
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation behaviors of SiOCN coatings with tunable carbon content on stainless steel at 800 °C in Ar, Ar + H2O, and air atmospheres†
Mohammad Hassan Shirani
Bidabadi
 a,
Hyeon Joon
Choi
b and
Kathy
Lu
*ab
a,
Hyeon Joon
Choi
b and
Kathy
Lu
*ab
aDepartment of Mechanical and Materials Engineering, University of Alabama at Birmingham, Birmingham, AL 35294, USA. E-mail: klu@uab.edu
bDepartment of Materials Science and Engineering, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA
First published on 17th April 2025
Abstract
This work investigates the influence of carbon content on the thermal stability of SiOCN coatings derived from polysilazanes on AISI 304 stainless steel after 100 h exposure at 800 °C to Ar, Ar + H2O, and air. Two polysilazanes with different ratios, including carbon-free (perhydropolysilazane, PHPS, (P)) and carbon-rich (Durazane 1800 (D)), were used as the coating materials. The carbon-free (P) coating exhibited superior oxidation resistance, forming a protective SiON layer. In contrast, the carbon-rich coating (D) resulted in duplex oxide nodules, indicating breakaway oxidation. The coatings with intermediate carbon contents showed a mixed behavior; areas with intact coatings adhered well to the substrate and provided significant protection, while localized spallation exposed the substrate to oxidation. The study highlights how PHPS and Durazane 1800 mixtures alter the precursor's behavior during pyrolysis and thermal treatment at 800 °C. These findings provide insight into the design of polymer-derived ceramic coatings for structural components in harsh environments such as power generation and aerospace.
1 Introduction
Damage resulting from metal oxidation at elevated temperatures in harsh environments leads to significant economic losses.1,2 This is particularly critical for applications such as gas turbines or components operating at high temperatures, such as 800 °C.3 Due to their excellent thermal and chemical stability, ceramic coatings have long played a crucial role in enhancing the stability of metals used at high temperatures. These coatings exhibit high resistance to aggressive atmospheres, strong adhesion to metal substrates, favorable microstructures, and resistance to cracking during high-temperature operation.4,5In recent years, polymer-derived ceramics (PDCs) have been the subject of extensive research due to their compositional versatility and easy-to-process nature, which enable them to replace traditional ceramic coatings.6–8 The general approach involves coating the substrate with a polymer precursor based on relatively inexpensive techniques such as spin coating, dip coating, and spray coating.9–14 The polymer coating can then be transformed into desired ceramics through pyrolysis at high temperatures. PDC coatings offer a range of adjustable properties, allowing for tuning and engineering of microstructures, phases, and properties through a selection of various coating synthesis conditions, pyrolysis atmospheres, thermal treatment temperatures, and precursors.1,7,15,16 Various polymer-derived ceramic coatings such as SiOC, SiCN, and SiOCN have been investigated to enhance the corrosion and oxidation resistance of metal substrates.6,7,13,15–18
Silicon oxycarbonitride (SiOCN), derived from the pyrolysis of polysilazane, finds wide applications in various fields, such as gas barrier coatings for food/medical packaging systems, and corrosion/oxidation-resistant coatings due to its high-temperature stability, mechanical robustness, and chemical durability. The compositions, microstructures, and properties of SiOCN coatings can be controlled by adjusting the compositions of the polymer precursors or by regulating the pyrolysis environment and temperature, which is crucial for optimizing performance in diverse environments.
Carbon-rich SiOCN ceramics consist of a nanocomposite composed of silicon oxycarbonitride (SiOxCyNz) and an excess of carbon, which is well known as free carbon (Cfree) in the literature.19,20 The characteristics of the final SiOCN coating, including composition and microstructure, influence high-temperature thermal stability. The presence of carbon within the SiOCN matrix can play a vital role in determining these properties. By adjusting the ratio, composition, and structure of carbon within the SiOCN matrix, the coating's thermal stability can be controlled. Silicon oxynitride (SiON) coatings have been reported to enhance oxidation resistance at high temperatures in various atmospheres.3,21 In the case of silicon carbonitride (SiCN), the presence of carbon not only improves coating oxidation resistance but also enhances resistance to carburization.7,13,22–24 For SiOCN, the form of free carbon (excess carbon) undergoes structural changes with temperature variations.25Fig. 1 shows a schematic representation of the structural evolution of free carbon within the SiOCN matrix at various temperatures. At low temperatures, excess carbon is present as carbon clusters (basic structural units, BSU). Between 600 °C and 900 °C, new C sp2 sites are formed at the expense of C sp3 atoms bonded to Si atoms, which are formed by Si–C bond cleavages.26 However, such sp2 C transforms into a graphitic carbon ribbon at a high temperature of >1000 °C.27
 | ||
| Fig. 1 Schematic representation of the structural evolution of free carbon (Cfree) within the SiOCN ceramic matrix with increasing temperature. | ||
This study investigates how the carbon content within the coatings influences the reactions during cross-linking (hydrosilylation, transamination, dehydrocoupling, and vinyl polymerization) and pyrolysis and ultimately the ceramic yield and thermal stability of SiOCN coatings. Additionally, oxidation products are analyzed by exposing the SiOCN-coated 304 stainless steel samples to different atmospheres (Ar, Ar + H2O, and air) at 800 °C for 100 hours.
2 Experimental
2.1 Coating preparation
AISI 304 stainless steel coupons with the composition given in Table S1,† cut to dimensions of 2.5 cm (L) × 1 cm (W) × 0.1 cm (H), were used as the substrate. Before applying any coating, the substrates were cleaned by sonication in acetone and ethanol for 15 minutes, and then dried at 120 °C for 2 h. Two commercially available polysilazanes (Fig. 2), perhydropolysilazane (PHPS, [SiH2NH]n, 20 wt% of PHPS in dibutyl ether, Iota Silicone Oil Co., Ltd, China) and Durazane 1800 ((SiCHCH2NHCH3)0.2n(SiHCH3NH)0.8n, Merck KGaA, Germany), were used as the coating precursors. Six different prepolymers were prepared by mixing these two materials in various ratios, as shown in Table S2.† | ||
| Fig. 2 Schematic structural representations of the two commercial polysilazane precursors used in this study: (a) Durazane 1800 and (b) perhydropolysilazane (PHPS). | ||
Dip coating was used to apply the polymer precursor mixture onto the steel substrate coupons. The coating thickness was influenced by both the withdrawal speed and the viscosity of the solution. To achieve a similar thickness (1 μm) across all mixtures, toluene was added to the mixtures (with a viscosity of 5 cp) to match the viscosity of pure PHPS (P10/D0). Subsequently, the cleaned substrates were dip-coated in the prepared solutions at a withdrawal speed of 3 mm s−1 in an argon (Ar) atmosphere glove box.28 All P/D coatings were crosslinked on a 250 °C hot plate inside the glove box for 24 h.
The crosslinked coating coupons were pyrolyzed in a tube furnace (1370-20 Horizontal Tube Furnace, CM Furnaces Inc., Bloomfield, NJ) in an Ar atmosphere at a rate of 1 °C min−1 to 800 °C, held at this temperature for 2 h, and then cooled back to room temperature at the same rate. After pyrolysis, pores were observed on the surface of the coatings with mixtures of the two polysilazane precursors. Those porous coatings were dip coated again at a withdrawal speed of 3 mm s−1 in an Ar atmosphere using a solution of 10 wt% PHPS in toluene, followed by the same crosslinking procedures performed for the first coating layer. Then, the crosslinked coating samples were pyrolyzed at 800 °C for 2 h in a nitrogen atmosphere with a heating and cooling rate of 1 °C min−1 using the same tube furnace. This top layer filled only the pores distributed throughout the first layer and did not form a separate layer. A detailed explanation of the application of this top layer can be found in our previous study.17 The total thickness of all formed coatings was measured using a profilometer (VK-3000, Keyence, Japan) and was in the range of 1.0 to 1.1 μm.
2.2 Thermal treatment
To evaluate the stability of the SiOCN coating at high temperatures, the coated substrates were thermally treated at 800 °C for 100 h in various atmospheres (Ar, Ar + 20% H2O, and air). The selected atmospheres mimic actual high-temperature oxidation conditions of stainless steel and provide insights for fundamental understanding of the coating behaviors. The thermal treatment in air was carried out in a muffle furnace (Thermo Scientific, Model No. F48015-60, Waltham, MA). In contrast, other thermal treatments were conducted in a tube furnace (1370-20 Horizontal Tube Furnace, CM Furnaces Inc., Bloomfield, NJ). The flow rate of the gas mixture was maintained at ∼1.2 L min−1 at 1 atm pressure. The Ar + 20% H2O mixture was obtained by heating a water container to 60 °C and then passing Ar gas through it. Afterwards, the Ar + 20% H2O mixture was flown through the tube furnace in the same way as the pure Ar gas.2.3 Characterization
The thermal profile of the coating materials was obtained by thermogravimetric analysis using a TGA (NETZSCH STA 449 F3 Jupiter). The coating materials (Table S2†) were crosslinked at 250 °C. Then, the samples (approximately 16 mg) were heated at a rate of 1 °C min−1 from room temperature to 800 °C under an ultra-high purity Ar atmosphere with a 50 ml min−1 flux and cooled down at a rate of 1 °C min−1. A scanning electron microscope (SEM, JEOL IT-500HR, JEOL Ltd., Japan) equipped with an energy-dispersive X-ray spectroscopy (EDS) detector was employed to evaluate the uniformity and composition of the coatings. SEM images were captured at an acceleration voltage of 5 kV. Grazing incidence X-ray diffraction (GIXRD) patterns were collected using an Empyrean diffractometer (Malvern Panalytical, Netherlands) with Cu Kα radiation (λ = 1.5406 Å), operated at 40 kV and 40 mA. The incident angle was fixed at 2°, with a scan rate of 2° min−1, a step size of 0.05°, and a 2θ range of 10–65°. The XRD pattern of 304 stainless steel was included as a substrate reference. The surface composition of the coatings was evaluated using a PHI 5000 VersaProbe II (Physical Electronics, USA) equipped with a monochromatic Al Kα X-ray source (1486.6 eV) operated at 25 W. The spectra were collected in the survey mode with a pass energy of 187.85 eV and in the high-resolution mode with a pass energy of 23.5 eV. All binding energies were calibrated with reference to the C 1s peak at 284.8 eV. The spectra were processed and deconvoluted using Multipak software (v9.7, Physical Electronics) with the Gaussian–Lorentzian peak shape. The carbon vibrational modes were determined using Raman spectroscopy (XploRA PLUS, Horiba, Ltd., Japan) with a wavelength of 532 nm within the spectral range of 500–3000 cm−1.For scanning transmission electron microscopy EDS (STEM-EDS) analysis, cross-sectional samples were prepared using a focused ion beam (FIB) lift-out technique (Helios 5 UX DualBeam, Thermo Fisher Scientific, USA). The STEM-EDS observations were performed using a Talos 200i TEM (Thermo Fisher Scientific, USA) with an operating voltage of 200 kV.
3 Results
3.1 Characteristics of the as-prepared coatings
Fig. 3 shows the SEM images of the as-made coatings obtained from carbon-free PHPS (P10/D0), carbon-rich Durazane 1800 (P0/D10), and a mixture of PHPS and Durazane 1800 (P4/D6) precursors. After pyrolysis, a crack-free, dense, and uniform coating layer was obtained for carbon-free (Fig. 3a) polymer precursors. As shown in Fig. 3b, small circular features were observed, which may be associated with minor outgassing during the pyrolysis process and the subsequent topological variations. Point EDS analysis of these specific regions revealed elemental compositions similar to the surrounding matrix, indicating they are not distinct phases or impurities. Pyrolysis of PHPS and Durazane 1800 mixtures (P2/D8, P4/D6, P6/D4, and P8/D2) resulted in porous coatings, although the coatings were still uniform. The average pore size was determined to be approximately 0.5 μm for the mixtures with low and high concentrations of one of the polysilazane precursors, i.e., P2/D8 and P8/D2, and 0.7 μm for intermediate PHPS or Durazane 1800 contents, i.e., P6/D4 and P6D4. Pore observations after pyrolysis for a mixture of carbon-free and carbon-rich precursors with different ratios were also reported by others.17,22 A top PHPS layer was applied to those porous coatings to fill/seal the residual pores, as shown in Fig. 3c and d, for P4/D6, as a representative example. | ||
| Fig. 3 SEM surface images of (a) P10/D0, (b) P0/D10, and (c) P6/D4 without the PHPS top layer and (d) P6/D4 with the PHPS top layer on the AISI 304 substrate after pyrolysis. | ||
Fig. S1† shows the XPS results for different coating samples. All were mainly composed of Si, O, C, and N elements. The XPS spectra (Fig. S1†) showed no significant difference for P6/D4 and P8/D2. Similar XPS spectra were also observed for P4/D6 and P2/D8. Four representative samples namely P0/D10, P4/D6, P6/D4, and P10/D0 were selected for the high-resolution XPS spectra. The near-surface elemental compositions for P10/D0, P6/D4, P4/D6, and P0/D10 are listed in Table 1. The highest N values were observed for P10/D0 (the highest proportion of PHPS), and the amount decreased as the content of Durazane 1800 increased. Although the total carbon content in sample P6/D4 is slightly higher than that in P0/D10, which was not initially expected, one can observe that the C/N atomic ratio decreases consistently with the addition of PHPS. This trend aligns with the expected outcome, as PHPS contributes more nitrogen. XPS detected carbon (2.7 at%) on the surface of the P10/D0 sample. Since PHPS does not contain carbon in its backbone, it might be attributed to a combination of adventitious carbon from the environment, residual dibutyl ether solvent, and possible trace contamination from the inert gas atmosphere during pyrolysis. Similar carbon signals have been reported in the literature for PHPS-derived ceramics, often attributed to environmental exposure and residual organic species.22
| Composition | P10/D0 | P6/D4 | P4/D6 | P0/D10 |
|---|---|---|---|---|
| Si | 34.62 | 29.73 | 31.33 | 30.83 |
| O | 57.59 | 56.45 | 64.51 | 60.32 |
| N | 5.08 | 3.97 | 1.19 | 1.68 |
| C | 2.71 | 9.85 | 2.97 | 7.17 |
The high-resolution Si 2p, O 1s, and N 1s XPS spectra of P0/D10, P4/D6, P6/D4, and P10/D0 are shown in Fig. 4. The binding energies of all deconvoluted peaks in the core-level regions, along with their relative areas for the representative samples, are summarized in Table 2. The Si 2p spectra for all samples showed a broad peak centered around 101–103 eV, corresponding to SiO2C2/SiOyN4−y, SiO3C, and SiO4 units29–31 (Fig. 4a1–a4). The O 1s spectra (Fig. 4b1–b4) of all samples can be divided into three peaks at around 531, 532, and 533 eV, corresponding to O–C, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and O–Si bonding,32–34 respectively. The peak centered around 398–401 eV in the N 1s spectra can be assigned to Si–N bonds in SiOyN4−y.30,35 In the C 1s spectra (Fig. 4d1–d4), the dominant peak is located at ∼284.7 eV, which corresponds primarily to C–C/CC bonding, likely arising from methyl and vinyl groups in the Durazane containing samples,22 while the same C 1s signal in the P10/D0 sample might be attributed to adventitious carbon.30,34 The smaller peaks at ∼286.5, 282.5, and 289 eV can be attributed to CO, C–Si, and O–CO/N–CO, respectively. The C 1s spectra in Fig. 4 and Table 2 suggest that the formation of SiC bonds becomes more visible by increasing the Durazane content from P10/D0 to P0/D10 samples. The results in Table 2 and Fig. 4 indicate oxidation of silicon species for all samples. Besides, Fig. 4 shows that nitrogen is bonded within the oxidized silicon, and its amount increases in samples with higher PHPS content. The XPS data suggest that the composition of P0/D10 is mainly SiOCN, and by increasing PHPS up to P10/D0, the coatings tend to form SiON.
C, and O–Si bonding,32–34 respectively. The peak centered around 398–401 eV in the N 1s spectra can be assigned to Si–N bonds in SiOyN4−y.30,35 In the C 1s spectra (Fig. 4d1–d4), the dominant peak is located at ∼284.7 eV, which corresponds primarily to C–C/CC bonding, likely arising from methyl and vinyl groups in the Durazane containing samples,22 while the same C 1s signal in the P10/D0 sample might be attributed to adventitious carbon.30,34 The smaller peaks at ∼286.5, 282.5, and 289 eV can be attributed to CO, C–Si, and O–CO/N–CO, respectively. The C 1s spectra in Fig. 4 and Table 2 suggest that the formation of SiC bonds becomes more visible by increasing the Durazane content from P10/D0 to P0/D10 samples. The results in Table 2 and Fig. 4 indicate oxidation of silicon species for all samples. Besides, Fig. 4 shows that nitrogen is bonded within the oxidized silicon, and its amount increases in samples with higher PHPS content. The XPS data suggest that the composition of P0/D10 is mainly SiOCN, and by increasing PHPS up to P10/D0, the coatings tend to form SiON.
 | ||
| Fig. 4 High resolution XPS spectra of P0/D10 (a1–d1), P4/D6 (a2–d2), P6/D4 (a3–d3), and P10/D0 (a4–d4): (a1–a4) Si 2p, (b1–b4) O 1s, (c1–c4) N 1s, and (d1–d4) C 1s spectra of the coating samples after pyrolysis at 800 °C. | ||
| P0/D10 | P4/D6 | P6/D4 | P10/D0 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Position (eV) | Area (%) | Position (eV) | Area (%) | Position (eV) | Area (%) | Position (eV) | Area (%) | ||
| Si 2p | SiO4 | 103.65 | 94.47 | 103.61 | 65.48 | 103.85 | 94.35 | 103.69 | 95.82 |
| SiO3C | 103.19 | 4.16 | 103.17 | 32.79 | 102.12 | 1.21 | 101.96 | 0.68 | |
| SiO2C2/SiOyN4−y | 102.0 | 1.36 | 101.68 | 1.73 | 101.92 | 4.44 | 101.86 | 3.51 | |
| O 1s | O–Si | 532.9 | 75.42 | 532.68 | 78.81 | 533.06 | 61.06 | 533.03 | 72.81 |
| OC |
533.46 | 20.42 | 533.28 | 18.8 | 533.07 | 33.77 | 533.37 | 23.11 | |
| O–C | 531.18 | 4.16 | 531.17 | 2.39 | 530.41 | 5.17 | 531.34 | 4.08 | |
| N 1s | SiOyN4−y | 398.92 | 78.97 | 398.38 | 88.5 | 398.69 | 98.1 | 398.74 | 97.58 |
| N–C | 401.14 | 21.03 | 399.28 | 11.5 | 400.03 | 1.9 | 400.45 | 2.42 | |
| C 1s | CC/C–C |
284.75 | 76.7 | 284.62 | 97.78 | 284.64 | 87.88 | 284.74 | 87.21 |
| CO |
286.47 | 13.39 | 287.47 | 0.46 | 286.82 | 5.24 | 287.05 | 9.46 | |
| C–Si | 282.63 | 4.8 | 281.8 | 1.59 | 282.73 | 1 | 281.24 | 0 | |
| O–CO/N–CO |
288.96 | 5.1 | 288.09 | 0.17 | 289.26 | 5.88 | 289.42 | 3.33 | |
Fig. 5 shows the XRD patterns (a) and Raman spectra (b) of selected coatings after pyrolysis. The XRD pattern of the P0/D10 sample reveals the formation of Cr-rich oxide phases, suggesting inward oxygen diffusion and subsequent selective oxidation of chromium during pyrolysis. This can be attributed to the low oxygen potential under the Ar atmosphere, allowing Cr to preferentially oxidize beneath the coating. In contrast, no such oxide phases are observed in the P6/D4 and P10/D0 samples, indicating that the addition of PHPS to the system may improve barrier properties and suppress oxygen diffusion. Similar trends were observed in other compositions; P2/D8 showed only weak Cr-rich oxide peaks, while P4/D6 and P8/D2 exhibited XRD patterns resembling those of P6/D4 and P10/D0, as shown in the ESI (Fig. S2).† The XRD peaks of 304 stainless steel are also included as a reference to identify substrate contributions and for comparison. The presence of the substrate peaks in all samples further confirms the thin nature of the coatings.
 | ||
| Fig. 5 (a) XRD patterns and (b) Raman spectra of the coated samples P0/D10, P6/D4, and P10/D0 after pyrolysis at 800 °C. | ||
The Raman spectra of P0/D10 and P6/D4 show prominent peaks at approximately 1350 cm−1 and 1588 cm−1, corresponding to the disorder-induced D and ordered graphite G bands of carbon, respectively. The absence of distinct D and G peaks in P10/D0 suggests low carbon content in this sample, in agreement with the XPS results (Table 1). The prominent peaks near 590 and 1000 cm−1, which are attributed to Si–O–Si stretching vibrations,36 are indicative of the presence of silicon oxide. The increase of the ID/IG intensity ratio from 1.1 for P0/D10 to 1.32 for P6/D4 suggests a higher degree of disorder in the carbon network, likely due to the presence of a more amorphous carbon phase in P6/D4 compared to P0/D10. Furthermore, the Raman shift observed in the 2100–2300 cm−1 region across all samples could stem from the absorption of atmospheric nitrogen, water vapor in the air, and dissolved atmospheric CO2 (known to produce a peak in the 2000–2325 cm−1 region36) because of sample storage conditions.
Fig. 6 shows representative results of the thermal stability of the crosslinked polymer coatings, which were subjected to thermogravimetric analysis (TGA) in an argon atmosphere. A slight mass loss of 1.5–2% for P0/D10 and P6/D4 samples and 0.5% for P10/D0 below 200 °C could be attributed to the evaporation of residual solvents for P0/D10 and P6/D4 and moisture loss for P10/D0. The P0/D10 sample showed a mass loss of about 5.6% from 200 to 440 °C due to the polymerization of the vinyl group, and the weight loss accelerated rapidly up to 550 °C because of dehydrogenation and transamination reactions (Fig. 7), in agreement with previous results reported for Durazane 1800.37 A mass loss of 2.6% between 550 and 700 °C suggested polymer-to-ceramic transformation, and the sample stabilized around 700 °C with a final mass retention of approximately 89%. The P6/D4 sample showed improved thermal stability compared to P0/D10, with a gradual mass loss starting around 250 °C. The final mass retention for P6/D4 was 92.6% at 800 °C, indicating 7.14% less mass loss. P10/D0 initially showed minimal mass loss (2.4%) up to around 450 °C, indicating its superior stability at lower temperatures to the other two compositions. However, from 450 to 800 °C, P10/D0 showed a substantial mass loss (10.8%), which might be due to the condensation of Si–OH groups.38,39
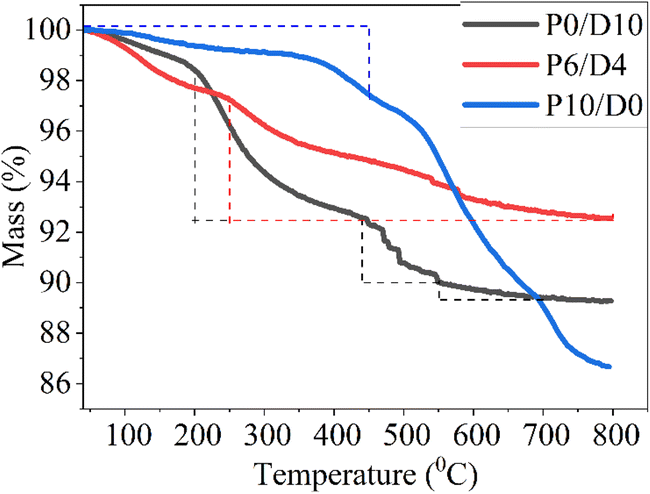 | ||
| Fig. 6 Mass change as a function of pyrolysis temperature for P10/D0, P6/D4, and P0/D10 samples in Ar with 1 °C min−1 heating rate, as measured by TGA. | ||
 | ||
| Fig. 7 Schematic illustration of the chemical transformations occurring during pyrolysis of the polymer precursors: (a) transamination, (b) dehydrogenation between Si–H groups, (c) dehydrogenation between Si–H and N–H groups, and (d) polymerization of the vinyl group. | ||
3.2 Oxidation behaviors at high temperatures
Photographs of the oxidized samples are shown in Fig. S3.†Fig. 8 shows the SEM surface morphology of the bare alloy and SiOCN-coated samples before and after oxidation in Ar, Ar + H2O, and air for 100 h. Uncoated samples formed uniform oxide layers in all oxidation environments. For all samples (regardless of the coating composition), the oxide surface was rougher in air and Ar–H2O compared to that in Ar, suggesting more oxide formation. This is due to air's higher oxygen partial pressure than Ar. The extensive oxide formation in Ar + H2O might be related to the dominant formation of volatile Cr-oxyhydroxide and hydrogen incorporation during oxidation, as reported in earlier studies.40–42 Additionally, findings reported in the literature43 indicate that the presence of water vapor in the atmosphere significantly increases the high-temperature oxidation rate of SiC + C ceramics. The P10/D0 sample (Fig. 8b) showed the smoothest surface with no scale spallation, suggesting that it is a protective material against oxidation of the steel substrate and is capable of effectively inhibiting the formation of volatile reaction products in H2O-containing environments, in agreement with our previous study.44 In both air and Ar + H2O environments, the P10/D0 and P8/D2 coatings showed similar surface morphologies (Fig. 8b and c), suggesting that adding Durazane up to 20 wt% does not significantly impact the oxidation resistance. However, coatings with higher Durazane contents, such as P6/D4, P4/D6, and P2/D8, with similar surface morphologies, showed clear signs of spallation, indicating that the effects of Durazane begin to manifest when the content reaches 40 wt%. Notably, no further significant differences were observed with Durazane additions beyond 40 wt%, up to 80 wt% (Fig. 8d–f). The pure Durazane sample (P0/D10) showed a different surface morphology with the appearance of oxide nodule formation, indicating a different oxidation behavior compared with the mixed precursor coatings (Fig. 8g). | ||
| Fig. 8 SEM surface morphologies of the samples before and after 100 h oxidation in Ar, Ar + H2O, and air at 800 °C: (a) uncoated 304 stainless steel, (b) P10/D0, (c) P8/D2, (d) P6/D4, (e) P4/D6, (f) P2/D8, and (g) P0/D10. | ||
We chose P10/D0 (as a representative for the P8/D2 and pure PHPS (P10/D0)), P6/D4 (as a representative for the P6/D4, P4/D6, and P2/D8 mixed coatings), and P0/D10 (pure Durazane) samples after oxidation, to further carry out microstructural analysis. Fig. S4† shows the SEM surface morphology combined with the EDS points (Table S3†) for P10/D0, P6/D4, and P0/D10 after 100 h oxidation in air. P10/D0 (Fig. S4a†) showed a relatively smooth surface morphology with some CrMn-rich oxide formation (point 7 in Fig. S4a and Table S3†). Since we did not observe any cracks or damage on the initial PHPS-based coating, these CrMn-rich oxides possibly formed in the areas where the coating had been damaged during thermal treatment. Others reported similar observations for the PHPS coating on 304 steel after oxidation at 900 °C.15 SEM-EDS analysis of the coating (point 8 in Fig. S4a and Table S3†) showed a significant amount of N (10.7 at%), with 32.6 at% silicon and 23.9 at% oxygen, suggesting the formation of SiON in this coating. Furthermore, the relatively low iron and chromium contents (7.8 at% Cr, 20.4 at% Fe) suggest that the oxide layer effectively protects the underlying alloy. The P6/D4 sample showed less oxidation resistance than P10/D0 due to localized regions of spallation. Nonetheless, the areas where the coating layer was still intact (point 6 in Table S3 and Fig. S4b†) provided some protection against further oxidation, as shown in the inset of Fig. S4b.† The spalled areas on the P6/D4 sample exposed the underlying material to the new oxidizing atmosphere, leading to the formation of a mixed oxide layer mainly containing Fe (24.6 at%) and Cr (17.8 at%), as shown in points 4 and 5 in Fig. S4b and Table S3.† Furthermore, a notable amount of Si (7.8 at%) suggests the formation of silicon-based oxides. The P0/D10 sample showed Fe-oxide nodule formation (point 1 in Fig. S4c and Table S3†) and spallation, indicative of substantial oxidation. The SEM-EDS results for points 2 and 3 (Fig. S4c and Table S3†) showed that the coating contained a notable amount of Fe (12.9 at%) and Cr (15.1 at%), suggesting interdiffusion between the alloy and the coating, which might cause the coating layer to be weakened.
XRD analysis (Fig. 9) of the coatings oxidized in air at 800 °C for 100 h reveals the formation of mixed oxide phases, including M2O3 with a corundum and M3O4 spinel structures where M = Fe, Cr, and/or FeCr. The most intense peaks for these oxides appear at the (104) and (311) planes, respectively. Notably, coatings with a higher PHPS content (e.g., P10/D0) show a systematic shift of these oxide peaks toward higher 2θ angles, indicating increased Cr incorporation into the oxide lattice. This Cr enrichment is known to enhance oxidation resistance by promoting the formation of stable and protective Cr-rich oxides. In contrast, carbon-rich coatings (e.g., P0/D10) exhibited less Cr-containing oxides, correlating with reduced oxidation resistance. Reflections from the underlying 304 stainless steel substrate are also evident in all patterns.
 | ||
| Fig. 9 XRD patterns of oxidized coatings (P10/D0, P6/D4, and P0/D10) and bare 304 stainless steel after exposure to air at 800 °C for 100 h. The (104) and (311) reflections, which are the most intense for M2O3 and M3O4, respectively, are highlighted in the inset for clarity. | ||
Fig. 10 shows the SEM cross-section images (obtained by FIB) after oxidation for the P0/D10 coating in Ar + H2O (Fig. 10a) and P6/D4 in air (Fig. 10b). P0/D10 formed thin Cr-rich (0.7 μm) layers and thick oxide nodules (2.8 μm), suggesting breakaway oxidation. The nodule oxides exhibited a duplex structure distinguished by SEM-EDS mapping: an outer Fe-rich region and an inner FeCr-rich region (Fig. 10a). Similar oxide morphology has been reported in the literature for stainless steel exposed to humid or carbon-rich environments.41,45 The FIB-SEM cross-section showed similar results for P0/D10 oxidized in air at 800 °C after 100 h. Fig. 10b shows the FIB-SEM cross-section of Fig. S4b† on spalled areas where a mixed FeCr oxide formed after oxidizing in air for 100 h. SEM-EDS mapping (Fig. 10b) of the spalled area revealed that the outermost layer contained Cr, Mn, Si, and Fe, while a Cr-rich oxide layer formed at the oxide/alloy interface.
 | ||
| Fig. 10 FIB-SEM cross-section images along with EDS maps after 100 h oxidation at 800 °C for (a) P0/D10 in Ar + H2O and (b) P6/D4 in air. | ||
Fig. 11 shows STEM-EDS analysis for the P0/D10 sample oxidized in air for 100 h. The STEM-HAADF image in Fig. 11a shows a clear separation between the coating and the substrate, suggesting a weak interface bonding formed on this sample. Fig. 11b shows the STEM-EDS mapping of region “A” in Fig. 11a, where Fe-rich oxide formation is observed beneath the coating. The EDS mapping shows that the coating mainly contains Si, O, and C with Cr within the coating and Mn accumulation at the top, indicating interdiffusion between the alloy and coating. This diffusion weakens the coating's protective capability by allowing oxygen to reach the substrate and form Fe-rich oxides. The high-resolution TEM-bright field (BF) images of regions “B” (within the coating) and “C” (Fe-rich oxide) with their corresponding selected area diffraction (SAD) patterns are shown in Fig. 11c–f. Fig. 11c and e indicate that the coating contains crystalline oxide phases within amorphous areas. The EDS analysis (Fig. 11g) shows that the substrate beneath the Fe-rich oxide layer is depleted in Cr. Furthermore, the EDS mapping in Fig. 11b and the EDS analysis in Fig. 11g confirm that the coating contains a high amount of carbon that diffuses into the alloy.
 | ||
| Fig. 11 P0/D10 coating sample treated at 800 °C in air for 100 h: (a) STEM high-angle annular dark-field (HAADF) image, (b) STEM HAADF with EDS mappings of the box “A” in (a), (c) high-resolution TEM bright field (BF) image of the box “B” in (b), (d) high-resolution TEM-BF image of the box “C” in (b), (e) the selected area diffraction (SAD) patterns from (c), (f) the SAD patterns from (d), and (g) EDS results from “1” marked in (a). | ||
The STEM-EDS mapping for the P6/D4 sample where the coating was still intact (see the area marked “6” in the inset of Fig. S4b†) is shown in Fig. 12. The outermost oxide layer above the coating is rich in Cr and Mn, indicating the diffusion of the steel alloy elements through the coating during the oxidation and reaction with the surrounding atmosphere. The coating contains C-, Si-, and Cr-rich regions. A protective Cr2O3 oxide layer formed beneath the coating. Carbide particles rich in Mo and Fe are also observed within the Cr2O3 oxide. The STEM-EDS results in Fig. 12 show a complex multi-phase; carbides plus protective oxides, due to the interdiffusion of the alloy and coating elements. The formation of protective Cr2O3 oxide layers, plus Si- and Cr-rich regions within the coating, prevents severe oxidation of the underlying substrate, although minor Fe diffusion from the substrate is observed, where there is an absence of Cr-rich, Mn-rich, or Si-rich zones.
 | ||
| Fig. 12 P6/D4 coating sample treated at 800 °C in air for 100 h: (a) BF and (b) HAADF STEM images with the corresponding EDS mappings. | ||
Fig. 13 shows TEM-EDS analysis for the P6/D4 sample where a mixed oxide layer formed on the spalled regions (see the area marked “4” in Fig. S4b†). An amorphous and partially crystalline (Am + PC)–Si oxide formed on top of an amorphous and partially crystalline (Am + PC)–SiCr-rich oxide layer, adjacent to which a Cr oxide formed, confirmed by the EDS mapping in Fig. 13b, high-resolution TEM-BF in Fig. 13c, SAD patterns in Fig. 13d and e, and EDS results in Fig. 13f and g for the areas marked “1” and “2” in Fig. 13a. It should be noted that the STEM/EDS analysis (not shown here) showed less carbon content (4.21 at%) than P0/D10 (79.4 at%) within the substrate beneath the coating, and the Cr content beneath the coating was 10.24 at%.
 | ||
| Fig. 13 (a) BF and (b) HAADF STEM images of the oxides formed on the top surface of the P6/D4 sample with the corresponding EDS mappings after 100 h oxidation at 800 °C in air, (c) high-resolution TEM-BF image of the box “A” in (a), (d and e) SAD patterns from the amorphous Si-rich oxide shown in (d) and the amorphous SiCr-rich oxide shown in (e), (f and g) EDS results from “1” marked in (a) shown in (f) and “2” marked in (a) shown in (g). Am + PC: amorphous and partially crystalline. | ||
The STEM-EDS images (Fig. 14) of the P10/D0 sample show that the coating was smooth and remained intact after the exposure, with no apparent delamination from the substrate. EDS mappings indicate that the coating is primarily SiON, with slight diffusion of Cr and Fe from the substrate into the coating at the coating/substrate interface. Nitrogen is present across the coating at a higher concentration compared to that in P0/D10 and P6/D4. The TEM results in Fig. 14 indicate that the SiON coating effectively acted as a diffusion barrier, preventing extensive oxidation of the underlying substrate.
 | ||
| Fig. 14 P10/D0 coating sample thermally treated at 800 °C in air for 100 h: (a) BF and (b) HAADF STEM images with the corresponding EDS mappings. | ||
4 Discussion
4.1 Coating formation
PHPS contains only Si, N, and H, with a –Si–N–Si– backbone, while Durazane 1800 contains vinyl and methyl groups and H attached to a –Si–N–Si– backbone (Fig. 2). Pyrolysis of carbon-rich (P0/D10) and carbon-free (P0/D10) coatings showed dense and crack-free layers. In contrast, mixtures of PHPS and Durazane 1800 (e.g., P4/D6) showed porosity after pyrolysis, and the porosity can be successfully mitigated by applying a PHPS top layer,17 ensuring a dense and uniform coating (Fig. 3). The composition of the pure PHPS (P10/D0) coating after pyrolysis showed SiON formation (in agreement with our previous work28) with higher Si and N values, indicative of enhanced nitrogen incorporation (Fig. 4, S1† and Table 1). Pyrolysis of the coatings including Durazane 1800 resulted in the formation of SiOCN with a higher C/Si ratio for P0/D10 than the coatings with precursor mixtures (e.g., P4/D6), as shown in the XPS results (Table 1). The decreasing C/Si ratio is likely related to increased N–CH3 bonds in the mixtures by adding PHPS. An NMR study of three polysilazanes by Gérardin et al.26 showed the breakdown of N–CH3 bonds during pyrolysis, leading to the release of CH4 gas and a corresponding reduction in the carbon content. This reaction also promotes the formation of new Si–N bonds at the expense of Si–C and Si–H bonds,26 which agrees with the decreasing C/Si ratios in the XPS results shown in Fig. 4, S1† and Table 1.Besides, the presence of carbon within pure Durazane 1800 and the mixtures of PHPS/Durazane coatings (even though less in the pure PHPS coating) is due to the vinyl groups in Durazane 1800.46 The lower ID/IG ratio for P0/D10 than P6/D4 in the Raman results (Fig. 5b) indicates a more ordered carbon network with predominantly sp2 hybridized carbon, which can be attributed to the high concentration of vinyl groups in Durazane 1800.47,48 During pyrolysis, these vinyl groups undergo polymerization at relatively low temperatures, forming carbon chains that transform into graphitic carbon.46 This process enhances the formation of sp2 carbon domains, contributing to the lower disorder observed in P0/D10. In contrast, the reduced vinyl group content in the P6/D4 mixture limits the extent of vinyl polymerization, while interactions between Si–H and Si–CH3 groups disrupt the formation of graphitic domains. This creates a more significant number of defects, as evidenced by the prominent D-band in the Raman spectra shown in Fig. 5b.
The ceramic yield of polysilazane precursors is influenced by their backbone structures and functional groups, which determine the reaction pathways during pyrolysis.49 In this study, the differences in ceramic yield among PHPS, Durazane 1800, and their mixtures highlight the impact of these structural variations (Fig. 6). For the pure PHPS (P10/D0), the pyrolysis reactions are likely dehydrocoupling and transamination involving the exchange of N–Si and redistribution of Si–H and Si–N bonds, resulting in the breaking and reforming of Si–N bonds.49,50 This process produces volatile fragments that evaporate during pyrolysis, lowering the ceramic yield. Despite these challenges, PHPS exhibits good thermal stability up to ∼350 °C, beyond which transamination reactions dominate, as reported in the literature.25,49 These combined effects explain the lower ceramic yield of PHPS despite its resistance to significant weight loss at moderate temperatures. When Durazane 1800 was added to PHPS, as in P6/D4, the introduction of vinyl and methyl functional groups significantly altered the pyrolysis behavior (Fig. 6 and 7). These functional groups stabilize the precursor by reducing the extent of transamination, thereby limiting the loss of oligomers. The vinyl groups, in particular, promote cross-linking and create a more robust polymer network. This cross-linked structure minimizes fragmentation and volatile loss during pyrolysis, improving ceramic yield.50 These observations align with the literature, which suggests that the vinyl group enhances cross-linking and stabilizes the structure under pyrolytic conditions.51 For pure Durazane 1800 (P0/D10), the predominant reaction at lower temperatures is hydrosilylation, involving Si–H and vinyl groups.25 This rapid reaction leads to the formation of carbosilane bridges, such as Si–C–Si or Si–C–C–Si. These bridges are highly stable and resist depolymerization mechanisms like transamination and redistribution, which are dominant in PHPS. As a result, P0/D10 showed minimal weight loss at higher temperatures. However, the early completion of hydrosilylation reactions and the subsequent loss of volatile species at lower temperatures contributed to a lower ceramic yield, particularly below 450 °C. Fig. 6 shows how carefully selecting precursor ratios and functional groups can effectively tailor the thermal and pyrolytic performance of polysilazane systems.
4.2 High temperature oxidation
High-temperature oxidation tests at 800 °C in Ar, Ar + H2O, and air reveal distinct oxidation mechanisms for the SiOCN coatings. In the inert Ar atmosphere, all coatings remained stable with minimal surface changes, highlighting the intrinsic thermal stability of the SiOCN matrix. However, oxygen and water vapor introduced reactive environments, significantly impacting the coatings' performance.The results of this study showed that the resistance of the coatings is primarily dictated by their carbon content. The carbon-free P10/D0 coating exhibited superior performance, forming an adhesive SiON oxide layer with a protective Cr-rich oxide formation in air and Ar + H2O (Fig. 8, 9, 14 and S4a†). The observed results in this study agree well with previously reported studies,1,15,44,52,53 which showed that the PHPS-derived coating on steels acts as a diffusion barrier layer against aggressive oxidizing species such as CO2, H2O, and O2. In contrast, the thermal stability and oxidation resistance of the SiOCN coatings were influenced by their carbon content, as evident in the behavior of the carbon-rich P0/D10 and intermediate-carbon P6/D4 coatings. Carbon-rich coatings (P0/D10) underwent breakaway oxidation, as evidenced by the formation of duplex oxide nodules with outer Fe- and inner FeCr-rich oxides (Fig. 9–13 and S4†). Besides, the formation of Fe oxides beneath the coating (Fig. 11) indicates that oxidant gases permeated through the coating during the thermal treatment; alloy elements diffused outward in the coating, and the oxidant gas diffused inward into the coating. At the coating surface, CO2/CO gas molecules might have built up through the following reactions:44,54–56
| CO2(g) + s(adsorption site on surface) → CO2–s | (1) |
| CO2–s → O–s + CO(g) | (2) |
| CO(g) → CO2(g) + C | (3) |
| C + H2O → CO + H2 | (4) |
| H2O + CO → CO2 + H2 | (5) |
| CO + H2 → H2O + C | (6) |
The self-diffusing alloy elements in their corresponding oxide (e.g., Cr in Cr2O3) might occur either via vacancy diffusion at the coating surface where oxygen activity is high or via interstitials at the coating/alloy interface where oxygen activity is low, as proposed by numerous authors.57–59 Thus, at the coating/steel interface, the metal interstitials are dominant defects, and the metal vacancies that arrive at this interface can be annihilated by the reaction in eqn (7):60,61
 | (7) |
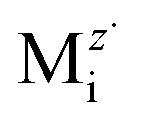 is the metal interstitial,
is the metal interstitial,  is the metal vacancy, MM is the metal on the normal metal lattice site in the oxide, and 0 ≤ Z ≤ higher oxidation state of the metal. Carbon within the coating might act as a sink for metal interstitials,62 reducing their mobility and thereby impeding the annihilation of metal vacancies at the interface.60,63 This accumulation of vacancies can coalesce into voids or micro-channels, weakening the coating and facilitating further ingress of oxidant gases. Such effects can be particularly pronounced in the P0/D10 coating, which contains a higher C/N ratio than P6/D4. The carbon released from eqn (1)–(7), or already present within the coating, diffuses into the underlying steel substrate, forming internal carbides. This carburization depletes chromium at the coating/substrate interface, lowering the chromium concentration below the critical threshold required to form a stable Cr2O3 protective layer. As the oxidation front progresses through these carburized regions, Cr within the carbides oxidizes, leaving behind Fe and Ni (Fig. 12). This localized depletion accelerates breakaway oxidation, leading to nodule formation, as observed for P0/D10, where Cr concentration at the interface was reduced to 1.71 at% (Fig. 11) after oxidation. The intermediate behavior observed for the P6/D4 coating can be attributed to its composition, which combines the properties of PHPS and Durazane 1800. The presence of Durazane 1800 introduces free carbon into the coating, which behaves similarly to the carbon-rich P0/D10 coating. This carbon promotes reactions that generate CO and CO2 gases and facilitates carburization and void formation at the coating/substrate interface. As a result, P6/D4 exhibits a mixed behavior, with intact regions forming protective Cr- and Si-rich (Fig. 13) oxide layers similar to PHPS-based coatings, while localized spallation and oxidation are driven by the carbon-related mechanisms observed in the Durazane-rich coatings. This balance between the protective effects of PHPS and the carbon-driven degradation from Durazane explains the intermediate performance of P6/D4.
is the metal vacancy, MM is the metal on the normal metal lattice site in the oxide, and 0 ≤ Z ≤ higher oxidation state of the metal. Carbon within the coating might act as a sink for metal interstitials,62 reducing their mobility and thereby impeding the annihilation of metal vacancies at the interface.60,63 This accumulation of vacancies can coalesce into voids or micro-channels, weakening the coating and facilitating further ingress of oxidant gases. Such effects can be particularly pronounced in the P0/D10 coating, which contains a higher C/N ratio than P6/D4. The carbon released from eqn (1)–(7), or already present within the coating, diffuses into the underlying steel substrate, forming internal carbides. This carburization depletes chromium at the coating/substrate interface, lowering the chromium concentration below the critical threshold required to form a stable Cr2O3 protective layer. As the oxidation front progresses through these carburized regions, Cr within the carbides oxidizes, leaving behind Fe and Ni (Fig. 12). This localized depletion accelerates breakaway oxidation, leading to nodule formation, as observed for P0/D10, where Cr concentration at the interface was reduced to 1.71 at% (Fig. 11) after oxidation. The intermediate behavior observed for the P6/D4 coating can be attributed to its composition, which combines the properties of PHPS and Durazane 1800. The presence of Durazane 1800 introduces free carbon into the coating, which behaves similarly to the carbon-rich P0/D10 coating. This carbon promotes reactions that generate CO and CO2 gases and facilitates carburization and void formation at the coating/substrate interface. As a result, P6/D4 exhibits a mixed behavior, with intact regions forming protective Cr- and Si-rich (Fig. 13) oxide layers similar to PHPS-based coatings, while localized spallation and oxidation are driven by the carbon-related mechanisms observed in the Durazane-rich coatings. This balance between the protective effects of PHPS and the carbon-driven degradation from Durazane explains the intermediate performance of P6/D4.
5 Conclusions
This work demonstrates the critical influence of carbon content on the high-temperature oxidation resistance and thermal stability of SiOCN coatings and on the oxidation behavior of AISI 304 stainless steel substrates at 800 °C in Ar, Ar + 20% H2O, and air environments for 100 h. Different SiOCN coatings were prepared from varying mixtures of perhydropolysilazane (PHPS) and Durazane 1800. Excellent oxidation resistance in the Ar atmosphere was observed for all coatings. However, they showed different oxidation behaviors in Ar–H2O and air depending on the PHPS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Durazane ratios. The carbon-free coatings (the lowest C/N atomic ratio) effectively protected stainless steel substrates by forming an adhesive SiON layer. In contrast, the carbon-rich coatings (the highest C/N atomic ratio) exhibited susceptibility to carburization and breakaway oxidation, limiting their protective capabilities. Intermediate compositions offered a balance between protection and degradation. The results highlight the importance of tailoring precursor composition to achieve desired coating properties for high-temperature applications. This study makes a significant contribution to the field of polymer-derived ceramics by elucidating the mechanisms governing the stability and oxidation resistance of SiOCN coatings in harsh environments, paving the way for improved designs in industrial applications such as gas turbines and high-temperature reactors.
:Durazane ratios. The carbon-free coatings (the lowest C/N atomic ratio) effectively protected stainless steel substrates by forming an adhesive SiON layer. In contrast, the carbon-rich coatings (the highest C/N atomic ratio) exhibited susceptibility to carburization and breakaway oxidation, limiting their protective capabilities. Intermediate compositions offered a balance between protection and degradation. The results highlight the importance of tailoring precursor composition to achieve desired coating properties for high-temperature applications. This study makes a significant contribution to the field of polymer-derived ceramics by elucidating the mechanisms governing the stability and oxidation resistance of SiOCN coatings in harsh environments, paving the way for improved designs in industrial applications such as gas turbines and high-temperature reactors.
Data availability
The datasets generated during the current study are available from the corresponding author upon reasonable request.Author contributions
Mohammad Hassan Shirani Bidabadi: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, writing – review and editing, data curation, visualization. Hyeon Joon Choi: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, writing – review and editing, visualization. Kathy Lu: resources, writing – review and editing, supervision, project administration, funding acquisition.Conflicts of interest
This manuscript has not been published elsewhere. All authors have approved the content and agreed to submit it for consideration for publication. There are no ethical/legal conflicts involved in the manuscript.Acknowledgements
This work was financially supported by the US Department of Energy (Grant Number DE-NE0008963).References
- M. Günthner, T. Kraus, A. Dierdorf, D. Decker, W. Krenkel and G. Motz, Advanced coatings on the basis of Si(C)N precursors for protection of steel against oxidation, J. Eur. Ceram. Soc., 2009, 29(10), 2061–2068 CrossRef.
- K. Wang, J. Unger, J. D. Torrey, B. D. Flinn and R. K. Bordia, Corrosion resistant polymer derived ceramic composite environmental barrier coatings, J. Eur. Ceram. Soc., 2014, 34(15), 3597–3606 CrossRef CAS.
- K. Bawane, K. Lu, Q. Li and R. Bordia, High temperature oxidation behaviors of SiON coated AISI 441 in Ar+O2, Ar+H2O, and Ar+CO2 atmospheres, Corros. Sci., 2020, 166, 108429 CrossRef CAS.
- K. N. Lee, Current status of environmental barrier coatings for Si-based ceramics, Surf. Coat. Technol., 2000, 133–134, 1–7 CAS.
- Y. Dong, K. Ren, Y. Lu, Q. Wang, J. Liu and Y. Wang, High-entropy environmental barrier coating for the ceramic matrix composites, J. Eur. Ceram. Soc., 2019, 39(7), 2574–2579 CrossRef CAS.
- K. Lu and D. Erb, Polymer derived silicon oxycarbide-based coatings, Int. Mater. Rev., 2017, 63(3), 139–161 CrossRef.
- M. Günthner, A. Schütz, U. Glatzel, K. Wang, R. K. Bordia, O. Greißl, W. Krenkel and G. Motz, High performance environmental barrier coatings, part I: passive filler loaded SiCN system for steel, J. Eur. Ceram. Soc., 2011, 31(15), 3003–3010 CrossRef.
- G. Barroso, Q. Li, R. K. Bordia and G. Motz, Polymeric and ceramic silicon-based coatings – a review, J. Mater. Chem. A, 2019, 7(5), 1936–1963 RSC.
- M. Fukushima, E. Yasuda, Y. Nakamura, H. Kita, H. Kawabata and Y. Tanabe, Preparation of a transition metal containing polymethylsilsesquioxane hybrids and silicon oxycarbide ceramics- the fabrication of coating and self-supported films, J. Ceram. Soc. Jpn., 2005, 113(1315), 210–215 CrossRef CAS.
- J. C. Pivin, P. Colombo, A. Martucci, G. D. Sorarù, E. Pippel and M. Sendova-Vassileva, Ion beam induced conversion of Si-based polymers and gels layers into ceramics coatings, J. Sol-Gel Sci. Technol., 2003, 26(1), 251–255 CrossRef CAS.
- F. Bauer, U. Decker, A. Dierdorf, H. Ernst, R. Heller, H. Liebe and R. Mehnert, Preparation of moisture curable polysilazane coatings: part I. Elucidation of low temperature curing kinetics by FT-IR spectroscopy, Prog. Org. Coat., 2005, 53(3), 183–190 CrossRef CAS.
- M. Günthner, Y. Albrecht and G. Motz, Polymeric and ceram1C-like coatings on the basis of SiN(C) precursors for protection of metals against corrosion and oxidation, in Advanced Ceramic Coatings and Interfaces: Ceramic Engineering and Science Proceedings, ed. D. Zhu, U. Schulz, A. Wereszczak and E. Lara-Curzio, The American Ceramics Society, 2006, ch. 26, pp. 277–284 Search PubMed.
- A. Klausmann, K. Morita, K. E. Johanns, C. Fasel, K. Durst, G. Mera, R. Riedel and E. Ionescu, Synthesis and high-temperature evolution of polysilylcarbodiimide-derived SiCN ceramic coatings, J. Eur. Ceram. Soc., 2015, 35(14), 3771–3780 CrossRef CAS.
- M. R. Mucalo, N. B. Milestone, I. C. Vickridge and M. V. Swain, Preparation of ceramic coatings from pre-ceramic precursors, J. Mater. Sci., 1994, 29(17), 4487–4499 CrossRef CAS.
- F. Riffard, E. Joannet, H. Buscail, R. Rolland and S. Perrier, Beneficial effect of a pre-ceramic polymer coating on the protection at 900 °C of a commercial AISI 304 stainless steel, Oxid. Met., 2017, 88(1), 211–220 CrossRef CAS.
- K. Wang, M. Günthner, G. Motz and R. K. Bordia, High performance environmental barrier coatings, part II: active filler loaded SiOC system for superalloys, J. Eur. Ceram. Soc., 2011, 31(15), 3011–3020 CrossRef CAS.
- H. J. Choi and K. Lu, A high compatibility SiOCN coating on stainless steel, J. Mater. Sci., 2023, 58(8), 3790–3801 CrossRef CAS.
- R. Bura, V. Girish C and R. M. Prasad, Enhanced corrosion protection performance using polysilazane-derived amorphous SiCN ceramic coating, Surf. Coat. Technol., 2024, 494, 131402 CrossRef CAS.
- H.-J. Kleebe, G. Gregori, Y. Blum and F. Babonneau, Evolution of C-rich SiOC ceramics part II. Characterization by high lateral resolution techniques: electron energy-loss spectroscopy, high-resolution TEM and energy-filtered TEM, Int. J. Mater. Res., 2006, 97, 710–720 Search PubMed.
- Y. D. Blum, D. B. MacQueen and H.-J. Kleebe, Synthesis and characterization of carbon-enriched silicon oxycarbides, J. Eur. Ceram. Soc., 2005, 25(2), 143–149 CrossRef CAS.
- M. Parchovianský, I. Petríková, G. Barroso, P. Švančárek, D. Galuskova, G. Motz and D. Galusek, Corrosion and oxidation behavior of polymer derived ceramic coatings with passive glass fillers on AISI 441 stainless steel, Ceram.-Silik., 2018, 62, 146–157 Search PubMed.
- A. Zambotti, M. Biesuz, R. Campostrini, S. M. Carturan, G. Speranza, R. Ceccato, F. Parrino and G. D. Sorarù, Synthesis and thermal evolution of polysilazane-derived SiCN(O) aerogels with variable C content stable at 1600 °C, Ceram. Int., 2021, 47(6), 8035–8043 CrossRef CAS.
- E. Zera, W. Nickel, S. Kaskel and G. D. Sorarù, Out-of-furnace oxidation of SiCN polymer-derived ceramic aerogel pyrolized at intermediate temperature (600–800 °C), J. Eur. Ceram. Soc., 2016, 36(3), 423–428 CrossRef CAS.
- H. J. Kleebe, H. Störmer, S. Trassl and G. Ziegler, Thermal stability of SiCN ceramics studied by spectroscopy and electron microscopy, Appl. Organomet. Chem., 2001, 15(10), 858–866 CrossRef CAS.
- P. Colombo, R. Riedel, G. D. Sorarù and H.-J. Kleebe, Polymer Derived Ceramics: From Nano-Structure to Applications, DEStech Publication, Inc., 2010 Search PubMed.
- C. Gérardin, F. Taulelle and D. Bahloul, Pyrolysis chemistry of polysilazane precursors to silicon carbonitride, J. Mater. Chem., 1997, 7(1), 117–126 RSC.
- P. Greil, Polymer Derived Engineering Ceramics, Adv. Eng. Mater., 2000, 2(6), 339–348 CrossRef CAS.
- H. J. Choi and K. Lu, Polysilazane-derived SiON coating on stainless steel weld for corrosion resistance, Mater. Chem. Phys., 2024, 315, 128988 CrossRef CAS.
- R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, 29Si NMR and XPS investigation of the structure of silicon oxycarbide glasses derived from polysiloxane precursors, MRS Online Proc. Libr., 1994, 346, 351 CrossRef CAS.
- J. Lu, J. Hu, H. Zhong, Y. Ren and L. Zhang, Synthesis of silicon oxycarbonitride nanosphere as cathode host for lithium–sulfur batteries, J. Alloys Compd., 2021, 860, 157903 CrossRef CAS.
- J. F. Moulder, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics, 1992, p. 261 Search PubMed.
- NIST X-ray Photoelectron Spectroscopy Database (SRD 20), Version 5.0, 2012, available, https://srdata.nist.gov/xps/ Search PubMed.
- A. Akkaya, B. Boyarbay, H. Çetin, K. Yıldızlı and E. Ayyıldız, A Study on the Electronic Properties of SiOxNy/p-Si Interface, Silicon, 2018, 10(6), 2717–2725 CrossRef CAS.
- H. Shi, A. Yuan and J. Xu, Tailored synthesis of monodispersed nano/submicron porous silicon oxycarbide (SiOC) spheres with improved Li-storage performance as an anode material for Li-ion batteries, J. Power Sources, 2017, 364, 288–298 CrossRef CAS.
- Y. K. Kim, H. S. Lee, H. W. Yeom, D.-Y. Ryoo, S.-B. Huh and J.-G. Lee, Nitrogen bonding structure in ultrathin silicon oxynitride films on Si(100) prepared by plasma nitridation, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70(16), 165320 CrossRef.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons Ltd, England, 3rd edn, 2004 Search PubMed.
- A. Qazzazie-Hauser, K. Honnef and T. Hanemann, Crosslinking behavior of UV-cured polyorganosilazane as polymer-derived ceramic precursor in ambient and nitrogen atmosphere, Polymers, 2021, 13(15), 1–20 CrossRef PubMed.
- I. S. Chuang and G. E. Maciel, A detailed model of local structure and silanol hydrogen bonding of silica gel surfaces, J. Phys. Chem. B, 1997, 101(16), 3052–3064 CrossRef CAS.
- N. Yang, W. Wang, W. Cai and K. Lu, Corrosion and tribocorrosion mitigation of perhydropolysilazane-derived coatings on low carbon steel, Corros. Sci., 2020, 177, 108946 CrossRef CAS.
- S. Chandra-ambhorn, T. Thublaor and P. Wiman, High temperature oxidation of AISI 430 stainless steel in Ar-H2O at 800 °C, Corros. Sci., 2020, 167, 108489 CrossRef CAS.
- W. J. Quadakkers, J. Żurek and M. Hänsel, Effect of water vapor on high-temperature oxidation of FeCr alloys, J. Miner. Met. Mater. Soc., 2009, 61(7), 44–50 CrossRef CAS.
- E. J. Opila, N. S. Jacobson, D. L. Myers and E. H. Copland, Predicting oxide stability in high-temperature water vapor, J. Miner. Met. Mater. Soc., 2006, 58(1), 22–28 CrossRef CAS.
- R. Naslain, A. Guette, F. Rebillat, S. Le Gallet, F. Lamouroux, L. Filipuzzi and C. Louchet, Oxidation mechanisms and kinetics of SiC-matrix composites and their constituents, J. Mater. Sci., 2004, 39(24), 7303–7316 CrossRef CAS.
- K. Bawane, K. Lu, Q. Li and R. Bordia, High temperature oxidation behaviors of SiON coated AISI 441 in Ar + O2, Ar+H2O, and Ar + CO2 atmospheres, Corros. Sci., 2020, 166, 108429 CrossRef CAS.
- T. D. Nguyen, J. Zhang and D. J. Young, Effects of Silicon on High Temperature Corrosion of Fe–Cr and Fe–Cr–Ni Alloys in Carbon Dioxide, Oxid. Met., 2014, 81(5), 549–574 CrossRef CAS.
- J. Seitz, J. Bill, N. Egger and F. Aldinger, Structural investigations of Si/C/N-ceramics from polysilazane precursors by nuclear magnetic resonance, J. Eur. Ceram. Soc., 1996, 16(8), 885–891 CrossRef CAS.
- K. Lu, D. Erb, K. Bawane and N. Yang, Comparison of traditional and flash pyrolysis of different carbon content silicon oxycarbides, J. Eur. Ceram. Soc., 2019, 39(10), 3035–3041 CrossRef CAS.
- M. G. Segatelli, A. T. N. Pires and I. V. P. Yoshida, Synthesis and structural characterization of carbon-rich SiCxOy derived from a Ni-containing hybrid polymer, J. Eur. Ceram. Soc., 2008, 28(11), 2247–2257 CrossRef CAS.
- E. Kroke, Y.-L. Li, C. Konetschny, E. Lecomte, C. Fasel and R. Riedel, Silazane derived ceramics and related materials, Mater. Sci. Eng., R, 2000, 26(4), 97–199 CrossRef.
- N. S. C. K. Yive, R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, Silicon carbonitride from polymeric precursors: thermal cross-linking and pyrolysis of oligosilazane model compounds, Chem. Mater., 1992, 4(1), 141–146 CrossRef.
- B. Boury, R. J. P. Corriu and W. E. Douglas, Poly(carbosilane) precursors of silicon carbide: the effect of cross-linking on ceramic residue, Chem. Mater., 1991, 3(3), 487–489 CrossRef CAS.
- I. Parchovianská, M. Parchovianský, D. Medveď and D. Galusek, Thermal shock behavior and high-temperature oxidation performance of PDC-based environmental barrier coatings on AISI 441 stainless steel, Surf. Coat. Technol., 2023, 474, 130074 CrossRef.
- W. Wang, S. Tamakloe, Z. Deng, L. Li, W. Cai and K. Lu, Effects of processing temperature on the corrosion and tribocorrosion resistance of perhydropolysilazane-derived coatings on AISI 304 steel, Surf. Coat. Technol., 2022, 439, 128463 CrossRef CAS.
- J. E. Antill, K. A. Peakall and J. B. Warburton, Oxidation of mild and low-alloy steels in CO2 based atmospheres, Corros. Sci., 1968, 8(9), 689–701 CrossRef CAS.
- N. M. Birks, G. H. Meier and F. S. Pettit, Introduction to the High Temperature Oxidation of Metals, Cambridge University Press, Cambridge, 2006 Search PubMed.
- D. J. Young, in High Temperature Oxidation and Corrosion of Metals, Elsevier, UK, 2nd edn, 2016, ch. 1, pp. 1–30 Search PubMed.
- M. Backhaus-Ricoult and R. Dieckmann, Defects and cation diffusion in magnetite (VII): diffusion controlled formation of magnetite during reactions in the iron-oxygen system, Ber. Bunsenges. Phys. Chem., 1986, 90(8), 690–698 CrossRef CAS.
- J. Töpfer, S. Aggarwal and R. Dieckmann, Point defects and cation tracer diffusion in (CrxFe1 − x)3 − δO4 spinels, Solid State Ionics, 1995, 81(3), 251–266 CrossRef.
- L. Martinelli and F. Balbaud-Célérier, Modelling of the oxide scale formation on Fe-Cr steel during exposure in liquid lead-bismuth eutectic in the 450–600 °C temperature range, Mater. Corros., 2011, 62(6), 531–542 CrossRef CAS.
- M. H. S. Bidabadi, C. Zhang, H. Chen and Z.-G. Yang, Temperature dependence of carbon deposits within oxide scale on CrMoV steel in atmospheric and supercritical CO2, Corros. Sci., 2022, 195, 109979 CrossRef CAS.
- S. Chandra-Ambhorn, T. Thublaor and C. Pascal, Thermodynamics and Kinetics of the High Temperature Oxidation of Stainless Steels, in High Temperature Corrosion of Stainless Steels: An Alain Galerie Festschrift, ed. S. Chandra-Ambhorn, Y. Wouters and W. Harnnarongchai, Trans Tech Publication Ltd, Switzerland, 1st edn, 2020, ch. 1, pp. 1–24 Search PubMed.
- S. Nishikawa, A. Tanaka and T. Yamaji, Reduction of transient boron diffusion in preamorphized Si by carbon implantation, Appl. Phys. Lett., 1992, 60(18), 2270–2272 CrossRef CAS.
- J. Robertson and M. I. Manning, Criteria for formation of single layer, duplex, and breakaway scales on steels, Mater. Sci. Technol., 1988, 4(12), 1064–1071 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08874c |
| This journal is © The Royal Society of Chemistry 2025 |