
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the origin of enhanced CO2 selectivity in amine-PIM-1 during mixed gas permeation†
Carmen
Rizzuto‡
 a,
Francesca
Nardelli‡§
b,
Marcello
Monteleone
a,
Lucia
Calucci
*bc,
C. Grazia
Bezzu
d,
Mariolino
Carta
d,
Elena
Tocci
*a,
Elisa
Esposito
a,
Giorgio
De Luca
a,
Bibiana
Comesaña-Gándara
e,
Neil B.
McKeown
f,
Bekir
Sayginer
g,
Peter M.
Budd
h,
Johannes C.
Jansen
a and
Alessio
Fuoco
*a
a,
Francesca
Nardelli‡§
b,
Marcello
Monteleone
a,
Lucia
Calucci
*bc,
C. Grazia
Bezzu
d,
Mariolino
Carta
d,
Elena
Tocci
*a,
Elisa
Esposito
a,
Giorgio
De Luca
a,
Bibiana
Comesaña-Gándara
e,
Neil B.
McKeown
f,
Bekir
Sayginer
g,
Peter M.
Budd
h,
Johannes C.
Jansen
a and
Alessio
Fuoco
*a
aInstitute on Membrane Technology (CNR-ITM), National Research Council of Italy, via P. Bucci 17/C, Rende, CS 87036, Italy. E-mail: alessio.fuoco@cnr.it; e.tocci@itm.cnr.it
bInstitute of Chemistry of Organometallic Compounds (ICCOM-CNR), via G. Moruzzi 1, Pisa, 56124, Italy. E-mail: lucia.calucci@pi.iccom.cnr.it
cCenter for Instrument Sharing, University of Pisa (CISUP), Lungarno Pacinotti 43/44, Pisa, 56126, Italy
dDepartment of Chemistry, Faculty of Science and Engineering, Swansea University, Swansea, SA2 8PP, UK
eIU CINQUIMA, University of Valladolid, Paseo Belén 5, Valladolid, 47011, Spain
fEaStCHEM, School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, Scotland EH9 3JF, UK
gDepartment of Chemistry, Faculty of Arts and Sciences, Kirsehir Ahi Evran University, Kirsehir, 40100, Turkiye
hDepartment of Chemistry, School of Natural Sciences, The University of Manchester, Manchester, M13 9PL, UK
First published on 28th April 2025
Abstract
Previously, it has been reported that amine-PIM-1, a polymer of intrinsic microporosity obtained by reduction of nitrile groups of PIM-1 to primary amine groups, shows enhanced CO2 selectivity during mixed gas permeation studies with respect to single gas measurements for gas pairs involving CO2. This distinct and potentially useful behaviour was ascribed to the affinity of CO2 for the polymer amine groups. Here, we demonstrate that enhanced selectivity originates from both CO2 physisorption and chemisorption. A combination of 13C and 15N solid-state NMR spectroscopic analyses of a CO2-loaded amine-PIM-1 membrane allowed the identification and quantitative determination of both chemisorbed and physisorbed species and the characterization of polymer-CO2 interactions. Experiments with 13C isotopically enriched CO2 unequivocally demonstrated the conversion of 20% of the NH2 groups into carbamic acids at 298 K and a CO2 pressure of 1 bar. Chemisorption was supported by the strong heat of CO2 adsorption for amine-PIM-1 that was estimated as 50 kJ mol−1. Molecular dynamics simulations with models based on the experimentally determined polymer structure gave a detailed description of intra- and interchain hydrogen bond interactions in amine-PIM-1 after chemisorption, as well as of the effect of chemisorption on polymer porosity and physisorption.
1. Introduction
Global warming poses an existential threat to humanity and Earth's ecosystem. This crisis demands urgent and transformative action to reduce greenhouse gas emissions and mitigate their catastrophic impact.1 Carbon capture and storage (CCS) is recognised as one of the most promising technologies capable of delivering net emissions reductions at the scale required to limit global temperature rise to below 2 °C.2 The intergovernmental panel on climate change estimates that CCS will need to contribute about one-sixth of the required CO2 emission reductions by 2050.3 CCS is particularly important for reducing emissions from industrial processes such as steel, cement, and chemicals production, which cannot be easily replaced by renewable technologies. These industries account for around 45% of the CO2 captured between 2015 and 2050 in a 2 °C scenario.3 The success of carbon capture depends on developing and optimising materials capable of selectively capturing, storing, and eventually releasing CO2. These materials must demonstrate high selectivity, capacity, and stability across various conditions.4 Key materials studied for carbon capture include adsorbents (e.g., metal–organic frameworks (MOFs) and activated carbon), which possess high surface area and porosity, enabling them to “physically” trap large quantities of CO2.5,6 Absorbents are also widely used for carbon capture. Typically, they are composed of amine-based solutions that can chemically react with CO2 to form a compound that can be later decomposed. Both these classes of materials are often functionalized to enhance their affinity for CO2.7Membranes for gas separation provide an alternative and potentially more energy-efficient technology for CCS.8,9 Permeable and selective materials can separate CO2 from gas mixtures through a combination of molecular sieving, realized through tailored pore sizes, and affinity for CO2, often achieved by incorporating carefully selected functional groups.10–12 However, developing membrane materials with enhanced sieving capabilities and affinity for CO2 is required to fulfil the promise of more efficient and cost-effective CO2 capture.11,13
Polymers of intrinsic microporosity (PIMs) are a class of amorphous glassy polymers characterised by a rigid and contorted molecular structure that inhibits efficient chain packing in the solid state. This typically results in the formation of high free-volume elements.14 The unique combination of microporosity and processability makes PIMs excellent candidate materials for energy storage, catalysis, and environmental applications.15–17 In particular, PIMs show potential as membranes for gas separation, with important applications such as carbon dioxide removal from natural gas (CO2/CH4) and flue gas (CO2/N2).18–20 Their molecular structures, combined with the potential to incorporate various functional groups into the polymeric backbone, enable PIMs to achieve a remarkable trade-off between permeability and selectivity that allows them to define and even surpass the Robeson upper bounds for various gas pairs.21 The continuous evolution and development of synthetic strategies has recently culminated in the publication of a series of PIMs used to establish the new upper bounds for CO2-based gas separations.22 PIMs performance can be further enhanced through strategies like chemical modification,23 crosslinking,24 and incorporation of MOFs and COFs for the efficient formation of mixed matrix membranes.25,26
PIM-1, the archetypal polymer of intrinsic microporosity and the best-studied,27 possesses two nitrile groups per repeat unit, which can be reduced to primary amine groups that have been shown to improve selectivity for Lewis acidic gases such as CO2.28 In our previous work, we found that amine-PIM-1 exhibits remarkably low ideal gas selectivity for the CO2/N2 and CO2/CH4 gas pairs.28,29 We attributed this behaviour to specific interactions between the functionalized polymer and CO2 on the basis of the unusual trend of the gas diffusion coefficients as a function of the squared effective diameter.30 In a more recent paper, the group led by Smith at MIT published intriguing research on functionalised PIM-1.31 They studied six different versions of PIM-1, including amine-PIM-1, where nitrile groups were converted into various functional groups to tune the backbone's affinity towards CO2. The permselectivity data were assessed using both single and mixed gases (with increasing CO2 concentration from 10% to 90% for CO2/CH4). It was found that, in some cases, the performance with mixed gases improved compared to that predicted by the single gases, especially after physical aging. This improvement was attributed to a mechanism known as “competitive gas sorption”, which allows the more permeable gas (CO2) to pass more favourably through the membrane, while the less permeable gas (either CH4 or N2) is blocked. An enhanced solubility-selectivity arises from the high affinity of the polymeric backbone for CO2, leading to an overall improvement in CO2/CH4 and CO2/N2 mixed-gas permselectivity. Especially important proved to be the conversion of the nitrile to amine groups to obtain amine-PIM-1 (or PIM-NH2), which showed an exceptional improvement of the selectivity with mixed gases up to 140% for CO2/CH4 and 250% for CO2/N2.
Here, we confirm the enhanced CO2 selectivity of amine-PIM-1 during mixed gas permeation, and we provide an in-depth experimental and computational study that explains the enhanced affinity of this polymer for CO2. By exploiting solid-state nuclear magnetic resonance (SSNMR), a powerful technique for identifying and quantifying physisorbed and chemisorbed CO2 species in amine-functionalized solid sorbents,32–37 we demonstrate that CO2 is partially chemisorbed on amine-PIM-1, forming carbamic acids by reaction with primary amine groups on the membrane surface (Scheme 1). Partial CO2 chemisorption is also supported by isosteric heat of adsorption. Molecular dynamics (MD) simulations on chemisorbed amine-PIM-1 models, prepared with a number of amine groups converted into carbamic acid groups as determined by SSNMR measurements, emphasize the complexity and critical role of hydrogen bonding in influencing the material's properties and interactions.
 | ||
| Scheme 1 Post-modification of PIM-1 to give amine-PIM-1 and formation of chemisorbed-amine-PIM-1 following CO2 loading. (i) borane dimethyl sulphide 5.0 M in diethyl ether. | ||
2. Experimental
2.1 Materials
13C labelled (99.0 atom %) carbon dioxide (13CO2) was purchased from Sigma Aldrich.Single gases for permeation measurements were supplied by Pirossigeno at a minimum purity of 99.9995%. Certified gas mixtures for permeation measurements were supplied by Sapio at a purity of ±0.01% from the certified concentration (CO2/CH4 mixture with 52.11 mol% CO2 and 47.89 mol% CH4, and N2/O2 mixture with 81.45 mol% N2 and 18.55 mol% O2).
2.2 Membrane preparation
The membranes for the permeation measurements were prepared as reported previously,29 while the powder for BET analysis and the membranes for SSNMR were prepared by a slightly different procedure to guarantee a conversion of nitrile to amine groups more than 95%, herein reported. Reduction of nitrile groups of PIM-1 membranes was carried out using borane-dimethyl sulphide complex in diethyl ether, a non-solvent for the PIM-1 precursor that enables the preparation of amine-PIM-1 membrane samples. Thus, PIM-1 membranes were placed in Petri dishes and then covered with 5.0 M borane-dimethyl sulphide complex in diethyl ether. Subsequently, the Petri dishes were lidded and, in turn, stowed inside a closed plastic container at room temperature to induce and maintain a saturated atmosphere. After one day of reaction time, membranes were collected and placed in Petri dishes of ethanol to remove the excess of borane-dimethyl sulphide complex. Subsequently, membranes were firstly soaked in Petri dishes of 1.0 M methanolic HCl overnight and then in Petri dishes of 5% aqueous NaOH solution for 3 hours. Finally, membranes were rinsed repeatedly in plenty of water and soaked in methanol for 24 h before being dried at 323 K under vacuum. The molecular structure and physical properties of the synthetized materials were confirmed by TGA, XRD and IR measurements (ESI Fig. S1–S3†).2.3 Gas adsorption measurements
N2 (77 K and 298 K) and CO2 (273 K, 298 K, and 308 K) adsorption/desorption measurements of polymer powders were made using an Anton Paar Nova 600. Samples were degassed over 8 h at 80 °C under high vacuum prior to analysis. The gases were supplied by BOC (N2 purity > 99.999%, CO2 purity > 99.995%). The data were analysed using the software Anton Paar Kaomi for NOVA, which is provided with the instrument. The Brunauer–Emmett–Teller (BET) surface area was calculated from N2 adsorption isotherms at a relative pressure P/P0 < 0.1. Non-local density functional theory (NLDFT) analysis on CO2 adsorption isotherms was performed to calculate the pore size distribution and volume, considering a carbon equilibrium transition kernel at 273 K based on a slit-pore model; the kernel is based on a common, one centre, Lennard-Jones model. Heats of adsorption were calculated from the CO2 curves measured at 273 K, 298 K, and 308 K. The data were analysed with the Anton Paar Kaomi software and fitted with the Langmuir–Freundlich equation and heat was calculated via the Clausius–Clapeyron equation.2.4 Gas permeation measurements
Pure and mixed gas permeation tests were carried out from 1 to 6 bar of feed pressure using a custom-made constant pressure/variable volume instrument, equipped with a quadrupole mass filter (HPR-20 QIC, Hiden Analytical). The experimental set-up and the procedures are described elsewhere.38,39 For mixed gas permeation tests, certified mixtures of CO2/CH4 (52.11/47.89 vol%), and N2/O2 (81.45/18.55 vol%) were used. The measurements were performed on an amine-PIM-1 membrane with 85% conversion and aged for 750 days to guarantee the membrane stability during the measurements. Aging of the membranes took place during their natural storage under atmospheric conditions between the individual measurements.2.5 SSNMR measurements
SSNMR experiments were carried out on a Bruker Avance NEO 500 spectrometer working at the Larmor frequency of 500.13, 125.77, and 50.68 MHz, for 1H, 13C, and 15N, respectively, equipped with a 4 mm double-channel (H/F-X) CP/MAS probe. 13C chemical shifts were referenced to the signal of adamantane at 38.48 ppm and calculated for the other nuclei using the unified scale recommended by IUPAC.40 All measurements were conducted at 298 K and under magic angle spinning (MAS) conditions at a frequency of 15 kHz using air as spinning gas, unless otherwise specified.13C quantitative direct excitation (DE) MAS spectra were acquired by applying high-power proton decoupling with a recycle delay of 100 s and 16 scans for the sample loaded with 13CO2 and a recycle delay of 60 s, using a flip angle of 60° and accumulating about 1600 scans, for the sample loaded with CO2.
1H–13C cross-polarization (CP) MAS experiments were carried out using a recycle delay of 2 s and accumulating 128–288 scans for the sample loaded with 13CO2 and 1000 scans for the unloaded sample and the sample loaded with CO2. Contact time values ranging from 0.5 to 8 ms were employed. 1H–13C CP MAS experiments were performed at different temperatures ranging from 263 to 303 K.
1H–15N CP MAS experiments were recorded using a recycle delay of 2 s and a contact time of 4 ms, accumulating 1600 scans; for these experiments the MAS frequency was 8 kHz.
For the sample loaded with 13CO2, the following experiments were also recorded. 1H–13C HETCOR experiments with FSLG homonuclear decoupling41 were acquired using a recycle delay of 2 s and contact time values from 0.5 to 3 ms, accumulating 16 scans for each of the 80 increments in the indirect dimension. 13C DE spectra were recorded with a MAS frequency of 2.5 kHz without applying proton decoupling, using a recycle delay of 1 s, and accumulating 6400 scans.
The analysis of the spinning side bands pattern of 13C DE MAS spectra recorded with the MAS frequency of 2.5 kHz was performed using the solid line shape analysis module (SOLA) included in the software Topspin by Bruker.
Samples for SSNMR measurements were prepared using a home-made cell provided with a mechanical lever operated from outside enabling the capping of the rotor without disturbing the cell atmosphere. In particular, the activated sample was prepared by heating overnight under vacuum (0.1 mbar) at the temperature of 423 K. The membrane was packed into the NMR rotor (4 mm external diameter) and the rotor was capped under N2 atmosphere. For the CO2-loaded samples, the activated membrane was loaded with either CO2 or 13CO2 (i.e.13C isotopically enriched CO2) at 1 bar pressure and the rotor was capped under the gas atmosphere after the equilibrium was reached.
2.6 Molecular modelling
Molecular dynamics simulations were performed using the BIOVIA software package.42 The COMPASS (condensed-phase optimized molecular potentials for atomistic simulations studies) force field43,44 was used, according to previous studies on PIM-1-like materials.29,45–48Molecular dynamics simulations were performed on amine-PIM-1 and on chemisorbed-amine-PIM-1.
The amine-PIM-1 polymer chain for the initial packing with the amorphous cell module consisted of 30 monomers (1860 atoms). Each chain contained 60 amines (see Scheme 1). Every packing model contained three polymer chains. A total of 5580 atoms were grown in a 3D model under periodic boundary conditions. Details on the preparation of the two simulation boxes can be found in our previous paper.29
For the simulations of the chemisorbed-amine-PIM-1, a chain of 30 monomer units is constructed with 12 carbamic acid moieties in accordance with the SSNMR measurements. The chain was grown with a torsion angle of 180° in an amorphous cell module. The simulation boxes contain three chains of amorphous polymer (5790 atoms), 300 argon atoms, and the number of CO2 corresponding to the adsorbate molecules that have not reacted with the amine groups of the polymer at the pressure of 1 bar. Two simulation boxes were selected and equilibrated following a procedure based on three steps: (1) deleting 100 Ar atoms for each step, (2) anneal dynamics run over the 300–700 K temperature range, (3) NPT-MD simulations (constant number of particles (N), pressure (P), and temperature (T)) at 1 bar and 298 K for 100 ps. The electrostatic and van der Waals interactions were studied using the group-based method49 with a cut-off distance of 12.5 Å. The unit cell parameters of the 3D-triclinic lattice were a = b = c = 41.78 Å, α = β = γ = 90°, cell volume = 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 962.9 Å3. NPT-MD simulations were performed at 298 K and 1 bar for a simulation time of 20 ns with a timestep of 1 fs. The initial velocities were randomly evaluated. Temperature and pressure were controlled using the Berendsen method.50 We have inserted 120 carbon dioxide (CO2) molecules into the simulation cells for the physisorbed system, corresponding to the amount adsorbed at 1 bar, as determined by experimental data. For the chemisorbed system, 36 CO2 molecules are incorporated into the polymer chains, while the remaining 90 molecules are free within the simulation box.
962.9 Å3. NPT-MD simulations were performed at 298 K and 1 bar for a simulation time of 20 ns with a timestep of 1 fs. The initial velocities were randomly evaluated. Temperature and pressure were controlled using the Berendsen method.50 We have inserted 120 carbon dioxide (CO2) molecules into the simulation cells for the physisorbed system, corresponding to the amount adsorbed at 1 bar, as determined by experimental data. For the chemisorbed system, 36 CO2 molecules are incorporated into the polymer chains, while the remaining 90 molecules are free within the simulation box.
The theoretical adsorption isotherms were evaluated by the grand canonical Monte-Carlo (GCMC) configurational bias method,51 implemented in the NVT ensemble (constant number of particles (N), volume (V), and temperature (T)) within the 0–5 bar fugacity range at 298 K.42
The fractional free volume (FFV) was calculated according to the Bondi equation (eqn (1)):52
 | (1) |
2.7 Radial distribution function
The radial distribution function, gAB(r), between particles of A and B is defined as follows (eqn (2)): | (2) |
local is the particle density of type B averaged over all spheres around particles A having radius rmax. The radial distribution function is employed to evaluate the specific interactions between the CO2 molecules and the polymer groups.
2.8 Calculated BET surface area
In the computational BET surface determination, the gas surface accessible area was determined using N2 as probe having Teplyakov-Meares’ radius (1.52 Å).53 The procedure calculates the accessible area described by the probe centre as it rolls over a scaled van der Waals surface. The gas surface accessible areas were also calculated using the Teplyakov-Meares’ radii for O2 (1.44 Å), CO2 (1.51 Å), and CH4 (1.59 Å).2.9 Pore size distribution analysis
The pore size distributions were determined using a geometry-based analysis on porous materials with a Voronoi decomposition of the space and lattice precision of 0.1 Å implemented in Zeo++ code.54,55 The pore size distributions were calculated by exploring the available void spaces with a probe of zero radius.3. Results and discussion
For amine-PIM-1, the pure and mixed gas permeabilities for the O2/N2 gas pair are closely correlated, which is consistent with the relatively low and similar solubility of both gases in the polymer. In both cases, a weak but significant decrease in permeability as a function of pressure is observed (ESI Fig. S4†), typical of samples exhibiting dual mode behaviour. The selectivity is slightly higher and the nitrogen permeability slightly lower in the gas mixture with respect to the pure gases, probably because of a small degree of competitive permeation. In contrast, as shown in Fig. 1a and b, the pressure dependence of CO2 and CH4 permeation properties in amine-PIM-1 exhibits a dramatic difference between the pure and the mixed gas measurements. This difference is much higher with respect to the O2/N2 gas pair for the same polymer (ESI Fig. S4†), as well as to the CO2/CH4 gas pair in the parent polymer PIM-1 (ESI Fig. S5†). In the literature, this behaviour is known as sorption-enhanced mixed gas transport31 and can be ascribed to specific CO2 affinity with the amine functional groups. | ||
| Fig. 1 Pressure dependence of permeability and selectivity in amine-PIM-1 for (a) pure CO2 and CH4, and (b) for a CO2/CH4 mixture (52.11/47.89 vol%). Filled symbols represent the pressure-increase steps and open symbols represent the subsequent pressure-decrease steps. | ||
Thus, isothermal gas adsorption studies were conducted on PIM-1 and amine-PIM-1 to evaluate the difference in their CO2 uptake and to better understand the increased affinity of this gas for the aminated PIM. Previous studies28,29 showed that converting the nitrile groups of PIM-1 into primary amine groups leads to stronger inter-chain hydrogen bonding and a drastic reduction in free volume, slowing down N2 adsorption kinetics at 77 K, typically used to assess the porosity and surface area of porous materials.56 However, our repeated studies show that amine-PIM-1 exhibits substantial nitrogen adsorption at 77 K, from which an apparent BET surface area (SABET) of 645 m2 g−1 can be calculated. Although lower than that of PIM-1 (SABET ∼750 m2 g−1), this value indicates that there is significant intrinsic microporosity available for the physisorption of CO2 (ESI Fig. S6†).
CO2 adsorption studies further revealed that, despite its lower porosity, amine-PIM-1 has significantly higher CO2 uptake than PIM-1 (ESI Fig. S6b†), confirming that converting nitriles into amines does indeed improve CO2 affinity. The pore size distribution (PSD) analysis, calculated via NLDFT from CO2 adsorption at 273 K, shows values for both polymers (peaks at 3.5–8.5 Å) that are typical for a PIM. Notably, the peak centred around 3.5 Å, indicating greater apparent ultra-microporosity, is much more intense for amine-PIM-1 than for PIM-1 (ESI Fig. S6c†), as expected for the greater CO2 adsorption at low pressure. CO2 uptake at different temperatures (273, 298, and 308 K) (ESI Fig. S7†) allowed the calculation of the isosteric heat of adsorption (Qst), which is crucial for understanding CO2 affinity and assessing adsorption mechanism in PIM-1 and amine-PIM-1 (ESI Fig. S6d†). Interestingly, the Qst for amine-PIM-1 (∼50 kJ mol−1) is almost twice that of PIM-1 (∼27 kJ mol−1). Physisorption in microporous materials is generally governed by van der Waals interactions with enthalpy values typically ranging between 10 and 50 kJ mol−1,57 while chemisorption involves stronger, often covalent or ionic interactions, leading to enthalpy values above this range,56 and thus a Qst value of 50 kJ mol−1 marks an estimated boundary between physisorption and chemisorption.58 Thus, the herein calculated value for amine-PIM-1 indicates that CO2 chemisorption occurs to some extent, similarly to what previously reported for an amine functionalized adsorbent59 and a MOF.60
This is further confirmed by the presence of weak hysteresis in the adsorption/desorption isotherms at all measured temperatures (ESI Fig. S8†). Thus, while physisorption may still be accounted as the primary adsorption mechanism, a non-negligible amount of CO2 is chemisorbed in the polymer matrix. To identify, quantify, and characterise physisorbed and chemisorbed CO2 species, various 1D and 2D SSNMR experiments were recorded on amine-PIM-1 membrane before and after its loading with CO2 or 13CO2 (1 bar at RT). The comparison of 13C magic angle spinning (MAS) spectra shown in Fig. 2 highlights the presence of physisorbed CO2 (peak at 125 ppm) and two different chemisorbed species (peaks at 158 and 161 ppm). Signals for these species are indeed present only in the spectra of the membrane loaded with CO2 or 13CO2 (Fig. 2b–d), together with the signals arising from the different membrane carbons,28,29 also observed in the spectrum of the membrane not exposed to CO2 (Fig. 2a). The signals with isotropic chemical shift (δiso) values of 158 and 161 ppm, typical of carbamic acid species differing in hydrogen bonding environment,61 clearly indicate that, at ambient temperature and atmospheric pressure, CO2 has access to and reacts with amine groups of amine-PIM-1. The integral areas of the membrane and CO2 carbon's peaks in the quantitative 13C direct excitation (DE) spectra of the CO2- or 13CO2-loaded membrane (Fig. 2b and c) allowed an estimate of the physisorbed and chemisorbed species: overall these species are present in a 1:1 molar ratio with the polymeric unit of amine-PIM-1, which is in agreement with the finding from CO2 uptake at 298 K (2 mmol g−1), while the molar ratio between physisorbed and chemisorbed species is 1:0.7 (ESI Fig. S9†). These results indicate that almost one out of five amine groups of amine-PIM-1 reacts with CO2 to form chemisorbed species, as sketched in Scheme 1.
 | ||
| Fig. 2 Quantitative 13C DE MAS spectra of: (a) amine-PIM-1; (b) amine-PIM-1 loaded with CO2; (c) amine-PIM-1 loaded with 13CO2. (d) 1H–13C CP MAS spectrum of amine-PIM-1 loaded with 13CO2 recorded with a contact time of 2 ms. All the spectra were acquired at 298 K at a spinning frequency of 15 kHz. | ||
As far as the identification of the chemisorbed species is concerned, the chemical shift values measured in the 13C spectra point to carbamic acid species differing in hydrogen bonding environment,61 although they are not decisive for a sound assignment of the signals, since δiso values between 153 and 168 ppm have been reported in the literature for both carbamic acid and ammonium carbamate carbons,33,62 two species that can form in amine-PIM-1 if CO2 reacts with one or two amine groups, respectively.63 Therefore, to undoubtedly identify the formed species, a combination of different experiments was adopted, including the acquisition of 1H–15N cross polarization (CP) MAS spectra and 2D 1H–13C HETCOR spectra, as well as the determination of the chemical shift tensor components for the two carbons resonating at 158 and 161 ppm.
Despite the low signal-to-noise ratio arising from the low natural abundance of 15N (0.36%), the 1H–15N CP MAS spectrum of CO2-loaded amine-PIM-1 clearly shows a signal at 83 ppm ascribable to the nitrogen of the amidic NH group of carbamic acids or carbamates, and a signal at 24 ppm, also observed in the spectrum of the pristine amine-PIM-1 membrane, associated to unreacted amine groups (Fig. 3).33,61,64,65 However, no signal is observed at 32–36 ppm, where the nitrogen of ammonium groups is expected to resonate,33,61,64,65 suggesting that only carbamic acid species are formed.
 | ||
| Fig. 3 1H–15N CP MAS spectra recorded with a contact time of 4 ms on: (a) pristine amine-PIM-1 membrane and (b) amine-PIM-1 membrane loaded with CO2 (i.e., chemisorbed-amine-PIM-1). The spectra were acquired at 298 K at a spinning frequency of 8 kHz. | ||
The 2D 1H–13C HETCOR spectrum recorded on the 13CO2-loaded amine-PIM-1 membrane with a short contact time of 0.5 ms (Fig. 4a and ESI Fig. S12a†) shows cross peaks between the 13C nucleus resonating at 158 ppm and 1H nuclei resonating at 5.2 (NH) and 10.4 (COOH) ppm, and between the 13C with δiso = 161 ppm and 1H nuclei resonating at 5.4 (NH) and 13.2 (COOH) ppm. The observed differences, especially that concerning the COOH 1H chemical shift, can be accounted for by a different arrangement of two forms of carbamic acid, the higher chemical shift value being associated to a form with the COOH group involved in a strong H-bond.
 | ||
| Fig. 4 Expansion of the 1H–13C HETCOR MAS spectra of chemisorbed-amine-PIM-1 loaded with 13CO2 recorded with a contact time of (a) 0.5 ms and (b) 1.5 ms. | ||
The attribution of the two chemisorbed species to carbamic acids involved or not/weakly involved in H-bonding was corroborated by the symmetry of the chemical shift tensors of the COOH carbons determined through the analysis of MAS spectra recorded at slow spinning frequency. According to Čendak et al.,66 an axial tensor is expected for protonated carbamic acid not/weakly involved in H-bonds, while a change towards an orthorhombic tensor is observed for carbamic acids involved in strong H-bonds and for the deprotonated carbamic acid (carbamate).67 Here, the principal components of the chemical shift tensors of the carbons resonating at 158 and 161 ppm were reconstructed from the analysis of the side band manifolds in the 13C MAS spectrum recorded at a MAS frequency of 2.5 kHz (Fig. 5). The chemical shift tensor of the species with δiso = 158 ppm has a nearly axial symmetry with principal components δ11 = 220 ppm, δ22 = 129 ppm, and δ33 = 125 ppm. On the other hand, the chemical shift tensor of the species with δiso = 161 ppm has an orthorhombic tensor with principal components δ11 = 212 ppm, δ22 = 157 ppm, and δ33 = 114 ppm; the tensor components were expressed using the Mehring notation, where δ11 ≥ δ22 ≥ δ33. From the tensor's principal components, values of A (A = δ11 + δ33 − δ22) and d (with d = δ11 − δiso when |δ11 − δiso| > |δ33 − δiso| and d = δ33 − δiso when |δ11 − δiso| < |δ33 − δiso|) were also determined. A is 216 and 169 ppm, and d is 62 and 51 ppm for carbons with δiso of 158 and 161 ppm, respectively. According to findings by Gu and McDermott on amino acids67 and by Čendak et al. on carbamic acids/carbamates,66 these parameters are associated to two protonated forms of carbamic acid, one not (or weakly) involved in H-bonds (δiso = 158 ppm) and the other involved in strong H-bonds (δiso = 161 ppm). Overall, the SSNMR data indicate that two types of carbamic acids form in the reaction of CO2 with amine groups of amine-PIM-1 in dry conditions, one strongly involved and one not (or weakly) involved in hydrogen bonding with a hydrogen bond acceptor.
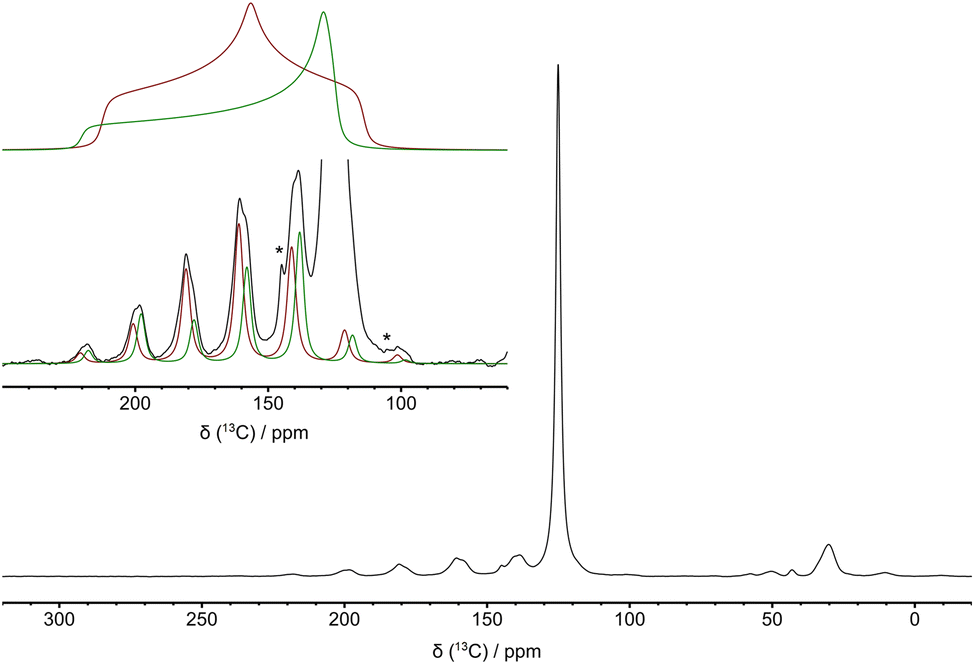 | ||
| Fig. 5 13C DE MAS spectrum of amine-PIM-1 loaded with 13CO2 recorded using a MAS frequency of 2.5 kHz. In the inset, simulations of the side band manifolds of signals of carbamic acids with δiso of 158 and 161 ppm (bottom) and the corresponding reconstructed chemical shift tensors (top) are shown in green and red, respectively. Asterisks indicate spinning side bands of physisorbed CO2 undergoing anisotropic motion. | ||
Further insights into hydrogen bonds between polymer moieties were achieved through MD calculations. Simulations on chemisorbed-amine-PIM-1 revealed complex inter- and intramolecular hydrogen bonding networks within and between the polymer chains, between amine/amine, amine/dioxin, and carbamic acid/dioxin groups (Fig. 6a–d). This confirms that the carbamic acids formed in the amine-PIM-1 are involved in different kinds of hydrogen bonds.
 | ||
| Fig. 6 Detailed snapshots of the interactions between (a) amine/amine, (b) dioxin/CH2–NH2, (c) amine/dioxin, and (d) amine/carbamic acid groups observed after the simulation time of 20 ns. In chemisorbed-amine-PIM-1, the H atom is white, the O atom is red, the N atom is blue, and the C atom is grey (settings: maximum hydrogen-acceptor distance 2.5 Å, minimum donor-hydrogen-acceptor angle 90°). Average pore size distribution of (e) amine-PIM-1 and (f) chemisorbed-amine-PIM-1, both calculated by exploring the available void spaces with a probe of zero radius. | ||
The intra- and inter-chain interactions play a pivotal role in the membrane's architecture, its free volume, and structural stabilization, and they commonly decrease the permeability and enhance the membranes' selectivity. Such packing effects were previously noted in PIM-2 (ref. 68) and various post-synthetically modified PIMs.69–75 However, to the best of our knowledge, this is the first time that this packing effect is deeply studied in the absence or presence of a molecule that changes the molecular structure of the polymer. The chemisorption of CO2 into amine-PIM-1, in the form of newly incorporated carbamic acid moieties, affects its morphology, leading to decreased spacing between chains in comparison to “pristine” amine-PIM-1. This results in a reduction of the BET surface area and the fractional free volume (FFV).
In fact, simulations on amine-PIM-1 molecular boxes calculate a BET surface area of 732.6 ± 0.8 m2 g−1, while a much lower BET surface area, i.e. 602.6 ± 0.2 m2 g−1, is calculated using the chemisorbed-amine-PIM-1 molecular model, showing that the presence of the carbamic acid has a strong influence on the polymer network, and specifically leads to a reduction of the FFV. The herein simulated FFV of amine-PIM-1 is 0.28 ± 0.02, which is larger with respect to that calculated for the chemisorbed-amine-PIM-1 (0.25 ± 0.01), again showing that the extra formed hydrogen bonds are leading to a lower FFV. This is further confirmed by the analysis of the accessible surface area of N2, O2, CO2, and CH4 (Table S1†), which shows that all the gases experience a lower accessible surface area in chemisorbed-amine-PIM-1 with respect to amine-PIM-1.
MD simulations reveal that amine-PIM-1 exhibits a broad range of pore sizes, from 1.1 to 14.1 Å, with predominant diameters between 6.0 and 6.1 Å (Fig. 6e). In contrast, chemisorbed-amine-PIM-1 shows a narrower distribution, ranging from 0.6 to 9.7 Å, with the most prevalent pore sizes around 4.3–4.4 Å (Fig. 6f), indicating a notable presence of smaller voids within the polymer structure. Considering that the simulated distributions capture a wider range of pore sizes due to the use of a zero-sized probe, results of both amine-PIM-1 and chemisorbed-amine-PIM-1 are consistent with the experimental pore size distribution analysis, calculated via NLDFT from CO2 adsorption at 273 K, which highlights peaks between 3.5 and 8.5 Å (ESI Fig. S6c†). The chemisorbed-amine-PIM-1 model, with its predominant pore sizes around 4.3–4.4 Å, aligns more closely with the experimental values than the amine-PIM-1 model, confirming that CO2 chemisorption and the formation of carbamic acid significantly alter the polymer's structure. The free volume and slice visualizations of chemisorbed-amine-PIM-1 and amine-PIM-1 are reported in ESI Fig. S10 and S11.†
1H–13C CP and HETCOR MAS spectra were also recorded on the 13CO2-loaded amine-PIM-1 membrane to highlight interactions between physisorbed CO2 and membrane moieties. In the 1H–13C CP MAS spectra (Fig. 2 and ESI Fig. S13†) the signal of physisorbed CO2 carbon at 125 ppm is observed together with signals from the membrane and carbamic acids' carbons at all contact time values employed (from 0.5 to 8 ms), indicating that adsorbed CO2 establishes interactions with the membrane hydrogens. In particular, the signal of physisorbed CO2 steadily increases with the contact time up to the longest interval explored (8 ms), suggesting that the magnetization transfer from 1H to 13C nuclei progressively occurs in time from hydrogens in molecular fragments increasingly distant from CO2. Correspondingly, a correlation peak is observed between the signal of physisorbed CO2 and the 1H signal of interacting amine groups at 4.7 ppm in the 1H–13C HETCOR MAS spectrum recorded with a contact time of 1.5 ms, while correlation peaks are observed with all membrane protons for a longer contact time of 3 ms (ESI Fig. 12b and c†). This evidence indicates that stronger dipolar interactions are established between CO213C and –NH21H nuclei, which are ascribable to a closer proximity of CO2 to amine groups in the membrane due to Lewis's acid–base interactions.
However, a comparison of the relative intensities of the signals from chemisorbed and physisorbed species in the quantitative 13C DE MAS spectra and in the CP MAS spectra (Fig. 2 and ESI Fig. S13†) suggests that only one part of physisorbed CO2 can get in close proximity to the membrane and establish dipolar interactions strong enough to allow magnetization transfer. The remaining physisorbed CO2 is located further from the membrane surface and/or undergoes fast dynamics. This can also be seen in the DE MAS spectrum recorded at the spinning frequency of 2.5 kHz shown in Fig. 5 where both features of CO2 undergoing isotropic (sharp peak at 125 ppm) and restricted anisotropic motions (side bands marked with asterisks) inside the membrane are observed at RT. However, a detailed analysis of the anisotropic CO2 signal is hampered by the very low intensity of the sidebands. The observed behavior is different from that reported for 13CO2-loaded PIM-1, tetrazole- (TZ-PIM), and methyl tetrazole-functionalized (MTZ-PIM) PIM-1, for which only an isotropic line is observed for CO2 in 13C static spectra at RT.76 Indeed, for PIM-1 an isotropic line shape resulting from the averaging of the CO213C CSA by translational hopping of CO2 among randomly oriented sites was already observed at 100 K, while it occurred at slightly higher temperatures for TZ-PIM (125 K) and MTZ-PIM (150 K). Our findings indicate stronger interactions of a part of physisorbed CO2 with amine-PIM-1 adsorption sites with respect to PIM-1 and tetrazole-functionalized PIM-1 membranes, which hamper fast isotropic motion in the membrane pores even at room temperature.
13C CP MAS spectra recorded at different temperatures between 263 and 303 K show a progressive decrease of the chemisorbed CO2 signal intensities (ESI Fig. S14†). Although these spectra are not quantitative, this observation could be ascribed to the decrease of chemisorbed species by increasing the temperature. These trends can be related to the decrease in CO2-polymer affinity and Langmuir site sorption capacity observed in a previous work,29 as well as to CO2 adsorption isotherms measured at different temperatures in the present work (ESI Fig. S8†).
Furthermore, the MD simulations indicated that physisorbed CO2 molecules establish hydrogen bonds with both the unreacted amine groups and carbamic acid groups of chemisorbed-amine-PIM-1. These interactions are identified using radial distribution functions, g(r), between the oxygen atoms of CO2 and selected atoms of the polymer (Fig. 7a). The first peak of the green curve obtained for the unreacted amine group, centered at 1.67 Å, indicates a strong direct hydrogen bond between the hydrogen of the unreacted amine group and the closest oxygen of CO2, while the second broad peak centered at 3.97 Å (Fig. 7a), and displayed at 3.81 Å in a snapshot (Fig. 7c), indicates the interaction between the second oxygen of the same CO2 molecule and the hydrogen of the amine group. The narrow first peaks of the curves related to –NH (blue) and –OH (red) of the carbamic acid are found at approximately 1.85–2.02 Å, while two broad and more intense peaks are observed, centered at 3.97 Å for the –NH and 5.07 Å for the –OH groups. The –NH of the unreacted amine groups (green curve) is in full overlap with the –NH of the carbamic acid, showing comparable patterns, while the –OH is at a close distance, helping to form a hydrogen bond network around CO2. In fact, all together the observed g(r) peaks correspond to weak hydrogen bonds between CO2 and different chains and/or different sections of the same chain in the chemisorbed-amine-PIM-1, indicating an organized structure hosting CO2 resembling a “pseudo-pocket”. Despite being located approximately 4–5 Å away from the sites, in accordance with the 1H–13C HETCOR MAS spectra (Fig. 4 and ESI Fig. 12†), CO2 coordinates simultaneously with both carbamic acid and unreacted amine groups. These results suggest that, on average, CO2 moves with restricted mobility when coordinated to capture sites, while uncoordinated CO2 has greater degree of freedom. This is confirmed by SSNMR studies, which reveal that CO2 undergoes both anisotropic motions, reflecting restricted mobility, and isotropic motions, indicating greater freedom.
 | ||
| Fig. 7 (a) Radial distribution function between the O atoms of CO2 and: the H atoms of the unreacted amine groups (green curve), the H atom of –NH– group of carbamic acid (blue curve), and the H atom of –OH group of carbamic acid (red curve) in chemisorbed-amine-PIM-1. (b) Radial distribution function between the O atoms of CO2 and the H atoms of the amine groups of amine-PIM-1 (violet curve). (c) Indicative hydrogen bonds between the O atoms of CO2 and the H atoms of unreacted amine groups corresponding to the two peaks of the green curve in a. (d) Examples of hydrogen bonds between the O atoms of CO2 and one H atom of unreacted amine (1.67 Å), the H atom of –NH– group (blue curve in a) of carbamic acid (3.97 Å), and the H atom of the –OH group (red curve in a) of carbamic acid (5.07 Å). The different polymeric chains are indicated in light blue and pink. Colour code: the C atom is grey, the O atom is red, the H atom is white, and the N atom is blue. | ||
A different situation is observed in models of amine-PIM-1 with physisorbed CO2, where only unreacted amine groups are present in the simulation box. In the g(r) function (Fig. 7b), a subtle first peak appears at 2.03 Å, indicating a specific interaction distance between the amine groups and CO2 molecules. A broader and more intense peak at 3.95 Å suggests a secondary interaction distance. Unlike the chemisorbed case, the first peak is not clearly distinguishable. Detailed analysis of our simulations shows that the number of CO2 molecules corresponds to an experimental pressure of 1 bar. At this pressure, the total number of CO2 molecules is 120, significantly higher than the 90 molecules used in the chemisorbed system (the remaining molecules being chemisorbed).
Due to the large number of CO2 molecules in the polymeric model, the interaction weakens with the formation of multiple shells, moving further from the membrane surface and undergoing faster dynamics, as confirmed by 13C DE MAS and CP MAS spectra. The strong hydrogen bonding with unreacted amine hydrogens is not visually apparent but rather emerges as a statistical effect. Recent analyses of physisorbed systems at different pressures and temperatures have revealed a strong relationship between the amine groups and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of CO2 molecules, particularly at pressures below 1 bar.29 As pressure and temperature increase, the g(r) distribution decreases, indicating a weaker interaction. At higher pressures, the interaction diminishes due to the large number of CO2 molecules in the polymeric model, causing the amine groups to “lose sight” of them due to faster CO2 dynamics. Similarly, at higher temperatures, the interaction between the amine groups and CO2 weakens as vibrational modes intensify.
O bonds of CO2 molecules, particularly at pressures below 1 bar.29 As pressure and temperature increase, the g(r) distribution decreases, indicating a weaker interaction. At higher pressures, the interaction diminishes due to the large number of CO2 molecules in the polymeric model, causing the amine groups to “lose sight” of them due to faster CO2 dynamics. Similarly, at higher temperatures, the interaction between the amine groups and CO2 weakens as vibrational modes intensify.
4. Conclusions
Our study confirmed the enhanced CO2 selectivity of amine-PIM-1 in mixed gas with respect to pure gas permeation for gas pairs involving CO2 and provided sound evidence of the strong CO2 affinity for this polymer through a combination of experimental and computational data. Adsorption isotherms revealed that amine-PIM-1 is a microporous polymer with higher CO2 adsorption capacity with respect to the archetypal PIM-1, and a considerably higher isosteric heat of adsorption (about 50 kJ mol−1 for amine-PIM-1 vs. 27 kJ mol−1 for PIM-1), which suggests the presence of chemisorption. Indeed, chemisorbed species were detected and quantified by combining different SSNMR experiments. Two carbamic acid species were found, characterized by different strength of hydrogen bond interactions with the polymer moieties, accounting for about 40% of the detected CO2 at 1 bar of pressure at RT, and resulting from the reaction of CO2 with about 20% of amine groups in dry conditions. The number of chemisorbed species was found to decrease by increasing the temperature. Remarkably, our study reveals that chemisorption occurs also in the absence of water, while the latter is fundamental to enhance the CO2 permselectivity in most facilitated transport membranes. Physisorbed CO2 was also revealed and characterized by SSNMR spectroscopy. Populations of CO2 in more and less restricted environments were found, undergoing anisotropic and isotropic dynamics, respectively. Molecular dynamics simulations supported both isothermal adsorption and SSNMR findings. Through the analysis of models for amine-PIM-1 before and after CO2 chemisorption (chemisorbed-amine-PIM-1), a detailed description of the interactions between polymer moieties was provided. A complex network of hydrogen bonds was found involving amine/amine, amine/dioxin, and amine/carbamic acid groups, which results in a decreased surface area and a narrower pore size distribution for chemisorbed-amine-PIM-1. Moreover, different states corresponding to different interactions with amine and carbamic acid moieties were identified for physiosorbed CO2. Coordination of CO2 in a pseudo-pocket environment was found in chemisorbed-amine-PIM-1 where CO2 strongly interacts with unreacted amine groups, indicating that chemisorption promotes physisorption with stronger interactions. In contrast to more common facilitated transport membranes that need water to favour CO2 transport by carbamate formation, the formation of carbamic acid under dry conditions in the voids of microporous polymers may be an interesting strategy to develop novel, highly selective membranes for CO2 capture. To summarize, while both sorption-driven affinity (physisorption/chemisorption) and pore-size modifications contribute to CO2 selectivity, Qst data and mixed-gas permeation trends highlight a synergistic relationship, where sorption-driven affinity is the predominant factor in CO2 uptake, while pore-size modifications via hydrogen bonding enhance overall selectivity by restricting CH4 and N2 permeability. This structural refinement reinforces CO2 permselectivity rather than directly increasing CO2 uptake. The combined influence of enhanced CO2 affinity and tailored polymer microstructure leads to the observed improvement in CO2-based gas separations. The elucidation of the transport properties in amine-PIM-1 may be relevant to the development of next generation membrane materials with enhanced CO2-philic behaviour.Data availability
The data supporting this article have been included as part of the ESI† and are available at Zenodo at https://zenodo.org/communities/dam4co2/.Author contributions
CR: conceptualization, data curation, formal analysis, investigation, methodology, resources, visualization, writing – original draft, writing – review & editing; FN: conceptualization, data curation, formal analysis, investigation methodology, resources, visualization, writing – original draft, writing – review & editing; MM: formal analysis, investigation, validation, visualization, writing – original draft; LC: conceptualization, formal analysis, methodology, project administration, supervision, writing – original draft, writing – review & editing; CGB: formal analysis, investigation, methodology, resources, writing – original draft, writing – review & editing; MC: conceptualization, formal analysis, funding acquisition, resources, supervision, writing – original draft, writing – review & editing; ET: conceptualization, data curation, formal analysis, methodology, project administration, supervision, writing – review & editing; EE: investigation, validation, writing – review & editing; GDL: validation, writing – review & editing; BCG: investigation, validation, writing – review & editing; NBM: funding acquisition, resources, supervision, writing – review & editing; BS: formal analysis, investigation, resources, writing – review & editing; PMB: funding acquisition, methodology, supervision, writing – review & editing; JCJ: formal analysis, methodology, project administration, supervision, writing – review & editing; AF: conceptualization, data curation, formal analysis, funding acquisition, methodology, project administration, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors are gratefully acknowledging the Ministry for Universities and Research (MUR) of Italy for financial support under the program PRIN 2020 under the project “doMino” (2020P9KBKZ) and the Grants TED2021-131170A-I00 and CNS2022-135430 funded by MICIU/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR. The research leading to these results has received funding from the European Union's Horizon Europe research and innovation programme under grant agreement No 101115488, project DAM4CO2 and by UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee, grant numbers 10083164 and 10091537.References
- T. M. Letcher, The Root Causes of Global Warming and the New Normal, Elsevier, 2024 Search PubMed.
- W. F. Lamb, T. Gasser, R. M. Roman-Cuesta, G. Grassi, M. J. Gidden, C. M. Powis, O. Geden, G. Nemet, Y. Pratama, K. Riahi, S. M. Smith, J. Steinhauser, N. E. Vaughan, H. B. Smith and J. C. Minx, Nat. Clim. Change, 2024, 14, 644–651 CrossRef CAS.
- A. C. Ruane, https://www.nationalgrid.com/stories/energy-explained/what-is-ccs-how-does-it-work, accessed June 2024.
- R. L. Siegelman, E. J. Kim and J. R. Long, Nat. Mater., 2021, 20, 1060–1072 CrossRef CAS PubMed.
- B. Chen, D. Fan, R. V. Pinto, I. Dovgaliuk, S. Nandi, D. Chakraborty, N. García-Moncada, A. Vimont, C. J. McMonagle, M. Bordonhos, A. Al Mohtar, I. Cornu, P. Florian, N. Heymans, M. Daturi, G. De Weireld, M. Pinto, F. Nouar, G. Maurin, G. Mouchaham and C. Serre, Adv. Sci., 2024, 11, 1–15 Search PubMed.
- A. Koli, A. K. Battu, R. K. Motkuri and S. Sabale, Biomass Convers. Biorefin., 2024, 14, 10177–10188 CrossRef CAS.
- X. Y. D. Soo, J. J. C. Lee, W. Y. Wu, L. Tao, C. Wang, Q. Zhu and J. Bu, J. CO2 Util., 2024, 81, 1–33 Search PubMed.
- E. Favre, R. Bounaceur and D. Roizard, J. Membr. Sci., 2009, 328, 11–14 CrossRef CAS.
- K. Xie, Q. Fu, G. G. Qiao and P. A. Webley, J. Membr. Sci., 2019, 572, 38–60 CrossRef CAS.
- Z. Dai and L. Deng, Sep. Purif. Technol., 2024, 335, 1–27 Search PubMed.
- A. Junaidi, U. Zulfiani, S. Khomariyah, T. Gunawan, N. Widiastuti, N. Sazali and W. N. W. Salleh, RSC Adv., 2024, 14, 2311–2319 RSC.
- P. Sherugar, A. M. Antony, N. A. H. M. Nordin, S. A. Patil and M. Padaki, Fuel, 2024, 361, 1–13 CrossRef.
- D. B. Gutierrez, E. B. Caldona, R. D. Espiritu and R. C. Advincula, MRS Commun., 2021, 11, 391–401 CrossRef CAS.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS.
- A. R. Antonangelo, N. Hawkins, E. Tocci, C. Muzzi, A. Fuoco and M. Carta, J. A. Chem. Soc., 2022, 144, 15581–15594 CrossRef CAS PubMed.
- M. O. Amin, E. Al-Hetlani, A. R. Antonangelo, H. Zhou and M. Carta, Appl. Water Sci., 2023, 13, 1–11 CrossRef.
- F. Topuz, M. H. Abdellah, P. M. Budd and M. A. Abdulhamid, Polym. Rev., 2024, 64, 251–305 CrossRef CAS.
- X. Feng, J. Zhu, J. Jin, Y. Wang, Y. Zhang and B. Van der Bruggen, Prog. Mater. Sci., 2024, 144, 1–56 CrossRef.
- H. Zhou, C. Rayer, A. R. Antonangelo, N. Hawkins and M. Carta, ACS Appl. Mater. Interfaces, 2022, 14, 20997–21006 CrossRef CAS PubMed.
- Y. Li, J. Brückel, M. Jereb, A. Zupanc, S. P. Hirvonen, S. Hietala, M. Kemell, Y. Wu, O. Fuhr, R. D. Jansen-van Vuuren, M. Carta and S. Bräse, Adv. Funct. Mater., 2024, 2401957, 1–8 Search PubMed.
- Y. Wang, B. S. Ghanem, Y. Han and I. Pinnau, Curr. Opin. Chem. Eng., 2022, 35, 1–9 Search PubMed.
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M. C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC.
- M. Tian, S. Rochat, H. Fawcett, A. D. Burrows, C. R. Bowen and T. J. Mays, Adsorption, 2020, 26, 1083–1091 Search PubMed.
- T. Joo, T. H. Lee, S. J. Kaser, W. N. Wu, S. Wi, J. Y. Yeo and Z. P. Smith, Chem. Mater., 2024, 36, 4275–4290 CrossRef CAS.
- E. Esposito, M. Carta, A. Fuoco, M. Monteleone, B. Comesaña-Gándara, E. Gkaniatsou, C. Sicard, S. Wang, C. Serre, N. B. McKeown and J. C. Jansen, J. Membr. Sci., 2024, 697, 1–14 CrossRef.
- F. Emamverdi, J. Huang, N. M. Razavi, M. J. Bojdys, A. B. Foster, P. M. Budd, M. Böhning and A. Schönhals, Macromolecules, 2024, 57, 1829–1845 CrossRef CAS PubMed.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 4, 230–231 RSC.
- C. R. Mason, L. Maynard-Atem, K. W. J. Heard, B. Satilmis, P. M. Budd, K. Friess, M. Lancì, P. Bernardo, G. Clarizia and J. C. Jansen, Macromolecules, 2014, 47, 1021–1029 CrossRef CAS PubMed.
- B. Satilmis, M. Lanč, A. Fuoco, C. Rizzuto, E. Tocci, P. Bernardo, G. Clarizia, E. Esposito, M. Monteleone, M. Dendisová, K. Friess, P. M. Budd and J. C. Jansen, J. Membr. Sci., 2018, 555, 483–496 CrossRef CAS.
- A. Fuoco, C. Rizzuto, E. Tocci, M. Monteleone, E. Esposito, P. M. Budd, M. Carta, B. Comesaña-Gándara, N. B. McKeown and J. C. Jansen, J. Mater. Chem. A, 2019, 7, 20121–20126 RSC.
- K. Mizrahi Rodriguez, F. M. Benedetti, N. Roy, A. X. Wu and Z. P. Smith, J. Mater. Chem. A, 2021, 9, 23631–23642 RSC.
- L. Mafra, T. Čendak, S. Schneider, P. V. Wiper, J. Pires, J. R. B. Gomes and M. L. Pinto, J. Am. Chem. Soc., 2017, 139, 389–408 CrossRef CAS PubMed.
- R. Afonso, M. Sardo, L. Mafra and J. R. B. Gomes, Environ. Sci. Technol., 2019, 53, 2758–2767 CrossRef CAS PubMed.
- D. Pereira, R. Fonseca, I. Marin-Montesinos, M. Sardo and L. Mafra, Curr. Opin. Colloid Interface Sci., 2023, 64, 1–19 CrossRef.
- S. M. Pugh and A. C. Forse, J. Magn. Reson., 2023, 346, 1–11 CrossRef PubMed.
- M. Ilkaeva, R. Vieira, J. M. P. Pereira, M. Sardo, I. Marin-Montesinos and L. Mafra, J. Am. Chem. Soc., 2023, 145, 8764–8769 CrossRef CAS PubMed.
- M. Sardo, T. Morais, M. Soares, R. Vieira, M. Ilkaeva, M. A. O. Lourenço, I. Marín-Montesinos and L. Mafra, Chem. Commun., 2024, 60, 4015–4035 RSC.
- M. Monteleone, E. Esposito, A. Fuoco, M. Lanč, K. Pilnáček, K. Friess, C. Bezzu, M. Carta, N. McKeown and J. C. Jansen, Membranes, 2018, 8, 73 CrossRef PubMed.
- S. C. Fraga, M. Monteleone, M. Lanč, E. Esposito, A. Fuoco, L. Giorno, K. Pilnáček, K. Friess, M. Carta, N. B. McKeown, P. Izák, Z. Petrusová, J. G. Crespo, C. Brazinha and J. C. Jansen, J. Membr. Sci., 2018, 561, 39–58 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483 CrossRef CAS PubMed.
- B.-J. Van Rossum, H. Förster, F. Förster and H. J. M. De Groot, J. Magn. Reson., 1997, 124, 516–519 CrossRef CAS.
- Accelrys Software Inc., P. U. G. P. S, BIOVIA Package; Ex Material Studio 7.0; Classical Simulation Theory Section; Sorption User Guide, Accelrys Software Inc., San Diego, CA, USA, 2013 Search PubMed.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- H. Sun, P. Ren and J. R. Fried, Comput. Theor. Chem., 1998, 8, 229–246 CAS.
- K.-S. Chang, K.-L. Tung, Y.-F. Lin and H.-Y. Lin, RSC Adv., 2013, 3, 10403 RSC.
- L. Zhao, D. Zhai, B. Liu, Z. Liu, C. Xu, W. Wei, Y. Chen and J. Gao, Chem. Eng. Sci., 2011, 68, 101–107 CrossRef.
- L. Zhang, W. Fang and J. Jiang, J. Phys. Chem. C, 2011, 115, 11233–11239 CrossRef CAS.
- O. Hölck, M. Böhning, M. Heuchel, M. R. Siegert and D. Hofmann, J. Membr. Sci., 2013, 428, 523–532 CrossRef.
- T. A. Halgren, J. Am. Chem. Soc., 1992, 114, 7827–7843 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 Search PubMed.
- R. L. C. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2013, 39, 1153–1164 CrossRef CAS.
- A. Bondi, J. Chem. Phys., 1964, 68, 442–451 Search PubMed.
- V. Teplyakov and P. Meares, Sep. Purif. Technol., 1990, 4, 66–74 CrossRef CAS.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- M. Pinheiro, R. L. Martin, C. H. Rycroft, A. Jones, E. Iglesia and M. Haranczyk, J. Mol. Graph. Model, 2013, 44, 208–219 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- M. M. Dubinin, Chem. Rev., 1960, 2, 235–241 CrossRef.
- L. W. Bruch, Surf. Sci., 1983, 125, 194–217 CrossRef CAS.
- W. Si, S. Ye, D. Zhang, B. Yang, Y. Hou, Z. Li, X. Zhang, J. Zhu and L. Lei, Can. J. Chem. Eng., 2019, 97, 697–701 CrossRef CAS.
- D. D. Zhou, X. W. Zhang, Z. W. Mo, Y. Z. Xu, X. Y. Tian, Y. Li, X. M. Chen and J. P. Zhang, Energy Chem., 2019, 1, 1–35 CAS.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS PubMed.
- M. L. Pinto, L. Mafra, J. M. Guil, J. Pires and J. Rocha, Chem. Mater., 2011, 23, 1387–1395 CrossRef CAS.
- K. Basaran, B. U. Topcubasi and T. Davran-Candan, J. CO2 Util., 2021, 47, 1–16 Search PubMed.
- C. H. Chen, D. Shimon, J. J. Lee, S. A. Didas, A. K. Mehta, C. Sievers, C. W. Jones and S. E. Hayes, Environ. Sci. Technol., 2017, 51, 6553–6559 CrossRef CAS PubMed.
- D. Shimon, C. H. Chen, J. J. Lee, S. A. Didas, C. Sievers, C. W. Jones and S. E. Hayes, Environ. Sci. Technol., 2018, 52, 1488–1495 CrossRef CAS PubMed.
- T. Čendak, L. Sequeira, M. Sardo, A. Valente, M. L. Pinto and L. Mafra, Chem.–Eur. J., 2018, 24, 10136–10145 CrossRef PubMed.
- Z. Gu and A. Mcdermott, J. Am. Chem. Soc., 1993, 115, 4282–4285 CrossRef CAS.
- A. Fuoco, B. Satilmis, T. Uyar, M. Monteleone, E. Esposito, C. Muzzi, E. Tocci, M. Longo, M. P. De Santo, M. Lanč, K. Friess, O. Vopička, P. Izák and J. C. Jansen, J. Membr. Sci., 2020, 594, 1–12 CrossRef.
- K. Mizrahi Rodriguez, A. X. Wu, Q. Qian, G. Han, S. Lin, F. M. Benedetti, H. Lee, W. S. Chi, C. M. Doherty and Z. P. Smith, Macromolecules, 2020, 53, 6220–6234 CrossRef CAS.
- R. Swaidan, B. S. Ghanem, E. Litwiller and I. Pinnau, J. Membr. Sci., 2014, 457, 95–102 CrossRef CAS.
- J. W. Jeon, D. G. Kim, E. H. Sohn, Y. Yoo, Y. S. Kim, B. G. Kim and J. C. Lee, Macromolecules, 2017, 50, 8019–8027 CrossRef CAS.
- N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372–375 Search PubMed.
- J. Weber, N. Du and M. D. Guiver, Macromolecules, 2011, 44, 1763–1767 Search PubMed.
- C. R. Mason, L. Maynard-Atem, N. M. Al-Harbi, P. M. Budd, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Macromolecules, 2011, 44, 6471–6479 CrossRef CAS.
- N. Du, G. P. Robertson, J. Song, I. Pinnau and M. D. Guiver, Macromolecules, 2009, 42, 6038–6043 CrossRef CAS.
- J. K. Moore, R. M. Marti, M. D. Guiver, N. Du, M. S. Conradi and S. E. Hayes, J. Phys. Chem. C, 2018, 122, 4403–4408 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08839e |
| ‡ These two authors contributed equally. |
| § Present address: Department of Chemistry and Industrial Chemistry, University of Pisa, via G. Moruzzi 13, 56124 Pisa, Italy. |
| This journal is © The Royal Society of Chemistry 2025 |