
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unlocking sustainable, aromatic, and versatile materials through transurethanization: development of non-isocyanate polyurethanes from lignins†
Nathan
Wybo
a,
Elise
Cherasse
ab,
Antoine
Duval
 *ab and
Luc
Avérous
*a
*ab and
Luc
Avérous
*a
aBioTeam/ICPEES-ECPM, UMR CNRS 7515, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France. E-mail: antoine.duval@unistra.fr; luc.averous@unistra.fr
bSoprema, 15 rue de Saint Nazaire, 67100 Strasbourg, France
First published on 7th March 2025
Abstract
Polyurethanes (PUs) are nowadays essential for a wide range of key applications, thanks to their exceptional and adaptable properties, which arise from their wide variety of macromolecular architectures. However, conventional PUs are predominantly fossil-based, synthesized from highly toxic isocyanates, and are difficult to recycle since most of them are thermosets. This study presents a novel approach to address these issues by developing bio-based, aromatic, and crosslinked non-isocyanate polyurethanes (NIPUs) derived from lignin, which are synthesized for the first time via transurethanization. This reaction was used as a simple, powerful and safe polymerization process. This method eliminates the need for lignin purification and requires only widely available compounds. A comprehensive analysis of the polymerization process and the resulting NIPU materials was conducted. It was observed that lignin could be introduced in the crosslinked architectures up to 32 wt%, and these aromatic NIPUs showed elastomeric behaviors with tunable properties. Additionally, two end-of-life management strategies based on mechanical and chemical recycling were also evaluated. These bio-based and aromatic materials offer a promising pathway toward sustainable and easy production of NIPU from lignin while adhering to multiple green chemistry principles, including the use of renewable feedstocks, safe reagents, limited synthesis steps, and waste prevention.
Introduction
Polyurethanes (PUs) are a key family of polymers with a large number of applications, including construction, transport, furniture, and appliances, because of their versatility and tunable physical properties.1 PUs can exhibit a large range of mechanical behaviors, bonding strength, impermeability, and chemical and abrasion resistance.2 This major polymer family accounts for 21 Mt per year, corresponding to around 5% of the annual worldwide plastic production in 2022.3 The PU market is dominated by flexible and rigid foams (65%), followed by coatings, adhesives, sealants, and elastomers.4However, conventional PUs face significant challenges regarding safety and sustainability throughout their life cycle. They are mainly derived from fossil-based monomers and involve the use of isocyanates. Isocyanates are toxic and sensitizing compounds,5,6 and their production relies on the use of highly toxic phosgene. Besides, the end of life of PUs is also questionable as most of them are thermosetting polymers with recycling issues. Consequently, their life cycle predominantly ends in landfills (50%) or incineration (45%), which leads to environmental hazards.2
Over the last two decades, extensive research has been done in the field of PUs to develop safer and more sustainable products. Among the recently developed strategies, the use of isocyanate alternatives and renewable feedstock7 and end-of-life management possibilities have brought tremendous improvements.2,8 Isocyanates are one of the main research targets in this field as their elimination would positively affect both safety and sustainability. In this regard, non-isocyanate polyurethanes (NIPUs) can be developed through various synthetic pathways,9,10 such as the very popular aminolysis between cyclocarbonates and amines (Fig. 1A) or transurethanization (Fig. 1B).10,11 The latter approach has been significantly less studied although it is a safe alternative to the conventional isocyanate–polyol reaction. Transurethanization is the urethane-pendant version of transesterification, which involves the condensation of a hydroxyl group with a carbamate (urethane). To generate NIPUs, a polycarbamate and a polyol are reacted (Fig. 1B). The release of a small alcohol compound drives the reaction to completion. Transurethanization reactants are also less toxic and more stable than isocyanates, making their storage and handling safer. More importantly, unlike the reactants required for NIPU synthesis by aminolysis (Fig. 1A), transurethanization reactants can be derived safely from common building blocks used in conventional PU synthesis and can be more easily implemented.2,12–14 Moreover, transurethanization-derived NIPUs have structures similar to conventional PUs, unlike polyhydroxyurethanes (PHUs) formed via aminolysis (Fig. 1A).
 | ||
| Fig. 1 Non-isocyanate pathways for the synthesis of NIPUs. (A) Aminolysis: the polyaddition of a polyamine and a polycyclocarbonate. (B) Transurethanization: the polycondensation of a polycarbamate and a polyol. | ||
The first report of polyurethane synthesis via transurethanization by Rokicki et al. dates back to 2002.15 Two main types of carbamates have been investigated in the literature: hydroxyethylcarbamates (HEC) and methylcarbamates (MC).16 MC and HEC can be formed using amines and safe carbonates.13–15 However, MC offer clear advantages over HEC. Side reactions occur with HEC through self-back-biting of hydroxyls, which creates hard segments in the polymer.12,17,18 Additionally, upon transurethanization, MC release a more volatile alcohol (methanol) that can be easily removed.12
In the past decade, a wide range of thermoplastic NIPUs have been prepared using MC. Aliphatic, cycloaliphatic, aromatic and bio-based monomers have been shown to perform well.19,20 Pr. Burel's team has probably published the most on this topic by preparing NIPUs from a variety of polyamines and polyols.12,21–26 They have used transurethanization to create NIPU oligomer precursors for photocrosslinked NIPUs,23,24,27 low-isocyanate PUs25 and epoxy-NIPU foams.26 Transurethanization is also well adapted for the synthesis of polycarbonate-NIPU28–30 and polyester-NIPU copolymers,31 in which the mechanical properties can be substantially improved thanks to aromaticity.31
However, the synthesis of thermoplastic NIPUs via transurethanization is still hindered by the relatively low molar masses achieved.16 Moreover, the large majority of PU applications necessitate thermoset materials with high thermal and mechanical performances. This drastically limits the applications and properties of the formed polymers. Thus, achieving the synthesis of sustainable, crosslinked and aromatic NIPUs would be valuable.
For this purpose, lignin can be used as a bio-based aromatic polyol crosslinker. Lignin is a low-cost and widely available second-generation (2G) biopolymer, which is a side-product of the pulp and paper industries and bioethanol production.32–34 Lignin is made of phenolic units, but the type of units, inter-unit linkages and their molar mass distribution vary depending on the botanical origin and extraction/fractionation process from lignocellulose. Owing to its aromaticity and high functionality, lignin has proven to be a particularly interesting bio-based aromatic building block for polymer synthesis,35–37 especially for PUs, in the past years.38–41
In this work, we report the first examples of crosslinked NIPUs and lignin-based NIPUs obtained via transurethanization. Hexamethylenediamine (HMDA) was reacted with dimethylcarbonate (DMC) to form hexamethylenedicarbamate (HMDC) through an optimized synthesis protocol. The kinetics and selectivity of transurethanization between HMDC and polyols were studied through FTIR, NMR and thermogravimetric analysis (TGA). Lignin was converted into liquid polyols (L-pOH) through a safe and green process.42 These L-pOH were then reacted with HMDC to form crosslinked NIPUs via transurethanization. The amount of lignin and the stoichiometry were varied to study their influence on the polymer properties. The obtained sustainable NIPU materials were studied by TGA, differential scanning calorimetry (DSC), dynamic mechanical analysis (DMA), tensile tests, solvent swelling and scanning electron microscopy (SEM). Finally, the mechanical and chemical recyclability of these bio-based thermoset NIPUs were assessed.
Experimental section
Materials
Birchwood organosolv lignin was kindly provided by TNO. It was obtained at a pilot scale through the aqueous acetone Fabiola™ process.43 Birchwood chips were fractionated in 50% w/w aqueous acetone and sulfuric acid at 140 °C. Lignin was recovered from the pulping liquor by precipitation after dilution in water. Dimethyl carbonate (DMC, 99%), pyridine, and ethylene carbonate (EC, 99%) were obtained from Alfa Aesar. Hexamethylenediamine (HMDA) was purchased from BASF. Polyethylene glycol with Mn 300 g mol−1 (PEG300) was bought from Acros Organics. Polyethylene glycol methyl ether with Mn 500 g mol−1 (PEG500MM) was purchased from Fluka analytical. Methanol (MeOH) and creosol were obtained from Fisher Scientific. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD, 98%), chromium(III) acetylacetonate (Cr(acac)3), cholesterol, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (Cl-TMDP, 95%) were bought from Sigma-Aldrich. Deuterated chloroform (CDCl3, 99.8%) and deuterated dimethylsulfoxide (DMSO-d6, 99.8%) were purchased from Eurisotop.Hexamethylenedimethylcarbamate (HMDC) synthesis
HMDA (1 eq.), DMC (10 eq.), and TBD (0.1 eq.) were agitated under an argon flux at 90 °C overnight. The product was cooled down to room temperature and then placed in an ice bath for 2 h for precipitation of the product. The precipitate was recovered by filtration, rinsed with water and dried at 40 °C under a vacuum overnight. HMDC was obtained as a white powder with an average yield of 86% (5 replicates). Mass balances can be found in Table S1 in ESI.†Liquid lignin-based polyol (L-pOH) synthesis
L-pOHs were synthesized according to a previously described procedure.42 Before each synthesis, PEG300 and EC were dried overnight at 40 °C under a vacuum. Lignin (for hydroxyl content and molar mass distribution, see Table S2 and Fig. S3 in ESI†), PEG300, EC (1.1 eq. with respect to the total hydroxyl content of the lignin) and the TBD catalyst (approximately 0.15 eq.) were placed in a round-bottom flask under mechanical agitation and reacted at 130 °C for 4 h. Different L-pOH samples were prepared with 30, 40 and 50 wt% lignin. The lignin-based polyols were obtained as a brown liquid at the end of the reaction and used without further purification. Mass balances can be found in Table S3 in ESI.†A series of L-pOH was synthesized with varying ratios of lignin/PEG300. The L-pOH samples are referred to as: “L-pOH-lignin wt%” (e.g.: L-pOH-30), and a batch letter is added as necessary.
Transurethanization kinetics
HMDC (1 eq.), PEG500MM (1 eq.) and TBD (0.1 eq.) were mixed. The mixture was stirred in a round-bottom flask under argon flux at 120, 140 or 160 °C. Aliquots were withdrawn at regular intervals and analyzed by 1H NMR in CDCl3. All aliquots were fully soluble in CDCl3. The conversion of HMDC to the urethane product was evaluated by comparing the integrations of the peaks at 4.14 (product, 4H) and 1.27 ppm (HMDC + product, 8H). Results of additional testing by TGA and using K2CO3 as the catalyst can be found in Fig. S7–S8 in ESI.†A 2D NMR analysis of the reaction mixture containing HMDC (1 eq.), PEG500MM (2 eq.) and TBD (0.1 eq.) was performed after 24 h at 160 °C under argon flux.
The kinetics of the reaction between HMDC and a model phenol (creosol) were also studied (Fig. S4 in ESI†).
Non-isocyanate polyurethane (NIPU) synthesis
L-pOH was mixed thoroughly in a mortar with HMDC (0.5 eq. with respect to OH groups in L-pOH) until a homogeneous mixture was obtained. No additional catalyst was added as L-pOH already contained TBD. The mixture was then placed in a PTFE plate and incubated in an oven under an argon flux at 140 °C for 3 h and then 160 °C for 2 h. The obtained material was then pressed into a sheet (100 × 100 × 1 mm) by compression molding. Pressing was done at 150 bar (15 tons) using a hydraulic press standard from Lab Tech Engineering Company Ltd (China). The materials were first preheated for 5 min at 160 °C, pressed and vented five times, and then finally pressed at 160 °C for 30 min. The reaction was repeated with different L-pOH and carbamate-to-hydroxyl (C/OH) molar ratios of 0.9, 0.95, 1.0 and 1.1. Mass balance for all the synthesis reactions can be found in Table S4 in ESI.†The NIPU materials are named “NIPU-lignin content in L-pOH wt%-C/OH molar ratio” (e.g.: NIPU-40-1).
NIPU thermo-mechanical recycling
For mechanical recycling tests, dry NIPU-40-0.9 sheets were ground finely in a small grinder. The powder was then pressed at 160 °C under 150 bar (15 tons) pressure from 30 min to 2 h. For specific tests, TBD (amount corresponding approximately to the initial wt% of TBD in the material) was added to the material during the grinding step. All experimental details can be found in Table S9 in ESI.†NIPU depolymerization with methanol
NIPU-40-1 was finely ground. Then, methanol was added (10 mL per gram of material), and the mixture was placed in a reactor. The reactor was flushed with argon before being sealed and heated at 140 °C under agitation for 24 h (the maximum pressure reached was 10 bar). After cooling down to room temperature, the brown homogeneous liquid obtained was then evaporated under a vacuum at 40 °C, yielding the depolymerized mixture (Depol-NIPU).For the repolymerization step, 2.2 wt% TBD was added to the mixture (the same quantity as in NIPU-40-1). The curing step was performed similar to that described for NIPU synthesis.
Characterization
Fourier transform infrared (FTIR) spectra were obtained using a Nicolet 380 spectrometer (Thermo Electron Corporation) equipped with an attenuated total reflectance (ATR) diamond module. Each spectrum was acquired with 32 scans in the range of 4000–400 cm−1 at room temperature.1H and 31P NMR spectra were acquired using a BRUKER Advance III HD-400 MHz instrument with a BBFO probe. 2D 1H–1H and 1H–13C NMR spectra were acquired on a Bruker 500 MHz spectrometer with a CryoProbe™ Prodigy. 1H NMR was performed in CDCl3 or DMSO-d6, and 16 scans of approximately 30 mg of each sample in 600 μL of the solvent were recorded. 2D NMR was performed on 100 to 200 mg of each sample in 600 μL of DMSO-d6, and 32 scans were obtained at room temperature.
Quantitative 31P NMR was performed by following a protocol described in the literature.42,44,45 About 30 mg of each sample was dissolved in 450 μL of a pyridine/CDCl3 1.6/1 (v/v) mixture. Then, 100 μL of a standard solution containing cholesterol (0.1 M) and Cr(acac)3 (5 mg mL−1) was added, followed by the addition of 75 μL of the phosphytilating reagent Cl-TMDP. The vessel was closed and stirred for 2 h before analysis. 128 scans were recorded with 15 s relaxation intervals at 25 °C.
Thermogravimetric analysis (TGA) was performed on a TA Instrument Hi-Res TGA Q5000. Approximately 2–3 mg of each was heated in a platinum crucible from room temperature to 600–700 °C at a rate of 20 °C min−1 under a N2 atmosphere (25 mL min−1). T5% is defined as the temperature at which 5% weight loss from the initial mass occurs. The TGA derivatives (DTG) were calculated to determine the main degradation temperatures at the maximum points of the peaks (Td).
Differential scanning calorimetry (DSC) measurements were recorded on a TA instruments Discovery DSC-25 apparatus under a 50 mL per min dry nitrogen flow. Typically, 2–3 mg samples were placed in aluminum pans, while an empty pan was used as the reference. To erase the thermal history, the samples were first heated to 160 °C for 10 min. They were then cooled down to −60 °C. Finally, the samples were heated up to 160 °C with a 10 °C min−1 ramp. The glass transition temperatures (Tg) were measured at the inflection points of heat flow during the last heating ramp.
Viscosity measurements were performed using a Discovery HR-3 Hybrid Rheometer by TA Instruments. The sample was placed in Peltier plates using 25 mm parallel plate geometry and tested for shear rates ranging from 0.1 to 1000 s−1 over a range of temperatures from 25 to 160 °C, and a Newtonian behavior was observed.
Dynamic Mechanical Analysis (DMA) was performed on a Discovery HR-3 Hybrid Rheometer by TA Instruments. A 2 cm × 1 cm sheet of sample was placed vertically in a rectangular torsion geometry. The sample were heated at 3 °C min−1 from −50 °C to 180 °C with a 0.01% strain at 1 Hz frequency. The alpha transition temperature (Tα) was measured at the maximum of tan(δ). The crosslinking density ν(mol cm−3) was obtained using eqn (1) based on the rubber elasticity theory.46 In this equation,  is the storage modulus (Pa) at the rubbery plateau (G′ independent of the temperature), Tα + T (K) is the minimum temperature of the rubbery plateau (chosen as 120 °C for most NIPUs), and R the gas constant (8.314 J mol−1 K−1).
is the storage modulus (Pa) at the rubbery plateau (G′ independent of the temperature), Tα + T (K) is the minimum temperature of the rubbery plateau (chosen as 120 °C for most NIPUs), and R the gas constant (8.314 J mol−1 K−1).
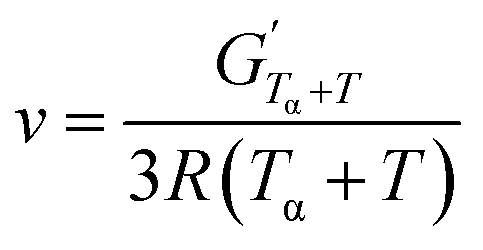 | (1) |
The density of the material was determined by weighing a disk of the sample measuring 25 mm in diameter. The molar mass between the crosslinks (Mc) in kg mol−1 was determined using eqn (2), where ρ (kg m−3) is the density of the material, and ν is the crosslinking density.
 | (2) |
Uniaxial tensile tests were performed using an INSTRON® 68TM-10 Universal Testing System equipped with a 10 kN load cell in a room set at 23 °C with a constant crosshead speed of 20 mm min−1 until failure. The experiments were performed using a set of 5 dumbbell-shaped samples with approximate dimensions of 45 × 5 × 1 mm3. The Young's modulus (E), stress at break (σ) and elongation at break (ε) were recorded.
The swelling ratios (SR) of the materials were measured on triplicate samples (200 mg) after immersion in water and acetone for 72 h. Eqn (3) was used to obtain SR, where mswelled is the mass of the sample after immersion, mfinal is the mass of the sample after drying at 40 °C under a vacuum overnight. The gel fractions (GF) were calculated using eqn (4).
 | (3) |
 | (4) |
Scanning electron microscopy (SEM) was carried out using a Jeol IT-100. The micrographs of the representative sections of frozen-fractured surfaces were obtained after metallic sputtering.
Stress-relaxation experiments were performed on a Discovery HR-3 Hybrid Rheometer from TA Instruments. A 25 mm diameter sample disk was placed in parallel plates. The sample was heated to the temperature under study, and a 1% strain was applied. Additional equations of the stress-relaxation experiments can be found in ESI (Fig. S24 and Table S8).†
Size-Exclusion Chromatography (SEC) was performed using an Acuity APC apparatus from waters with THF as the eluent (0.6 mL min−1) at 40 °C. Detection was performed using an Acquity ultraviolet transmission (UV) and refractive index (RID) detectors. Three 4.6 × 150 mm Acquity APC XT columns with 450, 200 and 45 Å pore sizes were connected. HMDC was analyzed without modification. The lignin was acetylated prior to analysis, as described in the literature, to maximize its solubility in THF and limit interactions with the columns.45 The L-pOH and depolymerized mixture was acetylated prior to analysis by following another protocol in order to observe PEG.47 The calibration was carried out based on polystyrene (PS) standards. The detector response was normalized based on the total integration of each signal.
Results and discussion
Preparation of dicarbamate and lignin-based polyols
Dimethylcarbamate was synthesized through a green procedure described previously in the literature (Fig. 2A).12,21,28 HMDC was synthesized via a solvent-free one-pot procedure. HMDA was reacted with a large excess of DMC to limit the formation of unwanted polyureas, and using TBD as the catalyst. The reaction was repeated several times with yields ranging from 85 to 90% and scaled up to 50 g of HMDA (Table S1 in ESI†). | ||
| Fig. 2 Preparation of HMDC. (A) Reaction scheme for HMDC synthesis. (B) Recovery and recycling of HMDC from the reaction media. | ||
The dicarbamate product precipitated at the end of the reaction and was recovered by simple filtration. The filtrate containing mostly excess DMC, the catalyst, and traces of methanol was recovered separately (Fig. 2B). The filtrate was then recycled and reacted with fresh HMDA, without adding catalyst, to produce a new batch of clean HMDC with similar yields (85%). Interestingly, the traces of methanol in the filtrate did not affect the production of HMDC. With one cycle of filtrate reuse, the E factor of the process consequently decreased from 5 to 3.3 (Table S1 in ESI†). Further improvement via cascade reuse of the reaction media might significantly reduce waste generation.
To synthesize NIPUs, HMDC must be reacted with a polyol. Lignin was selected as a low-cost, renewable and polyfunctional aromatic building block. Technical lignins are polyfunctional polymers in which the majority of reactive groups are phenols (Fig. 3A and Table S2 in ESI†).44,48 However, phenols are known to have poor reactivity towards isocyanates,49 and the same was also verified in this case with carbamates (Fig. S4 in ESI†). Thus, a chemical modification was performed to convert lignin phenols into aliphatic alcohols, allowing a more homogeneous distribution of the reactive groups. Common methodologies used for this functionalization involve oxypropylation with toxic and potentially explosive propylene oxide.35,38 To avoid the use of toxic chemicals, our team recently developed a solvent-free, purification-free, one-pot, and safe methodology to form lignin-based liquid polyols (L-pOH).42,46 Lignin was reacted with ethylene carbonate (EC) in polyethylene glycol (PEG300) using a catalyst, TBD in this case (Fig. 3B). PEG300 acts as a solvent for oxyalkylation of polyols while also acting as soft chains in the final material.
 | ||
| Fig. 3 L-pOH synthesis. (A) Representative technical lignin structure and a simplified representation with alcohols and phenols represented in green and grey, respectively. (B) Reaction scheme for L-pOH synthesis. | ||
Three different L-pOHs were produced with lignin contents between 30 and 50 wt%. The final OH contents of the L-pOHs were quantified by 31P NMR. For all L-pOH, complete conversion of the phenol groups was observed. The aliphatic hydroxyl contents of L-pOH-30, 40 and 50 were respectively 6.71, 5.22 and 6.07 mmol g−1 (Fig. S5 and Table S3 in ESI†). The viscosity of dry L-pOHs was measured to better understand their behavior during the synthesis of NIPUs (Fig. S6 and Table S3 in ESI†). The viscosity increased with the lignin content, measuring 29, 210 and 1900 Pa s at 25 °C for 30, 40, and 50 wt% lignin, respectively. The viscosity decreased dramatically with temperature and reached a low value of 0.23 Pa s for L-pOH-50 at 160 °C, thus allowing easier processing for the preparation of materials.
Kinetics and selectivity of the transurethanization reaction
Due to the lower reactivity of polycarbamates compared with isocyanates, their transurethanization requires stronger conditions than those needed for conventional PU synthesis. Typically, the reaction is performed using a catalyst for more than 4 h above 140 °C.12,19–21,28,31 Some commonly used catalysts for transurethanization are Bu2SnO, Ti(OBu)4, t-BuOK, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), K2CO3, and para-toluenesulfonic acid.19,21,50,51In order to estimate the optimal reaction conditions, the reaction was studied on a model system. HMDC was reacted with a monofunctional PEG (PEG500MM) using TBD as the catalyst (Fig. 4). The reaction was performed in bulk conditions to mimic polymer synthesis at 120, 140 and 160 °C, followed by 1H NMR analysis (Fig. S7 in ESI†). HMDC conversion via transurethanization was measured through the integration of the characteristic signal of the newly formed urethanes at 4.14 ppm (triplet). HMDC achieved full conversion after 6 h at 140 °C (Fig. 5). The reaction was slower as the temperature decreased. At 120 °C, the reaction did not reach completion within 6 h.
 | ||
| Fig. 4 Reaction scheme for the transurethanization of HMDC with PEG500MM using TBD as the catalyst. | ||
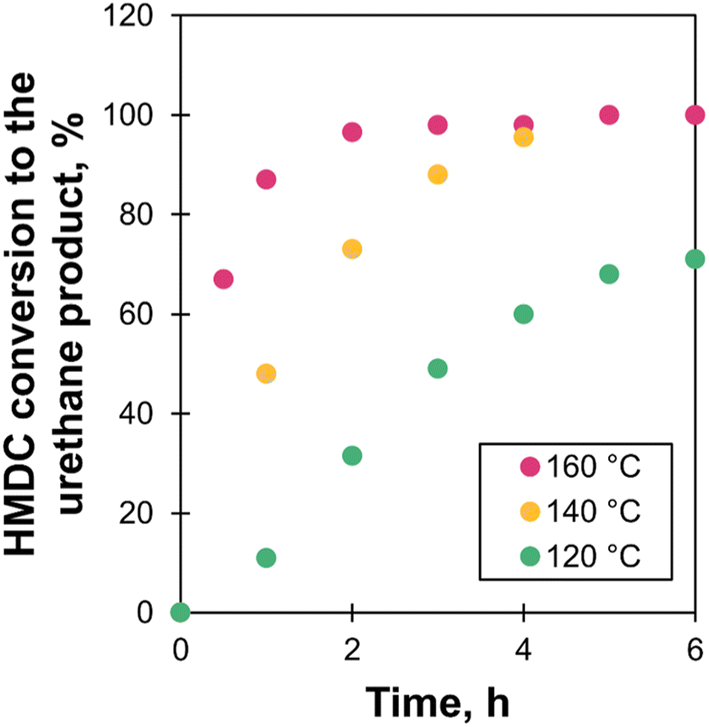 | ||
| Fig. 5 Evolution of the conversion of HMDC to the urethane product via transurethanization as measured by 1H NMR in CDCl3. | ||
The reaction mixture evolution was also followed by FTIR for up to 48 h at 160 °C (Fig. 6). Transurethanization was confirmed by the disappearance of the O–H band over 6 h (a1, 3500 cm−1) along with an enlargement of the N–H bands (a2 and b5, respectively 3330 cm−1 and 1525 cm−1), and a shift of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OHMDC band from 1690 (b2) to the COurethane band at 1715 cm−1 (b1). At extended reaction times, the COurethane and N–Hurethane bands (b1 and b5) were decreased. Additionally, the O–H band (a1, 3500 cm−1) increased, and a new band characteristic of urea bonds appeared at 1615 and 1575 cm−1 (b3 and b4).52 This may result from the self-condensation of the urethane structures in the presence of traces of water to form urea (Fig. 7A). The formation of other side products, such as allophanates and biurets, was also evidenced by the small peak at 1690 cm−1 after 24 h of reaction (Fig. 7B).29,53–55 Their presence was however limited, as confirmed by 2D 1H–13C NMR analysis of the reaction mixture after 24 h at 160 °C (Fig. S9 to S11 in ESI†).
OHMDC band from 1690 (b2) to the COurethane band at 1715 cm−1 (b1). At extended reaction times, the COurethane and N–Hurethane bands (b1 and b5) were decreased. Additionally, the O–H band (a1, 3500 cm−1) increased, and a new band characteristic of urea bonds appeared at 1615 and 1575 cm−1 (b3 and b4).52 This may result from the self-condensation of the urethane structures in the presence of traces of water to form urea (Fig. 7A). The formation of other side products, such as allophanates and biurets, was also evidenced by the small peak at 1690 cm−1 after 24 h of reaction (Fig. 7B).29,53–55 Their presence was however limited, as confirmed by 2D 1H–13C NMR analysis of the reaction mixture after 24 h at 160 °C (Fig. S9 to S11 in ESI†).
 | ||
| Fig. 6 FTIR spectra of the reaction mixture at different reaction times under TBD catalysis at 160 °C (0 to 48 h). (A) The 3000–3800 cm−1 region with a1 and a2 corresponding to the O–H and N–H vibration bands, respectively. (B) The 1400–1800 cm−1 region with the b1, b2, b3, b4, and b5 bands corresponding to COurethane, COHMDC/allophanate/biuret, COurea, N–Hurea, and N–Hurethane, respectively. | ||
 | ||
| Fig. 7 (A) Proposed side-reaction in the presence of water leading to urea (blue) formation. (B) Side-reaction of self-condensation of HMDC, forming allophanate (orange) and biuret (brown). | ||
Moreover, urea formation increased substantially when the reaction mixture contained traces of water, resulting in rapid and complete disappearance of the urethane band (1690–1715 cm−1). K2CO3 was also examined as a catalyst for the reaction and was found to exacerbate urethane disappearance (Fig. S12 in ESI†).
NIPU synthesis
NIPUs were produced in a green way by reacting HMDC with L-pOH in bulk conditions to avoid the use of solvents (Fig. 8). No additional catalyst was added as L-pOH already contained TBD from the previous step. Apart from TBD, all the reactants used here are potentially bio-based,56–58 which can lead to bio-based contents up to 97 wt% in the as-formed materials. | ||
| Fig. 8 Reaction scheme for the polymerization of L-pOH with HMDC (black arrow), and its depolymerization with methanol (blue arrow). | ||
Further, the dependency of NIPU properties on their lignin content and stoichiometry of the transurethanization reaction was studied. Due to its polyfunctional and polyaromatic structure, lignin can act both as a crosslinker and form hard segments in PUs, enhancing their rigidity and thermal stability.35,38,59 The lignin content of the NIPUs was varied by using L-pOH containing 30 to 50 wt% lignin, which resulted in NIPUs with final lignin contents varying from 18 to 32 wt% (Table 1).
| Material | L-pOH type | C/OH molar ratio | Lignin content in the material, wt% |
|---|---|---|---|
| NIPU-30-0.9 | L-pOH-30 | 0.90 | 18.7 |
| NIPU-30-1 | L-pOH-30 | 1.0 | 18.0 |
| NIPU-40-0.9 | L-pOH-40 | 0.90 | 27.2 |
| NIPU-40-0.95 | L-pOH-40 | 0.95 | 26.8 |
| NIPU-40-1 | L-pOH-40 | 1.0 | 26.3 |
| NIPU-40-1.1 | L-pOH-40 | 1.1 | 25.4 |
| NIPU-50-0.9 | L-pOH-50 | 0.90 | 32.3 |
| NIPU-50-1 | L-pOH-50 | 1.0 | 31.1 |
The carbamate-to-hydroxyl (C/OH) molar ratio was varied from 0.9 to 1.1 (Table 1). Lower ratios would result in a higher content of free hydroxyl groups that can participate in intra- and inter-molecular hydrogen bonds.60 Additionally, free hydroxyl groups can allow transurethanization reactions inside the cured NIPUs, turning the materials into covalent adaptable networks (CANs), such as vitrimers, in which thermoset architectures can be reprocessed at the end of their life by thermo-mechanical treatment.50,59,61 The stoichiometry of 1.1 allowed the study of the influence of excess amines, which could generate more ureas, allophanates and biurets. In total, eight different materials were prepared from the three initially synthesized L-pOH with four different C/OH ratios.
The L-pOHs were reacted with HMDC through optimized curing conditions obtained from the model study. A homogeneous liquid reaction medium was obtained after mixing (Fig. 9A and B). After curing, the obtained materials were solid and presented some trapped gas bubbles (Fig. 9C), confirming the release of methanol during transurethanization. The materials were then hot-pressed to yield different solid sheets of lignin-based NIPU materials (Fig. 9D). An FTIR analysis confirmed that the evolution of the reaction during curing was similar to that observed in the model study (Fig. S13 in ESI†). Notably, the transurethanization process was complete only after the pressing steps, as evidenced by the almost complete disappearance of the O–H band (a1). Additionally, no significant urea (b3 and b4) and allophonate/biuret (b2) bands could be detected, confirming that side reactions were minimized.
 | ||
| Fig. 9 Synthesis steps of NIPU-40-1. (a) Initial mixing of L-pOH and HMDC at room temperature. (b) Homogeneous reaction mixture before curing. (c) NIPU-40-1 after 3 h at 140 °C and 2 h at 160 °C. Trapped bubbles can be observed on the surface of the material. (d) Final product obtained after hot pressing at 160 °C for 30 min. | ||
Characterization of NIPUs
The thermal stability of the materials was analyzed by TGA (Table S5, Fig. S14 and S15 in ESI†). All NIPUs exhibited T5% values between 221 and 253 °C. The degradation occurred consistently across all the samples, with a single degradation peak at Td between 342 °C to 352 °C. These degradation temperatures are in the range of lignin degradation, as well as urethane linkage dissociation.12,62 Neither lignin content nor C/OH molar ratio influenced the degradation temperatures significantly.The thermal and thermo-mechanical behaviors of the materials were studied by DSC and DMA (Fig. 10, S16 to S19 and Table S5 in ESI†) in the stable temperature ranges identified by TGA. Tg ranged from −33 °C (NIPU-30-1) to −7 °C (NIPU-50-1). At room temperature, these materials existed in a rubbery state. The DSC curves did not show any inflection at the Tg of lignin at 141 °C (Table S5, Fig. S16 and S17 in ESI†), indicating the successful dispersion and inclusion of lignin into the macromolecular architecture. The Tg of the materials increased with the lignin content, as expected, but did not correlate directly to C/OH stoichiometry (Fig. 10A and S20 in ESI†). The Tα, values determined by DMA showed similar evolution and were very close to the Tg values, with an increase from −33 °C for NIPU-30-1 to −12 °C for NIPU-50-0.9 (Fig. 10B).
 | ||
| Fig. 10 Thermo-mechanical properties of NIPUs measured using DSC and DMA. (A) Glass transition temperatures (Tg) based on DSC curves. (B) Alpha transition temperatures (Tα) based on DMA. (C) Crosslinking density of the network (ν) based on DMA. In all the graphs, “NIPU-X-1” represents all the NIPUs with a C/OH ratio of 1, and “NIPU-40-Y” represents all the NIPUs produced with L-pOH-40. | ||
The crosslinking densities (ν) were determined from the DMA data (Fig. 10C and Table S6 in ESI†). All materials exhibited thermosetting behaviors, with ν from 46 to 294 mol m−3. The ν value increased with the lignin content, confirming the central role of lignin as a multifunctional crosslinker in the network. In contrast, the C/OH ratio had minimal influence on the crosslinking density in the studied range of 0.9 to 1.1 (Fig. S20 in ESI†).
Uniaxial tensile tests of the NIPUs were performed to evaluate their mechanical properties at room temperature (Fig. 11, S21, S22 and Table S6 in ESI†). The NIPU formulation had a significant impact on the uniaxial mechanical performance. The materials presented a broad spectrum of behaviors, with Young's modulus increasing from 5 to 58 MPa as the lignin content rose from 18 to 32 wt%. Following the same trend, the ultimate tensile strength increased from 0.7 to 3.8 MPa, while the maximum elongation decreased from 51 to 20%, indicating an obvious correlation between rigidity and lignin content, as expected given the rigid and aromatic structure and the crosslinking ability of lignin. As for DMA results, no clear relationship with the C/OH ratios could be established in the tested range, although some variability was observed at similar lignin levels (Fig. S23 in ESI†). Overall, the NIPUs exhibited thermo-mechanical properties comparable to vitrimer materials with the same lignin contents previously developed by our team using similar lignin-based polyols.46 The as-obtained proof-of-concept materials had, expectedly, rather low mechanical properties compared to conventional PUs. They exhibited a tensile strength in the range of PU elastomers (5–100 MPa) but with very low elongation at break compared to them (typically above 1000%).63,64 The materials also exhibited a lower range of mechanical properties than the bio-based NIPUs obtained by the more studied aminolysis reaction.65
 | ||
| Fig. 11 Mechanical properties of the materials as a function of lignin content, as obtained from uniaxial tensile tests. (A) Selected stress–strain curves of the NIPUs representative of average evolutions. (B) Young's modulus (E) of the NIPUs. (C) Maximum stress (σ) before the break of the NIPUs. (D) Maximum elongation (ε) before the break of the NIPUs. In (B)–(D) “NIPU-X-1” represents all the NIPUs with a C/OH ratio of 1, and “NIPU-40-Y” represents all the NIPUs produced with L-pOH-40. | ||
The chemical crosslinked structure of the NIPU networks was further analyzed by swelling ratio (SR) and gel fraction (GF) measurements (Fig. 12 and Table S7 in ESI†). Water and acetone were chosen as the protic and aprotic polar solvents because of their affinity to different chemical structures within the materials. PEG is soluble in water and acetone, while HMDC and lignin are more soluble in acetone. With both solvents, the GF increased with higher lignin content. In water and acetone, GF ranged from 66 to 81% and 47 to 85%, respectively. Similar values found with the two solvents might indicate that PEG chains constitute the majority of the soluble fraction. The increase in GF with lignin content is consistent with the role of lignin in enhancing crosslinking within the network. The relatively modest maximum GF values (81% and 85%) are likely due to the difunctional nature of PEG, which constitutes the majority of the L-pOH, leading to lower crosslinking densities, as well as possibly unreacted sites, as evidenced by the small remaining O–H band in the FTIR spectra of the NIPUs (Fig. S13†). Following the inverse trend to GF, SR decreased as the lignin content increased. This observation aligns with the rise in crosslinking density due to lignin polyfunctionality. The SR values were higher in water than in acetone, which can be explained by the hydrophilic structures in the materials derived from PEG and lignin.
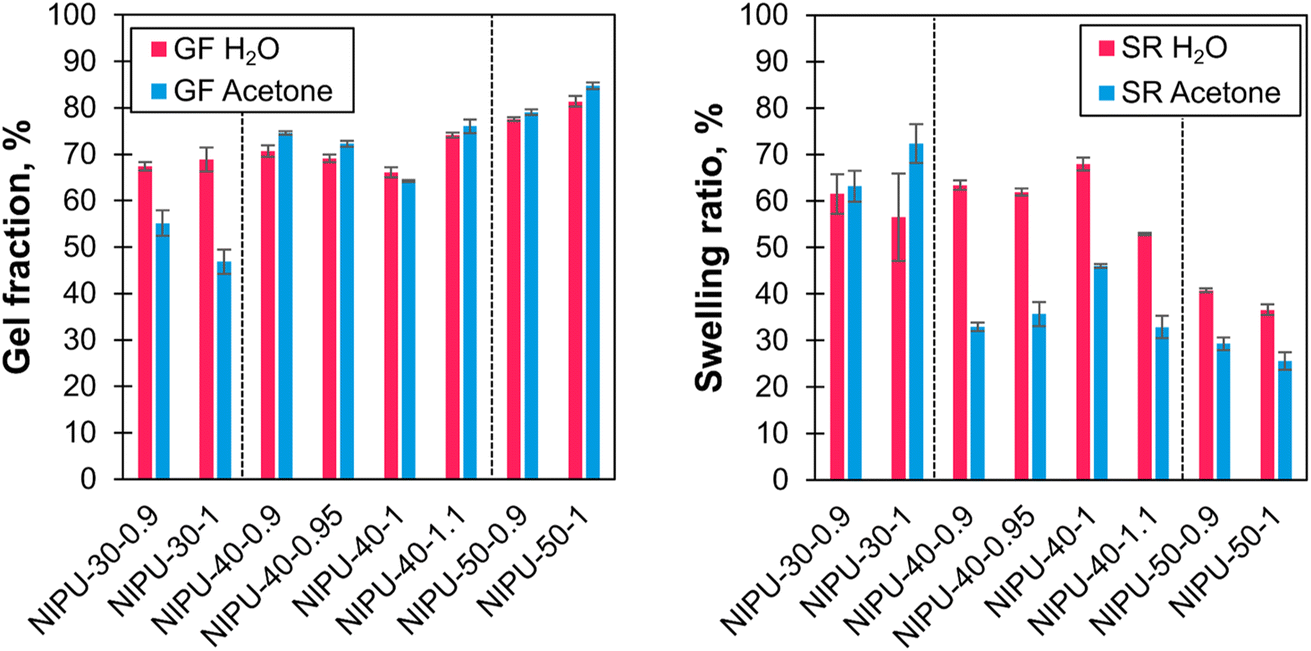 | ||
| Fig. 12 Gel fractions (GF) and swelling ratios (SR) of the NIPUs after soaking for three days in water (red) or acetone (blue). | ||
The microstructure of the NIPUs was examined using SEM imaging on frozen-fracture surfaces (Fig. S24 in ESI†). The micrographs revealed the effective incorporation and dispersion of lignin in the material, likely enhanced by the pre-modification of lignin to a liquid polyol. Some microscopic pores were also seen on the surfaces. Additional discussion on the origin of the porous structure and its influence on material properties can be found in ESI.†
NIPU recycling and dynamicity of the networks
The end-of-life management of thermoset PUs presents significant challenges due to their structure.66 The crosslinked structures can limit the options in terms of recycling. To address this bottleneck, which impacts the life cycle and the sustainability of the materials, two different strategies were examined for recycling the lignin-based NIPUs. Direct mechanical recycling by hot pressing was tested on ground materials, as for CANs. Another approach using chemical depolymerization was also investigated.Direct mechanical recycling of the thermoset-like NIPUs was explored through reprocessing without melting. Reprocessing or reshaping these materials relies on the possibility of performing urethane exchange reactions within the material.50 Stress-relaxation experiments were conducted on NIPU-40-0.9, NIPU-40-1 and NIPU-40-1.1 to assess the dynamicity of their network. The presence of TBD in the NIPUs facilitated transurethanization and allowed shape adaptation despite crosslinks. The stress-relaxation curves obtained for each NIPU demonstrated the network dynamicity between 140 °C and 160 °C (Fig. 13, S25 and Table S8 in ESI†). The average relaxation times (〈τ〉) and pseudo-activation energies (Ea) for bond exchange in the NIPUs were clearly linked with the C/OH ratio. NIPU-40-0.9, which has the highest amount of free OH groups, was the most dynamic, with a 〈τ〉 of 1200 s and an Ea of 138 kJ mol−1. These values are in the range observed for conventional PU CANs activated by DBTDL catalysis (100 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, and 100–180 kJ mol−1)50,67 and close to the values of dynamic PHU networks elaborated using different catalytic systems, such as DBTDL,61 DMAP,68 and tertiary amines.69 NIPU-40-1 and NIPU-40-1.1 were less dynamic, as expected, due to their lower free OH content. However, because of the limited thermal stability of TBD, which degrades above 160 °C,20,70 the thermal window for stress relaxation was relatively narrow as higher temperatures would disable its catalytic activity, and hence, impede the dynamicity of the network.
000 s, and 100–180 kJ mol−1)50,67 and close to the values of dynamic PHU networks elaborated using different catalytic systems, such as DBTDL,61 DMAP,68 and tertiary amines.69 NIPU-40-1 and NIPU-40-1.1 were less dynamic, as expected, due to their lower free OH content. However, because of the limited thermal stability of TBD, which degrades above 160 °C,20,70 the thermal window for stress relaxation was relatively narrow as higher temperatures would disable its catalytic activity, and hence, impede the dynamicity of the network.
 | ||
| Fig. 13 Normalized stress-relaxation curves of the NIPU-40-Y materials prepared with different C/OH ratios. (A) C/OH = 0.9 (NIPU-40-0.9). (B) C/OH = 1 (NIPU-40-1). (C) C/OH = 1.1 (NIPU-40-1.1). The data fitted to stretched exponential decay are represented by the dashed lines, as detailed in the ESI (eqn S1 in ESI).† (D) Average relaxation times at 140 °C (〈τ〉140°C) and pseudo-activation energies (Ea) determined using the pseudo-Arrhenius law. Additional details are given in ESI.† | ||
Thermo-mechanical recycling trials were performed using NIPU-40-0.9 by grinding and hot pressing. The ground particles exhibited a certain cohesion after hot pressing, but despite multiple attempts, the resulting materials were very brittle (Table S9 in ESI†). The FTIR analysis of the mechanically recycled material revealed a reduction in urethane bands (1715 and 1525 cm−1) and the formation of ureas, allophanate, and biurets (Fig. S26 in ESI†). This fast decrease in urethane content could be a result of water uptake and may be enhanced by TBD catalysis. TBD has been previously reported to promote side reactions during reprocessing of crosslinked NIPUs.61
Chemical depolymerization of the NIPUs was also investigated. Lignin-based PHUs have previously been depolymerized by hydroglycolysis to recover and reuse the lignin.71 Here, we used a transurethanization inverse reaction, namely the methanolysis of urethanes, to reform the initial MC and polyol reactants (Fig. 8). The reported example of this methodology involved the reaction in methanol/THF at 65 °C for 20 h using t-BuOK as the catalyst.72 To improve the viability of this depolymerization methodology, methanolysis was performed without a catalyst at 140 °C for 24 h in a closed vessel. This approach yielded a homogeneous liquid, which was analyzed by FTIR, SEC and NMR after the removal of excess methanol. The FTIR spectrum exhibited the reappearance of an O–H band (a1), indicating free alcohol groups (Fig. 14). The N–H bands (a2 and b4) were narrower, and the COurethane (b1) band was shifted to a sharp COHMDC band (b2), suggesting the successful re-formation of HMDC and polyol without side-reactions. The 1H NMR spectra confirmed the near-complete disappearance of urethanes and the presence of HMDC (Fig. S27 in ESI†). 31P NMR allowed the quantification of urethane cleavage by assessing the free hydroxyl group content (Fig. S28 in ESI†). The depolymerized mixture (Depol-NIPU) contained 3.0 mmol g−1 of alcohol groups compared with 3.2 mmol g−1 in the initial reaction mixture used for the NIPU synthesis, suggesting that around 90% of the urethane linkages were cleaved.
 | ||
| Fig. 14 FTIR Spectra of the (1) NIPU-40-1 initial reaction mixture, (2) NIPU-40-1, (3) depolymerized NIPU-40-1, and (4) NIPU-40-1-RC (recycled material). (A) The 3000–3800 cm−1 region with the a1 and a2 bands corresponding to O–H and N–H vibrations, respectively. (B) The 1400–1800 cm−1 region with b1, b2, b3, b4, and b5 bands corresponding to COurethane, COHMDC/allophanate/biuret, COurea, N–Hurea, and N–Hurethane, respectively. | ||
Depol-NIPU was then analyzed by SEC using two complementary detectors (Fig. 15). The refractive index (RID) detector was used to analyze the whole sample, while the UV detector only detected the lignin fractions.42 The comparison of the molar mass distribution of Depol-NIPU and the reactants in UV showed that it contained lignin with a similar molar mass distribution as L-pOH. The RID signals evidenced all the different constituents of Depol-NIPU. The higher molar mass structures visible only in the RID analysis could be residual HMDC-PEG polyurethane chains that had resisted urethane bond cleavage or the polyurea chains from HMDC self-condensation, as suggested by the band b3 in the FTIR spectrum. SEC thus confirmed that Depol-NIPU is particularly close to the initial reaction mixture. Further, 2D 1H–13C NMR analysis confirmed the structure of the depolymerized mixture (Fig. S29 to S31 in ESI†). The methanolysis approach was thus successful in depolymerizing NIPU-40-1, with the almost quantitative cleavage of urethane bonds, despite their known robustness.
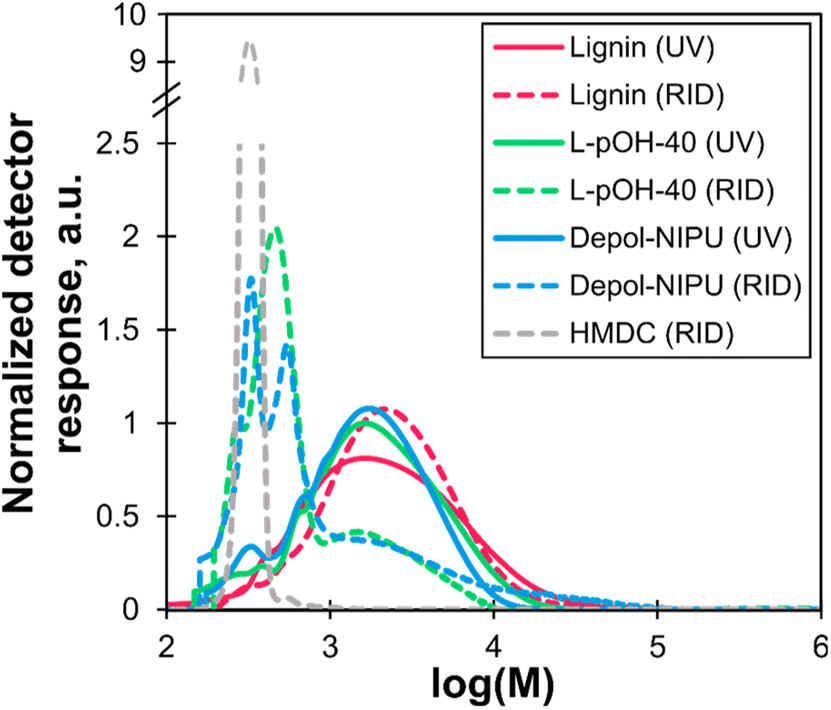 | ||
| Fig. 15 Apparent molar mass distribution of lignin, L-pOH-40, Depol-NIPU and HMDC from UV detector responses (solid lines) and RID detector responses (dashed lines). Solvent: THF. Calibration: polystyrene standards. | ||
Depol-NIPU contained the initial reactants and could potentially be used for the synthesis of new NIPUs. We thus attempted the synthesis of new materials directly from Depol-NIPU. The material obtained using this methodology, NIPU-40-1-RC, was rather brittle, and its FTIR spectrum presented the disappearance of the large urethane/carbamate bonds (b1 and b2) and the emergence of urea bonds (b3 and b4) (Fig. 14). This phenomenon is most likely due to water uptake by the depolymerized mixture, leading to urea formation in the presence of TBD. This hypothesis was further evidenced by the absence of the allophanate/biuret band b2 in the FTIR spectrum. Further investigations of different parameters, such as the drying process and the nature and the content of the catalyst, might help the wide application of this direct and complete chemical recycling strategy.
Conclusion
Transurethanization was successfully used for the synthesis of thermoset non-isocyanate polyurethanes (NIPUs) from lignin-based polyols. This new approach shows promising results in producing scalable and sustainable NIPUs from lignin according to the principles of green chemistry, including the use of renewable feedstocks, safe reagents, limitation of synthesis steps, and waste prevention.Lignin-based polyols and dicarbamate were prepared using safe and green procedures. The transurethanization polymerization was studied and optimized to minimize unwanted side reactions, such as urea formation. NIPUs with lignin contents of up to 32 wt% were successfully obtained. The analysis of the different aromatic materials obtained confirmed the crosslinked structure of the NIPUs. The integration of lignin into NIPUs significantly influenced their thermo-mechanical properties, with increasing lignin content leading to improved rigidity due to enhanced crosslinking density. In contrast, adjusting the carbamate-to-hydroxyl molar ratio in the range of 0.9–1.1 showed limited impact on the mechanical properties. The NIPUs exhibited elastomeric behavior at room temperature and moderate mechanical properties, with Young's modulus up to 58 MPa and maximum elongations ranging from 15 to 51%.
Efforts to recycle the NIPUs were partially successful. Chemical recycling through depolymerization by methanolysis showed promising results in recovering the initial reaction mixture. The dynamic nature of the crosslinked networks similar to CANs was confirmed by stress-relaxation experiments and could be enhanced using lower C/OH ratios. However, water uptake and the limited thermal stability linked to the choice of catalyst resulted in a narrow temperature window for reprocessing, which did not allow efficient material recycling. This can be achieved in the future using another catalyst system.
This study reports the first examples of lignin-based NIPU materials obtained via transurethanization, offering eco-friendly and high-performance materials. The as-obtained materials had potential bio-based content up to 97 wt% with their current formulation, aligning them with the growing demand for sustainable alternatives to polymers. Their tunable properties can unlock a wide range of potential applications. While the elastomers serve as a compelling proof of concept in this study, the versatility of these NIPUs positions them for use in heat-curing coatings or adhesives. This adaptability, combined with their sustainable nature, underscores the potential of lignin-based NIPUs to address the needs of diverse markets while advancing green chemistry principles. However, some challenges, such as the low gel content, moderate mechanical properties, and porosity of the materials, remain to be resolved. Optimization of the formulation (choice of carbamate and catalyst, and lignin content) and processing (reactant mixing under pre-heating, vacuum curing, dry conditions, and post-curing steps) will be the topic of future investigations.
Abbreviations
| PU | Polyurethane |
| NIPU | Non-isocyanate polyurethane |
| PHU | Polyhydroxyurethane |
| PEG | Polyethylene glycol |
| TBD | 1,5,7 Triazabicyclo[4.4.0]dec |
| DMC | Dimethyl carbonate |
| EC | Ethylene carbonate |
| HMDA | Hexamethylene diamine |
| HMDC | Hexamethylene dimethylcarbamate |
| L-pOH | Lignin-based polyol |
| C/OH | Carbamate-to-hydroxyl ratio |
| NMR | Nuclear magnetic resonance |
| HSQC | Heteronuclear single quantum coherence spectroscopy |
| HMBC | Heteronuclear multiple bond correlation |
| COSY | Correlated spectroscopy |
| FTIR | Fourier transform infrared |
| TGA | Thermogravimetric analysis |
| DSC | Differential scanning calorimetry |
| DMA | Dynamic mechanical analysis |
| SEM | Scanning electron microscopy |
| SR | Swelling ratio |
| GF | Gel fraction |
| a.u. | Arbitrary unit |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Nathan Wybo: investigation, data curation, conceptualization, methodology, visualization, writing – original draft, review & editing. Elise Cherasse: investigation, data curation, conceptualization, methodology, writing – review & editing. Antoine Duval: conceptualization, methodology, supervision, funding acquisition, writing – review & editing Luc Averous: supervision, funding acquisition, writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the Agence Nationale de la Recherche (France), ANR Biopoliol (ANR-21-CE43-0026) for financing this research. We thank Dr Arjan T. Smit (TNO, Petten, Netherlands) for kindly supplying the organosolv lignin used in this study. We acknowledge the Cronenbourg NMR Core Facility (CNRS/Université de Strasbourg, UMR 7042 LIMA, Strasbourg, France) and Grégoire Muller for the SEM measurements.References
- A. Das and P. Mahanwar, A Brief Discussion on Advances in Polyurethane Applications, Adv. Ind. Eng. Polym. Res., 2020, 3(3), 93–101, DOI:10.1016/j.aiepr.2020.07.002.
- A. Delavarde, G. Savin, P. Derkenne, M. Boursier, R. Morales-Cerrada, B. Nottelet, J. Pinaud and S. Caillol, Sustainable Polyurethanes: Toward New Cutting-Edge Opportunities, Prog. Polym. Sci., 2024, 151, 101805, DOI:10.1016/j.progpolymsci.2024.101805.
- Plastics – the fast Facts 2023, Plastics Europe, Plastics Europe, https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2023/, accessed 2024-09-16.
- J. Peyrton and L. Avérous, Structure-Properties Relationships of Cellular Materials from Biobased Polyurethane Foams, Mater. Sci. Eng. R Rep., 2021, 145, 100608, DOI:10.1016/j.mser.2021.100608.
- J. Elms, P. N. Beckett, P. Griffin and A. D. Curran, Mechanisms of Isocyanate Sensitisation. An in Vitro Approach, Toxicol. In Vitro, 2001, 15(6), 631–634, DOI:10.1016/S0887-2333(01)00078-9.
- H. Hamada, M. Bruze, E. Zimerson, M. Isaksson and M. Engfeldt, Sensitization and Cross-Reactivity Patterns of Contact Allergy to Diisocyanates and Corresponding Amines: Investigation of Diphenylmethane-4,4′-Diisocyanate, Diphenylmethane-4,4′-Diamine, Dicyclohexylmethane-4,4′-Diisocyanate, and Dicylohexylmethane-4,4′-Diamine, Contact Dermat., 2017, 77(4), 231–241, DOI:10.1111/cod.12809.
- H. Sardon, D. Mecerreyes, A. Basterretxea, L. Avérous and C. Jehanno, From Lab to Market: Current Strategies for the Production of Biobased Polyols, ACS Sustain. Chem. Eng., 2021, 9(32), 10664–10677, DOI:10.1021/acssuschemeng.1c02361.
- L. Polo Fonseca, A. Duval, E. Luna, M. Ximenis, S. De Meester, L. Avérous and H. Sardon, Reducing the Carbon Footprint of Polyurethanes by Chemical and Biological Depolymerization: Fact or Fiction?, Curr. Opin. Green Sustainable Chem., 2023, 41, 100802, DOI:10.1016/j.cogsc.2023.100802.
- A. Cornille, R. Auvergne, O. Figovsky, B. Boutevin and S. Caillol, A Perspective Approach to Sustainable Routes for Non-Isocyanate Polyurethanes, Eur. Polym. J., 2017, 87, 535–552, DOI:10.1016/j.eurpolymj.2016.11.027.
- C. Carré, Y. Ecochard, S. Caillol and L. Averous, From the Synthesis of Biobased Cyclic Carbonate to Polyhydroxyurethanes: A Promising Route towards Renewable NonIsocyanate Polyurethanes, ChemSusChem, 2019, 12(15), 3410–3430, DOI:10.1002/cssc.201900737.
- C. Carré, L. Bonnet and L. Avérous, Original Biobased Nonisocyanate Polyurethanes: Solvent- and Catalyst-Free Synthesis, Thermal Properties and Rheological Behaviour, RSC Adv., 2014, 4(96), 54018–54025, 10.1039/C4RA09794G.
- N. Kébir, S. Nouigues, P. Moranne and F. Burel, Nonisocyanate Thermoplastic Polyurethane Elastomers Based on Poly(Ethylene Glycol) Prepared through the Transurethanization Approach, J. Appl. Polym. Sci., 2017, 134(45), 44991, DOI:10.1002/app.44991.
- K. A. Alferov, Z. Fu, S. Ye, D. Han, S. Wang, M. Xiao and Y. Meng, One-Pot Synthesis of Dimethyl Hexane-1,6-Diyldicarbamate from CO2, Methanol, and Diamine over CeO2 Catalysts: A Route to an Isocyanate-Free Feedstock for Polyurethanes, ACS Sustain. Chem. Eng., 2019, 7(12), 10708–10715, DOI:10.1021/acssuschemeng.9b01345.
- P. Wang, Y. Fei, Q. Li and Y. Deng, Effective Synthesis of Dimethylhexane-1,6-Dicarbamate from 1,6-Hexanediamine and Dimethyl Carbonate Using 3-Amino-1,2,4-Triazole Potassium as a Solid Base Catalyst at Ambient Temperature, Green Chem., 2016, 18(24), 6681–6686, 10.1039/C6GC02509A.
- G. Rokicki and A. Piotrowska, A New Route to Polyurethanes from Ethylene Carbonate, Diamines and Diols, Polymer, 2002, 43(10), 2927–2935, DOI:10.1016/S0032-3861(02)00071-X.
- M. Włoch and J. Datta, Chapter 7 – Nonisocyanate Polyurethanes, in Polyurethane Polymers, ed. S. Thomas, J. Datta, J. T. Haponiuk, A. Reghunadhan, Elsevier, Amsterdam, 2017, pp. 169–202, DOI:10.1016/B978-0-12-804039-3.00007-5.
- S. Kotanen, P. Laurikainen, S. Lehtimäki, T. Harjunalanen, T. Laaksonen and E. Sarlin, The Role of Hard and Soft Segments in the Thermal and Mechanical Properties of Non-Isocyanate Polyurethanes Produced via Polycondensation Reaction, Int. J. Adhes. Adhes., 2024, 132, 103726, DOI:10.1016/j.ijadhadh.2024.103726.
- Y. Deng, S. Li, J. Zhao, Z. Zhang, J. Zhang and W. Yang, Crystallizable and Tough Aliphatic Thermoplastic Poly(Ether Urethane)s Synthesized through a Non-Isocyanate Route, RSC Adv., 2014, 4(82), 43406–43414, 10.1039/C4RA05880A.
- P. Deepa and M. Jayakannan, Solvent-Free and Nonisocyanate Melt Transurethane Reaction for Aliphatic Polyurethanes and Mechanistic Aspects, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(7), 2445–2458, DOI:10.1002/pola.22578.
- M. Unverferth, O. Kreye, A. Prohammer and M. A. R. Meier, Renewable Non-Isocyanate Based Thermoplastic Polyurethanes via Polycondensation of Dimethyl Carbamate Monomers with Diols, Macromol. Rapid Commun., 2013, 34(19), 1569–1574, DOI:10.1002/marc.201300503.
- C. Duval, N. Kébir, A. Charvet, A. Martin and F. Burel, Synthesis and Properties of Renewable Nonisocyanate Polyurethanes (NIPUs) from Dimethylcarbonate, J. Polym. Sci., Part A: Polym. Chem., 2015, 53(11), 1351–1359, DOI:10.1002/pola.27568.
- N. Kébir, M. Benoit and F. Burel, Elaboration of AA-BB and AB-Type Non-Isocyanate Polyurethanes (NIPUs) Using a Cross Metathesis Polymerization between Methyl Carbamate and Methyl Carbonate Groups, Eur. Polym. J., 2018, 107, 155–163, DOI:10.1016/j.eurpolymj.2018.07.045.
- P. Boisaubert, N. Kébir, A.-S. Schuller and F. Burel, Photo-Crosslinked Non-Isocyanate Polyurethane Acrylate (NIPUA) Coatings through a Transurethane Polycondensation Approach, Polymer, 2020, 206, 122855, DOI:10.1016/j.polymer.2020.122855.
- P. Boisaubert, N. Kébir, A.-S. Schuller and F. Burel, Photo-Crosslinked Coatings from an Acrylate Terminated Non-Isocyanate Polyurethane (NIPU) and Reactive Diluent, Eur. Polym. J., 2020, 138, 109961, DOI:10.1016/j.eurpolymj.2020.109961.
- P. Boisaubert, N. Kébir, A.-S. Schuller and F. Burel, Polyurethane Coatings from Formulations with Low Isocyanate Content Using a Transurethane Polycondensation Route, Polymer, 2022, 240, 124522, DOI:10.1016/j.polymer.2022.124522.
- V. Valette, N. Kébir, F. B. Tiavarison, F. Burel and L. Lecamp, Preparation of Flexible Biobased Non-Isocyanate Polyurethane (NIPU) Foams Using the Transurethanization Approach, React. Funct. Polym., 2022, 181, 105416, DOI:10.1016/j.reactfunctpolym.2022.105416.
- A. Martin, L. Lecamp, H. Labib, F. Aloui, N. Kébir and F. Burel, Synthesis and Properties of Allyl Terminated Renewable Non-Isocyanate Polyurethanes (NIPUs) and Polyureas (NIPUreas) and Study of Their Photo-Crosslinking, Eur. Polym. J., 2016, 84, 828–836, DOI:10.1016/j.eurpolymj.2016.06.008.
- D. Wołosz, P. G. Parzuchowski and A. Świderska, Synthesis and Characterization of the Non-Isocyanate Poly(Carbonate-Urethane)s Obtained via Polycondensation Route, Eur. Polym. J., 2021, 155, 110574, DOI:10.1016/j.eurpolymj.2021.110574.
- Z. Shen, J. Zhang, W. Zhu, L. Zheng, C. Li, Y. Xiao, J. Liu, S. Wu and B. Zhang, A Solvent-Free Route to Non-Isocyanate Poly(Carbonate Urethane) with High Molecular Weight and Competitive Mechanical Properties, Eur. Polym. J., 2018, 107, 258–266, DOI:10.1016/j.eurpolymj.2018.08.006.
- K. Zhang, K. Shuai, Z. Ni, T. Kaneko, W. Dong, M. Chen and D. Shi, Synthesis of Non-Isocyanate Polyurethanes with High-Performance and Self-Healing Properties, J. Appl. Polym. Sci., 2023, e54899, DOI:10.1002/app.54899.
- S. Ye, X. Xiang, S. Wang, D. Han, M. Xiao and Y. Meng, Nonisocyanate CO2-Based Poly(Ester-Co-Urethane)s with Tunable Performances: A Potential Alternative to Improve the Biodegradability of PBAT, ACS Sustain. Chem. Eng., 2020, 8(4), 1923–1932, DOI:10.1021/acssuschemeng.9b06294.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Lignin Valorization: Improving Lignin Processing in the Biorefinery, Science, 2014, 344(6185), 1246843, DOI:10.1126/science.1246843.
- M. Broda, D. J. Yelle and K. Serwańska, Bioethanol Production from Lignocellulosic Biomass—Challenges and Solutions, Molecules, 2022, 27(24), 8717, DOI:10.3390/molecules27248717.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, A Concise Review of Current Lignin Production, Applications, Products and Their Environmental Impact, Ind. Crops Prod., 2019, 139, 111526, DOI:10.1016/j.indcrop.2019.111526.
- S. Laurichesse and L. Avérous, Chemical Modification of Lignins: Towards Biobased Polymers, Prog. Polym. Sci., 2014, 39(7), 1266–1290, DOI:10.1016/j.progpolymsci.2013.11.004.
- S. Bertella and J. S. Luterbacher, Lignin Functionalization for the Production of Novel Materials, Trends Chem., 2020, 2(5), 440–453, DOI:10.1016/j.trechm.2020.03.001.
- E. Khadem, M. Ghafarzadeh, M. Kharaziha, F. Sun and X. Zhang, Lignin Derivatives-Based Hydrogels for Biomedical Applications, Int. J. Biol. Macromol., 2024, 129877, DOI:10.1016/j.ijbiomac.2024.129877.
- M. Alinejad, C. Henry, S. Nikafshar, A. Gondaliya, S. Bagheri, N. Chen, S. K. Singh, D. B. Hodge and M. Nejad, Lignin-Based Polyurethanes: Opportunities for Bio-Based Foams, Elastomers, Coatings and Adhesives, Polymers, 2019, 11(7), 1202, DOI:10.3390/polym11071202.
- H. Li, Y. Liang, P. Li and C. He, Conversion of Biomass Lignin to High-Value Polyurethane: A Review, J. Bioresour. Bioprod., 2020, 5(3), 163–179, DOI:10.1016/j.jobab.2020.07.002.
- X. Ma, J. Chen, J. Zhu and N. Yan, Lignin-Based Polyurethane: Recent Advances and Future Perspectives, Macromol. Rapid Commun., 2021, 42(3), 2000492, DOI:10.1002/marc.202000492.
- A. Shafiq, I. Ahmad Bhatti, N. Amjed, M. Zeshan, A. Zaheer, A. Kamal, S. Naz and T. Rasheed, Lignin Derived Polyurethanes: Current Advances and Future Prospects in Synthesis and Applications, Eur. Polym. J., 2024, 209, 112899, DOI:10.1016/j.eurpolymj.2024.112899.
- A. Duval, D. Vidal, A. Sarbu, W. René and L. Avérous, Scalable Single-Step Synthesis of Lignin-Based Liquid Polyols with Ethylene Carbonate for Polyurethane Foams, Mater. Today Chem., 2022, 24, 100793, DOI:10.1016/j.mtchem.2022.100793.
- A. T. Smit, M. Verges, P. Schulze, A. van Zomeren and H. Lorenz, Laboratory- to Pilot-Scale Fractionation of Lignocellulosic Biomass Using an Acetone Organosolv Process, ACS Sustain. Chem. Eng., 2022, 10(32), 10503–10513, DOI:10.1021/acssuschemeng.2c01425.
- A. Granata and D. S. Argyropoulos, 2-Chloro-4,4,5,5-Tetramethyl-1,3,2-Dioxaphospholane, a Reagent for the Accurate Determination of the Uncondensed and Condensed Phenolic Moieties in Lignins, J. Agric. Food Chem., 1995, 43(6), 1538–1544, DOI:10.1021/jf00054a023.
- N. Wybo, A. Duval and L. Avérous, Benign and Selective Amination of Lignins towards Aromatic Biobased Building Blocks with Primary Amines, Angew. Chem., Int. Ed., 2024, 63(41), e202403806, DOI:10.1002/anie.202403806.
- L. Sougrati, A. Duval and L. Avérous, From Lignins to Renewable Aromatic Vitrimers Based on Vinylogous Urethane, ChemSusChem, 2023, 16(23), e202300792, DOI:10.1002/cssc.202300792.
- A. Duval and L. Avérous, Characterization and Physicochemical Properties of Condensed Tannins from Acacia Catechu, J. Agric. Food Chem., 2016, 64(8), 1751–1760, DOI:10.1021/acs.jafc.5b05671.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. D. Peinder, R. Boelens, D. S. V. Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, New Insights into the Structure and Composition of Technical Lignins: A Comparative Characterisation Study, Green Chem., 2016, 18(9), 2651–2665, 10.1039/C5GC03043A.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-Isocyanate Polyurethane, Chem. Rev., 2013, 113(1), 80–118, DOI:10.1021/cr300195n.
- C. Bakkali-Hassani, D. Berne, V. Ladmiral and S. Caillol, Transcarbamoylation in Polyurethanes: Underestimated Exchange Reactions?, Macromolecules, 2022, 55(18), 7974–7991, DOI:10.1021/acs.macromol.2c01184.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Non-Isocyanate Polyurethanes: Synthesis, Properties, and Applications, Polym. Adv. Technol., 2015, 26(7), 707–761, DOI:10.1002/pat.3522.
- J. Reignier, F. Méchin and A. Sarbu, Chemical Gradients in PIR Foams as Probed by ATR-FTIR Analysis and Consequences on Fire Resistance, Polym. Test., 2021, 93, 106972, DOI:10.1016/j.polymertesting.2020.106972.
- Z. Shen, L. Zheng, C. Li, G. Liu, Y. Xiao, S. Wu, J. Liu and B. A. Zhang, Comparison of Non-Isocyanate and HDI-Based Poly(Ether Urethane): Structure and Properties, Polymer, 2019, 175, 186–194, DOI:10.1016/j.polymer.2019.05.010.
- Z. Shen, L. Zheng, D. Song, Y. Liu, C. Li, J. Liu, Y. Xiao, S. Wu, T. Zhou, B. Zhang, X. Lv and Q. Mei, A Non-Isocyanate Route to Poly(Ether Urethane): Synthesis and Effect of Chemical Structures of Hard Segment, Polymers, 2022, 14(10), 2039, DOI:10.3390/polym14102039.
- T. Stern, Conclusive Chemical Deciphering of the Consistently Occurring Double-Peak Carbonyl-Stretching FTIR Absorbance in Polyurethanes, Polym. Adv. Technol., 2019, 30(3), 675–687, DOI:10.1002/pat.4503.
- A. B. Dros, O. Larue, A. Reimond, F. D. Campo and M. Pera-Titus, Hexamethylenediamine (HMDA) from Fossil- vs. Bio-Based Routes: An Economic and Life Cycle Assessment Comparative Study, Green Chem., 2015, 17(10), 4760–4772, 10.1039/C5GC01549A.
- P. P. Pescarmona, Cyclic Carbonates Synthesised from CO2: Applications, Challenges and Recent Research Trends, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457, DOI:10.1016/j.cogsc.2021.100457.
- H.-Z. Tan, Z.-Q. Wang, Z.-N. Xu, J. Sun, Y.-P. Xu, Q.-S. Chen, Y. Chen and G.-C. Guo, Review on the Synthesis of Dimethyl Carbonate, Catal. Today, 2018, 316, 2–12, DOI:10.1016/j.cattod.2018.02.021.
- P. K. Karoki, S. Zhang, Y. Pu and A. J. Ragauskas, Lignin-Based Vitrimers: Valorization and Utilization of Lignin in High-Value Applications, Mater. Adv., 2024, 5(18), 7075–7096, 10.1039/d4ma00281d.
- Y. Ecochard and S. Caillol, Hybrid Polyhydroxyurethanes: How to Overcome Limitations and Reach Cutting Edge Properties?, Eur. Polym. J., 2020, 137, 109915, DOI:10.1016/j.eurpolymj.2020.109915.
- C. Bakkali-Hassani, D. Berne, P. Bron, L. Irusta, H. Sardon, V. Ladmiral and S. Caillol, Polyhydroxyurethane Covalent Adaptable Networks: Looking for Suitable Catalysts, Polym. Chem., 2023, 14(31), 3610–3620, 10.1039/D3PY00579H.
- Y. Cao, Z. Liu, B. Zheng, R. Ou, Q. Fan, L. Li, C. Guo, T. Liu and Q. Wang, Synthesis of Lignin-Based Polyols via Thiol-Ene Chemistry for High-Performance Polyurethane Anticorrosive Coating, Composites, Part B, 2020, 200, 108295, DOI:10.1016/j.compositesb.2020.108295.
- C. Qiao, X. Jian, Z. Gao, Q. Ban, X. Zhang, H. Wang and Y. Zheng, Tough Polyurethane Elastomers with High Strength and Rapid Healing Ability, Mater. Adv., 2023, 4(7), 1711–1719, 10.1039/D2MA01021F.
- X. Wang, J. Xu, Y. Zhang, T. Wang, Q. Wang, Z. Yang and X. Zhang, High-Strength, High-Toughness, Self-Healing Thermosetting Shape Memory Polyurethane Enabled by Dual Dynamic Covalent Bonds, Polym. Chem., 2022, 13(23), 3422–3432, 10.1039/D2PY00564F.
- Y. Qin, Y. Wang, Y. Wang, J. Zhao, J. Cheng and J. Zhang, Bio-Based Poly(Hydroxyurethane)s of Good Thermal/Mechanical Properties upon Insertion of Polyamide Segments, Mater. Chem. Phys., 2025, 130522, DOI:10.1016/j.matchemphys.2025.130522.
- G. Rossignolo, G. Malucelli and A. Lorenzetti, Recycling of Polyurethanes: Where We Are and Where We Are Going, Green Chem., 2024, 26(3), 1132–1152, 10.1039/D3GC02091F.
- L. Sougrati, A. Duval and L. Avérous, Biobased and Aromatic Covalent Adaptable Networks: When Architectures Meet Properties, within the Framework of a Circular Bioeconomy, Mater. Sci. Eng. R Rep., 2024, 161, 100882, DOI:10.1016/j.mser.2024.100882.
- S. Hu, X. Chen, M. A. Bin Rusayyis, N. S. Purwanto and J. M. Torkelson, Reprocessable Polyhydroxyurethane Networks Reinforced with Reactive Polyhedral Oligomeric Silsesquioxanes (POSS) and Exhibiting Excellent Elevated Temperature Creep Resistance, Polymer, 2022, 252, 124971, DOI:10.1016/j.polymer.2022.124971.
- A. Hernández, H. A. Houck, F. Elizalde, M. Guerre, H. Sardon and F. E. Du Prez, Internal Catalysis on the Opposite Side of the Fence in Non-Isocyanate Polyurethane Covalent Adaptable Networks, Eur. Polym. J., 2022, 168, 111100, DOI:10.1016/j.eurpolymj.2022.111100.
- A. Basterretxea, E. Gabirondo, C. Jehanno, H. Zhu, I. Flores, A. J. Müller, A. Etxeberria, D. Mecerreyes, O. Coulembier and H. Sardon, Polyether Synthesis by Bulk Self-Condensation of Diols Catalyzed by Non-Eutectic Acid–Base Organocatalysts, ACS Sustainable Chem. Eng., 2019, 7(4), 4103–4111 Search PubMed.
- J. Sternberg and S. Pilla, Chemical Recycling of a Lignin-Based Non-Isocyanate Polyurethane Foam, Nat. Sustain., 2023, 6(3), 316–324, DOI:10.1038/s41893-022-01022-3.
- L. Zhao and V. Semetey, Recycling Polyurethanes through Transcarbamoylation, ACS Omega, 2021, 6(6), 4175–4183, DOI:10.1021/acsomega.0c04855.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08582e |
| This journal is © The Royal Society of Chemistry 2025 |