C2 hydrocarbon formation, further validating the CO dimerization pathway as the primary route for C2 product formation.
Tafel slope analysis (Fig. 7D) provides additional mechanistic insights by revealing distinct rate-determining steps (RDSs) for ethane and ethylene. Specifically, the Tafel slopes for ethane (92.3 mV dec−1) and ethylene (108.8 mV dec−1) on the CTC-65 catalyst indicate efficient charge transfer kinetics but distinct RDSs for each C2 product. The lower Tafel slope for ethane suggests that the RDS is the protonation of CO dimers, which aligns with a hydrogenation pathway. This is facilitated by Cl ions that stabilise CO intermediates, enhancing the local CO concentration and promoting hydrogenation to ethane.58 Conversely, the higher Tafel slope for ethylene suggests that C–C coupling is the RDS, involving CO dimerization followed by dehydrogenation. This aligns with the role of Sn species, which enhance CO binding strength and promote C–C coupling through electronic modifications of the Cu surface.59,60
These observations indicate a CO dimerization mechanism facilitated by Cl ions, which stabilise CO intermediates and enhance local CO concentration, promoting C–C coupling. The presence of Sn species modifies the electronic structure of Cu, influencing CO binding strength and promoting hydrogenation to ethane. The distinct Tafel slopes for ethane and ethylene emphasize different rate-determining steps, confirming that ethane formation is governed by the protonation of CO dimers, while ethylene formation involves C–C coupling followed by dehydrogenation. The synergistic effects of Cu, Sn, and Cl enable efficient CO stabilisation, C–C coupling, and selective hydrogenation, leading to enhanced selectivity and efficiency for C2 hydrocarbon formation on the CTC-65 catalyst. This coherent mechanistic pathway not only elucidates the role of CO dimerization in driving C2 hydrocarbon formation but also underlines the strategic interplay between adsorbed Cl− ions and Sn species in modulating the electronic and catalytic properties of the CuSnCl-based system.
Fig. 9 compares the Projected Density of States (PDOS) of the CTC electrode under three conditions: without CO, with adsorbed CO, and with CO and additional potassium (K) from the KOH electrolyte, revealing key electronic structure modifications relevant to CO2 reduction. In the absence of CO, the PDOS is dominated by the d-orbitals of Cu and the p-orbitals of Sn and Cl near the Fermi level, indicating good electron transfer capabilities but lacking sharp features indicative of strong adsorbate interactions (Fig. 9A). Upon CO adsorption, from Fig. 9B, significant shifts occur in the density of states, with new peaks emerging from the p-orbitals of CO, interacting with the d- and p- orbitals of Cu and Cl, respectively, signifying strong binding and electronic restructuring. The addition of K further modifies the PDOS, introducing new states near the Fermi level and enhancing CO stabilization, which is crucial for promoting hydrocarbon formation (Fig. 9C). This electronic modulation facilitates selective C–C coupling and hydrogenation, optimizing the catalytic efficiency of the CuSnCl electrode for CO2 electroreduction toward ethylene and ethane.
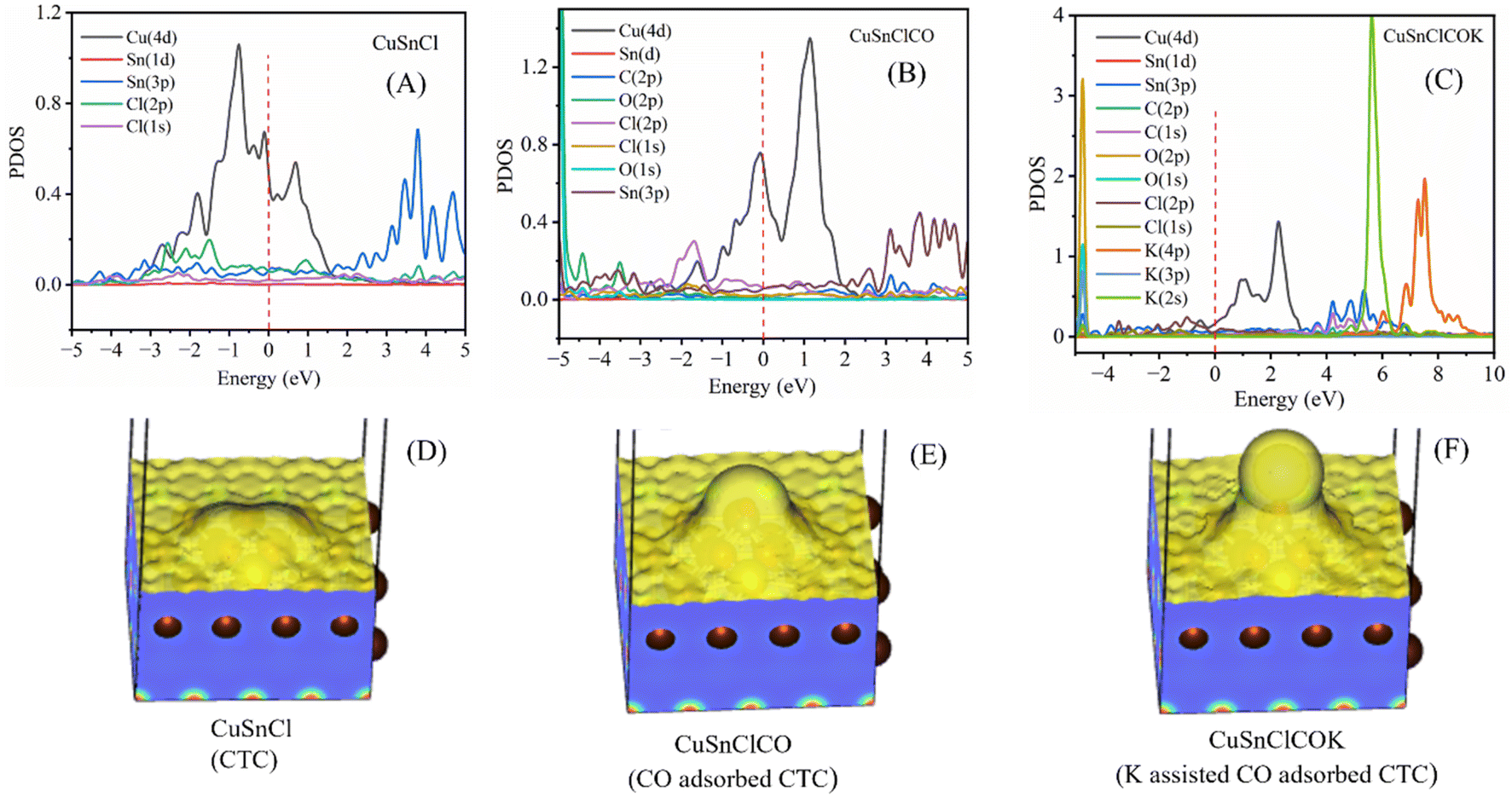 | ||
| Fig. 9 PDOS spectra of CTC (A), CTC-CO (B), and CTC–CO–K (C); and charge density profiles of CTC (D), CTC-CO (E), and CTC–CO–K (F). | ||
The charge densities for CuSnCl without adsorbates, with adsorbed CO, and with both CO and K from the KOH electrolyte reveal a progression in electron density localization, indicating the role of each adsorbate in enhancing the catalytic activity of CuSnCl for CO2 reduction. In the bare CuSnCl profile (Fig. 9D), the electron density is uniformly distributed across the surface, indicating no specific regions of high charge concentration, which suggests limited interaction sites for catalytic intermediates. With the addition of CO (Fig. 9E), a pronounced increase in charge density is observed around the adsorbed CO molecule, indicating strong charge transfer between CO and the CuSnCl (CTC) surface. This localized electron density around CO stabilizes it as an intermediate, essential for further reduction steps. However, in Fig. 9F, where both CO and K are adsorbed on the CTC surface, an even greater concentration of electron density appears around the CO molecule and the K–CO interaction region. The presence of K enhances the stabilization of CO by further increasing electron density at the active site, which favors subsequent hydrogenation and C–C coupling steps. This sequential increase in charge density from CTC alone to CTC with CO and then to CTC with CO and K demonstrates how CO and K together create an optimized electronic environment, crucial for effective CO2 reduction to hydrocarbons on the catalyst surface.
From Fig. 10, the stability test for the CTC-65 electrode demonstrates remarkable durability and consistent catalytic performance for CO2 electroreduction to ethane over a 24 hour period. As shown in Fig. 10, Jtotal remains stable at approximately −75 mA cm−2 without significant fluctuations, indicating sustained catalytic activity under continuous electrolysis conditions. Simultaneously, the FE for ethane remains consistently above 30%, fluctuating slightly between 31% and 35% throughout the 24 hour duration. This steady FE suggests that the active sites on the CTC-65 electrode are stable and resist deactivation, thereby maintaining high selectivity toward ethane formation.
 | ||
| Fig. 10 24 hour stability and ethane faradaic efficiency of CTC-65 GDE at −1 V vs. RHE. | ||
Collectively, the presence of Cu2O, SnO2, SnCl2·2H2O, CuCl, and Sn(OH)Cl from XRD indicates that the electrode consists of multiple phases, each contributing uniquely to the catalytic process.
The XPS data complement these findings by confirming the presence of both Sn2+ and Sn4+ oxidation states and a predominance of Cu+ in Cu2O. The electronic structure likely reflects these oxidation states, showing how they contribute to the overall catalytic mechanism. Tin's dual oxidation states might explain the initial preference for single-carbon products like CO and formate (HCOOH) at less negative potentials, as these states can stabilize different reaction intermediates through electronic interactions with Cu and Cl. As the potential becomes more negative, the electronic states shift, favoring the formation of C–C bonds, leading to increased hydrocarbon production, as observed in the faradaic efficiency data.
4. Conclusion
In conclusion, this work demonstrates the significant potential of SnCl4 Lewis acid-modified Cu2O as a highly efficient and selective catalyst for electrochemical CO2 reduction in a flow cell electrolyser configuration. By introducing chloride ions directly onto the Cu2O surface, the synergistic interactions between Sn, Cl, and Cu active sites were optimized, facilitating the stabilization of CO intermediates and promoting C–C bond formation for the selective production of C2 hydrocarbons. The system achieved a peak faradaic efficiency of 34.8% for ethane and 11.3% for ethylene at −1.0 V vs. RHE, with a stable total current density of 74.8 mA cm−2. Supported by detailed mechanistic insights from PDOS, Raman, XPS, and EIS analyses, the study highlights the critical role of chloride ions in enhancing the catalytic performance. Using a Fumasep bipolar membrane and a Pt foil anode in the flow cell electrolyser, this scalable and economically viable catalyst design paves the way for sustainable CO2 utilization and renewable hydrocarbon production, offering a promising pathway for carbon-neutral energy systems.Data availability
All data generated during this study are included in the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project has received funding from the Engineering & Physical Sciences Research Council – EP/X527257/1, Royce at Manchester Reference: MCAP072.References
- P. De Luna, et al. , Science, 2019, 364, 350 Search PubMed.
- AR6 Synthesis Report: Climate Change 2023, 2023 Search PubMed.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C. J. Yang and K. Caldeira, et al. , Science, 2018, 360(6396), eaas9793 CrossRef PubMed.
- Climate.gov, Climate Change: Atmospheric Carbon Dioxide, 2024 Search PubMed.
- European Environment Agency, Climate Change Mitigation: Reducing Emissions, 2024 Search PubMed.
- D. L. McCollum, W. Zhou, C. Bertram, H.-S. de Boer, V. Bosetti, S. Busch, J. Després, L. Drouet, J. Emmerling, M. Fay, O. Fricko, S. Fujimori, M. Gidden, M. Harmsen, D. Huppmann, G. Iyer, V. Krey, E. Kriegler, C. Nicolas, S. Pachauri, S. Parkinson, M. Poblete-Cazenave, P. Rafaj, N. Rao, J. Rozenberg, A. Schmitz, W. Schoepp, D. van Vuuren and K. Riahi, Nat. Energy, 2018, 3, 589–599 Search PubMed.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CAS.
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CAS.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C. T. Dinh, P. D. Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S. C. Lo, A. Ip, Z. Ulissi and H. S. Edward, Nature, 2020, 581, 178–183 CAS.
- S. Man, W. Jiang, X. Guo, O. Ruzimuradov, S. Mamatkulov and S. Mamatkulov, Chem. Mater., 2024, 36, 1793–1809 CAS.
- D. W. Green and R. H. Perry, Perry's Chemical Engineer's Handbook, McGraw-Hill Education, 8th edn, 2007 Search PubMed.
- A. D. Handoko, K. W. Chan and B. S. Yeo, ACS Energy Lett., 2017, 2, 2103–2109 CrossRef CAS.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 Search PubMed.
- A. Dutta, M. Rahaman, N. C. Luedi and P. Broekman, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 Search PubMed.
- J. Hussain, H. Jonsson and E. Skulason, ACS Catal., 2018, 8, 5240–5249 Search PubMed.
- A. Vasileff, Y. Zhu, X. Zhi, Y. Zhao, L. Ge, H. M. Chen, Y. Zheng and S. Qiao, Angew. Chem., Int. Ed., 2020, 59, 19649–19653 Search PubMed.
- P. Chen, P. Zhang, X. Kang, L. Zheng, G. Mo, R. Wu, J. Tai and B. Han, J. Am. Chem. Soc., 2022, 144, 14769–14777 CAS.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 Search PubMed.
- T. Chen, J. Hu, K. Wang, K. Wang, G. Gan and J. Shi, Energy Fuels, 2021, 35, 17784–17790 CAS.
- M. Zhao, H. Tang, Q. Yang, Y. Gu, H. Zhu, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 4565–4571 Search PubMed.
- J. J. Masana, B. Peng, Z. Shuai, M. Qiu and Y. Yu, J. Mater. Chem. A, 2022, 10, 1086–1104 CAS.
- M. Cho, J. T. Song, S. Back, Y. Jung and J. Oh, ACS Catal., 2018, 8, 1178–1185 CrossRef CAS.
- S. Li, X. Dong, Y. Zhao, J. Mao, W. Chen, A. Chen, Y. Song, G. Li, Z. Jiang, W. Wei and Y. Sun, Angew. Chem., 2022, 134, e202210432 Search PubMed.
- M. Zhao, H. Tang, Q. Yang, Y. Gu, H. Zhu, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 4565–4571 CrossRef CAS PubMed.
- Y. C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- D. Gao, F. Scholten and B. R. Cuenya, ACS Catal., 2017, 7, 5112–5120 CrossRef CAS.
- T. Yuan, T. Wang, G. Zhang, W. Deng, D. Cheng, H. Gao, J. Zhao, J. Yu, P. Zhang and J. Gong, Chem. Sci., 2022, 13, 8117–8123 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- (a) P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. Fabris, G. Fratesi, S. De Gironcoli, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed; (b) F. Molani, S. Jalili and J. Schofield, J. Mol. Model., 2015, 21, 4 CrossRef PubMed.
- T. Zou, F. L. P. Veenstra, E. I. Alé, R. G. Muelas, G. Zichittella, A. J. Martín, N. López and J. P. Ramírez, Cell Rep. Phys. Sci., 2023, 4, 101294 CrossRef CAS.
- Q. Zhang, L. Huang, S. Kang, C. Yin, Z. Ma, L. Cui and Y. Wang, RSC Adv., 2017, 7, 43642 CAS.
- H. Xiang, S. Rasul, B. Hu, J. Portoles, P. Cumson and E. H. Yu, ACS Appl. Mater. Interfaces, 2020, 12, 601–608 CAS.
- C. Akyil, G. Akdas, P. Afsin and M. Urgen, Mater. Chem. Phys., 2019, 221, 263–271 CrossRef CAS.
- T. Wang, J. Chen, X. Ren, J. Zhang, J. Ding, Y. Liu, K. H. Lim, J. Wang, X. Li, H. Yang, Y. Huang, S. Kawi and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202211174 CrossRef CAS PubMed.
- J. Wang, S. Liu, X. Cao, Z. Wang, Y. Guo, X. Li, C. Liu, W. Jiang, H. Wang, N. Wang, S. Wu, H. Tao and W. Ding, Appl. Phys. A, 2020, 126(44), 1–7 Search PubMed.
- M. Chmielová, J. Seidlerová and Z. Weiss, Corros. Sci., 2003, 45, 883–889 CrossRef.
- A. F. Shihada, A. S. Abushamlesh and F. Weller, Z. Anorg. Allg. Chem., 2004, 630, 841–847 Search PubMed.
- X. D. Liu, X. G. Zheng, D. D. Meng, X. L. Xu and Q. X. Guo, J. Phys.: Condens. Matter, 2013, 25, 256003 CrossRef PubMed.
- I. M. Chou, R. Wang and J. Fang, Geochem. Perspect. Lett., 2021, 20, 1–5 Search PubMed.
- C. S. Gopinath, J. Chem. Soc., Faraday Trans., 1996, 92, 3605–3610 Search PubMed.
- Y. A. Teterin, M. I. Sosulnikov, V. I. Ozhogin, T. M. Senchenkova, N. S. Tolmacheva and L. D. Shustov, Phys. C, 1991, 185–189, 837–838 CrossRef CAS.
- D. Ferrah, A. R. Haines, R. P. Galhenage, J. P. Bruce, A. D. Babore, A. Hunt, I. Waluyo and J. C. Hemminger, ACS Catal., 2019, 9, 6783–6802 CrossRef CAS.
- M. Li, X. Tian, S. Garg, T. E. Rufford, P. Zhao, Y. Wu, A. J. Yago, L. Ge, V. Rudolph and G. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 22760–22770 CrossRef CAS PubMed.
- E. Castillo, A. F. Pasha, Z. I. Larson and N. Dimitrov, Mater. Adv., 2024, 5, 2285–2295 Search PubMed.
- S. Go, W. Kwon, D. Hong, T. Lee, S. H. Oh, D. Bae, J. H. Kim, S. Lim, Y. C. Joo and D. H. Nam, Nanoscale Horiz., 2024, 9, 2295–2305 RSC.
- L. Yu, X. Ba, M. Qiu, Y. Li, L. Shuai, W. Zhang, Z. Ren and Y. Yu, Nano Energy, 2019, 60, 576–582 CrossRef CAS.
- B. Sankeerthana, R. Shanmugam and G. R. Rao, J. Mater. Chem. A, 2019, 7, 3757–3771 Search PubMed.
- G. Cunge, B. Pelissier, O. Joubert, R. Ramos and C. Maurice, Plasma Sources Sci. Technol., 2005, 14, 599–609 CrossRef CAS.
- G. H. Yang, Y. Zhang, E. T. Kang and K. G. Neoh, J. Phys. Chem. B, 2003, 107, 2780–2787 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- A. Pramanik, A. G. Manche, R. Clulow, P. Lightfoot and A. R. Armstrong, Dalton Trans., 2022, 51, 12467–12475 Search PubMed.
- A. L. S. Eh, J. Chen, S. H. Yu, G. Thangavel, X. Zhou, G. Cai, S. Li, D. H. C. Chua and P. S. Lee, Adv. Sci., 2020, 7, 1903198 CrossRef CAS PubMed.
- J. O'M. Bockris, B. E. Conway and E. Yeager, Comprehensive Treatise of Electrochemistry, Plenum Press, New York, 1980 Search PubMed.
- O. van der Heijden, S. Park, R. E. Vos, J. J. J. Eggebeen and M. T. M. Koper, ACS Energy Lett., 2024, 9, 1871–1879 CrossRef CAS PubMed.
- N. B. Watkins, Z. J. Schiffer, Y. Lai, C. B. Musgrave III, H. A. Atwater, W. A. Goddard III, T. Agapie, J. C. Peters and J. M. Gregoire, ACS Energy Lett., 2023, 8, 2185–2192 CrossRef CAS.
- M. K. Birhanu, B. Ü. Abdioglu and A. Uçar, Catal. Sci. Technol., 2025, 15, 262–317 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 3D drawing of the CO2 electrolyser, synthesis and faradaic efficiencies of Cu3Sn and Cu2O, XRD, XPS, Raman, and SEM analyses of Cu2O and post-CO2 electrolysed CTC-65, and simulated band structures. See DOI: https://doi.org/10.1039/d5ta00176e |
| This journal is © The Royal Society of Chemistry 2025 |