
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gel electrolyte interdigitation enables stable high areal capacity cycling of the 3D Zn electrode†
Yuan
Shang
a,
Ravindra
Kokate
a,
Patrick
Tung
 b,
Haoyin
Zhong
c,
Erlantz
Lizundia
de,
Francisco J.
Trujillo
a,
Priyank
Kumar
a and
Dipan
Kundu
*a
b,
Haoyin
Zhong
c,
Erlantz
Lizundia
de,
Francisco J.
Trujillo
a,
Priyank
Kumar
a and
Dipan
Kundu
*a
aSchool of Chemical Engineering, UNSW Sydney, Kensington, NSW 2052, Australia. E-mail: d.kundu@unsw.edu.au
bResearch Technology Services (ResTech), UNSW Sydney, Kensington, NSW 2052, Australia
cDepartment of Materials Science and Engineering, National University of Singapore, 117575, Singapore
dLife Cycle Thinking Group, Department of Graphic Design and Engineering Projects, Faculty of Engineering in Bilbao, University of the Basque Country (UPV/EHU), 48013 Bilbao, Spain
eBCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, Leioa, 48940, Spain
First published on 27th February 2025
Abstract
Inferior rechargeability of the metallic zinc anode, especially under high areal capacities and moderate to high current densities, remains a significant bottleneck for developing scalable aqueous zinc-ion batteries. While three-dimensional (3D) porous zinc anodes can mitigate high local current density and enhance deposition kinetics, limited electrolyte percolation within the 3D framework with conventional liquid electrolyte-soaked separator leads to surface-concentrated ion flux, which confines zinc nucleation and growth to the uppermost surface. This results in early short-circuit events mediated by dendrites. Here, electrolyte-interdigitation with a biopolymer hydrogel electrolyte is presented as a facile strategy, which by design enables complete electrolyte percolation within the porous and tortuous structure of the 3D zinc and uniform mass transport across the whole electrode structure. The increased accessible surface area and interconnected transport pathways effectively regulate zinc plating/stripping, thus maintaining the structural integrity upon cycling. As a result, the integrated design enables extended zinc rechargeability and a cumulative cycling capacity of 1680 mA h cm−2 under demanding 5 mA cm−2–5 mA h cm−2 conditions. Suppressed corrosion and dendrite inhibition for the interdigitated anode also leads to excellent rate capability and stability of the full-cell, highlighting a significant advance in the field of 3D zinc anode design.
Introduction
Over the past few years, aqueous Zn-ion batteries (AZIBs) have come to the forefront of research on next-generation batteries, particularly for stationary energy storage, owing to the promise of tremendous safety, significantly low cost, high theoretical capacity of the zinc anode (820 mA h g−1), and potential for extended cyclability and excellent power capability.1–5 Yet, the successful transition of AZIBs from research to translation requires addressing several critical fundamental issues hindering zinc anode and cathode rechargeability under demanding current and capacity conditions.6,7 For zinc, its extreme vulnerability to dendritic deposition and (electro)chemical corrosion (hydrogen evolution reaction (HER) and metal corrosion) often leads to premature cell failure, especially when a thin separator and a limited zinc anode are employed.8–10Diverse strategies have been proposed to address the Zn reversibility issue, including electrolyte modification11–13 and electrolyte–anode interface engineering.14–16 While these strategies can ensure relatively homogeneous Zn deposition and mitigate side reactions, and thus prolong the Zn metal anode cycling lifespan, the conventional Zn foil-type anode with limited active sites presents a kinetic limit for Zn plating/stripping, especially at high current densities.17 Consequently, many of the strategies developed so far toward enabling the improved zinc anode cyclability in AZIBs are predominantly realized under relatively low current densities and areal capacities.9,18,19 In this context, three-dimensional (3D) anodes, such as metallic foams20–22 or conductive 3D scaffolds23,24 can be employed to alleviate the high local current density and facilitate fast zinc deposition kinetics through enhanced specific surface area and thus additional nucleation sites.25–27 However, the enlarged accessible surface area of such 3D electrodes also offers more reactive sites for zinc corrosion, which in turn can result in a significant accumulation of “dead” zinc. As a result, the structural integrity of the 3D anodes and Zn reversibility are compromised.
In addition, complete electrolyte infiltration within the 3D structure, necessary for uniform zinc deposition/stripping across the electrode, can be difficult to ensure with a soaked separator membrane. The active 3D structure inevitably increases the electrode thickness with tortuous channels,28 which makes it challenging to properly infiltrate conventional liquid electrolytes (e.g., aqueous ZnSO4) within the electrode structure unless in a flooded electrolyte system, making the majority of the electrode electrochemically inaccessible. The resulting superficial electrolyte wetting yields a surface-concentrated ion flux that confines Zn nucleation and growth to the uppermost surface.29,30 Consequently, 3D porous electrodes can suffer from early short-circuit events under moderate to high-current conditions.17,31 Additionally, achieving effective interfacial contact can also be challenging when using a gel electrolyte with a porous electrode due to the gel's quasi-solid nature.32–34 Therefore, there is an imperative need for an integrated design that can facilitate the complete electrolyte infiltration and enable a uniform ion flux so long-term cycling stability for 3D structured zinc electrodes can be realized under high current density (≥5 mA cm−2) and areal capacity (≥5 mA h cm−2) conditions.
Herein, we present an electrolyte interdigitated 3D Zn electrode design with an agarose-based hydrogel electrolyte to achieve controlled and uniform Zn deposition throughout the entire 3D structure of the electrode. Agarose, a polysaccharide of marine origin, has been selected as a model electrolyte material because of its double helical aggregation into a gel structure, functional ether (R–O–R) and hydroxyl (–OH) groups, and chemo-mechanical stability.35 The zinc foam–agarose interlocked system (denoted as ZF–agarose), prepared by Zn foam immersion into an aqueous agarose solution, followed by ambient temperature curing (Fig. 1a), allows complete percolation of the gel electrolyte within the tortuous and porous structure of Zn foam and provides 3D ion transport channels. Thus, the ZF–agarose electrode effectively homogenizes the electric field distribution, Zn2+ flux, and local current density, preventing dendrite-mediated short-circuit. Moreover, by precluding direct contact between the Zn foam and the liquid electrolyte, the side reactions, often exacerbated by the enlarged specific surface area of 3D electrodes, are significantly curtailed. As a result, the ZF–agarose electrode shows excellent zinc rechargeability, highlighted by a cumulative cycling capacity of 1680 mA h cm−2 under a demanding 5 mA cm−2 and 5 mA h cm−2 condition, against rapid short circuit for the standard 3D Zn foam electrode. Suppressed corrosion and improved transport across the large accessible surface area for the interdigitated 3D anode also lead to high rate capability and stability of the Zn0.25V2O5·nH2O-based full-cell.
 | ||
| Fig. 1 Fabrication and characterization of the interdigitated 3D electrode. (a) Schematic showing the preparation of ZF–agarose electrode. (b) FTIR spectra of agarose gel before and after soaking into 3 M ZnSO4 electrolyte. (c) A representative scanning electron microscope (SEM) image of the soaked agarose gel surface. (d) A cross-section SEM image of the ZF–agarose electrode. (e) The comparison of ionic conductivity between agarose hydrogel and aqueous 3 M ZnSO4 electrolyte. (f) Schematic demonstration of hydrogel-foam interdigitated electrode for rapid Zn2+ transport. | ||
Result and discussion
The fabrication process of the interdigitated ZF–agarose electrode is summarized in Fig. 1a. Firstly, agarose powder was mixed with H2O at a concentration of 6 wt%. The mixture was subsequently heated up to 90 °C under stirring until dissolution to obtain a transparent solution. Then, the cleaned Zn foam was submerged in the solution under stirring and heating for 5 min to ensure a complete infiltration of the solution throughout the Zn foam matrix. The as-prepared electrode was cured at room temperature and soaked into a 3 M ZnSO4 electrolyte to obtain the ZF–agarose electrode. During cooling, a physical cross-linking of agarose occurs into a single-helix or double-helix structure. The water molecules remain captured and anchored by hydrophilic –OH groups in agarose, facilitating bonding between each helical segment.36,37 Fourier transform infrared (FTIR) spectroscopy of the agarose gel before electrolyte soaking (Fig. 1b) presents characteristic peaks at 1638 and 1374 cm−1 attributed to the H–O–H and H–C–H vibrations of agarose, respectively.38 Furthermore, the peaks at 1073 and 933 cm−1 are associated with the vibration of C–O–C in glycosidic linkage and 3,6-anhydro galactose, corroborating the hydrogel formation.38 After immersion in the 3 M ZnSO4 electrolyte, the characteristic peak corresponding to SO42− at 1072 cm−1 appears to overlap with the band arising from the C–O–C of agarose. No discernible peak shift is observed, suggesting a negligible interaction of dissolved Zn2+ and SO42− with agarose.39Raman spectroscopy was conducted to further confirm the appearance of ZnSO4 (Fig. S1†). A clear peak at 982 cm−1 originating from SO42− is seen. Before electrolyte infusion, the vibration bands at 3209 cm−1, and 3412 cm−1 (Fig. S2a†) are seen due to the intermolecular hydrogen bonds and asymmetric stretching vibration in H2O, respectively.40 Owing to the diffusion of Zn2+ into agarose gel after soaking, the intermolecular hydrogen bonds and asymmetric stretching vibration shift to 3252 cm−1 and 3439 cm−1, respectively (Fig. S2b†). This indicates a strong hydration of Zn2+, which substantially attenuates the hydrogen bonds in the agarose gel. As observed by SEM in Fig. 1c, the ZF–agarose presents a smooth and compact agarose gel surface with no cracks or wrinkles, and by design, a 40 μm gel layer sticks out on the foam surface, as is apparent from Fig. 1d. Electrolyte ionic conductivity is a crucial parameter that dictates the zinc deposition kinetics. This was determined from the Nyquist data (Fig. S3†) obtained by electrochemical impedance spectroscopy (EIS). Although the agarose hydrogel electrolyte exhibits an ionic conductivity (10.4 mS cm−1) that is slightly inferior to the aqueous 3 M ZnSO4 electrolyte (17.3 mS cm−1, Fig. 1e), it is still considerably high. It furnishes a continuous 3D framework throughout the entire porous structure of the zinc foam, which can function as a protective layer and effectively regulate Zn2+ flux for homogeneous deposition (Fig. 1f).
To evaluate the efficacy of the interdigitated design, the electrochemical zinc cycling performance of the ZF–agarose electrode was investigated vis-à-vis the pristine Zn foam (ZF) electrode (Whatman GF/A and GF/D was used for agarose–ZF and ZF symmetric cell, respectively, to balance the inter-electrode distance). The incorporation of agarose gel has a negligible effect on the Zn2+/Zn redox potential (Fig. S4†). However, it exhibits a lower current response than the ZF electrode, which is in agreement with the reduced ionic conductivity of the agarose gel (Fig. 1d). The rechargeability of the ZF–agarose and ZF electrodes was then evaluated by long-term galvanostatic cycling in a symmetric cell configuration. While the ZF electrode affords a porous 3D structure with a high specific surface area, insufficient electrolyte infiltration within the 3D structure and direct exposure to the aqueous electrolyte leads to pronounced upper surface-limited stripping/deposition and corrosion, thereby resulting in an inferior cyclability (290 h, Fig. 2a) at 2 mA cm−2 and 2 mA h cm−2. To note, here and throughout the manuscript, the specific current and the capacity refer to the geometric area of the electrode. In comparison to the ZF electrode with liquid electrolyte, the 3D electrolyte percolation in the ZF–agarose electrode, coupled with the surface protective hydrogel, renders the ZF–agarose symmetric cell with an extended zinc rechargeability of over 1000 h (Fig. 2a). The increased accessible surface area and improved mass transport in the ZF–agarose also result in appealing performance under high current density and areal capacity conditions. As shown in Fig. 2b, the electrode stably operates for 672 h at a current density of 5 mA cm−2 and an areal capacity of 5 mA h cm−2. This corresponds to a cumulative Zn plating capacity of 1680 mA h cm−2, which is significantly improved in comparison to that of a pristine ZF electrode (32 h or 80 mA h cm− 2). To further rationalize the benefits of interdigitated electrode design, agarose hydrogel, prepared by curing 6 wt% agarose solution onto a Petri-dish and subsequently soaked in 3 M ZnSO4 electrolyte for one day, was directly tested with ZF and Zn foil electrodes (denoted as agarose@ZF and agarose@Zn, respectively). These electrodes were assembled into symmetric cells and cycled at 5 mA–5 mA h cm−2. While benefiting from the protective effect of the agarose hydrogel, agarose@ZF and agarose@Zn electrodes enjoy extended zinc rechargeability of 380 and 290 h (Fig. S5 and S6†), respectively, this still falls short of the agarose–ZF electrode. This observation demonstrates the impact of the 3D interconnected mass transport channels rendered by the hydrogel-integrated electrode design.
 | ||
| Fig. 2 The electrochemical evaluation of the integrated ZF–agarose anode. The long-term galvanostatic cycling of symmetric Zn cells with bare ZF and ZF–agarose electrode at (a) 2 mA–2 mA h cm−2, and (b) 5 mA–5 mA h cm−2. (c) The coulombic efficiency (CE) assessment of Zn plating/stripping for ZF and ZF–agarose electrodes in Ti‖Zn asymmetric configuration. The EIS spectra of rested (d) ZF–agarose and (e) ZF symmetric cell during cycling at 2 mA–2 mA h cm−2. The inset in (d) shows the equivalent circuit model used to fit the impedance data. Further information is provided in the experimental details. (f) LSV response curve for the 2 M Na2SO4 electrolyte and agarose hydrogel electrolyte (after soaking in 2 M Na2SO4 solution) 1 mV s−1. (g) Comparison of the Zn cycling performance against literature on 3D structured electrodes to boost the Zn anode cyclability.17,26,28,41–45 | ||
The coulombic efficiency (CE) for Zn plating/stripping for both ZF and ZF–agarose electrodes was assessed in asymmetric Ti–Zn configuration. As illustrated in Fig. 2c, owing to the suppression of side reactions,46 the ZF–agarose cell demonstrates an improved Zn reversibility with an average CE of 99.1% over 250 cycles at 2 mA–2 mA h cm−2 compared to the average CE of 98.9% for the ZF electrode. As expected, the ZF electrode suffers from an early failure due to short-circuit. In an effort to assess the consequences of corrosion, the symmetric cells were rested for 24 h, and the evolution of the charge transfer kinetics, which gets impacted by corrosion-mediated byproduct accumulation on the zinc surface, was monitored by EIS. During the resting period, the bare ZF electrode shows a pronounced growth in impedance, particularly within the initial 3 hour rest, where the ohmic component of the impedance increases from 37 Ω (t = 0 h) to 58 Ω (t = 3 h) – as obtained by regression of the impedance data (Fig. S7a†). As time progresses, the impedance continues to increase to reach 85 Ω after 24 h, corroborating the dynamic corrosion reactions between the porous Zn foam and the aqueous 3 M ZnSO4 electrolyte. Notably, the impedance observed here for the 3D zinc electrode is strikingly low compared to what is typically observed for the foil-type zinc anode.47 This can be explained by the large surface area of the porous 3D electrode, which leads to an accelerated charge-transfer kinetics and, hence, a much smaller charge-transfer impedance, as observed here, even under aggressive corrosion. Nevertheless, the direct liquid electrolyte–electrode interaction is subdued in the ZF–agarose electrode, as evidenced by a relatively smaller impedance rise (71 Ω after 24 h, Fig. S8a†) and stabilization during resting.
Interestingly, when the rested electrodes are subsequently cycled at 2 mA cm−2–2 mA h cm−2, the ZF–agarose cell exhibits a stable voltage profile (Fig. S8b†) with a slightly increased charge transfer impedance (Fig. 2d), but the ZF cell delivers a much larger overpotential at the onset of cycling leading to soft shorts and quick cell failure (Fig. S7b and 2e†). The greater extent of corrosion for the ZF cell likely exacerbates the uneven Zn deposition, increasing the propensity for dendrite-mediate short circuits. For the ZF–agarose cell, the impedance increase during resting and follow-up cycling, albeit smaller than that for the ZF cell, suggests that the corrosion is not entirely inhibited, which explains the slow rise in polarization for the ZF–agarose cell during cycling (Fig. 2a and b) and the not dramatically higher average CE compared to the ZF cell. This issue can be addressed with a modified hydrogel electrolyte, e.g., by incorporating a zinc corrosion-suppressing additive. However, this route was not pursued as the focus was not on designing an improved hydrogel electrolyte but on investigating the efficacy of electrolyte-integrated 3D electrode design. Anyhow, it is evident from Fig. 2f that the HER activity is significantly suppressed in agarose hydrogel with lower current responses (0 V to −0.15 V vs. Zn2+/Zn range), compared to that for the ZF electrode. To eliminate interference from Zn plating, saturated Na2SO4 (∼2 M) was used as the reference electrolyte, and agarose gel electrolyte was soaked in the saturated Na2SO4 solution. The effective suppression of dendrites and HER and associated side reactions in the ZF–agarose electrode leads to excellent Zn reversibility, confirming the effectiveness of the integrated design. It should be noted that the current density and capacity used here to evaluate the ZF–agarose symmetric cell are significantly higher than previously reported studies of 3D structured electrodes (typically ≤5 mA cm−2 and 5 mA h cm−2, Fig. 2g). Moreover, the cumulative cycling capacity (1680 mA h cm−2) rendered by the ZF–agarose electrode is well above those reported in the literature, highlighting a significant advance in the field of 3D electrode design.
The electrodeposited zinc morphology typically correlates with the cycling performance of the metal anode, but the morphology evolution is expected to be unique for a 3D electrode with a porous architecture and high surface area. SEM was employed first to assess the surface topography of the bare ZF electrode. As shown in Fig. 3a and b, before cycling, the ZF anode displays a homogeneously distributed porous framework coupled with a planar and densely packed zinc surface. The corresponding X-ray computed tomography (CT) image in Fig. 3c and S9a, reveals homogeneous porosity distribution throughout the electrode for the pristine ZF. After 10 cycles at 2 mA–2 mA h cm−2, under SEM, the ZF–agarose electrode (treated in a 90 °C water bath for agarose gel removal) shows retention of the porous architecture (Fig. 3d) and exhibits a uniform Zn deposition (Fig. 3e). According to X-ray CT, the cycled anode maintains uniform pore distribution (Fig. 3f and S9b†) and showcases a similar porosity (44.5%, Fig. 3j) as the pristine ZF (44.8%). Unsurprisingly, the 3D interconnected transport pathways inherent to the ZF–agarose electrode effectively regulate the Zn2+ transport and plating/stripping, thus maintaining the 3D structure integrity.
 | ||
| Fig. 3 The Zn deposition and corresponding tomographic characterization. Representative SEM images of (a and b) pristine ZF electrode and cycled (d and e) ZF–agarose and (g and h) ZF electrode after 10 cycles at 2 mA–2 mA h cm−2 with corresponding tomographic images (c, f and i) revealed by X-ray CT. (j) The comparison of porosity and structure (the volume of metallic Zn) for pristine ZF, cycled ZF–agarose, and cycled ZF electrode obtained by X-ray CT. | ||
For typical 3D electrodes, the intrinsic complexity of their 3D architecture often impedes the complete permeation of liquid electrolytes, especially when using lean electrolytes. This results in surface-limited nucleation and growth, which ultimately clogs the porous surface, as observed here for the cycled, unmodified ZF electrode under SEM (Fig. 3g). When examined more closely (Fig. 3h), flaky zinc deposition and a substantial byproduct accumulation also become evident. Such uncontrolled zinc growth and side reactions also result in structural deterioration (∼2.5% structure loss compared to the pristine one, Fig. 3j). X-ray CT images in Fig. 3i and S9c† reveal that the closed pores tend to arise at the edge of the cycled ZF electrode, which can be attributed to the differential electrolyte wetting due to non-uniform pressure distribution. Besides, in the absence of protective agarose gel, the ZF electrode exhibits pronounced corrosion, as revealed by the unique X-ray diffraction signature of the layered zinc hydroxy sulfate (Fig. S10†). Clearly, the simple combination of 3D structure with a liquid electrolyte not only causes restricted electrolyte infiltration and mass transport limited unstable deposition but also enhances corrosion and undermines the inherent merits of 3D electrodes due to blocked surface porosity.
The morphology of agarose@ZF and agarose@Zn electrodes were also probed by SEM after 10 cycles under the same condition for comparison. Compared to the cycled ZF electrode with liquid electrolyte (Fig. 3g and h), the agarose@Zn presents a more compact surface (Fig. S11a†), coupled with some needle and flake-like zinc surface features (Fig. S11b†). The suppressed dendritic growth and corrosion, enabled by the agarose hydrogel electrolyte, align with the enhanced cyclability observed for the agarose@Zn symmetric cell (Fig. S6†). Similarly, for the agarose@ZF electrode, the agarose hydrogel enables a smoother morphology with reduced Zn4(OH)6SO4·xH2O byproduct accumulation, but the surface-confined deposition still blocks the pores after cycling (Fig. S12†). These observations further validate that the incorporation of agarose gel into the bulk structure of the Zn foam facilitates 3D mass transport, enabling the complete engagement of 3D structured electrodes in the electrochemical process.
COMSOL simulation was performed to rationalize the Zn2+ transport at the electrode–electrolyte interface for the bare ZF and ZF–agarose electrodes. Firstly, the pristine ZF and ZF–agarose in Fig. S13,†4a and b represent the 2D geometries of the base model, which defines the top boundary as the cathode (electrolyte/cathode interface) and the bottom boundary as the anode (electrolyte/anode interface). In the pristine ZF model, owing to limited infiltration, the electrolyte wets the foam interior as a thin layer, whereas in the ZF–agarose model, the interdigitated design facilitates the electrolyte percolation through the entire pore. Regarding the above, the 3D simulation model is constructed on the premise of the 2D model. With respect to the ZF electrode, the electrolyte is restricted to permeating inward along the inner walls of the electrode pores (Fig. 4c and e). This is illustrated by the confined transport route within the model. In contrast, the ZF–agarose electrode features bottom-to-top channels that allow Zn2+ to access the entire interior of the electrode via the bulk-incorporated agarose gel electrolyte, as shown in Fig. 4d and f. According to the COMSOL simulations, the limited Zn-ion transport in the bulk of the ZF electrode creates a significant flux gradient between the electrolyte/anode interface and the interior of the electrode (Fig. 4c). This pronounced ion flux accumulation at the interface will cause Zn2+ to preferentially nucleate and grow on the surface framework, which can eventually block the pores. For the ZF–agarose electrode, the facile and interconnected transport path enables uniform Zn2+ migration and distribution (Fig. 4d). As a result, the abundant channels established by the integrated gel electrolyte network in ZF–agarose also ensure homogeneous electric field distribution throughout the interior of the electrode (Fig. 4e), in contrast to the ZF electrode (Fig. 4f). The ion flux and electric field uniformity facilitate uniform zinc plating/stripping throughout the porous 3D structure. The 2D simulation (Fig. S13†) also presents ion flux and electric field distribution similar to the 3D model among the ZF–agarose and ZF electrodes. This distinct zinc deposition behavior for the electrolyte-integrated 3D Zn electrode vis-à-vis the conventional 3D electrode with liquid electrolyte is illustrated in Fig. 4g. As depicted, in contrast to the surface-confined deposition in conventional 3D electrode–electrolyte assembly, which blocks the porous surface structure and undermines the benefits of the high specific surface area of the 3D matrix, the integrated design enables uniform zinc cycling across the whole 3D structure, which helps maintain the porous and high surface area electrode architecture. To further demonstrate the feasibility of the ZF–agarose electrode, full Zn-ion cells were assembled with Zn0.25V2O5·nH2O48 (ZVO, Fig. S14 and S15†) cathode (termed as ZVO‖ZF–agarose cell). From the cyclic voltammetry (CV) curves in Fig. 5a, it is apparent that the ZVO‖ZF–agarose cell shows a similar current–potential profile as the reference ZVO‖ZF cell with several reduction and oxidation peaks, confirming the multistep redox behavior during charge and discharge.48 However, the ZVO‖ZF–agarose cell displays a smaller polarization and higher current response. The comparison of rate performance between the two cells in Fig. 5b shows that the integrated anode yields specific capacities of 290, 255, 197, 113 mA h g−1 at 0.5, 1, 2, 4C (1C = 250 mA h g−1; in a narrow 0.5–1.4 V window), respectively. In agreement with the CV observation, the capacity of the ZVO‖ZF cell at different rates is significantly inferior to that of the ZF–agarose electrode, especially at high rates (2C, 4C). In fact, the ZVO‖ZF cell maintains a capacity of only 18 mA h g−1 at a 4C rate and struggles to recover the original capacity after the current rate is reduced. The smaller polarization and notably improved rate capability for the ZVO‖ZF–agarose cell can primarily be attributed to uniform zinc dissolution/deposition, which leads to uniform pressure distribution across the electrode interface. Besides, reduced zinc corrosion for the ZF–agarose electrode also benefits the full-cell performance.
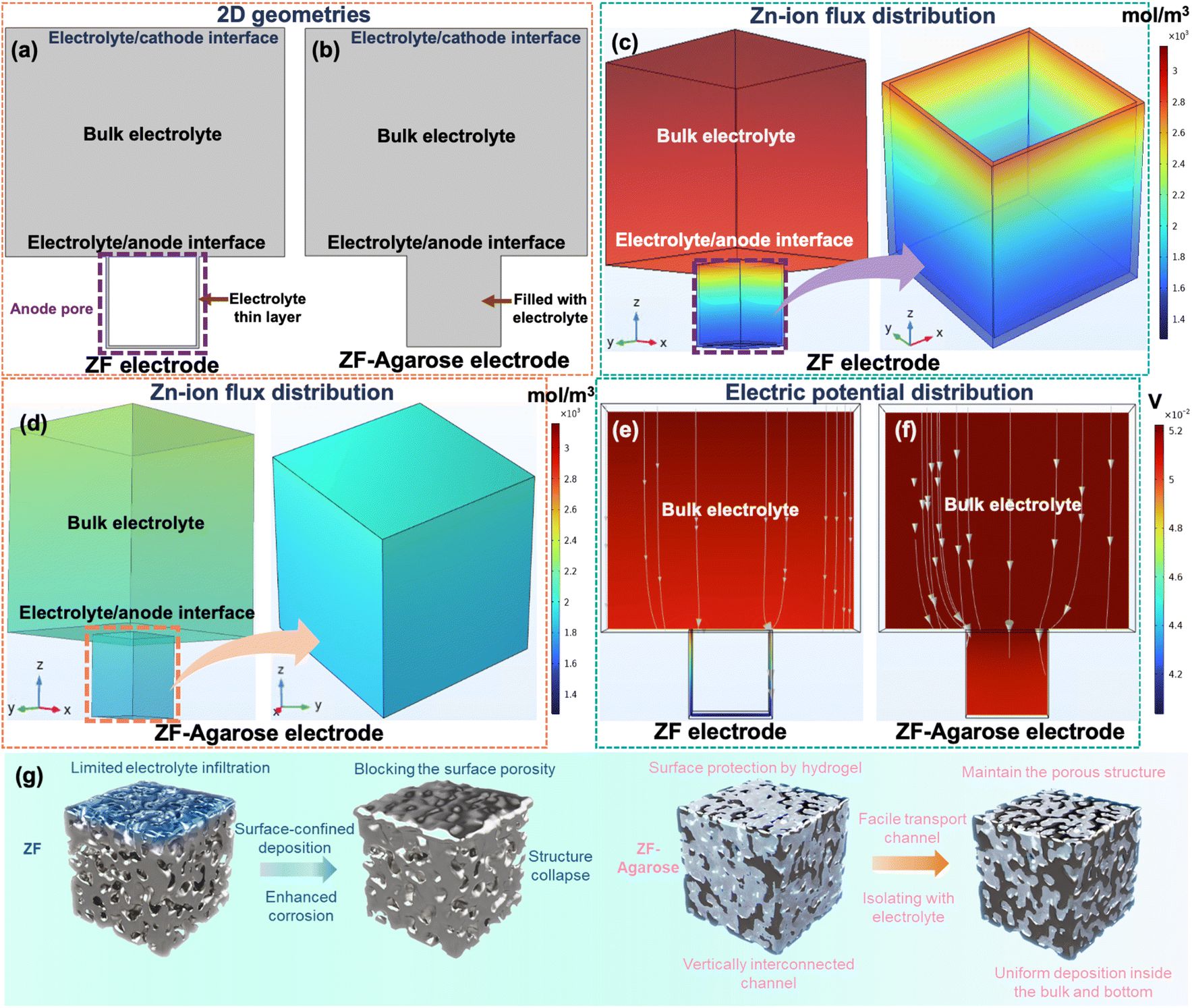 | ||
| Fig. 4 The COMSOL simulation of ion flux and electric field distribution and schematic representation of deposition regimes. The basic 2D simulation model for (a) ZF and (b) ZF–agarose electrodes. The Zn-ion flux distribution of (c) ZF and (d) ZF–agarose electrode. The electric potential distribution of (e) ZF and (f) ZF–agarose electrodes. (g) Schematic diagram of ZF and ZF–agarose electrode for illustration of different Zn-deposition manners. | ||
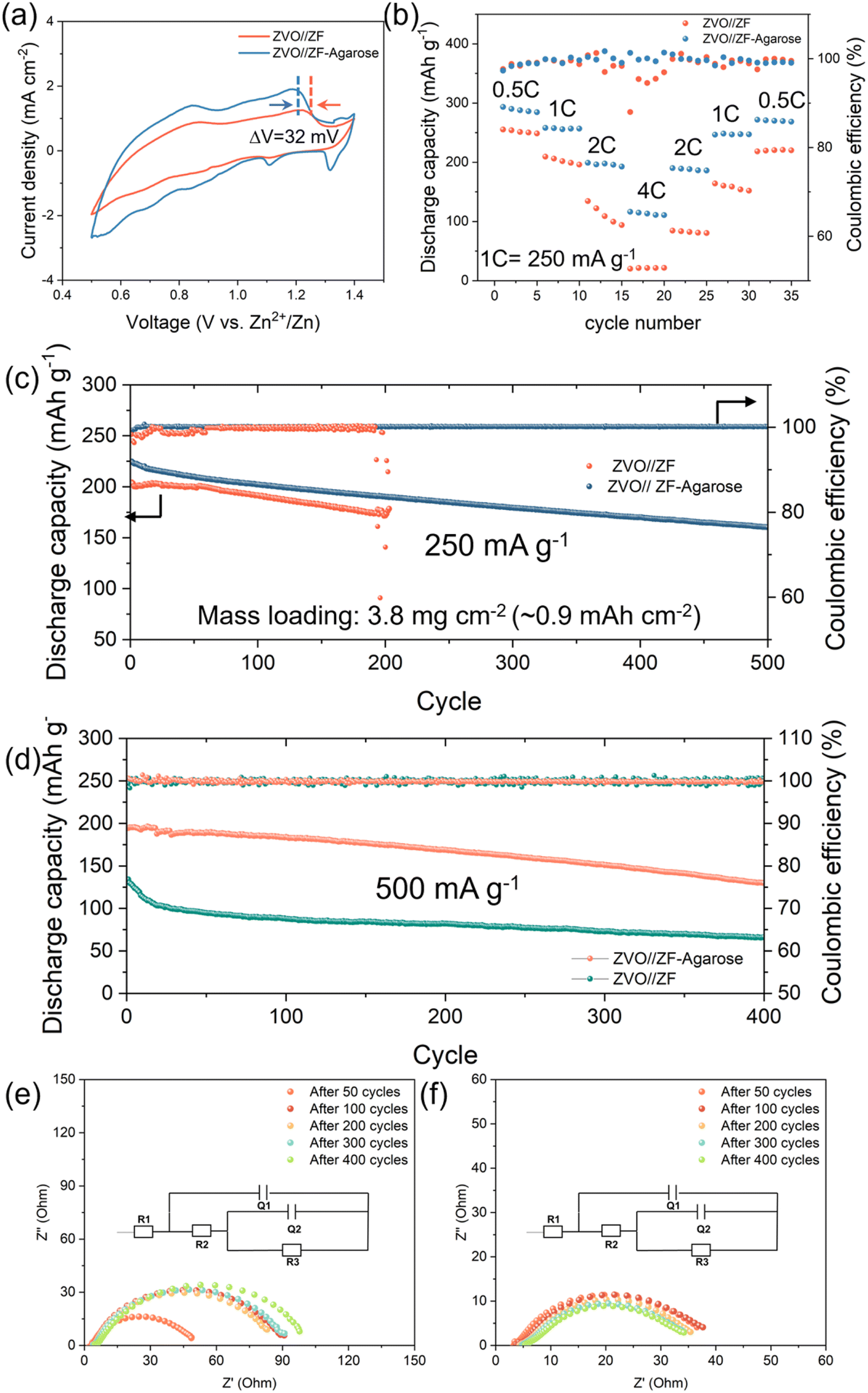 | ||
| Fig. 5 The comparison of full-cell performance for the two electrodes. (a) The CV profiles of ZVO‖ZF–agarose and ZVO‖ZF full-cell at a scan rate of 0.5 mV s−1. (b) Rate performance of ZVO cathode with ZF and ZF–agarose electrode under variable current rates. (c) The long-term cyclability for ZVO‖ZF–agarose cell and ZVO‖ZF cell at (c) 250 mA g−1 and (d) 500 mA g−1. The evolution of EIS spectra for (e) ZVO‖ZF full-cell and (f) ZVO‖ZF–agarose full-cell obtained at 500 mA g−1. The inset in (d and e) shows the equivalent circuit model used to fit the impedance data. Further information is provided in the experimental details. | ||
Further assessment was done by long-term cyclability testing with a moderately high active ZVO loading (∼4–5 mg cm−2) corresponding to a starting capacity of ∼1 mA h cm−2. As shown in Fig. 5c and S16, the ZVO‖ZF–agarose cell displays a high initial specific capacity of 225 mA h g−1 at a 1C rate and notably high cycling stability with 161 mA h g−1 after 500 cycles at an average CE of 99.9%. However, the cell with the bare ZF electrode suffers from a rather fast capacity loss (Fig. S17†) with a low average CE (98.1%) during cycling and experiences short-circuit failure only after 204 cycles (Fig. 5c). This is consistent with the half-cell cycling data, albeit with an extended cycling time stemming from a lower (∼1 mA h cm−2) cycling capacity. When the current density is increased to 500 mA g−1 (Fig. 5d), the ZVO‖ZF cell displays a starting capacity of 140 mA h g−1 – significantly lower than that of the ZVO‖ZF–agarose cell (195 mA g−1) – and experiences a pronounced capacity fading within the initial 20 cycles due to pore closure and corrosion at the anode and resultant sluggish kinetics (Fig. 5e). Conversely, by leveraging the facile 3D mass transport and protective effects of the agarose in the integrated ZF–agarose anode, the ZVO‖ZF–agarose cell demonstrates excellent long-term cycling performance, accompanied by stable impedance evolution during cycling (Fig. 5f).
Conclusion
Moving away from the regular 3D zinc anode–liquid electrolyte-soaked separator membrane configuration, which cannot avail the benefit of high surface area 3D zinc, electrolyte interdigitation is introduced as a facile strategy for a more effective 3D anode design. Using agarose as a model biopolymer, its hydrogel-integrated zinc foam anode is demonstrated to enable complete electrolyte percolation within the porous and tortuous structure, which ensures 3D mass transport and uniform zinc plating/stripping involving the whole structure as opposed to the uppermost surface limited zinc cycling for the regular design. Thus, while the regular designs leads to the blockage of the surface pores and rough morphology evolution with cycling and hence inflict hastened short-circuit events, the electrolyte-interdigitated design maintains the porosity and structural integrity, resulting in prolonged cycling stability. Suppressed zinc corrosion by the integrated hydrogel also contributes to the enhanced deposition kinetics and extended rechargeability of the 3D zinc and AZIB full-cells under demanding currents/capacities, which is relevant for translation development.It is reasonable to infer that the interdigitated 3D zinc anode design can be achieved with most hydrogel and 3D zinc structure or current collectors, and improving the hydrogel's zinc-ion conductivity and its zinc corrosion inhibition capability can further boost the stability of the 3D anode.
Experimental details
Materials
Agarose (low electroendosmosis), zinc acetate (≥98%), ZnSO4·7H2O (99.5%), and V2O5 (99%) were all obtained from Sigma-Aldrich.Preparation of ZF–agarose electrode
Agarose powder was mixed with H2O at a concentration of 6 wt%, which was then heated up to 90 °C under stirring for 1 h to obtain a transparent pre-hydrogel solution. The Zn foam (diameter: 11 mm) was cleaned by ultrasonication in acetone and soaked into the pre-hydrogel solution under stirring and heating for 5 min. Finally, the as-prepared electrode was removed from the solution and cooled to room temperature for 30 min. Then, it was immersed into the 3 M ZnSO4 electrolyte for 24 h.Preparation of agarose@Zn and agarose@ZF electrode
Once the transparent pre-hydrogel solution was formed, it was cast onto a Petri dish and cured at room temperature for 30 min to obtain the hydrogel. The agarose hydrogel was then soaked into a 3 M ZnSO4 electrolyte for 24 h, and punched out to obtain the agarose hydrogel electrolyte. Finally, the hydrogel electrolyte was directly used for Zn foil and Zn foam, namely in agarose@Zn and agarose@ZF, configurations.Preparation of ZVO cathode
20 mg reduced graphene oxide (rGO) was dispersed in 50 mL of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/acetone (volume) mixture by ultrasonication. Then, 2 mmol V2O5 and 1.3 mmol of zinc acetate were directly added to the former solution and stirred for 1 h. After that, the obtained solution was transferred into a Teflon-lined stainless-steel autoclave (100 mL; Parr Instrument) and held at 180 °C for 48 h. The final products were collected by filtration and washed further with water and ethanol three times.
:1 water/acetone (volume) mixture by ultrasonication. Then, 2 mmol V2O5 and 1.3 mmol of zinc acetate were directly added to the former solution and stirred for 1 h. After that, the obtained solution was transferred into a Teflon-lined stainless-steel autoclave (100 mL; Parr Instrument) and held at 180 °C for 48 h. The final products were collected by filtration and washed further with water and ethanol three times.
Electrochemical measurement
The ZVO cathode was mixed with conductive carbon (Super P®, Timcal) and styrene butadiene rubber/sodium carboxyl methyl cellulose (SBR/CMC) binder at a weight ratio of 70:25:5 in DI water (solvent). The cathode mixture was ultrasonicated for 1.5 h, and free-standing electrode discs were obtained by vacuum filtration. After 12 h vacuum drying at 80 °C, 1 cm2 electrode coins with an average active ZVO loading of ∼2–4 mg cm−2 were punched out for full-cell testing.
The zinc plating/stripping studies were performed in a Swagelok® type cell in Zn‖Zn symmetric configuration by using a binder-free glass fiber filter paper (GF/A and GF/D was used for agarose–ZF and ZF symmetric cell, respectively, to balance the inter-electrode distance of ∼300 μm after pressing) with 50 μL 3 M ZnSO4 electrolyte. Cell assembly and all other procedures were carried out in ambient air. The galvanostatic cycling studies were performed on the LAND CT3002AU cycler. The electrochemical impedance spectroscopy (VMP-3, BioLogic) was performed in a symmetric (Ti‖Ti or Zn‖Zn) configuration in the 1 MHz to 0.1 Hz frequency range by applying an AC amplitude of 10 mV. The electrolyte resistance was acquired from the intercept of the Nyquist plot on the real (impedance, Z) axis and the ionic conductivity was calculated according to the following equation:
 | (1) |
Physicochemical characterization
The corrosion byproduct formation in different electrolytes was investigated by X-ray diffraction (PANalytical Xpert Multipurpose X-ray Diffraction System (MPD)) with Cu Kα radiation. The patterns were recorded in a 2θ window of 5 and 80° at a scanning speed of 0.04° s−1. The morphology of the Zn foils before/after cycling and resting in electrolytes was probed using FEI Nova NanoSEM 230 field emission scanning electron microscope and corresponding tomography information was obtained by X-ray CT (HeliScan Micro CT-2). Fourier transform infrared (FTIR) spectra of all electrolytes were acquired in the range 4000–600 cm−1 using a Tensor 27 FTIR spectrometer. Raman spectroscopy data of various electrolytes were collected on a Renishaw inVia Raman spectrometer equipped with a 532 nm argon ion laser.Computational methods
In this study, we employed the finite element method using COMSOL Multiphysics 6.2 software for simulations. We developed two models featuring different anode types: pristine ZF (Zn foam) and ZF–agarose, in both 2D and 3D geometries (refer to Fig. S13† and 4). Each model designated the top boundary as the cathode, the bottom boundary as the anode (highlighting a single pore on the Zn anode surface), and the middle section as the electrolyte. In the pristine ZF model, the electrolyte partially fills the anode pore with a thin layer, while in the ZF–agarose system, the electrolyte uniformly occupies the entire pore; the uniform distribution is critical for controlling Zn ion flux and local current density. This approach allowed a detailed examination of electrochemical interactions between the electrode and electrolyte. The simulation utilized the Nernst–Planck equation to track electrodeposition accurately and analyze Zn-ion flux distribution. Boundary conditions were governed by the Butler–Volmer equation, describing the electrochemical reaction kinetics for Zn deposition. The specified key material properties include the diffusion coefficients of Zn2+ and SO42− in the non-agarose system, which are 6.4459 × 10−8 m2 s−1 and 9 × 10−8 m2 s−1, respectively, with an initial Zn2+ ion concentration set at 3000 mol m−3. In the agarose system, the diffusion coefficients of Zn2+ and SO42− are 6.02 × 10−8 m2 s−1 and 9 × 10−9 m2 s−1, respectively, and the initial concentration of 2100 mol m−3. Our investigation focused on analyzing the initial Zn ion flux and corresponding electrolyte current density distribution at the anode/electrolyte interface. In the non-agarose scenario (see Fig. 4a and c), simulations revealed significant variations in Zn ion concentration and electrolyte potential along the trench length, potentially leading to irregular Zn deposition, dendrite formation, corrosion, and material depletion. In contrast, in the ZF–agarose model (see Fig. 4b and d), minimal changes in Zn ion flux and electrolyte potential during deposition indicated controlled and uniform Zn deposition. This model effectively homogenized electric field distribution, Zn2+ ion flux, and local current density, thereby reducing risks of short-circuit failures from dendritic growth and corrosion reactions. Consequently, the ZF–agarose electrode exhibited significant Zn reversibility. However, our simulation, though insightful, simplifies the electrodeposition process and does not encompass real-world complexities such as potential side reactions.Data availability
All the data relevant for the current submission entitled “Electrolyte Interdigitation Enables Stable High Areal Capacity Cycling of the 3D Zn Electrode” are available upon reasonable request directly from the authors via email to the corresponding author Dipan Kundu.Author contributions
Yuan Shang: conceptualization, methodology, investigation, data curation, formal analysis, validation, and writing – original draft; Ravindra Kokate: methodology, investigation, and writing – original draft; Patrick Tung: investigation and formal analysis; Haoyin Zhong: visualization; Erlantz Lizundia: conceptualization, methodology, and writing – review & editing; Francisco J. Trujillo: resources, methodology, and supervision; Priyank Kumar: methodology, supervision, writing – review & editing, and funding acquisition; Dipan Kundu: conceptualization, methodology, resources, data analysis, validation, writing – original draft and review & editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. K. and P. K. acknowledge the Australian Government – Dept. of Education's support through the AEA seed grant (AE230100320). We also acknowledge the Mark Wainwright Analytical Center at UNSW for access and support with the characterization work presented here.References
- J. Cao, D. Zhang, X. Zhang, Z. Zeng, J. Qin and Y. Huang, Energy Environ. Sci., 2022, 15, 499–528 RSC.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 Search PubMed.
- P. Yadav, N. Sanna Kotrappanavar, P. B. Naik, H. K. Beere, K. Samanta, N. S. Reddy, J. S. Algethami, M. Jalalah, F. A. Harraz and D. Ghosh, ACS Appl. Energy Mater., 2023, 6, 1799–1809 CrossRef CAS.
- D. Singh, Y. Hu, S. S. Meena, R. Vengarathody, M. Fichtner and P. Barpanda, Chem. Commun., 2023, 59, 14391–14394 RSC.
- Y. Li, B. Liu, J. Ding, X. Han, Y. Deng, T. Wu, K. Amine, W. Hu, C. Zhong and J. Lu, Batteries Supercaps, 2021, 4, 60–71 CrossRef CAS.
- Y. Shang and D. Kundu, Joule, 2023, 7, 244–250 CrossRef.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- W. Du, E. H. Ang, Y. Yang, Y. Zhang, M. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- Q. Yang, Q. Li, Z. Liu, D. Wang, Y. Guo, X. Li, Y. Tang, H. Li, B. Dong and C. Zhi, Adv. Mater., 2020, 32, 2001854 Search PubMed.
- C. Wang, X. Zeng, J. Qu, J. M. Cairney, Q. Meng, P. J. Cullen and Z. Pei, Matter, 2023, 6, 3993–4012 CrossRef CAS.
- Y. Wang, T. Wang, S. Bu, J. Zhu, Y. Wang, R. Zhang, H. Hong, W. Zhang, J. Fan and C. Zhi, Nat. Commun., 2023, 14, 1828 Search PubMed.
- X. Zhang, Y. Liu, S. Wang, J. Wang, F. Cheng, Y. Tong, L. Wei, Z. Fang and J. Mao, Energy Storage Mater., 2024, 70, 103471 CrossRef.
- S. Liu, J. (Pimm) Vongsvivut, Y. Wang, R. Zhang, F. Yang, S. Zhang, K. Davey, J. Mao and Z. Guo, Angew. Chem., Int. Ed., 2023, 135, e202215600 CrossRef.
- Q. Zhang, J. Luan, X. Huang, Q. Wang, D. Sun, Y. Tang, X. Ji and H. Wang, Nat. Commun., 2020, 11, 3961 CrossRef CAS PubMed.
- W. Zhang, Q. Zhao, Y. Hou, Z. Shen, L. Fan, S. Zhou, Y. Lu and L. A. Archer, Sci. Adv., 2021, 7, eabl3752 CrossRef CAS PubMed.
- H. He, L. Zeng, D. Luo, J. He, X. Li, Z. Guo and C. Zhang, Adv. Mater., 2023, 35, 2211498 CrossRef CAS PubMed.
- J. Zheng, Z. Huang, Y. Zeng, W. Liu, B. Wei, Z. Qi, Z. Wang, C. Xia and H. Liang, Nano Lett., 2022, 22, 1017–1023 CrossRef CAS PubMed.
- S.-B. Wang, Q. Ran, R.-Q. Yao, H. Shi, Z. Wen, M. Zhao, X.-Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef CAS PubMed.
- W. Liu, Z. Chen, G. Zhou, Y. Sun, H. R. Lee, C. Liu, H. Yao, Z. Bao and Y. Cui, Adv. Mater., 2016, 28, 3578–3583 CrossRef CAS PubMed.
- C. Li, X. Shi, S. Liang, X. Ma, M. Han, X. Wu and J. Zhou, Chem. Eng. J., 2020, 379, 122248 CrossRef CAS.
- X. Fan, H. Yang, X. Wang, J. Han, Y. Wu, L. Gou, D.-L. Li and Y.-L. Ding, Adv. Mater. Interfaces, 2021, 8, 2002184 CrossRef CAS.
- M. Zhang, P. Yu, K. Xiong, Y. Wang, Y. Liu and Y. Liang, Adv. Mater., 2022, 34, 2200860 CrossRef CAS PubMed.
- Y. Zeng, X. Zhang, R. Qin, X. Liu, P. Fang, D. Zheng, Y. Tong and X. Lu, Adv. Mater., 2019, 31, 1903675 CrossRef PubMed.
- Y. Mu, Z. Li, B. Wu, H. Huang, F. Wu, Y. Chu, L. Zou, M. Yang, J. He, L. Ye, M. Han, T. Zhao and L. Zeng, Nat. Commun., 2023, 14, 4205 CrossRef CAS PubMed.
- G. Zhang, X. Zhang, H. Liu, J. Li, Y. Chen and H. Duan, Adv. Energy Mater., 2021, 11, 2003927 CrossRef CAS.
- J. Zhou, F. Wu, Y. Mei, Y. Hao, L. Li, M. Xie and R. Chen, Adv. Mater., 2022, 34, 2200782 CrossRef CAS PubMed.
- Q. Cao, Z. Pan, Y. Gao, J. Pu, G. Fu, G. Cheng and C. Guan, Adv. Funct. Mater., 2022, 32, 2205771 CrossRef CAS.
- S. Ni, S. Tan, Q. An and L. Mai, J. Energy Chem., 2020, 44, 73–89 CrossRef.
- J. Yun, B.-K. Park, E.-S. Won, S. H. Choi, H. C. Kang, J. H. Kim, M.-S. Park and J.-W. Lee, ACS Energy Lett., 2020, 5, 3108–3114 CrossRef CAS.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- Y. Zuo, K. Wang, S. Zhao, M. Wei, X. Liu, P. Zhang, Y. Xiao and J. Xiong, Chem. Eng. J., 2022, 430, 132996 Search PubMed.
- N. Shang, K. Wang, M. Wei, Y. Zuo, P. Zhang, H. Wang, Z. Chen, D. Zhong and P. Pei, Adv. Funct. Mater., 2023, 33, 2303719 CrossRef CAS.
- Y. Zuo, K. Wang, M. Wei, P. Zhang, S. Zhao, P. Pei, H. Wang, Z. Chen and N. Shang, Chem. Eng. J., 2023, 452, 139301 CrossRef CAS.
- E. Lizundia and D. Kundu, Adv. Funct. Mater., 2021, 31, 2005646 CrossRef CAS.
- A. Rüther, A. Forget, A. Roy, C. Carballo, F. Mießmer, R. K. Dukor, L. A. Nafie, C. Johannessen, V. P. Shastri and S. Lüdeke, Angew. Chem., Int. Ed., 2017, 129, 4674–4678 CrossRef.
- K. Bertula, L. Martikainen, P. Munne, S. Hietala, J. Klefström, O. Ikkala and Nonappa, ACS Macro Lett., 2019, 8, 670–675 CrossRef CAS PubMed.
- G. K. Mehta, S. Kondaveeti and A. K. Siddhanta, Polym. Chem., 2011, 2, 2334–2340 RSC.
- P. Sun, W. Liu, D. Yang, Y. Zhang, W. Xiong, S. Li, J. Chen, J. Tian and L. Zhang, Electrochim. Acta, 2022, 429, 140985 CrossRef CAS.
- H. Yang, Z. Chang, Y. Qiao, H. Deng, X. Mu, P. He and H. Zhou, Angew. Chem., Int. Ed., 2020, 59, 9377–9381 Search PubMed.
- L. Zeng, H. He, H. Chen, D. Luo, J. He and C. Zhang, Adv. Energy Mater., 2022, 12, 2103708 CrossRef CAS.
- Q. Cao, Y. Gao, J. Pu, X. Zhao, Y. Wang, J. Chen and C. Guan, Nat. Commun., 2023, 14, 641 Search PubMed.
- B. Wu, B. Guo, Y. Chen, Y. Mu, H. Qu, M. Lin, J. Bai, T. Zhao and L. Zeng, Energy Storage Mater., 2023, 54, 75–84 CrossRef.
- Z. Bie, Q. Yang, X. Cai, Z. Chen, Z. Jiao, J. Zhu, Z. Li, J. Liu, W. Song and C. Zhi, Adv. Energy Mater., 2022, 12, 2202683 CrossRef CAS.
- Q. Cao, H. Gao, Y. Gao, J. Yang, C. Li, J. Pu, J. Du, J. Yang, D. Cai, Z. Pan, C. Guan and W. Huang, Adv. Funct. Mater., 2021, 31, 2103922 CrossRef CAS.
- Z. Wu, Y. Li and J. Liu, Small Methods, 2024, 8, 2300660 CrossRef CAS PubMed.
- Y. Shang, V. Kundi, I. Pal, H. N. Kim, H. Zhong, P. Kumar and D. Kundu, Adv. Mater., 2024, 36, 2309212 CrossRef CAS PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 1–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta09006c |
| This journal is © The Royal Society of Chemistry 2025 |